

Abstract
Breast cancer metastasis and disease recurrence are hypothesized to result from residual cancer stem cells, also referred to as tumor-initiating cells, which evade initial treatment. Using both syngeneic mouse and human xenograft models of triple-negative breast cancer, we have demonstrated that a subpopulation enriched in cancer stem cells was more resistant to treatment with 6 gray of ionizing radiation than the bulk of the tumor cells, and accordingly their relative proportion increased 48 to 72 hours after ionizing radiation treatment. In contrast, we achieved a larger reduction in tumor size without a concomitant increase in the percentage of cancer stem cells by treating with local hyperthermia for 20 minutes at 42°C after ionizing radiation using intravenously administered, optically activated gold nanoshells. Forty-eight hours after treatment, cells derived from the tumors treated with ionizing radiation plus hyperthermia exhibited both a marked decrease in tumorigenicity and a more differentiated phenotype than mock- and ionizing radiation–treated tumors. Thus, we have confirmed that these cancer stem cells are responsible for accelerated repopulation in vivo and demonstrated that hyperthermia sensitizes this cell population to radiation treatment. These findings suggest that local hyperthermia delivered by gold nanoshells plus radiation can eliminate radio-resistant breast cancer stem cells.
Introduction
Despite the significant advances made in breast cancer biology over the past decade, the etiology of breast cancer remains poorly understood. Although most breast cancer recurrences occur within the first 5 years of diagnosis, they are also often preceded by a prolonged disease-free interval, indicating that there may be a population of treatment-resistant cells with an extremely long half-life (1). Metastatic disease and recurrence have been hypothesized to result from residual cancer stem cells (CSCs), also referred to as tumor-initiating cells, which evade initial treatment (2). CSCs are thought to have the ability to recapitulate the heterogeneity of the parental tumor, to self-renew, and to maintain their self-renewing potential over long periods of time. Therapeutic targeting of the CSC population, in addition to differentiated malignant cells, may be critical for the successful treatment and elimination of residual disease in human breast cancer.
Hyperthermia (HT) therapy acts as a cancer treatment by direct cell killing, radiosensitization, and promotion of tumor reoxygenation [reviewed in (3, 4)]. Thermal radiosensitization is believed to work by heat-induced inhibition of repair of radiation-induced double-strand breaks (DSBs) or alterations in how DSBs are recognized or processed (5), possibly through the effect of heat on nuclear protein unfolding. Early clinical trials lacked adequate thermometry or had suboptimal regimens and inadequate delivery systems, which led to negative results, virtually halting investigations of clinical HT, except in a few centers [reviewed in (6)]. After the adoption of international quality control guidelines, results from phase 3 clinical trials were generally positive (7–9). A meta-analysis of data from five randomized control trials in breast cancer with thorough thermometry documentation demonstrated an improved response rate and duration of local control, particularly among previously irradiated patients with limited therapy options (10). Thus, therapeutic gains from HT therapy are possible with improved technology.
Advances in nanoparticle technology have enabled temperature control in a site-specific, noninvasive manner to further optimize HT treatment (11). Nanoshells are optically tunable nanoparticles composed of a silica core with an ultrathin gold layer (12). The nanoshells in this experiment have been designed to absorb light in the near-infrared (NIR) spectrum, which provides optimal light penetration of tissue (13). The nanoshells passively concentrate within the tumor as a result of an enhanced permeability and retention (EPR) effect and thereby allow for specific noninvasive heating (14, 15).
Here, we used two different preclinical breast cancer models to study radiation resistance with and without thermal enhancement. The p53-null genetically engineered mouse (GEM) mammary tumor model mimics subsets of human breast cancers more closely than many other mouse models, as assessed by histological and gene expression analyses (16). The mammary tumors in these mice harbor a Lineagenegative (Lin−) subpopulation of cells that is characterized by high CD29 and CD24 expression, termed CD29HighCD24High (Lin−CD29HCD24H), which are enriched ∼65-fold in tumor formation ability (17) and likely represent an enriched subpopulation of CSCs.
We also used primary human breast cancer xenografts to investigate the effect of radiation with HT treatment on CSCs. These xenograft models were chosen on the basis of their similarity to triple-negativ breast cancers, a subtype of breast cancer that does not express estrogen receptor (ER), progesterone receptor (PR), or human epidermal growth factor receptor 2 (HER2). This subtype of breast cancer is more aggres sive and less responsive to standard treatments, including radiation, and is associated with poor overall patient prognosis (18–20). Cells positiv for aldehyde dehydrogenase (ALDH+) in this model are enriched in CSCs (21).
We have investigated, both in p53-null GEM mice and in xenograft of triple-negative human breast cancers, whether gold nanoparticle used for HT can enhance the radiation sensitivity of CSCs.
Results
In vitro HT increases radiation sensitivity of CSCs in p53-null GEM mammary tumors
Using the in vitro mammosphere assay, we performed our initial studie to establish the radiation resistance and sensitivity to HT of p53-nul GEM mammary tumors. In the mammosphere assay, single cells ar cultured in low-attachment plates, with no serum but with a medium enriched in growth factors, and serially passaged by trypsin dissociation. We tested single cells isolated from T7, a solid, poorly differentiated ER− adenocarcinoma developed from the p53-null GEM mammary model. When passaged as mammospheres, the cells become significantly enriched for the Lin−CD29HCD24H subpopulation, which represent less than 15% of the initial total cells (17). We observed no effect of ionizing radiation (IR) treatment up to 6 Gy (gray) on proliferation, survival, or mammosphere formation from single cells derived from dissociated T7 mammospheres (Fig. 1, A to C). However, 1 hour of HT (42°C) immediately after radiation (IR+HT) significantly decreased cellular proliferation, causing the cells to begin to die 48 hours after treatment, whereas the cells subjected to mock or IR treatment were viable and grew at similar rates (Fig. 1A). The delay in cell death observed in Fig. 1A in the IR+HT treatment group is likely a result of the cells' inability to repair DSBs (5). In addition to decreasing survival and proliferation, IR+HT also caused a decrease in mammosphere formation efficiency (MSFE), indicating a loss of self-renewal potential (Fig. 1, B and C).Nochange inmammosphere formation was seen with HT-only treatment (Fig. 1C).
Fig. 1.
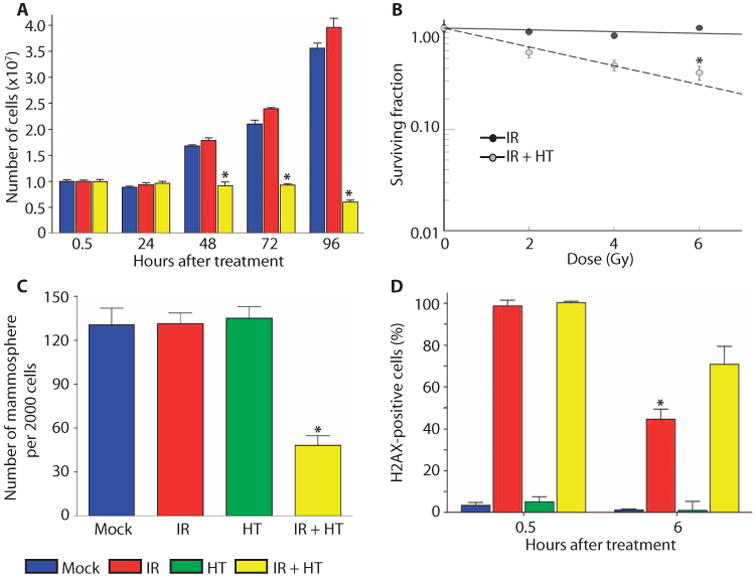
IR+HT alters DNA damage repair in mammospheres. Serially passaged trypsin-digested mammospheres from tumor T7 were replated as 10th-generation mammospheres. (A) A viability assay indicates that cells treated with IR+HT (1 hour; yellow columns) do not begin to die until after 48 hours, when IR (red columns) and mock treatment (blue columns) cells have begun to divide. (B) IR+HT (gray circles) results in a significant decrease in mammosphere survival as compared with IR (black circles). (C) Mock treatment (blue columns), IR treatment (red columns), and HT (green columns) had a significantly higher MSFE compared to IR+HT (yellow columns). (D) Significant repair of DNA damage, measured with γ-H2AX FACS, was observed 6 hours after IR (red columns) compared to IR+HT (yellow columns). HT (green columns) and mock treatment (blue columns) did not have significant DNA damage. These data are representative of three tumors from different mice with six technical replicates performed for each tumor. (*P < .01)
IR causes DNA DSBs that recruit the phosphorylated form of histone H2AX (γ-H2AX), which is detectable in nuclear foci that correlate in number with the amount of unrepaired DNA damage(22). Accordingly, we analyzed γ-H2AX by fluorescence-activated cell sorting (FACS) in cells treated with IR and IR+HT. Six hours after treatment, there was a significantly lower percentage of γ-H2AX–positive cells after IR treatment as compared with IR+HT (Fig. 1D). HT alone did not result in a significant difference in the percentage of positive γ-H2AX cells detectable by FACS analysis when compared to mock treatment (Fig. 1D).
In vivo HT radiosensitizes CSCs in p53-null tumors
To determine whether the Lin−CD29HCD24H subpopulation was radiation-resistant in vivo, we treated p53-null GEM tumors in situ with a single dose of 6-Gy radiation. The tumors tested in these experiments were T1, a highly keratinized squamous ER− adenocarcinoma, and T7. Mice with tumors ∼1 cm in diameter (500 mm3 volume) were exposed to a single dose of 6 Gy, and tumors were processed for CSC analysis by FACS 48 hours after treatment. IR treatment resulted in no significant difference in T1 tumor volume compared to mock treatment, but in T7 tumors, IR treatment decreased tumor volume more than mock treatment (Fig. 2, A and B). However, experimental and clinical evidence suggest that a decrease in tumor size may not correlate with a decrease in the CSC subpopulation (23–25). In both of the tumor types, the Lin−CD29HCD24H subpopulations were increased (by 30 or 100%) 48 hours after IR treatment (Fig. 2, C and D). These results suggest that the proportion of tumor cells with CSC properties actually increased after treatment with IR alone, despite the overall decrease in tumor size.
Fig. 2.
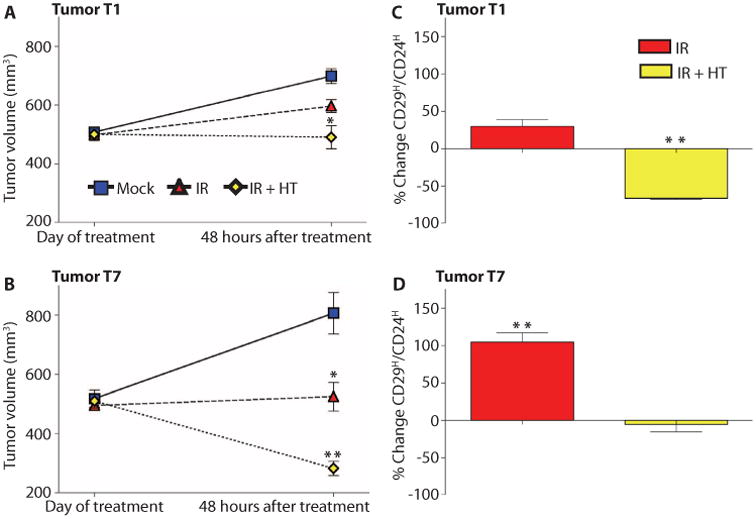
Thermal enhancement with gold nanoshells in combination with radiation prevents the relative increase in Lin−CD29HCD24HCSCs seen with radiation. (A and B) In both T1 and T7, IR+HT (yellow diamonds) caused a significant decrease in tumor volume. The percent of the Lin−CD29HCD24H population in the mock treatment group was averaged to calculate the percent change. (C) In T1, IR (red columns) increased and IR+HT (yellow columns) significantly decreased the percentage of the Lin−CD29HCD24H cells. (D) In T7, IR (red columns) significantly increased and IR+HT (yellow columns) prevented the increase in the percent of Lin−CD29HCD24H cells (*P < 0.01; **P < 0.001). Sample FACS profiles are shown in fig. S2. These data are representative of six tumors from different mice per treatment.
We next determined whether HT in vivo would sensitize the resistant-enriched CSC subpopulation to radiation treatment as observed in vitro with the mammosphere assays. In vivo HT was performed with NIR illumination of gold nanoshells that had accumulated in tumors after intravenous administration with thermocouple measurements to monitor tumor temperature. This system was previously validated with real-time noninvasive magnetic resonance thermal imaging measurements, which were consistent with the thermocouple measurements (26). Tumors reached the optimal temperature of 42°C within 5 min of activating the laser and likewise returned to baseline temperature within 5 min of laser inactivation (fig. S1). Using this approach, we determined whether HT sensitized the resistant-enriched CSC subpopulation to radiation treatment. Mice with tumors 1 cm in diameter (500 mm3 volume) were exposed to a single dose of 6 Gy plus 20 min at 42°C (IR+HT), and tumors were processed for CSC analysis by FACS 48 hours after treatment. In both T1 and T7 tumors, IR+HT caused a greater regression of tumor size than did IR treatment alone (Fig. 2, A and B). In the T1 tumor, IR+HT resulted in a significantly lower percentage of Lin−CD29HCD24H cells than IR treatment, suggesting that the combination treatment adversely affected the Lin−CD29HCD24H cells in this tumor (Fig. 2C). In T7, IR+HT also resulted in a lower percentage of Lin−CD29HCD24H cells than did IR alone but did not decrease their proportion below that of mock treatment, suggesting that in T7, IR+HT killed all tumor cells equally (Fig. 2D). HT alone had noeffect on tumor volume or on the proportion of cells in the CD29HCD24H subpopulations (fig. S2). Thus, thermal enhancement of radiation therapy effectively decreased tumor size without increasing the relative proportion of Lin−CD29HCD24H CSCs.
In vivo treatment alters tumor characteristics in limiting dilution transplants
To determine whether the treated tumors retained their tumorigenic properties after IR+HT treatment, we digested tumors 48 hours after treatment, and transplanted the cells as single cells in limiting dilution into syngeneic recipients (Table 1). Cells from T1 and T7 tumors after IR treatment were more tumorigenic than were mock-treated cells (P = 0.023, Wald test). However, a significant decrease (P = 0.002, Wald test) in tumor formation was observed for cells isolated from both the T1 and the T7 tumors after receiving IR+HT compared to mock treatment (Table 1). The differences in tumorigenicity between IR and IR+HT were significant (P < 0.0001, Wald test).
Table 1.
Limiting dilution transplantation after 48 hours of treatment with IR demonstrates a significant increase in tumor initiation, and IR+HT demonstrates a marked reduction in the tumor initiation. T1 palpable tumors formed between 2 and 4 weeks after mock and IR, and between 3 and 5 weeks with IR+HT. T7 palpable tumors formed after all treatments in 2 to 7 weeks.
| No. of cells injected | 10,000 | 1000 | 100 | 10 |
|---|---|---|---|---|
|
| ||||
| No. of tumors formed/total no. of transplants | ||||
| Mock T1 | 6/6 | 6/6 | 1/6 | 0/8 |
| Mock T7 | 6/6 | 6/6 | 1/6 | 0/6 |
| Mock total | 12/12 | 12/12 | 2/12 | 0/14 |
| IR T1 | 6/6 | 6/6 | 4/6 | 3/8 |
| IR T7 | 6/6 | 6/6 | 2/6 | 1/6 |
| IR total | 12/12 | 12/12 | 6/12 | 4/14* |
| IR+HT T1 | 6/6 | 3/6 | 0/6 | 0/8 |
| IR+HT T7 | 6/6 | 3/6 | 0/6 | 0/6 |
| IR+HT total | 12/12 | 6/12 | 0/12 | 0/14† |
P = 0.02
P = 0.002
T1 is a well-differentiated infiltrating ductal carcinoma with squamous metaplastic tubular features (Fig. 3A). The tumors that developed from the limiting dilution assay after IR therapy converted to a poorly differentiated carcinoma in three of six of the tumors analyzed. These tumors demonstrated a significant loss of the tubular architectural pattern and a more aggressive undifferentiated solid or nested phenotype (Fig. 3A). These results indicate that either the well-differentiated component or the progenitors producing the well-differentiated cells were sensitive to IR. In contrast, none of the six tumors resulting fromThe tumors that arose after transplantation from cells treated with IR+HT were histologically less aggressive, suggesting that they may be more sensitive to subsequent treatments.
Fig. 3.
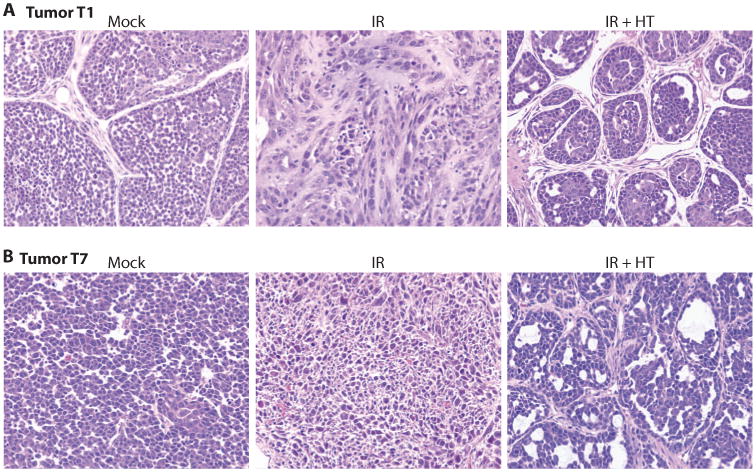
Tumor phenotype alterations following limiting dilution transplantation 48 hours after treatment. Hematoxylin and eosin staining of paraffin-embedded sections of tumors that grew after treatment. (A) T1 mock treatment, T1 48 hours after IR, and 8 hours after IR with HT. IR alone decreases the tubular architectural pattern and results in a more aggressive undifferentiated phenotype. (B) T7 mock treatment, T7 48 hours after IR, and T7 48 hours after IR+HT. IR+HT generated a tumor with a more differentiated phenotype.
T7 is a poorly differentiated ductal carcinoma (Fig. 3B), and four of five tumors developed a more differentiated phenotype after treatment with IR+HT and transplantation (Fig. 3B, IR+HT). Along with the observed decrease in the well-differentiated phenotype after radiation therapy, one tumor in each of the T1 and T7 groups showed pathologic signs of pleomorphic, histologically aggressive tumors (Fig. 3, A and B, IR). Together, these data suggest that IR+HT decreases the tumorigenicity of residual tumor cells and that the surviving cells form tumors that have pathologic signs of histological differentiation with a less aggressive phenotype than either the mock-treated tumors or the tumors treated with sublethal doses of radiation.
In vivo HT sensitizes CSCs to radiation treatment in human breast cancer xenografts
We next determined whether the results obtained in the p53-null GEM model were reproducible in primary human breast cancers. In these studies, pretreatment biopsy specimens of human breast tumors were transplanted directly into epithelium-free “cleared” fat pads of recipient severe combined immunodeficient (SCID)/beige mice and then serially transplanted into SCID/beige mice. The human xenograft tumors digested into single cells were sorted by FACS with the ALDEFLUOR assay, which measures ALDH enzymatic activity. These xenografts contained a cell subpopulation expressing high levels of ALDH, representing less than 5% of the total cell population. ALDH+ cells are more tumorigenic (by a limiting dilution transplantation assay) and exhibit a greater MSFE than do ALDH− cells (27). When human breast cancer xenograft cells were cultured as mammospheres, they were enriched for ALDH+cells, a result similar to that observed for the p53-null GEM mammary tumors and the CD29HCD24H subpopulation of cells (17) (fig. S4).
The patient breast cancer biopsies were ER/PR/HER2-negative in both xenograft models tested. The cell line BCM-2665A was established from a patient with bilateral breast cancer, and MC1 was established from a pleural effusion in a patient with metastatic breast cancer (21). Following a similar protocol as was used with the mouse models, mice with tumors ∼1 cm in diameter were exposed to a single dose of 6-Gy radiation, and tumors were collected and analyzed by FACS 72 hours after treatment. One dose of radiation resulted in cessation of tumor growth in MC1, whereas in BCM-2665A, a significant regression in size was observed (Fig. 4, A and C). The patient from whom the BCM-2665A biopsy was collected also initially exhibited a clinical response to doxorubicin therapy, which may explain its marked sensitivity to radiation treatment as a tumor xenograft. As with the mouse models, both xenografts showed a similar response to radiation treatment, with about a 100 to 150% enrichment in the ALDH+ subpopulation (Fig. 4, B and D). Although xenograft BCM-2665A was responsive to radiation treatment and exhibited a large decrease in tumor volume, it displayed a similar increase in the percentage of ALDH+ cells as the xenograft MC1, suggesting that the CSCs were not affected by the initial treatment of 6 Gy. Next, we performed in vivo HT experiments with IR+HT therapy using gold nanoshells. Seventy-two hours after treatment, tumors were measured and processed for CSC analysis by FACS (Fig. 4, A and C). In both xenografts, the IR+HTwhen compared to IR, suggesting that the combination treatment eliminated the ALDH+ cells in both tumors (Fig. 4, B and D).
Fig. 4.
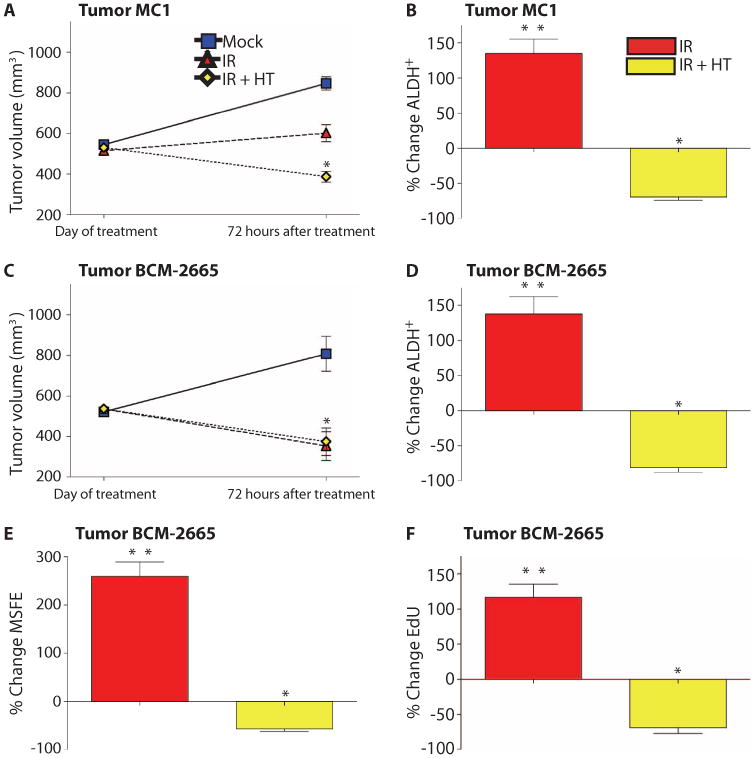
Thermal enhancement with gold nanoshells in combination with radiation prevented the increase in ALDH+ seen with radiation. (A and C) Tumor volume decreased significantly with both the IR (red triangle) and the IR+HT (yellow diamond). The percent of ALDH+in the mock treatment was averaged to calculate the percent change. (B and D) IR treatment (red columns) significantly increased the percent of ALDH+, and IR+HT (yellow columns) prevented the increase in ALDH+. These data are representative of six tumors from different mice per treatment. (E) The MSFE of mock treatment was averaged to calculate the percent change. Secondary MSFE was significantly higher in IR-treated cells (red columns) and significantly decreased in the IR+HT treatment group (yellow columns). (F) EdU incorporation was significantly increased in IR-treated cells (red columns) and significantly decreased in IR+HT (yellow columns). Sample FACS profiles in fig. S5. These data are representative of three tumors from different mice with six technical replicates performed for each tumor (*P < 0.05; **P < 0.001).
To test the self-renewal potential of the treated tumors, we performed mammosphere assays on the freshly digested BCM-2665A tumors. A significant increase in secondary MSFE from tumors treated with IR was observed (Fig. 4E), suggesting that the increase in the ALDH+ subpopulation correlated with an increase in mammosphere formation. However, when tumors were given a combination treatment of IR+HT therapy, mammosphere formation was decreased (Fig. 4E). Proliferation in the whole tumor after treatment was measured with the Click-iT EdU system. BCM-2665A tumors treated with IR exhibited more proliferation than did mock-treated tumors, whereas IR+HT treatment resulted in decreased proliferative capacity (Fig. 4F). Cells that survived sublethal IR treatment grew back at a greater rate than did mock-treated cells, as reported previously with fractionated radiation (23). Thus, these results obtained with the breast cancer xenografts corroborate those obtained in the p53-null GEM mammary tumor model.
CSC populations have increased DNA DSB repair
To explore the mechanisms by which HT affects the DNA damage response (DDR), and to address whether there are differences in DDR in the CSC subpopulation compared to other cells in these breast cancers, we investigated DNA damage markers after treatment. FACS-sorted Lin−CD29HCD24H cells from the p53-null GEM mammary tumors, as well as ALDH+ cells from human xenografts, were immunostained with antibodies directed against 53BP1, a cellular marker of DSB formation (28). The number of cells with 53BP1 foci was quantified to determine the response to DNA damage within the different treatment groups. Tumors were collected at different time points after mock, IR, and IR+HT treatments. As expected, 0.5 hours after both IR and IR+HT treatments, tumors displayed strongly positive 53BP1 foci in the CD29HCD24H andALDH+ subpopulations (Fig. 5, A, a and d, B, C, a and d, D). Consistent with the results obtained previously by γ-H2AX FACS, by 6 hours after treatment, a significant proportion of the cells had repaired the DSBs in the Lin−CD29HCD24H and ALDH+ CSC subpopulations (Fig. 5, A, b, B, C, b, and D). These data are consistent with the CSC subpopulations Lin−CD29HCD24H and ALDH+ having more efficient DNA repair than did the other subpopulations within the tumor (figs. S5 and S6). In all sub-populations, IR+HT treatment prevented distinct 53BP1 foci formation. Instead, a diffuse positive staining pattern was observed, in contrast to the discrete foci seen with IR treatment (Fig. 5, A, f, B, C, f, and D). This disorganized state of 53BP1 was maintained in most cells at both 48 and 72 hours after IR+HT treatment. These data suggest that the efficient DNA repair observed in the tumorigenic CSC-enriched subpopulation was prevented by IR+HT treatment.
Fig. 5.

Altered DNA damage repair with IR+HT. (Aa, Ad and B; Ca, Cd and D) Thirty minutes after IR (red columns) and IR+HT (yellow columns) treatment, all cells have small 53BP1 foci with a nondistinct staining pattern (checkered columns). (Ab and B; Cb and D) A significant number of cells have repaired foci or have distinct foci remaining 6 hours after IR (red columns). (Ac and B; Cc and D) A significant number of cells have repaired DSBs and lost most foci by 48 hours (B) or 72 hours (D) after IR (red columns). (Ae and B; Ce and D) With IR+HT (yellow columns), a significant number of cells have not repaired foci or have formed distinct foci at 6 hours after IR+HT. (Af and B; Cf and D) No significant change from the 6-hour time point was observed 48 hours (B) or 72 hours (D) after IR with HT. Cells were treated with IR or IR+HT in vitro after sorting populations (a, b, d, and e), and with IR or IR+HT in vivo (c and f). Scale bars, 20 μm (A), 15 μm (C). (B and D) IR (red columns), damage is significantly reduced at 6 hours after IR treatment and repair continues at 48 and 72 hours (D) hours. IR+HT (yellow columns), a significant percentage of cells with non-distinct foci (checkered columns) at 6 hours with an inability to form or repair foci at 48 hours (B) and 72 hours (D) (*P < 0.01).
Heat shock proteins may account for differences in DNA repair and tumor phenotype after treatment
Heat shock proteins (HSPs) are traditionally expressed at a higher level in cancer compared to normal cells and have been linked to treatment resistance (29). Inadequate chaperoning of unfolded nuclear proteins after HT has also been implicated in the mechanism of cytotoxicity (30). Accordingly, HSP27, HSP40, HSP60, HSP70, and HSP90α, along with HSP client proteins AKT and phospho-AKT (pAKT), were analyzed by multiplex analysis of FACS-sorted Lin−CD29HCD24H cells isolated from the p53-null GEM mammary tumors, as well as the ALDH+ cells isolated from the human breast cancer xenografts. In xenograft BCM-2665A, an increase in HSP27, HSP40, HSP60, and HSP70 was observed with IR treatment, but treatment with IR+HT prevented the increase in HSP40, HSP60, and HSP70 (Fig. 6A). HSP27 has been reported to be inversely related to cell proliferation (31). Moreover, analysis of breast cancer biopsies has suggested that overexpression of HSP27 may be involved in reduced cell growth and increased differentiation (32). After treatment with IR+HT, HSP27 expression was increased by more than twice as much than in cells treated with IR alone in xenograft BCM-2665A, suggesting that HSP27 might be partially responsible for the change in tumor characteristics observed after IR+HT treatment. The significance of these changes is further illustrated by comparing these results to the HSP expression in the total tumor (fig. S7). In both IR- and IR+HT-treated tumors, specific HSP expression in the total tumor was different from that in the ALDH+ subpopulation of tumor cells.
Fig. 6.

HSPs and pAKT were activated in CSC subpopulations after treatment with IR, but not with IR+HT. The percent of ALDH+in the mock treatment was averaged to calculate the percent change in (A) and (B). (A) HSP40, HSP60, HSP70, and HSP90α have increased expression after IR (red columns) and decreased expression after IR+HT (yellow columns) 72 hours after treatment. HSP27 was increased in both treatments compared to mock. (B) The relative expression of pAKT is slightly increased after IR (red columns) and significantly decreased below mock treatment in IR+HT (yellow columns). (C) HSP90α is slightly increased after IR (red columns), but significantly decreased in IR+HT (yellow columns), compared with mock treatment (blue columns) in tumor T1. The percent of the Lin−CD29HCD24H population in the mock treatment group was averaged to calculate the percent change. (D) The relative expression of pAKT is significantly increased after IR (red columns) and is similar to mock treatment in IR+HT (yellow columns). (*P < 0.01; **P < 0.001)
HSP90α was the only HSP that was affected, with similar changes observed in all of the tumors analyzed. In the T1 mouse tumor model, the expression of HSP90α was increased after treatment with IR only and decreased after IR+HT treatment at 48 hours. These results suggest that HSP90α may be a critical factor in mediating the increased radiation sensitivity of CSCs observed with HT treatment (Fig. 6C). Similar to HSP90α, a significant increase in pAKT was seen after IR in the Lin−CD29HCD24H subpopulation of T1, and IR+HT treatment prevented this increase (Fig. 6D). In BCM-2665A, a slight increase in pAKT was also observed after IR, but after IR+HT treatment, there was a significant decrease in pAKT as compared with mock-treated tumors (Fig. 6B).
Discussion
Despite improvement in overall survival with adjuvant radiation and chemotherapy in the treatment of breast cancer, some patients experience treatment failure, developing local tumor recurrence or metastatic disease after an initial response. Local recurrence and metastatic disease have been attributed to CSCs resistant to initial treatment. Here, CSCs were detected by FACS on the basis of cell surface marker expression in the p53-null GEM model (Lin−CD29HCD24H) or ALDH expression in the primary human breast cancer xenograft model. Using these markers to isolate CSC-enriched subpopulations, we determined that thermal enhancement with gold-coated nanoshells as a radiation sensitizer significantly decreased the radiation resistance of these CSC-enriched subpopulations. Our in vivo studies in mice indicated that sublethal radiation plus HT therapy reduced the tumorigenic subpopulation within the residual cancer burden compared to radiation therapy. Limiting dilution transplantation 48 hours after treatment functionally demonstrated the ability of IR+HT treatment to decrease the frequency of CSCs. Although residual disease and subsequent tumor development after therapy was observed in every treatment group, as expected, the tumors formed after IR+HT treatment tended to display more differentiated characteristics, hallmarks of better prognosis after radiation treatment. In contrast, in the IR treatment group, some of the resulting tumors displayed pathological characteristics of aggressive tumors, suggesting that inadequate therapy can alter, and perhaps even worsen, the phenotype of the surviving tumor cells.
Various endpoints have been used in tumors to study the effects of HT, for example, tumor regression, growth delay, and cell survival. Previous clinical trials in which these endpoints were used have reported variable outcomes (33). However, contemporary phase 3 HT clinical trials incorporating rigorous standardized quality assurance practices show improved response rate, duration of local control, and survival for a number of tumor sites (7–9). Targeted HT was achieved in our study with gold-silica nanoshells that convert electromagnetic energy into heat. This method of treating cancer allows for site-specific HT and offers potential as a noninvasive treatment (11). Currently, this technology is in a phase 1 clinical trial of thermal ablation with head and neck cancer.
On the basis of previously reported evidence of CSC radioresistance (34, 35), we examined markers of DNA damage after radiation treatment and observed enhanced radiation resistance and reduced residual DNA damage in the CSC-enriched subpopulations. We demonstrated that HT in combination with radiation prevented the formation of distinct 53BP1 foci. It has previously been shown that HT will prevent the recruitment of 53BP1 in a heat-dependent manner (36). This delay in recruitment of 53BP1, part of the necessary repair machinery, could be responsible for the heat-treated cells' inability to repair DSBs. The delay in recruitment was evident 6 hours after treatment, a time when the CSC-enriched subpopulations from all of the tumor cells tested displayed decreased 53BP1 foci formation and increased diffuse staining.
HSPs function as chaperones within the cell and protect cells from hyperthermic damage by stabilizing unfolded proteins to prevent aggregation [reviewed in (37)]. Previous studies have also indicated that HSPs play a critical role in differentiation and apoptosis of tumor cells [reviewed in (38)]. However, clinical data to support this latter hypothesis are controversial and dependent on the tumor type studied. In investigating the role of HSPs in tumor cells, our data indicate that it may be critical to examine the tumorigenic subpopulations within the remaining tumor. Analysis of the levels of the HSPs in CSCs may help clarify some of the current confusion in the interpretation of clinical data (39). Several HSPs protect cells from apoptosis after DNA damage and may not function properly after IR+HT treatment. We were surprised that in the ALDH+ subpopulation at 72 hours after IR+HT treatment, HSP40, HSP60, HSP70, and HSP90α showed a significant decrease compared to their response to IR treatment. Although most tumor cells experienced apoptosis after IR+HT treatment, the remaining cells appeared more differentiated. Interference with HSP function after radiation treatment may alter tumor cell fate, promoting differentiation instead of apoptosis. They may consequently be more sensitive to subsequent treatments, although this has not been formally tested.
HSP90 and the AKT pathway were both inhibited after treatment with IR+HT. The AKT pathway, which has been linked to HSP90, was previously shown to be activated in the Lin−CD29HCD24H subpopulation. Similar to the effects of HT, inhibition with the AKT inhibitor perifosine was able to radiosensitize the radiation-resistant Lin−CD29HCD24H subpopulation (40).
Although the pleiotropic mechanisms of heat-induced cell death with radiation remain to be elucidated, the inability to repair DNA damage and alterations in HSPs must play critical roles. Radiation and HT may be effective in part because this combination simultaneously affects multiple cellular components by altering protein function, influencing multiple survival pathways, and delaying the DDR. Tumor cell death may also be enhanced in vivo by hyperthermic nanoparticle effects on the tumor microenvironment, leading to the disruption of the tumor CSC niche. The preclinical data presented here suggest that mid-range HT may be effective to sensitize resistant CSCs to radiation treatment, even in highly resistant triple-negative breast cancer. We propose that these effects may contribute to the observed clinical improvements seen in contemporary HT trials.
Materials and Methods
Antibodies
R-phycoerythrin (R-PE)–conjugated rat antibody to mouse CD44, fluorescein isothiocyanate (FITC)– and R-PE–conjugated rat antibody to mouse CD24, biotin-conjugated mouse lineage panel (anti-CD3e, CD11b, CD45R/B220, Ly-6G/C, and TER-119), biotin-conjugated rat antibody to mouse CD31, streptavidin-allophycocyanin conjugate antibody, and their corresponding isotype controls [FITC-conjugated rat immunoglobulin G2a (IgG2a), R-PE–conjugated rat IgG2, and R-PE– H2K[d]] were all purchased from BD Biosciences. FITC antibody to mouse/rat CD29 and FITC Armenian hamster IgG isotype control were from BioLegend. Biotin-conjugated CD140a was purchased from eBioscience. Biotinylated goat secondary antibodies to rabbit or to rat lgG were obtained from Vector Laboratories Inc. Texas red– and Alexa Fluor 488–conjugated secondary antibodies for immunofluorescence were from Molecular Probes. H2A.X Phosphorylation Assay kit was purchased from Millipore. Rabbit polyclonal antibody to 53BP1 was purchased from Novus Biologicals. The ALDEFLUOR assay was performed with an ALDEFLUOR kit from StemCell Technologies, according to the manufacturer's protocols.
Preparation of single mammary tumor cells
Mice were maintained in accordance with the National Institutes of Health Guide for the Care and Use of Experimental Animals with approval from the Baylor College of Medicine Institutional Animal Care and Use Committee. The p53-null mammary tumors were generated as described (41). Single mammary tumor cells were prepared as described (17) with the following modifications: Briefly, after tumors were removed, they were minced with Vibratome McIIwain Tissue Chopper and digested in 10 ml of digestion medium per 1 g of tissue [digestion buffer containing Dulbecco's modified Eagle's medium (DMEM)/F12, gentamicin (100 mg/ml), antibiotic-antimycotic (Invitrogen), and collagenase type III (225 U/ml; Worthington Biochemical)] at 37°C. Samples were disrupted by pipetting every 15 min for 1 to 2 hours while being shaken on a rotary shaker at 125 rpm. If cells were to be used for in vitro studies, they were washed with Hanks' balanced salt solution before culturing.
Flow cytometry
Mouse cells were labeled with antibodies at a concentration of 10 million cells/ml under optimized conditions (1:200 for CD29-FITC, and 1:100 for CD24-PE), and dead cells were excluded with Sytox Red (Invitrogen). Human cells were stained with the ALDEFLUOR assay (performed with an ALDEFLUOR kit, 1:100 for H2K[d]-PE); dead cells were isolated with propidium iodide (PI). All cells were subjected to FACS analysis and sorting on BD FACSAria II cell-sorting system. The H2A.X Phosphorylation Assay kit was modified with herring sperm DNA with the antibodies, with an overnight incubation at 4°C. Data analysis was performed on BD FACSDiva software version 6.1.3.
Transplantation into the cleared fat pad tumor study
Clearance of mammary fat pads and transplantation procedures were performed as described previously (42). Cells from T1 and T7 (primary tumors) were freshly digested 48 hours after treatment. Cells were washed with 1× phosphate-buffered saline (PBS) and transplanted into the cleared fat pad of 3-week-old BALB/c female mice with a decreasing number of cells assayed as shown. Mice were monitored until tumors were observed, or up to 6 months if no tumors were detected.
Mammosphere assays
Mammosphere assays were performed as described (17) with the following modifications: Briefly, dissociated single cells digested from mammospheres were irradiated (0, 2, 4, or 6 Gy), and half were then immediately heated to 42°C and grown in six-well ultralow-attachment plates (Corning) with 2 ml of serum-free mammosphere medium. Mouse cell media were composed of DMEM/F12 with basic fibroblast growth factor (20 ng/ml), epidermal growth factor (20 ng/ml), B27, gentamicin (100 mg/ml), and antibiotic-antimycotic (all from Invitrogen). The human tissue culture media was MEGM (Lonza Walkersville Inc.) instead of DMEM/F12. Medium (2 ml) was added 3 days after culturing and counted on day 5. After counting mammospheres, mock-treated cells were passaged with 0.05% trypsin/0.53 mM EDTA-4Na. Dissociated cells (2000 per well) were replated in 2 ml of mammosphere medium for up to 10 passages thereafter. Mammospheres were counted with a Leica dissecting scope and analyzed with Image-Pro Plus software (Media Cybernetics Inc.).
Viability assay
Single dissociated cells were treated with 6 Gy, 6 Gy + 42°C for 1 hour, or mock treatment. The cells were plated in mammosphere media in 24-well low-attachment plates. Single wells were digested according to a previous protocol and counted at 0, 0.5, 24, 48, 72, and 96 hours after treatment. Cells were combined with 10 ml of trypan blue and counted with Invitrogen Countess. Viability, cell concentration, and size were recorded for each sample.
EdU staining and analysis
Sorted H2K[d] negative cells from in vivo treatment in xenografts were plated in mammosphere conditions with 5% fetal bovine serum (SAFC Bioscience) and allowed to recover for 48 hours. After 48 hours, proliferation was determined with the Click-iT EdU Flow Cytometry Assay kit (Invitrogen) with PI incorporation and analyzed on an Aria II. Staining and secondary antibody incubation were performed according to the suggested manufacturer's protocol and reagents.
Localized HT with gold nanoshells
The protocol for gold nanoshell injection has been described (26), with the modifications listed below. Nanospectra Biosciences Inc. provided all of the gold nanoshells for these experiments. All mice were injected with gold nanoshells 24 hours before treatment. Mice were treated with IR, IR+HT ± 1°C, HT ± 1°C, or mock treatment. Whole-body temperature was monitored with an axillary probe. Tumors were removed 48 or 72 hours after treatment as described above. A fraction of the tumor was fixed in 4% paraformaldehyde for paraffin embedding and digested for FACS analysis or transplantation as described above. Six independent tumors from each treatment were included for each tumor type.
Histological analysis
Hematoxylin and eosin staining was performed with standard protocols and analyzed by a pathologist (A.C.) specializing in breast cancer.
Microscopy analysis
Microscopic analysis was done with ×40 or ×63 magnification with 1 × 1 bin at 1024 resolution on a DeltaVision fluorescence microscope (Applied Precision) at 0.1 or 0.2. In addition, microscopic analysis was performed with an Olympus BX50 fluorescence microscope (Applied Precision) at ×20 magnification with 2 × 2 bin for quantitation of various samples. The images were processed with Spot Imaging software and Adobe Photoshop. At least 200 cells in a minimum of four fields were recorded for each tumor at each time repeated in both T1 and T7 tumors and BCM-2665A and MC1 xenografts.
HSP multiplex assay
Protein was isolated according to HSP/Chaperone 8-Plex MultiBead kit protocol (Assay Designs Inc.) with the modifications listed below. Cells were sorted and analyzed at the time of FACS analysis after treatment. Cells were washed in PBS and protein was isolated in radioimmunoprecipitation assay solution with Protease inhibitor cocktail and Phosphatase inhibitor cocktail (Sigma-Aldrich). The remaining procedures were according to MultiBead kit protocol. Standard curves were set up for AKT, HSP27 (phospho-Ser82), HSP27 (phospho-Ser15), HSP40, HSP60, HSP70, HSP90α, and pAKT (phospho-Ser473), with fit >95%. Protein concentrations were determined with MultiBead Analysis software (Assay Designs).
Statistical analysis
Descriptive statistical analysis was generated for all repeated quantitative data, with inclusion of means and SE, with SigmaPlot (Systat Software Inc.) and GraphPad Prism version 4.00 for Macintosh (GraphPad Software). The significance values of data comparing two data points were assessed with Student's t tests. The significance values of data sets with more than two data points were calculated with a one-way analysis of variance (ANOVA) model followed by Bonferroni pairwise comparisons between groups. Data are represented in graphs as means ± SEM. Statistical analyses for limiting dilution were performed in the core bio-statistical facility of the Dan L. Duncan Cancer Center at Baylor College of Medicine (S.G.H.). Limiting dilution transplantation data were analyzed with a generalized linear model with a complementary log-log transformation after Hu and Smyth (43). Preliminary analysis found no evidence of interaction between tumors (T1 versus T7) and treatment (mock versus IR and IR+HT), and the final model included terms for main effects of log dose of cells (continuous), tumor, and treatment. Overall fit for the single-hit Poisson model was verified by testing whether the regression coefficient for log(dose) is different from 0.0 (P < 0.0001, likelihood ratio test), and not different from 1.0 (P = 0.74, likelihood ratio test).
Supplementary Material
Fig. S1. Sample temperature profile.
Fig. S2. Hyperthermia alone does not change tumor volume or Lin−CD29HCD24H subpopulation.
Fig. S3. Thermal enhancement with gold nanoshells in combination with radiation prevented relative increase in Lin−CD29HCD24H seen with radiation.
Fig. S4. Mammospheres enrich in ALDH+ cells.
Fig. S5. Thermal enhancement with gold nanoshells in combination with radiation prevented relative increase in ALDH+ seen with radiation.
Fig. S6. Altered DNA damage repair in non-CD29HCD24H subpopulations.
Fig. S7. Altered DNA damage repair in ALDH− cells.
Fig. S8. HSPs and pAKT were alternatively activated in total tumor subpopulations compared to ALDH+ subpopulations.
Acknowledgments
We thank the Cytometry and Cell Sorting Core Facility at Baylor College of Medicine for flow cytometry assistance, core funding from the NIH (National Center for Research Resources grant S10RR024574, National Institute of Allergy and Infectious Diseases grant AI036211, and National Cancer Institute grant P30CA125123), and also the Baylor Integrated Microscopy Core for help with deconvolution microscopy. We thank Nanospectra Biosciences Inc. for providing the gold nanoshells for these experiments. We thank M. Wicha from the University of Michigan for providing the MC1 xenograft line. We also thank H. LaMarca and S. Greene for constructive criticisms on this manuscript.
Funding: These studies were supported by grants from Diana Helis Henry Medical Research Foundation and National Cancer Institute grants R01 CA112305 (J.C.C.), R37 CA16303 (J.M.R.), and P30 CA125123. W.A.W. was supported by NIH grant R01CA138239-01, the State of Texas Grant for Rare and Aggressive Cancers, the University of Texas Health Sciences Center grant KL2RR024149, and the American Airlines Komen Foundation Promise grant KGO81287.
Footnotes
Author contributions: R.L.A., W.A.W., S.K., J.C.C., and J.M.R. designed the research; R.L.A., M.Z., and P.D. performed the research; R.L.A., M.Z., S.P., A.C., S.G.H., W.A.W., J.C.C., and J.M.R. analyzed the data; and R.L.A., J.C.C., and J.M.R. wrote the paper
Competing interests: The authors declare that they have no competing interests.
Accession numbers: The clinical trial mentioned in the Discussion can be found at ClinicalTrials.gov identifier NCT00848042.
References
- 1.Li Y, Rosen JM. Stem/progenitor cells in mouse mammary gland development and breast cancer. J Mammary Gland Biol Neoplasia. 2005;10:17–24. doi: 10.1007/s10911-005-2537-2. [DOI] [PubMed] [Google Scholar]
- 2.Rosen JM, Jordan CT. The increasing complexity of the cancer stem cell paradigm. Science. 2009;324:1670–1673. doi: 10.1126/science.1171837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dewhirst MW, Vujaskovic Z, Jones E, Thrall D. Re-setting the biologic rationale for thermal therapy. Int J Hyperthermia. 2005;21:779–790. doi: 10.1080/02656730500271668. [DOI] [PubMed] [Google Scholar]
- 4.Horsman MR, Overgaard J. Hyperthermia: A potent enhancer of radiotherapy. Clin Oncol (R Coll Radiol) 2007;19:418–426. doi: 10.1016/j.clon.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 5.Roti Roti JL. Cellular responses to hyperthermia (40–46°C): Cell killing and molecular events. Int J Hyperthermia. 2008;24:3–15. doi: 10.1080/02656730701769841. [DOI] [PubMed] [Google Scholar]
- 6.Jones EL, Marks LB, Prosnitz LR. Point: Hyperthermia with radiation for chest wall recurrences. J Natl Compr Canc Netw. 2007;5:339–344. doi: 10.6004/jnccn.2007.0029. [DOI] [PubMed] [Google Scholar]
- 7.Franckena M, Lutgens LC, Koper PC, Kleynen CE, van der Steen-Banasik EM, Jobsen JJ, Leer JW, Creutzberg CL, Dielwart MF, van Norden Y, Canters RA, van Rhoon GC, van der Zee J. Radiotherapy and hyperthermia for treatment of primary locally advanced cervix cancer: Results in 378 patients. Int J Radiat Oncol Biol Phys. 2009;73:242–250. doi: 10.1016/j.ijrobp.2008.03.072. [DOI] [PubMed] [Google Scholar]
- 8.Gabriele P, Ferrara T, Baiotto B, Garibaldi E, Marini PG, Penduzzu G, Giovannini V, Bardati F, Guiot C. Radio hyperthermia for re-treatment of superficial tumours. Int J Hyperthermia. 2009;25:189–198. doi: 10.1080/02656730802669593. [DOI] [PubMed] [Google Scholar]
- 9.Jones EL, Oleson JR, Prosnitz LR, Samulski TV, Vujaskovic Z, Yu D, Sanders LL, Dewhirst MW. Randomized trial of hyperthermia and radiation for superficial tumors. J Clin Oncol. 2005;23:3079–3085. doi: 10.1200/JCO.2005.05.520. [DOI] [PubMed] [Google Scholar]
- 10.Vernon CC, Hand JW, Field SB, Machin D, Whaley JB, van der Zee J, van Putten WL, van Rhoon GC, van Dijk JD, González González D, Liu FF, Goodman P, Sherar M. Radiotherapy with or without hyperthermia in the treatment of superficial localized breast cancer: Results from five randomized controlled trials. International Collaborative Hyperthermia Group. Int J Radiat Oncol Biol Phys. 1996;35:731–744. doi: 10.1016/0360-3016(96)00154-x. [DOI] [PubMed] [Google Scholar]
- 11.Cherukuri P, Glazer ES, Curley SA. Targeted hyperthermia using metal nanoparticles. Adv Drug Deliv Rev. 2010;62:339–345. doi: 10.1016/j.addr.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O'Neal DP, Hirsch LR, Halas NJ, Payne JD, West JL. Photo-thermal tumor ablation in mice using near infrared-absorbing nanoparticles. Cancer Lett. 2004;209:171–176. doi: 10.1016/j.canlet.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 13.Weissleder R. A clearer vision for in vivo imaging. Nat Biotechnol. 2001;19:316–317. doi: 10.1038/86684. [DOI] [PubMed] [Google Scholar]
- 14.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: The key role of tumor-selective macromolecular drug targeting. Adv Enzyme Regul. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 15.Maeda H, Fang J, Inutsuka T, Kitamoto Y. Vascular permeability enhancement in solid tumor: Various factors, mechanisms involved and its implications. Int Immunopharmacol. 2003;3:319–328. doi: 10.1016/S1567-5769(02)00271-0. [DOI] [PubMed] [Google Scholar]
- 16.Aldaz CM, Hu Y, Daniel R, Gaddis S, Kittrell F, Medina D. Serial analysis of gene expression in normal p53 null mammary epithelium. Oncogene. 2002;21:6366–6376. doi: 10.1038/sj.onc.1205816. [DOI] [PubMed] [Google Scholar]
- 17.Zhang M, Behbod F, Atkinson RL, Landis MD, Kittrell F, Edwards D, Medina D, Tsimelzon A, Hilsenbeck S, Green JE, Michalowska AM, Rosen JM. Identification of tumor-initiating cells in a p53-null mouse model of breast cancer. Cancer Res. 2008;68:4674–4682. doi: 10.1158/0008-5472.CAN-07-6353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin Cancer Res. 2007;13:4429–4434. doi: 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- 19.Onitilo AA, Engel JM, Greenlee RT, Mukesh BN. Breast cancer subtypes based on ER/PR and Her2 expression: Comparison of clinicopathologic features and survival. Clin Med Res. 2009;7:4–13. doi: 10.3121/cmr.2009.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parikh RR, Housman D, Yang Q, Toppmeyer D, Wilson LD, Haffty BG. Prognostic value of triple-negative phenotype at the time of locally recurrent, conservatively treated breast cancer. Int J Radiat Oncol Biol Phys. 2008;72:1056–1063. doi: 10.1016/j.ijrobp.2008.02.066. [DOI] [PubMed] [Google Scholar]
- 21.Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, Jacquemier J, Viens P, Kleer CG, Liu S, Schott A, Hayes D, Birnbaum D, Wicha MS, Dontu G. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chowdhury D, Xu X, Zhong X, Ahmed F, Zhong J, Liao J, Dykxhoorn DM, Weinstock DM, Pfeifer GP, Lieberman J. A PP4-phosphatase complex dephosphorylates g-H2AX generated during DNA replication. Mol Cell. 2008;31:33–46. doi: 10.1016/j.molcel.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lagadec C, Vlashi E, Della Donna L, Meng Y, Dekmezian C, Kim K, Pajonk F. Survival and self-renewing capacity of breast cancer initiating cells during fractionated radiation treatment. Breast Cancer Res. 2010;12:R13. doi: 10.1186/bcr2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, Hilsenbeck SG, Pavlick A, Zhang X, Chamness GC, Wong H, Rosen J, Chang JC. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst. 2008;100:672–679. doi: 10.1093/jnci/djn123. [DOI] [PubMed] [Google Scholar]
- 25.Woodward WA, Bristow RG. Radiosensitivity of cancer-initiating cells and normal stem cells (or what the Heisenberg uncertainly principle has to do with biology) Semin Radiat Oncol. 2009;19:87–95. doi: 10.1016/j.semradonc.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Diagaradjane P, Shetty A, Wang JC, Elliott AM, Schwartz J, Shentu S, Park HC, Deorukhkar A, Stafford RJ, Cho SH, Tunnell JW, Hazle JD, Krishnan S. Modulation of in vivo tumor radiation response via gold nanoshell-mediated vascular-focused hyperthermia: Characterizing an integrated antihypoxic and localized vascular disrupting targeting strategy. Nano Lett. 2008;8:1492–1500. doi: 10.1021/nl080496z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Charafe-Jauffret E, Ginestier C, Iovino F, Wicinski J, Cervera N, Finetti P, Hur MH, Diebel ME, Monville F, Dutcher J, Brown M, Viens P, Xerri L, Bertucci F, Stassi G, Dontu G, Birnbaum D, Wicha MS. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 2009;69:1302–1313. doi: 10.1158/0008-5472.CAN-08-2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foster ER, Downs JA. Histone H2A phosphorylation in DNA double-strand break repair. FEBS J. 2005;272:3231–3240. doi: 10.1111/j.1742-4658.2005.04741.x. [DOI] [PubMed] [Google Scholar]
- 29.Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]
- 30.Roti Roti JL. Heat-induced alterations of nuclear protein associations and their effects on DNA repair and replication. Int J Hyperthermia. 2007;23:3–15. doi: 10.1080/02656730601091759. [DOI] [PubMed] [Google Scholar]
- 31.Kindas-Mügge I, Herbacek I, Jantschitsch C, Micksche M, Trautinger F. Modification of growth and tumorigenicity in epidermal cell lines by DNA-mediated gene transfer of Mr 27,000 heat shock protein (hsp27) Cell Growth Differ. 1996;7:1167–1174. [PubMed] [Google Scholar]
- 32.Vargas-Roig LM, Fanelli MA, López LA, Gago FE, Tello O, Aznar JC, Ciocca DR. Heat shock proteins and cell proliferation in human breast cancer biopsy samples. Cancer Detect Prev. 1997;21:441–451. [PubMed] [Google Scholar]
- 33.Field SB. 1985 Douglas Lea memorial lecture. Hyperthermia in the treatment of cancer. Phys Med Biol. 1987;32:789–811. doi: 10.1088/0031-9155/32/7/001. [DOI] [PubMed] [Google Scholar]
- 34.Phillips TM, McBride WH, Pajonk F. The response of CD24-/low/CD44+ breast cancer– initiating cells to radiation. J Natl Cancer Inst. 2006;98:1777–1785. doi: 10.1093/jnci/djj495. [DOI] [PubMed] [Google Scholar]
- 35.Woodward WA, Chen MS, Behbod F, Alfaro MP, Buchholz TA, Rosen JM. WNT/β-catenin mediates radiation resistance of mouse mammary progenitor cells. Proc Natl Acad Sci USA. 2007;104:618–623. doi: 10.1073/pnas.0606599104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Laszlo A, Fleischer I. Heat-induced perturbations of DNA damage signaling pathways are modulated by molecular chaperones. Cancer Res. 2009;69:2042–2049. doi: 10.1158/0008-5472.CAN-08-1639. [DOI] [PubMed] [Google Scholar]
- 37.Calderwood SK, Ciocca DR. Heat shock proteins: Stress proteins with Janus-like properties in cancer. Int J Hyperthermia. 2008;24:31–39. doi: 10.1080/02656730701858305. [DOI] [PubMed] [Google Scholar]
- 38.Lanneau D, de Thonel A, Maurel S, Didelot C, Garrido C. Apoptosis versus cell differentiation: Role of heat shock proteins HSP90, HSP70 and HSP27. Prion. 2007;1:53–60. doi: 10.4161/pri.1.1.4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ciocca DR, Calderwood SK. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones. 2005;10:86–103. doi: 10.1379/CSC-99r.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang M, Atkinson RL, Rosen JM. Selective targeting of radiation-resistant tumor-initiating cells. Proc Natl Acad Sci USA. 2010;107:3522–3527. doi: 10.1073/pnas.0910179107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Medina D, Kittrell FS, Shepard A, Stephens LC, Jiang C, Lu J, Allred DC, McCarthy M, Ullrich RL. Biological and genetic properties of the p53 null preneoplastic mammary epithelium. FASEB J. 2002;16:881–883. doi: 10.1096/fj.01-0885fje. [DOI] [PubMed] [Google Scholar]
- 42.Welm AL, Kim S, Welm BE, Bishop JM. MET and MYC cooperate in mammary tumori-genesis. Proc Natl Acad Sci USA. 2005;102:4324–4329. doi: 10.1073/pnas.0500470102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu Y, Smyth GK. ELDA: Extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Sample temperature profile.
Fig. S2. Hyperthermia alone does not change tumor volume or Lin−CD29HCD24H subpopulation.
Fig. S3. Thermal enhancement with gold nanoshells in combination with radiation prevented relative increase in Lin−CD29HCD24H seen with radiation.
Fig. S4. Mammospheres enrich in ALDH+ cells.
Fig. S5. Thermal enhancement with gold nanoshells in combination with radiation prevented relative increase in ALDH+ seen with radiation.
Fig. S6. Altered DNA damage repair in non-CD29HCD24H subpopulations.
Fig. S7. Altered DNA damage repair in ALDH− cells.
Fig. S8. HSPs and pAKT were alternatively activated in total tumor subpopulations compared to ALDH+ subpopulations.