

SUMMARY
Most potent protein kinase inhibitors act by competing with ATP to block the phosphotransferase activity of their targets. However, emerging evidence demonstrates that ATP-competitive inhibitors can affect kinase interactions and functions in ways beyond blocking catalytic activity. Here, we show that stabilizing alternative ATP-binding site conformations of the mitogen-activated protein kinases (MAPKs) p38α and Erk2 with ATP-competitive inhibitors differentially, and in some cases divergently, modulates the abilities of these kinases to interact with upstream activators and deactivating phosphatases. Conformation-selective ligands are also able to modulate Erk2’s ability to allosterically activate the MAPK phosphatase DUSP6, highlighting how ATP-competitive ligands can control noncatalytic kinase functions. Overall, these studies underscore the relationship between the ATP-binding and regulatory sites of MAPKs and provide insight into how ATP-competitive ligands can be designed to confer graded control over protein kinase function.
INTRODUCTION
Protein kinases are one of the largest protein families encoded by the human genome and major constituents of most intracellular signaling cascades (Manning et al., 2002b),(Manning et al., 2002a). These signaling enzymes play important roles in countless cellular pathways, and the proper regulation of their activity is essential for normal cellular behavior. Aberrant kinase function is linked to numerous diseases, and a number of kinases are promising targets for the development of small molecule-based therapies (Cohen and Alessi, 2013). Currently, a majority of potent and selective kinase inhibitors block phosphotransferase activity by competing with ATP (Zhang et al., 2009). While many of these inhibitors are able to interact with the ATP-binding clefts of kinases in an active conformation, a subset of inhibitors are conformation-selective, in that they only bind to their targets if conserved catalytic residues have been displaced from a catalytically competent conformation. Many kinases can be inhibited by ATP-competitive ligands with different binding modes, due to the conformational plasticity of their active sites.
Over the last five years it has become apparent that ATP-competitive inhibitors can affect kinases in ways beyond blocking their phosphotransferase activity. For example, the activation loop of the serine/threonine (S/T) kinase Akt becomes hyper-phosphorylated when its ATP-binding site is occupied by small molecule inhibitors (Chan et al., 2011; Okuzumi et al., 2009). Additionally, it has been shown that many inhibitors of the S/T kinase Raf promote trans-dimer auto-activation (Hatzivassiliou et al., 2010; Poulikakos et al., 2010), which may contribute to undesired drug responses in the clinic (Cichowski and Janne, 2010). Importantly, there is emerging evidence that it is possible to produce different, in some cases divergent, effects by varying the active site interactions made by ATP-competitive inhibitors. For example, conformation-selective inhibitors are able to either activate or inactive the RNase domain of the bifunctional kinase/RNase Ire1α, depending on whether they stabilize an active or inactive ATP-binding site conformation (Wang et al., 2012). We have also demonstrated that different classes of ATP-competitive inhibitors can divergently modulate the regulatory domain accessibility of Src-family kinases (Krishnamurty et al., 2013).
While the above examples demonstrate that it is possible for different classes of ATP-competitive inhibitors to differentially modulate interactions and functions outside of kinase active sites, the overall generality of these phenomena to the rest of the kinome is unclear. We were particularly interested in whether these observations can be extended to the mitogen-activated protein kinase (MAPK) family because these kinases are central components of numerous signaling pathways, and a number of noncatalytic MAPK functions have been reported (Rodriguez and Crespo, 2011). Because MAPKs have no regulatory domains and devote much of their exposed surface area to interacting with other proteins, there is the intriguing possibility that their noncatalytic functions can be modulated by ligands that stabilize different ATP-binding site conformations.
Here we report that conformation-selective ATP-competitive inhibitors are able to differentially modulate the regulatory interactions of MAPKs (Figure 1). We show that the specific conformations stabilized by these ligands dictate the behavior of MAPKs towards their activators (MAPK kinases) and inactivators (dual specificity phosphatases). We also demonstrate that ATP-competitive ligands can modulate MAPK functions that are independent of phosphotransferase activity. The examples presented herein provide compelling evidence that ATP-binding site ligands can differentially modulate a diverse number of protein kinase interactions.
Figure 1. Modulation of kinase regulation and function by stabilizing alternative active site conformations.

Protein kinases, like Erk2, are regulated by multiple phosphorylation and dephosphorylation events. Furthermore, many kinases are known to have a number of noncatalytic functions, such as scaffolding and allosteric activation. Conformation-selective inhibitors can potentially be used to modulate the regulatory and noncatalytic interactions of protein kinases.
RESULTS AND DISCUSSION
Conformation-selective inhibitors prevent the in vitro activation of Erk2
Two recurrent ATP-binding site conformations are characterized by the orientations of protein kinase activation loops, specifically, the conserved Asp-Phe-Gly (DFG) motif, which is near the base of this flexible structural element (Figure 2A). In the active conformation (active, DFG-in), the DFG motif and all of the catalytic residues in the ATP-binding site are aligned for catalysis. In contrast, many kinases can also adopt an inactive conformation (inactive, DFG-out) in which the DFG motif is translocated from an active conformation to accommodate a conformation-selective inhibitor. Regardless of the conformation stabilized, both modes of ATP-competitive inhibition lead to a loss of phosphotransferase activity. However, Sullivan et al. reported that when the MAPK p38α is stabilized in the DFG-out conformation with ligands, such as pyrazolourea 1 (Figure 2B), its activation loop cannot be phosphorylated by its upstream MAPK kinase (MEK) (Sullivan et al., 2005). Since activation loop phosphorylation is necessary for efficient catalytic activity, these inhibitors prevent the activation of p38α. However, when stabilized in the active DFG-in conformation by the p38α inhibitor SB203580, the phosphorylation (and subsequent activation) of p38α proceeds uninhibited. Still unknown, however, is whether this effect is specific to p38α or more broadly applicable to other MAPKs. We were particularly interested in Erk2 due to the substantial number of noncatalytic functions that are mediated by the activation-loop phosphorylated form of this kinase (Rodriguez and Crespo, 2011). If Erk2 must be activated to interact with certain proteins, then inhibitors that block its phosphorylation will also hinder these noncatalytic functions. However, wild type Erk2 (Erk2WT) is very weakly inhibited by SB203580 and ligands, like 2, that stabilize the DFG-out inactive conformation (Figures 3A, B, and E). To overcome this limitation, we utilized two drug sensitive Erk2 mutants (ERK2DS1 = Q103T/C164L and ERK2DS2 = Q103A/C164L) that are highly sensitive to inhibitors 2 and SB203580 (Hari et al., 2013).
Figure 2. MAPK active site conformational plasticity.

A. Superimposed structures of Erk2 in the active DFG-in (blue, DFG motif in teal) (PDB ID: 1PME) and inactive DFG-out (yellow, DFG motif in magenta) (PDB ID: 4I5H) conformations. The DFG motif phenylalanine residues for both structures are shown in stick form, and the distance between their α-carbons is 9.6 Å. Portions of the p-loops are hidden for clarity. B. Inhibitors used in this report that stabilize the active DFG-in (SB203580) and inactive DFG-out (1 and 2) conformations. Crystal structures of these ligands stabilizing specific kinase conformations can be found in the Protein Data Bank under accession codes 1A9U (SB203580), 2BAJ (1), and 4I5H (2).
Figure 3. The effects of conformation-selective ligands on MAPK activation by upstream MEKs.

A. Schematic of the assay used to determine Erk2 inhibitory constants (Kis). B. Inhibition of Erk2WT and Erk2DS1 by 2 and SB203580. Data for 2 were reported previously (Hari et al., 2013). C. Schematic of the assay used to determine activation loop phosphorylation inhibition of Erk2. D. Inhibition of Erk2WT and Erk2DS1 activation loop phosphorylation by 2 and SB203580. The amount of bis-phosphorylated Erk2 produced at each inhibitor concentration was determined by ELISA. E. Enzymatic inhibition (Ki) and activation inhibition (EC50) values for Erk2WT and Erk2DS1 against ligands 2 and SB203580. Ki values were reported previously (Hari et al., 2013). aPartial inhibition (20–30%) at saturating inhibitor concentrations. Error bars represent standard error of the mean (SEM) for three replicate measurements. See also Figure S1 and Table S1.
First, the effects of ligands 2 and SB203580 on the activation of Erk2 by its upstream kinase, MEK2, were probed using an in vitro activation assay (Figures 3C, D, and E). Varying concentrations of each inhibitor (2 or SB203580) were incubated with a fixed concentration of Erk2DS1 and MEK2 in the presence of ATP. The amount of bis-phosphorylated Erk2 produced in the reaction was then quantified by ELISA. These data indicate that ligand 2 potently inhibits the dual phosphorylation of Erk2’s activation loop (Figures 3D and E). In contrast, SB203580 had only a minimal effect on the ability of MEK2 to activate Erk2. These results closely resemble the behavior observed for MEK6-mediated activation of p38α first reported by Sullivan et al. (Sullivan et al., 2005) and confirmed presently (Table S1A). To rule out the possibility that ligands 2 and SB203580 directly inhibit MEK2, which would also lead to decreased levels of phospho-Erk2, the assays were repeated with Erk2WT, which is insensitive to both compounds (Figure 3B and Table S1B). As expected, neither 2 nor SB203580 lead to a significant decrease in the amount of activation loop phosphorylated Erk2WT (Figure 3D and Table S1B), strongly suggesting that stabilization of the DFG-out conformation of Erk2 is the primary factor behind activation inhibition.
Full activation of MAPKs requires both residues in the TxY motif on the activation loop to be phosphorylated. Therefore, we determined whether 2 inhibits the phosphorylation of Thr183, Tyr185, or both of these residues on the Erk2’s TxY motif. To do so, Erk2DS1 was activated by MEK2 in the presence of DMSO, ligand 2, or SB203580, and the phosphorylated isoforms of the kinase were separated by PhosTag-acrylamide PAGE (Kinoshita et al., 2004). This analysis revealed that inhibitor 2 completely prevents the formation of any bis-phosphoryated Erk2 but permits some mono-phosphorylated product formation (Figure S1A). In contrast, comparable levels of bis-phosphorylation were observed for apo Erk2DS1 and SB203580-bound Erk2DS1. Interestingly, we found that stabilizing p38α in the inactive DFG-out conformation with ATP-competitive inhibitor 1 completely prevents activation loop phosphorylation by MEK6 (Figure S1B). This result builds on the data reported by Sullivan et al. (Sullivan et al., 2005) through the analysis of individual phospho-isoforms of p38α to show that neither site on the TxY motif is appreciably phosphorylated in the presence of inhibitor 1. Therefore, there appear to be some subtle differences in how conformation-selective inhibitors influence the ability of MEKs to interact with and activate p38α and Erk2.
Conformation-selective inhibitors prevent Erk2 activation in cells
Having demonstrated the ability of ligand 2 to potently block Erk2DS1 activation in vitro, we were interested in whether this effect could be observed in a cellular environment. Therefore, GFP-tagged Erk2 (WT or DS1) was transiently transfected into HEK293 cells, which were then incubated with either 2 or SB203580. The cells were then briefly stimulated with EGF to activate the Erk pathway (Robinson et al., 2002). Immunoblot analysis showed that addition of 2 prevented Erk2DS1 activation upon stimulation, whereas SB203580 had no effect (Figure 4). Consistent with the in vitro insensitivity of Erk2WT to 2 and SB203580, the cellular activation of this kinase was not affected by the presence of either inhibitor.
Figure 4. Cellular inhibition of Erk2 activation.

GFP-tagged Erk2WT or Erk2DS1 were transiently transfected into HEK293 cells. After serum starvation, the cells were incubated with DMSO (lanes 1 and 2), ligand 2 (lanes 3–6 [1000, 500, 300, and 100 nM]), or SB203580 (lane 7 [1000 nM]). Epidermal growth factor (EGF) was then added (lanes 2–7), followed by cell lysis, SDS-PAGE, and immunoblotting.
Characterization of an alternative inactive ATP-binding site conformation of Erk2
Although upstream MEKs are unable to activate p38α and Erk2 that are bound to ligands that stabilize the DFG-out inactive conformation, it is unclear whether this effect is due to the substrate MAPK being in the DFG-out conformation or whether this is due to these kinases’ inability to access their active form. Therefore, we conducted a screen in order to identify inhibitors of Erk2 that stabilize the other most frequently observed inactive ATP-binding site conformation in kinases, the Src/Cdk-like or helix αC-out inactive conformation (Jura et al., 2011). In this inactive form, the DFG motif in the activation loop adopts a nearly active conformation, but movement of helix αC in the N-terminal lobe disrupts a key salt bridge between the catalytic lysine and a conserved glutamic acid (Figure 5B). While no ligands that stabilize this inactive form of wild type Erk2 have been identified, we felt that our drug sensitive Erk2 mutants may be susceptible to inhibition by a series of previously characterized conformation-selective Src inhibitors (Krishnamurty et al., 2013). Indeed, inhibitor 3 (Figure 5A), which contains a bulky benzyl group that is predicted to be sterically incompatible with the active conformation of helix αC, inhibits Erk2DS2 (Ki = 133 nM).
Figure 5. Erk2DS2 stabilized in a modified helix αC-out conformation.

A. Structure of ligand 3. B. Overall topology of 3-bound Erk2DS2. Helix αC is shown in yellow, the activation loop in magenta, and 3 in green. C. Superimposed structures of 3-bound Erk2DS2 (gray and yellow with magenta activation loop), SB203580-bound pentamutant Erk2 (cyan) (PDB ID: 1PME), and 2-bound Erk2DS2 (blue) (PDB ID: 4I5H). The DFG motif phenylalanine residues for all three structures are represented in stick form. The distances from the α-carbon of the phenylalanine residue in 2-bound Erk2DS2 from those of 3- and SB203580-bound Erk2 are 11.1 and 9.6 Å, respectively. D. Superimposed structures of 3-bound Erk2DS2 (gray and yellow), SB203580-bound pentamutant Erk2 (cyan) (PDB ID: 1PME), and 2-bound Erk2DS2 (blue) (PDB ID: 4I5H). Glu-69 and Lys-52 for all three structures are represented in stick form. The distances between these residues for 3-, 2-, and SB203580-bound Erk2 are 7.6, 2.7, and 2.7 Å, respectively. See also Figure S2 and Table S2.
To test whether 3 stabilizes the helix αC-out conformation of Erk2 as predicted, the crystal structure of 3-bound Erk2DS2 was determined (Figure 5B and Table S2). This structure is very similar to that of Erk2 bound to the inhibitor SB203580, with an overall root-mean-square deviation (RMSD) of 0.854 Å. Equally alike are the positions of their activation loops; both are stabilized in the DFG-in conformation, which starkly contrasts with that of 2-bound Erk2DS2 (Figure 5C). As expected, the benzyl group of 3 is directed towards helix αC, disrupting the salt bridge between Glu-69 and the catalytic lysine (Lys-52). Somewhat surprisingly, helix αC does not rotate and move away from the active site to accommodate the benzyl moiety of ligand 3; instead, Glu-69 on helix αC simply assumes a more compact rotamer (Figure 5D). Therefore, while the active site movements induced by ligand 3 still disrupt the salt bridge between Glu-69 and Lys-52, which is the salient feature of the helix αC-out inactive conformation, they do so without changing the position of helix αC. Given that a close analogue of 3 causes a rotation and translocation of Src’s helix αC (Krishnamurty et al., 2013), it is unclear why 3 does not induce a similar change in Erk2 (Figure S2A). Even the inactive form of the S/T kinase Cdk2, which is ~50% similar in sequence to Erk2, presents the more canonical helix αC-out conformation (Schulze-Gahmen et al., 1996). However, superimposing cyclin-bound active Cdk2 with 3-bound Erk2 suggests that Erk2’s C-terminal extension (L16), which is not present in Src or CDK2, behaves like cyclin to keep helix αC packed in the active site (Figure S2B). However, more work is required to validate this hypothesis.
To determine the effect of 3 on MEK2-mediated phosphorylation of Erk2DS2, ligand 3 was titrated into a fixed concentration of Erk2DS2, after which ATP and MEK2 were added. The amount of bis-phosphorylated Erk2 generated was quantified by ELISA. While 3 directly inhibits MEK2, this inhibitor does not appear to prevent activation loop phosphorylation through its interaction with Erk2DS2 (Table S1B). Therefore, the inhibition of activation loop phosphorylation that is observed in the presence of 2 is most likely due to stabilization of Erk2 in the DFG-out inactive conformation.
Divergent modulation of MAPK dephosphorylation with conformation-selective inhibitors
Since conformation-selective inhibitors can prevent MAPK activation loop phosphorylation, we predicted that their cognate dephosphorylation would also be sensitive to these ligands (Figure 6A). MAPKs are selectively dephosphorylated at both residues on their TxY motifs in the activation loop by dual-specificity phosphatases (DUSPs) (Sun et al., 1993). The specificities of DUSPs vary, but DUSP6 is reported to target Erk MAPKs (Muda et al., 1996). For this reason, DUSP6 was used to dephosphorylate activation loop phosphorylated Erk2DS2 in the presence of inhibitors 2, 3, or SB203580, and this reaction was monitored by following the decrease of activated Erk2 by ELISA. Similar to its ability to prevent the phosphorylation of Erk2, 2 limits the ability of DUSP6 to dephosphorylate the TxY motif (Figure 6B and Table S3). Interestingly, both 3 and SB203580 enhanced the phosphatase activity of DUSP6, resulting in increased dephosphorylation of phospho-Erk2DS2 relative to the apo enzyme. To exclude the possibility of the direct enzymatic activation or inhibition of DUSP6, the experiment was repeated with phospho-Erk2WT; as expected, none of the inhibitors affected the ability of DUSP6 to dephosphorylate this kinase. Therefore, 2, 3, and SB203580 affect DUSP6 activity by modulating the conformation of its substrate, phospho-Erk2.
Figure 6. The effects of conformation-selective inhibitors on MAPK dephosphorylation.
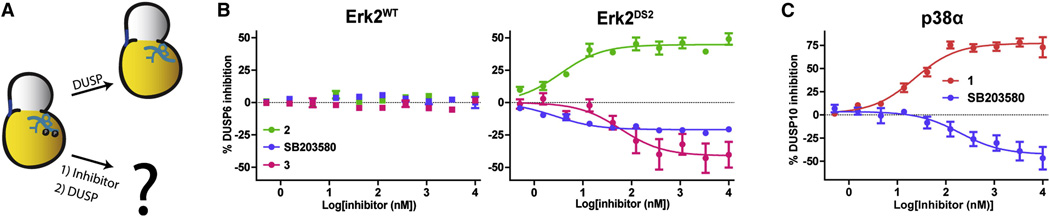
A. Schematic for MAPK dephosphorylation. B. Effect of ligands 2, 3, and SB203580 on DUSP6-mediated dephosphorylation of phospho-Erk2WT and phospho-Erk2DS2. C. Effect of ligand 1 or SB203580 on DUSP10-mediated phospho-p38α dephosphorylation. Error bars represent standard error of the mean (SEM) for three replicate measurements. See also Table S3.
To see if this effect applied to other MAPKs, the same experiment was performed with p38α and its specific phosphatase DUSP10 (Figure 6C) (Tanoue et al., 1999). Similar to what was observed for Erk2, SB203580 and 1 enhanced and reduced, respectively, the dephosphorylation of phospho-p38α. Hence, the combined results from p38α and Erk2 reveal divergent regulation of signal transduction through the use of inhibitors that stabilize distinct ATP-binding site conformations. While the exact mechanism of this regulation is not yet fully established, it is logical that the ~10 Å translocation undergone by the activation loop (Figure 2A), which is characteristic of the DFG-out inactive conformation, would influence enzymes (such as MEKs and DUSPs) that closely interact with this motif. Less clear, though, is how ligands such as 3 and SB203580, which stabilize the activation loop in the DFG-in conformation, enhance the ability of MAPKs to be acted upon by these enzymes.
Conformation-selective inhibitors modulate Erk2’s ability to allosterically activate a phosphatase
Having demonstrated that conformation-selective inhibitors can modulate the post-translational regulation of Erk2, we wondered whether these same ligands are also able to influence the noncatalytic roles of this kinase. One of these interesting noncatalytic phenomena also involves DUSP6. Camps et al. reported that the catalytic activity of this phosphatase for exogenous substrates is increased by non-covalent interaction with Erk2 (Fig. 7A) (Camps et al., 1998). To test the effects of our conformation-selective inhibitors on DUSP6 activation, the abilities of the drug-sensitive Erk2 mutants to activate DUSP6 were first determined by following the turnover of 4-methylumbelliferyl phosphate (4-MUP), which yields a fluorescent product upon dephosphorylation (Figure 7A). As expected, both drug sensitive Erk2 mutants activate DUSP6 to the same extent as Erk2WT (Figure S3A).
Figure 7. Conformation-selective inhibitors affect MAPK activation of DUSP6.

A. Schematic of Erk2-mediated activation of DUSP6. B. Ligands 2, 3, or SB203580 were added to unphosphorylated Erk2WT or Erk2DS2. DUSP6 was then added to the reaction, followed by 4-MUP. The phosphatase reaction was measured by monitoring the fluorescence increase. Background subtraction was performed using samples without kinase. The asterisks indicate p-value < 0.001. C. Titration of Erk2-mediated DUSP6 activation. Kact[Erk2] values are 280 ± 20 nM and 500 ± 140 nM for apo Erk2DS1 and the 2-Erk2DS1 complex, respectively. Error bars represent standard error of the mean (SEM) for three replicate measurements. See also Figure S3.
Next, how 2, 3, and SB203580 influence the ability of Erk2 to activate DUSP6 was tested by incubating these ligands with DUSP6, 4-MUP, and Erk2DS1 or Erk2DS2 (Figure 7B and S3B). Interestingly, 2 almost completely suppressed the activation of DUSP6 by the drug-sensitive Erk2 mutants. In contrast, neither 3 nor SB203580 demonstrated a comparable inhibitory effect, with both inhibitor-bound Erk2 complexes showing similar activation as apo Erk2. Again, none of these ligands affected the ability of Erk2WT to activate DUSP. In summary, these results suggest that Erk2 is interacting with DUSP6 on or close to the activation loop, and stabilizing this kinase in the DFG-out inactive conformation is sufficient to disrupt this interaction.
To gain mechanistic insight into this effect, apo Erk2DS1 or the 2-Erk2DS1 complex were titrated into DUSP6, and the consumption of 4-MUP was measured (Figure 7C). Interestingly, the concentrations of apo Erk2DS1 and 2-Erk2DS1 necessary to achieve 50% maximal activation of DUSP6 are similar. Therefore, it appears that 2 does not notably disrupt the interaction of Erk2 with DUSP6. Instead, Erk2’s interaction with 2 decreases the ability of bound Erk2 to activate phosphatase activity. Furthermore, 2 does not appear to affect the ability of DUSP6 to act upon its substrates, as the DUSP6/Erk2DS1 and DUSP6/Erk2DS1-2 complexes have similar Michaelis constants (Km) for 4-MUP (Figure S3C). A model by Zhou et. al. based on HX experiments proposes that the substrate-binding domain of Erk2 is critical for DUSP6 activation (Zhou et al., 2006). The positions of the residues that form the substrate-binding domain are largely unperturbed when Erk2 is in the DFG-out conformation (Figure S3D), which explains why ligand 2 does not change the affinity of Erk2 for DUSP6. Our results suggest that an interaction between the activation loop of Erk2 and DUSP6 is necessary for allosteric phosphatase activation. The ability to control the conformation of Erk2’s activation loop with conformation-selective ligands presents the opportunity to modulate a number of noncatalytic functions of this kinase.
SIGNIFICANCE
The conventional mode of action of protein kinase inhibitors is to block phosphotransferase activity, but a growing body of evidence strongly indicates that these inhibitors can also affect regions distal to the active site. It is worthwhile to consider that these changes may lead to effects of therapeutic relevance.
We have demonstrated that conformation-selective inhibitors have differential effects on the regulatory interactions of MAPKs. Activation, deactivation, and noncatalytic allosteric enzyme activation can be modulated by the conformations stabilized by these inhibitors. As MAPKs are implicated in a number of noncatalytic functions in the cell, such as scaffolding and transcriptional regulation, it may be possible to control diverse cellular processes through the ATP-binding sites of MAPKs. Specifically, perturbation of the activation loop using a specific class of ATP-competitive inhibitors consistently alters MAPK post-translational modification and noncatalytic function. While the results in this study highlight the importance of the activation loop in modulating these effects, it remains to be seen how general this observation is in other kinase families. The increasing availability of conformation-selective ligands will facilitate these studies.
Therefore, while the generality of the phenomena reported here is still unknown, given the growing number of identified noncatalytic protein kinase functions, it is highly possible that targeting the active site to control functions that do not rely on catalytic activity will become a viable strategy for therapeutic development. The assays developed for this study could easily be adapted to high-throughput formats, and we have demonstrated that cell-based experiments are also possible. Thus, as more noncatalytic functions are identified, it is both important and experimentally feasible to develop biochemical assays that can be incorporated into the kinase inhibitor discovery pipeline to test these functions.
EXPERIMENTAL PROCEDURES
General information
All assays were calibrated to ensure linearity of signal with respect to both substrate and enzyme. Protein concentrations were determined by absorbance at 280 nm.
Statistical analysis
Error bars represent standard error of the mean (SEM) for three replicate measurements.
Small molecule synthesis
Detailed procedures and commercial suppliers are provided in the Supplemental Text.
Cloning and protein expression
Detailed information is provided in the Supplemental Text.
Activation inhibition assays (ELISA)
These assays were performed based on the work of Sullivan et al (Sullivan et al., 2005). Briefly, kinase was incubated in phosphorylation buffer (50 mM MOPS [pH 7.4], 10 mM MgCl2, 1 mM DTT, 0.001% (v/v) Tween 20) and BSA (0.1 mg/mL) with ATP (5 µM) and titrated inhibitor (2% in DMSO) for 30 min at room temperature. Upstream kinase (MEK2 for Erk2, MEK6 for p38) was then added, and the reactions were incubated at room temperature. Downstream and upstream kinase concentrations as well as incubation times varied by kinase based on substrate linearity. The reactions were then quenched with 20 mM EDTA and bound to polystyrene plates (Costar 96-well EIA/RIA #3590) overnight at 4 °C. Subsequent ELISA was performed using primary antibodies specific for phosphorylated kinase (p38: Cell Signaling #4511; Erk2: Santa Cruz Biotechnology #16982) and α-rabbit HRP-linked secondary antibody (Cell Signaling #7074). EC50s were determined by nonlinear regression using GraphPad Prism software.
Activation inhibition assays (Immunoblot)
These assays were performed essentially as described above, but ten times the amount of MEK2 was added in the case of Erk2. The reactions were quenched with SDS loading buffer without EDTA, then separated by SDS-PAGE gels cast with 50 µM PhosTag-acrylamide (Wako) and 100 µM MnCl2.
In-cell activation inhibition
Erk2WT or Erk2DS1 kinase genes in pcEGFP-LIC were transfected into low-passage HEK293 cells using Fugene HD transfection reagent (Roche) and split the next day into a 96-well plate. Cells were serum starved for 24 h, after which inhibitor was added (final 1% DMSO in culture media) and incubated for 1 h at 37 °C. Human EGF (Cell Signaling, 50 ng/mL) was then added to each well and incubated at 37 °C for 5 min. Cells were lysed by boiling in SDS loading buffer, and cleared lysates were separated by SDS-PAGE. Transferred membranes were immunoblotted with mouse α-GFP and rabbit α-ppErk (Cell Signaling, #2955 and #4370, respectively) and imaged with a Licor Odyssey scanner.
Phosphatase inhibition
Phosphorylated kinase (20 nM) was pretitrated with inhibitor for 30 min in buffer (50 mM HEPES [pH 7.4], 300 mM NaCl, 5 mM DTT, 0.1 mg-mL−1 BSA) before incubation with DUSP for 1 h. Amount of DUSP added varied by preparation and was tested for linearity before use. Reactions were quenched with 8 mM Na3VO4 and bound to polystyrene plates (Costar 96-well EIA/RIA #3590) overnight at 4 °C. Subsequent ELISA was performed using primary antibodies specific for phosphorylated kinase (p38: Cell Signaling #4511; Erk2: Santa Cruz Biotechnology #16982) and α-rabbit HRP-linked secondary antibody (Cell Signaling #7074). EC50s were determined by nonlinear regression using GraphPad Prism software.
DUSP6 activation
Either no kinase, Erk2WT, Erk2DS1, or Erk2DS2 (250 nM) was incubated with DUSP6 (140 nM) for 1 h at room temperature, after which 4-MUP (250 µM) was added. Samples were read at (Ex/Em: 355/460 nm) after 2 h at room temperature. Samples without either DUSP6 or Erk2 were used for background correction.
DUSP6 activation inhibition (single concentration)
Either no kinase, Erk2WT, or Erk2DS2 (1 µM) was incubated with inhibitor (1% in DMSO, 5 µM) in buffer (50 mM HEPES [pH 7.4], 300 mM NaCl, 5 mM DTT, 0.1 mg-mL−1 BSA) for 30 min at room temperature. DUSP6 (140 nM) was added, and the solutions were incubated for 1 h at room temperature, after which 4-MUP (250 µM) was added. Samples were read at (Ex/Em: 355/460 nm) after 2 h at room temperature. Samples without Erk2 were used for background correction.
DUSP6 activation inhibition (titration)
Erk2 (100 nM) was preincubated with titrated inhibitor (1% in DMSO) in buffer ((50 mM HEPES [pH 7.4], 300 mM NaCl, 5 mM DTT, 0.1 mg-mL−1 BSA) for 30 min. DUSP6 (140 nM) was then added and allowed to incubate for 1 h at room temperature, after which 4-MUP (250 µM) was added. The reaction proceeded for 2 h at room temperature and then read at (Ex/Em: 355/460 nm). EC50s were determined by nonlinear regression using GraphPad Prism software.
DUSP6 kinetics (Erk2)
Titrated amounts of Erk2DS1 (starting at 3 µM) were preincubated with inhibitor (10 µM, >9.9% bound) for 30 min in buffer (50 mM HEPES [pH 7.4], 300 mM NaCl, 5 mM DTT, 0.05 mg-mL−1 BSA), then incubated with DUSP6 (140 nM) for 1 h before addition of 4-MUP (250 µM). Samples were read at (Ex/Em: 355/460 nm) after 2 h at room temperature. Michaelis constants were determined by nonlinear regression using GraphPad Prism software.
DUSP6 kinetics (4-MUP)
Erk2DS1 (2 µM) was preincubated with inhibitor (10 µM, >9.9% bound) for 30 min in buffer (50 mM HEPES [pH 7.4], 300 mM NaCl, 5 mM DTT, 0.05 mg-mL−1 BSA), then incubated with DUSP6 (140 nM) for 1 h before addition of titrated 4-MUP. Samples were read at (Ex/Em: 355/460 nm) after 2 h at room temperature. Due to the excessive fluorescence of 4-MUP, separate wells for each concentration of 4-MUP without Erk2 or DUSP6 were prepared for background correction. Michaelis constants were determined by nonlinear regression using GraphPad Prism software.
Crystallography
Detailed procedures are provided in the Supplemental Text. The atomic coordinates and structure factors for 3-bound Erk2DS2 have been deposited in the Protein Data Bank under accession code 4N4S.
Supplementary Material
HIGHLIGHTS.
ATP-competitive kinase inhibitors can affect the regulatory interactions of MAPKs
Conformation-selective inhibitors allow differential modulation of MAPK regulation
MAPK regulation is sensitive to the conformation of their ATP-binding sites
A noncatalytic function of Erk2 can be mediated by ATP-competitive inhibitors
ACKNOWLEDGEMENTS
We thank S. Leonard for providing inhibitor 3 and S. Turley for assisting with crystallographic data collection. This work was supported by the NIH (R01 GM086858) (D.J.M.), Alfred P. Sloan and Camille and Henry Dreyfus Foundations (D.J.M.), and a predoctoral fellowship from the American Heart Association (S.B.H.).
Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the U.S. Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Camps M, Nichols A, Gillieron C, Antonsson B, Muda M, Chabert C, Boschert U, Arkinstall S. Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science. 1998;280:1262–1265. doi: 10.1126/science.280.5367.1262. [DOI] [PubMed] [Google Scholar]
- Chan TO, Zhang J, Rodeck U, Pascal JM, Armen RS, Spring M, Dumitru CD, Myers V, Li X, Cheung JY, et al. Resistance of Akt kinases to dephosphorylation through ATP-dependent conformational plasticity. Proc. Natl. Acad. Sci. U.S.A. 2011;108:E1120–E1127. doi: 10.1073/pnas.1109879108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cichowski K, Janne PA. Drug discovery: inhibitors that activate. Nature. 2010;464:358–359. doi: 10.1038/464358a. [DOI] [PubMed] [Google Scholar]
- Cohen P, Alessi DR. Kinase drug discovery--what's next in the field? ACS Chem. Biol. 2013;8:96–104. doi: 10.1021/cb300610s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hari SB, Merritt EA, Maly DJ. Sequence determinants of a specific inactive protein kinase conformation. Chem. Biol. 2013;20:806–815. doi: 10.1016/j.chembiol.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJ, Stokoe D, Gloor SL, Vigers G, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- Jura N, Zhang X, Endres NF, Seeliger MA, Schindler T, Kuriyan J. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol. Cell. 2011;42:9–22. doi: 10.1016/j.molcel.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita E, Takahashi M, Takeda H, Shiro M, Koike T. Recognition of phosphate monoester dianion by an alkoxide-bridged dinuclear zinc(II) complex. Dalton Trans. 2004:1189–1193. doi: 10.1039/b400269e. [DOI] [PubMed] [Google Scholar]
- Krishnamurty R, Brigham JL, Leonard SE, Ranjitkar P, Larson ET, Dale EJ, Merritt EA, Maly DJ. Active site profiling reveals coupling between domains in SRC-family kinases. Nat. Chem. Biol. 2013;9:43–50. doi: 10.1038/nchembio.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G, Plowman GD, Hunter T, Sudarsanam S. Evolution of protein kinase signaling from yeast to man. Trends Biochem. Sci. 2002a;27:514–520. doi: 10.1016/s0968-0004(02)02179-5. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002b;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Muda M, Theodosiou A, Rodrigues N, Boschert U, Camps M, Gillieron C, Davies K, Ashworth A, Arkinstall S. The dual specificity phosphatases M3/6 and MKP-3 are highly selective for inactivation of distinct mitogen-activated protein kinases. J. Biol. Chem. 1996;271:27205–27208. doi: 10.1074/jbc.271.44.27205. [DOI] [PubMed] [Google Scholar]
- Okuzumi T, Fiedler D, Zhang C, Gray DC, Aizenstein B, Hoffman R, Shokat KM. Inhibitor hijacking of Akt activation. Nat. Chem. Biol. 2009;5:484–493. doi: 10.1038/nchembio.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson FL, Whitehurst AW, Raman M, Cobb MH. Identification of novel point mutations in ERK2 that selectively disrupt binding to MEK1. J. Biol. Chem. 2002;277:14844–14852. doi: 10.1074/jbc.M107776200. [DOI] [PubMed] [Google Scholar]
- Rodriguez J, Crespo P. Working without kinase activity: phosphotransfer-independent functions of extracellular signal-regulated kinases. Sci. Signal. 2011;4:re3. doi: 10.1126/scisignal.2002324. [DOI] [PubMed] [Google Scholar]
- Schulze-Gahmen U, De Bondt HL, Kim SH. High-resolution crystal structures of human cyclin-dependent kinase 2 with and without ATP: bound waters and natural ligand as guides for inhibitor design. J. Med. Chem. 1996;39:4540–4546. doi: 10.1021/jm960402a. [DOI] [PubMed] [Google Scholar]
- Sullivan JE, Holdgate GA, Campbell D, Timms D, Gerhardt S, Breed J, Breeze AL, Bermingham A, Pauptit RA, Norman RA, et al. Prevention of MKK6-Dependent Activation by Binding to p38α MAP Kinase. Biochemistry. 2005;44:16475–16490. doi: 10.1021/bi051714v. [DOI] [PubMed] [Google Scholar]
- Sun H, Charles CH, Lau LF, Tonks NK. MKP-1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylates MAP kinase in vivo. Cell. 1993;75:487–493. doi: 10.1016/0092-8674(93)90383-2. [DOI] [PubMed] [Google Scholar]
- Tanoue T, Moriguchi T, Nishida E. Molecular cloning and characterization of a novel dual specificity phosphatase, MKP-5. J. Biol. Chem. 1999;274:19949–19956. doi: 10.1074/jbc.274.28.19949. [DOI] [PubMed] [Google Scholar]
- Wang L, Perera BG, Hari SB, Bhhatarai B, Backes BJ, Seeliger MA, Schurer SC, Oakes SA, Papa FR, Maly DJ. Divergent allosteric control of the IRE1alpha endoribonuclease using kinase inhibitors. Nat. Chem. Biol. 2012;8:982–989. doi: 10.1038/nchembio.1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer. 2009;9:28–39. doi: 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- Zhou B, Zhang J, Liu S, Reddy S, Wang F, Zhang ZY. Mapping ERK2-MKP3 binding interfaces by hydrogen/deuterium exchange mass spectrometry. J. Biol. Chem. 2006;281:38834–38844. doi: 10.1074/jbc.M608916200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.