

A classifier was recently reported to predict with 70% accuracy if an individual has an Autism Spectrum Disorder (ASD) using 237 single nucleotide polymorphisms (SNPs) (Skafidas et al. 2012). Biomarkers, genetic or otherwise, that would facilitate earlier ASD diagnosis are crucial, so these results warrant careful scrutiny. One potential confounder of such genetic studies is bias when cases and controls have different ancestral origins. Here, we show that the largest components of this classifier's autism risk score distinguish populations but do not separate cases from controls. In short, the frequencies of reported risk and protective alleles do not differ between related individuals with or without autism in independent data sets; instead they reflect ancestral origin. Specifically, cases have more diverse ancestral origins within Europe than controls. The putative risk alleles are more common in Northeastern Europe than in Northwestern European, while the putative protective alleles reflect the opposite trend. Likewise, we find that the autism risk scores based on the strongest SNPs do not differ between people with and without autism in an independent dataset, but that they do differ between European populations.
The classifier was originally trained using case genotype data from the Autism Genetics Resource Exchange (AGRE) (Geschwind et al. 2001; Lajonchere et al. 2010). Although only the top 15 ‘risk’ SNPs and the top 15 ‘protective’ SNPs were provided (Skafidas et al. 2012), even those 30 SNPs were reportedly sufficient for 58% accurate discrimination between controls (the Western and Northern European CEU population in HapMap3) and cases (the CEU-like AGRE cases).We thus applied the autism risk classifier to 379 cases and 472 related controls that had been added to AGRE after development of the classifier. Of the 30 SNPs comprising the classifier, 19 were genotyped in the new cohort. To match the original publication, we limited analysis to the individuals more similar to CEU than to any other HapMap3 population. The resulting distributions of autism risk scores were not significantly different between cases and controls (Figure 1; two-sided two-sample Kolmogorov-Smirnov [K-S] test, p = 0.68). Likewise, we found no differences in the minor allele frequencies of any of the 30 putatively discriminative SNPs, neither at the level of an individual SNP (Fisher's exact test) nor when the p-value distributions were considered (K-S test). Furthermore, we found no difference between cases and controls in the same minor allele frequency comparisons within the Simons Simplex Collection (SSC; http://sfari.org/sfari-initiatives/simons-simplex-collection).
Figure 1. The most predictive SNPs in the classifier are correlated with ancestry within Europe, but not with autism.
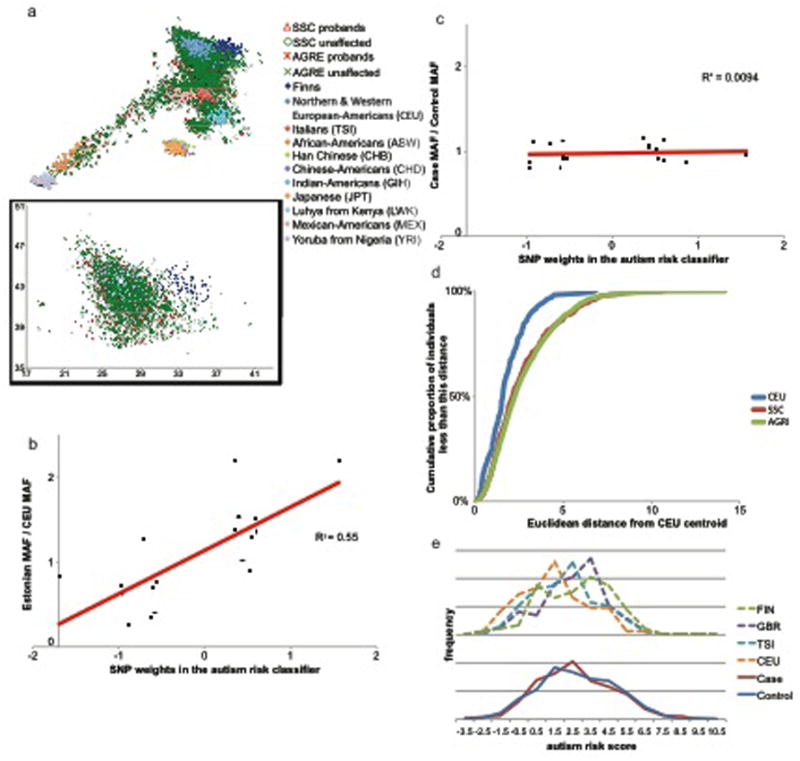
(a) The cases and controls used for training and validation were drawn from different European population distributions, in a manner consistent with the differences in minor allele frequencies of the top SNPs in the classifier. Plotted on rotated and inverted geographic axes of European variation (Yang et al. 2012) are individuals from the control population CEU (light blue diamonds representing Northern and Western European-Americans), and individuals with and without autism from the training and replication case datasets (AGRE probands as red asterisks and unaffected relatives as green Xs; SSC probands as red triangles and unaffected relatives as green circles). The eleven HapMap3 populations and a reference Northeastern European population (Finns from the 1000 Genomes Project) are denoted by diamonds. To reduce population stratification bias, Skafidas and colleagues only used individuals nearer CEU than any other HapMap population to train the classifier. However, we highlight considerable differences in population structure between cases and controls even in this subset (represented in the inset box, the red cross indicates the centroid of CEU). (b) The weights of the top reported SNPs (x-axis) are correlated with the minor allele frequency in Estonians (a Northeastern European population) divided by that of CEU (y-axis) (p= 4×10-4). While 1000 Genomes data were not available for Estonians, the Estonian minor allele frequencies are based on considerably more chromosomes, allowing for more accuracy. (c) In contrast, the weights of the top reported SNPs (x-axis) are uncorrelated with the odds ratios among an independent set of individuals in AGRE nearest CEU (p = 0.997). (d) The case sets are more diverse than the control sets (p=6.2×10-5 for CEU vs AGRE or p=4.6×10-8 for CEU vs SSC, two-sided Kolmogorov-Smirnov test). Cumulative proportion of AGRE, SSC and CEU individuals plotted against Euclidean distance from the CEU centroid, for those individuals nearer the CEU centroid than that of any other HapMap population in the inset of panel a. (e) Frequency distributions of autism risk scores for the independent set of AGRE cases and controls and four populations sequenced in the 1000 Genomes Project (FIN – Finns, GBR – British, TSI – Italians, and CEU – Northwestern Europeans).All classifier scores were calculated using the 19 SNPs that had data in all of these sets and were also in the top 30 most discriminative SNPs in Skafidas et al (2012). Neither the distributions of cases and controls, nor the proportion above the threshold level differ significantly from one another. In contrast, different populations have different distributions. Finns differ from neither independent AGRE cases nor independent AGRE controls, while other European populations have lower scores.
We then asked if the cases and controls have different ancestral origins. If so, population structure would be correlated with autism in the sample, leading to the faulty conclusion that genetic variants that differentiate populations instead mediate autism risk. Much of the genetic diversity among Europeans reflects geography (Yang et al. 2012). To attempt to control for population structure, the classifier's authors excluded individuals whose genomes better-reflected HapMap3 populations other than CEU (a Western and Northern European population). For example, because an Italian population (TSI) was included in HapMap3, their removal reduced bias that could be introduced from different Northern and Southern origins in cases and controls within Europe. In both training and validation sets, the cases were European Americans who have diverse ancestral origins, whereas the controls were explicitly intended to represent populations in Northwestern Europe (CEU and a British birth cohort). This raised the concern that genetic differences between Eastern and Western Europeans could be a major confound.
To investigate this possibility, we compared the allele frequencies of the reported discriminative SNPs between CEU (Sherry et al. 2001), representing Northwestern Europe, and Estonians (Kidd et al. 2003), reflective of Northeastern Europe. Eighteen of the thirty SNPs were genotyped in both of these studies. Of these, all but one ‘risk’ SNP and one ‘protective’ SNP differed in the direction one would expect if the allele frequency distributions were due to population structure (Table S1). The differences were striking: the mean and median odds ratios were 1.49 and 1.38 for Estonians and 0.66 and 0.69 for CEU (p=3×10-4, 2-tailed t-test on the odds ratios).
To more directly confirm that cases and controls were taken from different populations, we plotted the two case sets and the training control set on the geographical axes of genetic variation in Europe (Figure 1; Yang et al. 2012). As expected, cases had more diverse ancestral origins than controls. While we did not have immediate access to the validation control set, it is a 1958 British birth cohort that is by definition Northwestern European. So, although all of the SNPs in the classifier are not publicly available, the properties of the SNPs that are provided are most consistent with confounding due to population substructure. It was previously noted that the classifier was useless for individuals clustered with the Chinese population (Skafidas et al. 2012), as one would expect if it were a spurious artifact of local European population structure. Further, the classifier was considerably less accurate for individuals clustering with the Italian HapMap group (Skafidas et al. 2012). The poor performance among Southern Europeans may reflect differences between the East-West genetic gradients across Southern Europe and Northern Europe. Even the previously reported distributions of autism risk score of AGRE individuals with and without the disorder (Skafidas et al. 2012) are consistent with this explanation (Supplementary Data).
Because we found that autism risk scores based on the publicly available SNPs did not distinguish independent cases from controls, we asked if these score distributions differed between European populations. CEU (the control group used to train the classifier) had the lowest median and mean autism risk scores of these European populations (1.3 and 1.4) while Finns, a representative Northeastern European population, had the highest median and mean autism risk scores (2.8 and 2.7), as would be expected if the classifier were confounded by population structure. Their overall distributions also differed (two-sample Kolmogorov-Smirnov test, p = 0.0005).
In the publication describing the classifier, an autism risk score cutoff of 3.93 was used to predict affectation status. We examined the properties of our populations using this cutoff, although we note that since we only had data on 19 of the 30 SNPs, it is an approximation of the results based on the 30 SNP classifier (Skafidas et al. 2012). Importantly, the proportion of Finns above this autism risk score cutoff (29%) differed neither from AGRE cases (28%) nor AGRE controls (31%) (two-tailed Fisher's exact tests p = 0.89 and p = 0.81, respectively). In contrast, more Finns were classified as autistic than the training HapMap3 population CEU (12%; two-tailed Fisher's exact test p = 0.0054), the independent 1000 Genomes British population GBR (17%; two-tailed Fisher's exact test p = 0.055), and the HapMap3 Italian population TSI (16%; two-tailed Fisher's exact test p = 0.039). These analyses lead to the conclusion that the autism risk scores based on the publicly available SNPs effectively separate European populations from one another, but do not separate cases from controls. Moreover, since Northeastern Europeans generally had higher scores than Western or Southern Europeans, this would result in inflated measures of accuracy in the previously reported independent validation that used diverse European Americans as cases and Northwestern Europeans as controls (Skafidas et al. 2012).
While these strongest contributors to the classifier are more consistent with artifacts of population structure than with true ASD signal, it remains possible that there are some true signals differentiating cases and controls, particularly among the 207 weaker SNPs that are not currently publicly available. However, until more evidence can be provided, we favor the more conservative interpretation that these associations are due to previously unobserved population stratification in the cases and controls and do not contribute meaningfully to a diagnostic classifier.
Supplementary Material
Footnotes
All other authors declare no financial conflicts of interest.
Supplementary information is available at Molecular Psychiatry's website.
References
- Geschwind DH, et al. Am J Hum Genet. 2001;69(2):463–466. doi: 10.1086/321292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd KK, et al. Am J Phys Anthropol. 2003;(Suppl S36):128. Annual Meeting Issue. [Google Scholar]
- Lajonchere CM AGRE Consortium. Neuron. 2010;68(2):187–191. doi: 10.1016/j.neuron.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, et al. Nucleic Acids Res. 2001;29(1):308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skafidas E, et al. Mol Psychiatry. 2012 doi: 10.1038/mp.2012.126. e-pub ahead of print 11 September 2012. [DOI] [Google Scholar]
- Yang WY, et al. Nature. 2012;44:725–731. doi: 10.1038/ng.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.