

Abstract
During the past two decades the first sequencing of the human genome was performed showing its high degree of inter-individual differentiation, as a result of large international research projects (Human Genome Project, the 1000 Genomes Project International HapMap Project, and Programs for Genomic Applications NHLBI-PGA). This period was also a time of intensive development of molecular biology techniques and enormous knowledge growth in the biology of cancer. For clinical use in the treatment of patients with colorectal cancer (CRC), in addition to fluoropyrimidines, another two new cytostatic drugs were allowed: irinotecan and oxaliplatin. Intensive research into new treatment regimens and a new generation of drugs used in targeted therapy has also been conducted. The last 20 years was a time of numerous in vitro and in vivo studies on the molecular basis of drug resistance. One of the most important factors limiting the effectiveness of chemotherapy is the primary and secondary resistance of cancer cells. Understanding the genetic factors and mechanisms that contribute to the lack of or low sensitivity of tumour tissue to cytostatics is a key element in the currently developing trend of personalized medicine. Scientists hope to increase the percentage of positive treatment response in CRC patients due to practical applications of pharmacogenetics/pharmacogenomics. Over the past 20 years the clinical usability of different predictive markers has been tested among which only a few have been confirmed to have high application potential. This review is a synthetic presentation of drug resistance in the context of CRC patient chemotherapy. The multifactorial nature and volume of the issues involved do not allow the author to present a comprehensive study on this subject in one review.
Keywords: Pharmacogenetics, Pharmacogenomics, Drug resistance, Colorectal cancer, Chemoresistance, Individualized medicine
Core tip: Insufficient effectiveness of chemotherapy is still the most important factor limiting the successful treatment of patients with colorectal cancer (CRC). Drug resistance in anticancer therapy has been recognized virtually from the very beginning, as cytostatic drugs were first used in oncology practice. Intensive research on the causes of low sensitivity in colorectal cancer cells to such drugs as fluoropyrimidines, irinotecan and oxaliplatin, has resulted in evidence on the importance of genetic factors in phenotype conditioning of drug resistance. This review is a synthetic presentation of drug resistance in the context of its role in chemotherapy, and the potential clinical use of different biomarkers in individualization of CRC patient treatment.
RESEARCH ON THE EFFECTIVENESS OF CYTOTOXIC ANTINEOPLASTIC DRUGS FOR THE TREATMENT OF COLORECTAL CANCER
Since the beginning of the 21st century, very rapid development of high-throughput research techniques described by the term ‘‘omics’’ (genomics, transcriptomics, proteomics and metabolomics) has been observed. Pharmacogenomics uses advanced research techniques “omics”, which allow researchers to identify the genetic basis of inter-individual differences in the pharmacodynamics and pharmacokinetics of drugs[1,2]. An important objective of this research is to identify biomarkers for predicting treatment outcomes, as well as avoiding the toxic effects arising during the course of pharmacotherapy (prognostic and predictive markers)[3]. The terms pharmacogenetics and pharmacogenomics are closely related and are often used interchangeably, although there are some historical differences between them. Today, pharmacogenomics is commonly used synonymously with “individualized” or “personalized” medicine, although the latter term is often understood to stratify medical treatment by the use of genomic biomarkers rather than to treat an individual. Accordingly, the Personalized Medicine Coalition defined personalized medicine as “the application of genomic and molecular data to better target the delivery of health care, facilitate the discovery and clinical testing of new products, and help determine a person’s predisposition to a particular disease or condition”[4,5].
Environmental factors such as age, sex or health condition of the patient are the classic factors which affect treatment outcomes and have been studied for decades. The influence of genetic factors on response variability is far greater than sex, age, or interactions with other drugs. Therefore, it seems advisable to determine the basis of all abnormal body reactions in relation to the treatment used. It should also be noted that the distribution frequency of correct responses to drug usage in a population is far from a normal distribution, which means that the presence of treatment non-responders and over-responders (increased toxicity) is much more common than has been assumed so far[6]. The first studies on pharmacogenomics and colorectal cancer (CRC) outcome were conducted and published approximately 20 years ago[7]. Since then, hundreds of possible biodeterminants have been studied with many expectations. The technology, and its spread, has improved incredibly, and the importance with which this subject is regarded by many research groups throughout the world has grown relentlessly. The reproducibility of some results was, initially, promising, as were some confirmatory clues derived from deeper biological studies, but the final step of clinical validation has remained an unmet objective for almost all putative biomarkers[8].
Treatment options in CRC have systematically advanced over the last several years with the introduction of effective chemotherapeutic and targeted drugs. However, providing individual treatment with low toxicity and significant benefit is still an unsolved problem[9]. This part of the review focuses on pharmacogenomic knowledge of substances routinely administered in patients with CRC: fluoropyrimidines, irinotecan (CPT-11), and oxaliplatin (OX).
5-FLUOROURACIL AND FLUOROPYRIMIDINES
In 1957 Heidelberger et al[10] reported the antitumour activity of 5-fluorouracil (5-FU). Charles Heidelberger synthesized 5-FU as a result of experiments which showed the ability of tumour cells to acquire uracil for DNA synthesis[11]. Fifty years after the first synthesis of 5-FU it is still a standard component of adjuvant and palliative therapy having a proven impact on survival time in patients with CRC[12]. Experimental studies have shown that 5-FU is converted to an active metabolite, FdUMP (fluorodeoxyuridine monophosphate), which is a potent inhibitor of DNA synthesis (Figure 1). FdUMP forms a ternary complex together with thymidylate synthase enzyme (TS) and 5,10-methylenetetrahydrofolate (CH2THF) cofactor, responsible for the catalytic conversion of deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP). dTMP is a substrate for deoxythymidine triphosphate (dTTP) necessary for the process of DNA synthesis (Figure 2). Furthermore, on the basis of fundamental and clinical research it has been proven that the addition to an exogenous therapy a source of folic acid, such as leucovorin (LV) increases the degree of inhibition of TS supporting the formation of active complexes of 5-FU with the enzyme[13]. 5-FU/LV combination therapy in patients with diagnosed CRC is much more effective than monotherapy with 5-FU[14].
Figure 1.
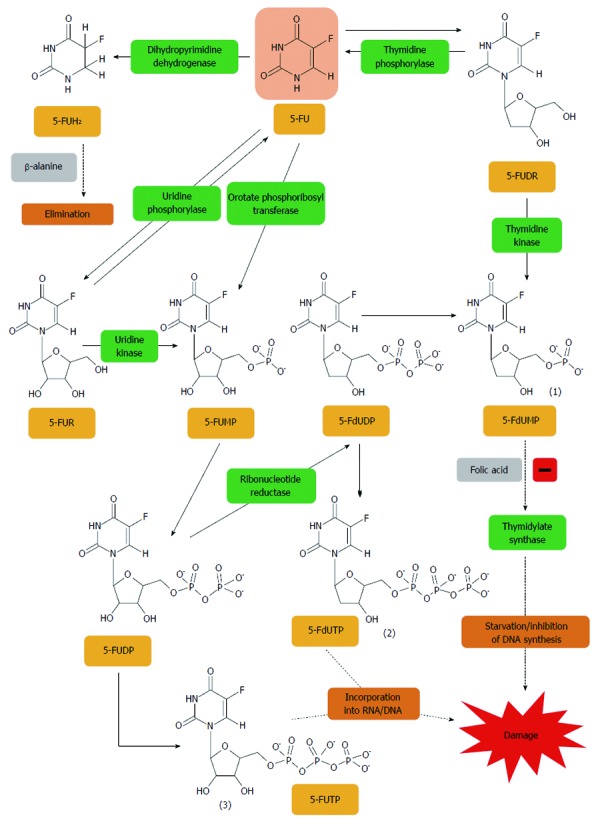
5-fluorouracil is converted to three major active metabolites. (1) fluorodeoxyuridine monophosphate (FdUMP); (2) fluorodeoxyuridine triphosphate (FdUTP); and (3) fluorouridine triphosphate (FUTP). The main mechanism of 5-fluorouracil (5-FU) activation is conversion to fluorouridine monophosphate (FUMP) either directly by orotate phosphoribosyl transferase (OPRT), or indirectly via fluorouridine (FUR) through the sequential action of uridine phosphorylase and uridine kinase. FUMP is then phosphorylated to fluorouridine diphosphate (FUDP), which can be either further phosphorylated to the active metabolite fluorouridine triphosphate (FUTP), or converted to fluorodeoxyuridine diphosphate (FdUDP) by ribonucleotide reductase. In turn, FdUDP can either be phosphorylated or dephosphorylated to generate the active metabolites FdUTP and FdUMP, respectively. An alternative activation pathway involves the thymidine phosphorylase catalyzed conversion of 5-FU to 5-fluoro-2’-deoxyuridine (5-FUDR), which is then phosphorylated by thymidine kinase to the thymidylate synthase inhibitor, FdUMP. Dihydropyrimidine dehydrogenase (DPD)-mediated conversion of 5-FU to dihydrofluorouracil (DHFU) is the rate-limiting step of 5-FU catabolism in normal and tumour cells[401].
Figure 2.
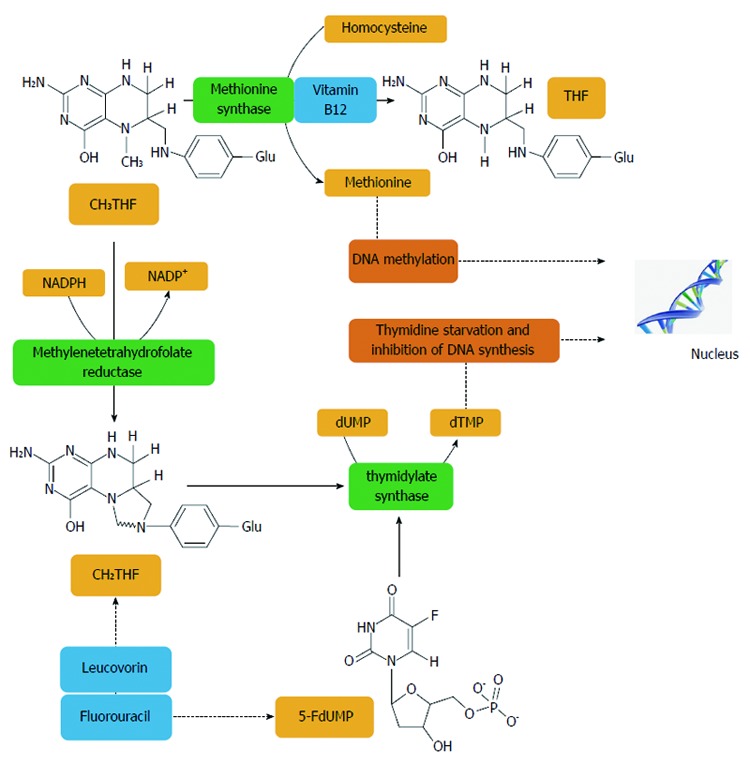
Methylentetrahydrofolate reductase plays an important role in the action of 5-fluorouracil, an inhibitor of thymidylate synthase. Methylentetrahydrofolate reductase (MTHFR) catalyses a unidirectional reaction that lowers the levels of 5,10-methylenetetrahydrofolate (CH2THF) by increasing levels of 5-methyltetrahydrofolate (CH3THF) which is used for biological methylation. Other factors, such as vitamin B12 and homocysteine, are involved in biological methylation processes. The addition of folinic acid (leucovorin) to 5-FU improves the response rates and survival of CRC patients. Thymidylate synthase (TS) catalyses the reductive methylation of deoxyuridine monophosphate (dUMP) to deoxythymidine monophosphate (dTMP) with the reduced folate, CH2THF, as the methyl donor. This reaction provides the sole de novo source of thymidylate, which is necessary for DNA replication and repair. TS contains a nucleotide-binding site and a binding site for CH2THF. The 5-FU metabolite, FdUMP, binds to the nucleotide-binding site of TS, forming a stable ternary complex with the enzyme and CH2THF which blocks binding of the normal substrate dUMP, thereby inhibiting dTMP synthesis. Inhibition of thymidylate synthesis causes disruption of nucleotide levels that results in DNA damage[402].
The purpose of individualized therapy is to choose the most effective treatment and the optimal dosage for each patient, while minimizing toxicity and side effects of the therapy. This objective is particularly important in the case of the new generation of anticancer drugs which include expensive targeted therapies such as the antibodies cetuximab and bevacizumab. The much cheaper 5-FU therapy can also be individualized in a selection of CRC patients with potentially best response to the administration of 5-FU which appears to be justified medically and financially. Despite significant progress in understanding the 5-FU activity mechanisms, the identification of molecular markers potentially clinically useful in predicting 5-FU treatment efficacy is still the subject of research.
TS
TS is an important enzyme involved in the metabolism of folic acid and catalyzes dUMP methylation to dTMP, which is a critical reaction in maintaining the balance of available deoxynucleotides (dNTPs) in cells, substrates necessary for the synthesis and repair of DNA. The interaction with TS is the main aim of such cytostatic drugs as 5-FU, and the level of TYMS gene expression and TS protein is a prognostic marker in the treatment of several types of cancer. Thus, the 5-FU cell sensitivity profile may be affected by genetic variants of the TYMS gene, expression level of TYMS/TS gene/ -protein, and intracellular concentration of dNTP and CH2THF[15]. Expression of TS as a sensitivity determinant for fluoropyrimidines has been shown in vitro[16] as well as in vivo, where intratumour TS expression level was associated with the chemosensitivity of tumour tissue exposed to 5-FU. The most important data collected during the past few years indicate that TS expression varies considerably between different types of cancers and that the degree of tumour response to 5-FU treatment is inversely proportional to the measured level of intratumour mRNA and protein expression[17]. Leichman et al[18] first proved that there is an inverse relationship between intratumoural TYMS gene expression and the degree of response to 5-FU treatment. CRC patients with low levels of TYMS gene expression had a significantly higher rate of response to therapy and longer median survival compared to patients with higher TYMS expression in tumour tissue (13.6 mo vs 8.2 mo, P = 0.02)[19]. A meta-analysis of 13 clinical trials of patients with advanced CRC (total number of patients: 887 cases) carried out by Popat et al[20] showed that patients with low TS expression had longer overall survival (OS) than patients with higher TS expression in tumour tissue. Recently, a meta-analysis including 24 clinical trials with more than 1100 CRC patients was also published[21]. The pooled relative risk of overall response rate (ORR) indicated that the group with lower TS expression had greater sensitivity to fluoropyrimidine-based chemotherapy than patients with high TS expression level[21]. Numerous studies were also carried out to investigate different TS expression levels in tissue derived from primary tumours and metastases[22,23]. Analysis of the two subgroups it was demonstrated that predictive TS expression levels determined in tissue derived from metastases were more pronounced than those determined in primary tumours[21]. Furthermore, during the assessment of the predictive values of TS expression level, the results obtained using RT-PCR techniques were statistically more significant than those in which the expression was determined using immunohistochemistry (IHC) techniques[21].
These results indicated that low TS expression in CRC patients with advanced tumours was associated with increased individual sensitivity to 5-FU therapy[7,17,19,24-39]. Furthermore, in vitro studies using cell lines and tumour tissues demonstrated that 5-FU therapy contributes to the induction of TS expression[40,41]. This increase in TS expression upon 5-FU exposure seems to be a result of a negative feedback loop in which ligand-free TS binds to its own mRNA and inhibits its own translation[42]. When stably bound by FdUMP, TS can no longer bind its own mRNA and suppress translation, resulting in increased protein expression. This constitutes a potentially important resistance mechanism, as acute increases in TS would facilitate recovery of enzyme activity[41].
Although, the reason for ontogenetic variation in TS expression is still not clear, one of the main examined hypotheses is the possible influence of TYMS gene polymorphisms on TS expression. As it is now known, some of the described polymorphisms affect inter-individual differences in patient sensitivity to 5-FU treatment (Figure 3 and Table 1)[43-52]. Polymorphism of the variable number of tandem repeats (VNTR) located in the TYMS gene sequence is one of the studied genetic variants that may have clinical relevance as a predictive marker for the effectiveness of 5-FU treatment. Horie et al[53] reported a 28-nucleotide sequence in the 5’-region of the TYMS gene, which occurs in the population with a variable number of iterations: two (2R) or three (3R). According to the classification proposed by Kawakami and Watanabe, it is assumed that VNTR in this region is responsible for the occurrence of two alleles, 2R and 3R, and three different genotypes (2R/2R, 2R/3R and 3R/3R)[54]. The results of various studies suggest that the 3R allele is responsible for four times higher mRNA level of the TYMS gene observed in tissue tumours obtained from patients with metastatic CRC compared to patients who were carriers of the 2R variant (P < 0.004)[55]. Homozygous patients having both alleles with a double repeat (2R/2R) showed a significantly higher percentage of favourable response to 5-FU treatment as compared to those who had the 3R/3R genotype (50% vs 9%, P = 0.04)[55]. In addition to the predictive values for 5-FU chemotherapy, retrospective studies have demonstrated that this polymorphism also has the properties of a toxicity marker for fluoropyrimidine-based chemotherapy. Patients who are carriers of the 3R/3R genotype exhibited reduced toxicity as compared to patients with the 2R variant. A high TS expression level related to the presence of 3R/3R genotype accounts for less effective inhibition of TS, which contributes to both an increased likelihood of survival of cancer cells (drug resistance), and a reduced loss of healthy cells and less toxic therapy[55]. Moreover, a single nucleotide polymorphism (SNP) of guanine instead of cytosine (G/C) in 3R determines two different alleles (3C or 3G)[55]. Based on the presence of this polymorphism two different groups of patients can be distinguished with two levels of TS expression: a high expression group with (2R/3G, 3C/3G and 3G/3G genotype carriers) and a low expression group (2R/2R, 2R/3C and 3C/3C genotypes). Taking into account the study results published by Mandola et al[56], it is believed that the presence of the 28-bp G>C SNP within the second repeat of the 3R allele TYMS promoter enhancer region (TSER) is associated with a weaker bond in the promoter region of USF-1 transcription factor leading to a decreased transcriptional activity of TYMS gene. A lower transcription rate of the TSER 3RC allele in vitro is also observed when compared with TSER 3RG, comparable with the TSER 2R/2R genotype[56,57]. These results may, at least partly, explain why some patients with 3R/3R genotype have low TS expression and a good response to 5-FU chemotherapy.
Figure 3.
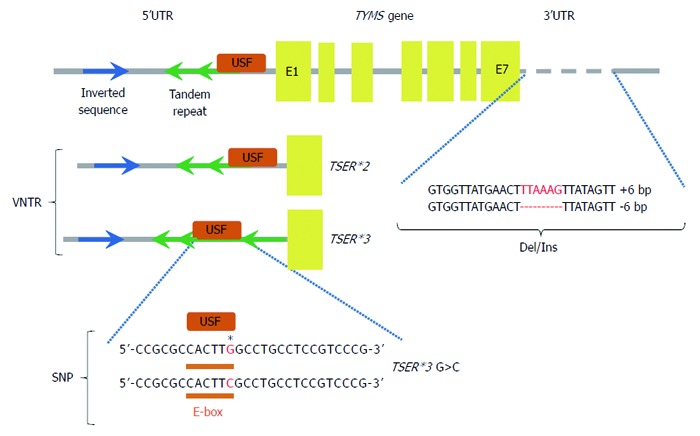
Some of the described polymorphisms affect inter-individual differences in patient sensitivity to 5-fluorouracil treatment. Polymorphisms in the thymidylate synthase gene (TYMS gene), 5’ and 3’ untranslated regions (5’UTR and 3’UTR), exons (E1-E7), binding site for upstream stimulating factor (USF), variable number tandem repeats (VNTR), single nucleotide polymorphism (SNP), deletion/insertion polymorphism (Del/Ins), two-tandem repeats (TSER*2), three-tandem repeats (TSER*3), TSER*3 G>C (single nucleotide polymorphism of TSER*3). Regulation of TYMS gene expression. TSER polymorphism (TS 2R/3R repeat) is a tandem repeat upstream of the TYMS translational start site containing either double (2R) or triple (3R) repeats of 28-bp sequences. These tandem repeats regulate transcription and translation of TYMS. Additional functional variants of the TYMS gene have been identified and TSER 2R/3R repeat is now studied together with a G to C SNP within the second repeat of the 3R allele. TSER 3RC/3RC genotype causes lower transcriptional activity of TYMS, comparable with the TS 2R/2R genotype. TS 1494del6bp is another functional variant of the TYMS gene and has been shown to decrease RNA stability and therefore influence TS mRNA and TS protein expression in vitro[52].
Table 1.
Some common polymorphisms of genes TYMS, MTHFR, DPYD and UMPS and their potential impact on the functioning of proteins associated with the pharmacology of 5-fluorouracil
| dbSNP rs cluster ID | Type of polymorphism | Function | Ref. |
| Thymidylate synthase (TYMS) (OMIM # 188350) | |||
| rs45445694 | VNTR | [43-51,68,409-413] | |
| TSER*2/ TSER*3 | TSER polymorphism (TS 2R/3R repeat) is a tandem repeat upstream of the TYMS translational start site containing either double (2R) or triple (3R) repeats of 28-bp sequences | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 45445694 | |||
| rs34743033 | SNP | [44-46,49,50,414] | |
| TSER*3G>C | TSER*2/*3 repeat is studied together with a G to C SNP within the second repeat of the TSER*3 allele | ||
| TSER*3C allele = decrease transcriptional activity of TYMS | |||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 34743033 | |||
| rs151264360 | Del/Ins | [44,46,49,51,72,415] | |
| TS 1494del6bp | -6-bp deletion, decreased stability of TS mRNA | ||
| +6-bp insertion, increased stability of TS mRNA | |||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 151264360 | |||
| Methylenetetrahydrofolate reductase (MTHFR) (OMIM # 607093) | |||
| rs1801133 | SNP | [66-69,72,313,316,362] | |
| 677C>T | At codon 222 in exon 4 (Ala → Val) | ||
| Reduces enzymatic activity and increased thermolability | |||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 1801133 | |||
| rs1801131 | SNP | [67-69,72,313,316] | |
| 1298A>C | At codon 429 in exon 7 (Glu → Ala) | ||
| Reduces MTHFR activity | |||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 1801131 | |||
| rs4846051a | SNPs | [71,416] | |
| 1305T>C | At codon 435 (synonymous), effect unknown | ||
| rs201095365b | 1798G>A | At codon 600 (Glu → Lys), effect unknown | |
| ahttp://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 4846051 | |||
| bhttp://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 201095365 | |||
| Dihydropyrimidine dehydrogenase (DPYD) (OMIM # 612779) | |||
| rs3918290 | SNP | [88,412,417,418] | |
| IVS14+1G>A | Exon 14 is skipped as a result of the G → A translocation at intron 14, inactive enzyme is formed | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 3918290 | |||
| rs75017182 | SNP | [92,419] | |
| c.1129– 5923C>G | Cryptic splice donor site leads to a 44 bp fragment of intron 10 insert in mrna, frameshift and premature stop codon in exon 11 | ||
| Associated with toxicity | |||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 75017182 | |||
| - | SNPs | [92,417] | |
| IVS5+18G>A | G → A translocation at intron 5, effect unknown | ||
| IVS6+139G>A | G → A translocation at intron 6, effect unknown | ||
| IVS9–51T>G | T → G translocation at intron 9, effect unknown | ||
| rs1801265 | SNP | [85,420-424] | |
| 85T>C | At codon 29 in exon 2 (Cys → Arg) | ||
| Decreased expression | |||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 1801265 | |||
| rs2297595 | SNP | [420,421,424-427] | |
| 496A>G | At codon 166 in exon 6 (Met → Val) | ||
| Significantly conserved site close to the Fe-S motif, may disrupt electron transport | |||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 2297595 | |||
| rs1801159 | SNP | [421,424,427-430] | |
| 1627A>G | At codon 543 in exon 13 (Ile → Val) | ||
| Decreased expression | |||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 1801159 | |||
| rs55886062 | SNP | [92,422,431-434] | |
| 1679T>G | At codon 560 in exon 13 (Ile → Ser) | ||
| Might destabilize FMN (flavine mononucleotide) binding domain | |||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 55886062 | |||
| rs1801160 | SNP | [424,428] | |
| 2194G>A | At codon 732 in exon 18 (Val → Ile) | ||
| Decreased expression | |||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 1801160 | |||
| rs67376798 | SNP | [92,412,417,422,425,426, 432,435-437] | |
| 2846A>T | At codon 949 in exon 22 (Asp → Val) | ||
| Significantly conserved site near the Fe-S motif, may disrupt cluster formation and electron transport and lead to lower DPD activity | |||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 67376798 | |||
| Uridine monophosphate synthetase (UMPS) [OMIM #613891] | |||
| rs121917890a | SNPs | [122-126] | |
| 213A>G | At codon 96 (Arg → Gly), effect unknown | ||
| rs121917892b | 326T>G | At codon 109 (Val → Gly), effect unknown | |
| rs1801019c | 638G>C | At codon 213 (Gly → Ala), increase activity | |
| rs2291078d | 1050T>A | At codon 350 (synonymous), effect unknown | |
| rs121917891e | 1285G>C | At codon 429 (Gly → Arg), effect unknown | |
| rs3772809f | 1336A>G | At codon 446 (Ile → Val), effect unknown | |
| ahttp://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 121917890 | |||
| bhttp://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 121917892 | |||
| chttp://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 1801019 | |||
| dhttp://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 2291078 | |||
| ehttp://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 121917891 | |||
| fhttp://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 3772809 | |||
SNP: Single nucleotide polymorphism.
The third described polymorphism is an insertion/deletion of hexanucleotide TTAAAG sequence at 1494 position on the 3’-UTR (1494del6)[58]. This polymorphism may contribute to stability changes of secondary mRNA structure as has been demonstrated for alterations of the 3’-region in other genes[59]. Ulrich et al[58] analysed the mRNA expression level of TYMS gene in 43 patients and showed that homozygous patients with 6-bp deletion had a steady-state TS mRNA level three times lower than patients who were homozygous for the 6-bp insertion alleles (P = 0.017). Furthermore, it was shown that homozygous patients with deletion (del/del) had significantly lower mRNA levels of the TYMS gene which was also associated with greater sensitivity to 5-FU-based therapy as compared to homozygous patients with (ins/ins) insertion (P = 0.017)[57,60]. There is a need for further analyses to allow identification of TYMS transcription regulatory mechanisms including the role played by combinations of different genetic variants, such as polymorphisms, SNPs and VTNR in TYMS/TS expression variability in populations.
A major limitation of correlational research on the pharmacogenetic importance of polymorphisms and TYMS/TS expression is an increasing proportion of patients who are treated with combination therapy, for which 5-FU is not the only component in the chemotherapy. Therefore, it is often difficult to determine whether the observed greater sensitivity in a small number of patients to a treatment is associated with the presence of genetic determinants (e.g., 2R/2R homozygous status, 6 bp- /6 bp- 3’-UTR, allele G of the G>C SNP) or is a result of drugs other than 5-FU used in the combination therapy[50].
Methylenetetrahydrofolate reductase
The use of folic acid in combination with 5-FU has been standard in the treatment of advanced CRC for more than 30 years[61]. The intracellular metabolic balance of folic acid is regulated by methylenetetrahydrofolate reductase (MTHFR), a critical enzyme in the folic acid pathway catalysing irreversible conversion of CH2THF to 5-methyltetrahydrofolate (CH3THF) (Figure 2). 677C>T is one of numerous polymorphisms of the MTHFR gene described in the literature, which may contribute to activity changes in this enzyme. 677TT genotype is responsible for a 30% reduction in enzymatic activity compared to 677CC genotype associated with reduced thermolability observed in vitro[62], which results in a decreased erythrocyte concentration of CH3THF and accumulation of CH2THF[63]. The frequency of specific genetic variants of MTHFR for SNP 677C>T is ethnically diverse. Analyses of Caucasian and Asian populations suggest that the prevalence of 677TT genotype oscillates between 12% and 15% with a frequency of 677CT homozygotes at the 50% level. Whereas, in a population of African-Americans there was a very low frequency of 677TT genotype[64]. An important consequence of the presence of MTHFR 677T variant is the possibility of accumulation of CH2THF in the cells, which may have a significant effect on the pharmacological efficacy of 5-FU. This is due to the fact that the effect of 5-FU is largely dependent on the concentration of foliants. The 5-FU-5-FdUMP metabolite irreversibly forms a stable complex with TS and CH2THF. Creation of this complex inhibits the activity of TS, which leads to an intracellular drop in dTMP concentration and finally inhibition of DNA synthesis. Increased concentration of CH2THF as a consequence of the presence of the MTHFR 677C>T polymorphism may therefore contribute to changes in the chemosensitivity of cancer cells exposed to 5-FU by increasing the amount and stability of CH2THF-TS-FdUMP ternary complex, and thus a stronger inhibition of DNA synthesis. Sohn et al[65] in both in vitro and in vivo studies observed that the presence of 677T allele of the MTHFR gene is responsible for greater chemosensitivity in colon cancer cells, suggesting that the genetic variant 677C>T may be a pharmacogenetic factor used to assess the effectiveness of 5-FU-based chemotherapy. However, clinical studies published in recent years have led to contradictory and inconsistent conclusions[64]. In advanced CRC patients undergoing 5-FU-based therapy, in three published studies the presence of the 677T variant of the MTHFR gene was associated with a higher percentage of positive responses[66-68], while the results of another study did not confirm the existence of such a relationship (Table 1)[69].
Another frequent polymorphism of the MTHFR gene is SNP 1298A>C, which results in substitution of glutamine amino acid by alanine an in enzyme protein sequence[70,71]. Similar to SNP 677C>T, 1298A>C polymorphism contributes to the reduction in enzymatic activity of MTHFR, but has no connection with the thermolabile proteins. The observed frequency of the mutated 1298C allele is approximately 33%[70,71]. Some of the published studies on SNP 1298A>C suggest that the presence of the 1298C variant of the MTHFR gene has no impact on the percentage of positive responses to 5-FU treatment[68,69,72], while two studies suggest that it is associated with significantly decreased patient survival time[67,73]. Thus, contrary conclusions concerning both polymorphic variants of 677C>T and 1298A>C of the MTHFR gene call into question their practical application as response predictors in 5-FU-based therapy[74]. However, recent reports suggest that the simultaneous assessment of several markers, such as MTHFR 1298A>C and TYMS 3’UTR ins/del polymorphisms makes it possible to obtain accurate assessments to predict the toxic effects of 5-FU treatment in CRC patients[75]. Large-scale and well-planned clinical trials are necessary to determine if the practical application of MTHFR 677C>T and 1298>C gene polymorphisms would be possible to predict treatment efficacy. It is also necessary to assess whether these SNPs may be used as prognostic markers in patients undergoing CRC treatment based on 5-FU.
Dihydropyrimidine dehydrogenase
5-FU as a prodrug, in order to achieve its intracellular cytotoxic activity, requires metabolic activation (with over 80% of the administered dose of 5-FU degrading rapidly)[76]. Considering 5-FU metabolic pathways in cells, it seems important to conduct pharmacogenetic analysis of the molecular factors that are associated with biotransformation of the drug. Inter-individual variability in the response of patients to 5-FU treatment may in fact be associated with a decrease in the activity of enzymes responsible for catabolism of the drug, which will result in an increase in drug concentration and longer half-life, and thus an increased risk of serious toxic effects[77]. Dihydropyrimidine dehydrogenase (DPD) acts as a regulatory enzyme in the 5-FU catabolic pathway responsible for conversion of 5-FU to 5-fluorodihydrouracil (5-FUH2). After this conversion, 5-FUH2 is further metabolized to its final metabolite, 5-fluoro-β-alanine, which is excreted in the urine (Figure 1)[78].
Partial DPD activity deficiency in the general population is about 5%, and its total loss is very rare, about 0.2%[79]. Partial or total loss of DPD activity may be associated with the presence of genetic determinants influencing the function of the DPYD gene including SNPs[80], deletion mutations[81,82] and methylation[83]. DPD deficit was first described in an autosomal recessive disease in patients with various neurological symptoms and an accumulation of uracil and thymine in the urine[84]. In recent years, several research groups have investigated the genetic variations present in the DPYD gene, and DPD expression levels in tumour cells with respect to their use as predictive markers for predicting both the effectiveness and toxicity of 5-FU treatment[85]. So far, more than 15000 genetic polymorphisms have been recorded in NCBI dbSNP in the coding, intronic and untranslated 3’ and 5’ regions of DPYD. Conditions resulting in a mutant DPYD allele include base substitutions, splicing deficits and frameshift mutations[85-87]. Taking into account the effect of catabolic processes on the pharmacokinetics of 5-FU and toxicity resulting from dosage, patients with low DPD activity are at an increased risk of serious or even fatal side effects when using the standard 5-FU dose. Also, case reports of severe and fatal toxicity in patients with markedly low DPD activity and treated with capecitabine suggest that DPD deficiency increases the risk of toxicity after oral administration of 5-FU[88].
Meinsma et al[89] described the molecular basis of observed DPD activity deficiency by testing the phenotype and genotype of patients with no DPD activity. Among the analysed cases, there was no 165 nucleotide fragment of mRNA sequence as a result of ejection of one of the exons, moreover, no enzyme DPD protein was detected in these patients[89]. Wei et al[90] identified a heterozygous deletion of 165 nucleotides in a British cancer patient, in whom there was no partial DPD activity and who had serious toxicity following administration of 5-FU. They found that a G to A transition within the 5’ splice site of intron 14 resulted in exon skipping and an inactive DPYD allele (IVS14+1G>A, DPYD*2A) (Figure 4)[90]. Other rare (frequency < 0.1%) polymorphisms and mutations have also been identified (85T>C, 496A>G, 1627A>G, 2194G>A, and 2846G>T) as factors possibly affecting the appearance of toxic symptoms after standard 5-FU treatment (Table 1). DPD activity deficiency is observed in approximately 60% of cases occurring in patients with severe toxicity, and DPYD*2A polymorphism is found in 50% of patients with the 4th stage of neutropenia as a result of 5-FU treatment[91]. In total, more than 40 DPYD polymorphisms were described to have potential use in 5-FU treatment prediction. In addition to single polymorphism changes it has also been demonstrated that the presence of a haplotype consisting of three new intronic SNPs (IVS5+18G>A, IVS6+139G>A, and IVS9-51T>G), and synonymous mutation (1236G>A) may be associated with a decrease in DPD activity[92]. Moreover, hypermethylation of the promoter region of the DPYD gene is described as a possible mechanism of variable DPD activity[83,93]. It is believed that only a few of the reasons listed above are responsible for drug resistance and/or toxicity of fluoropyrimidines[94].
Figure 4.
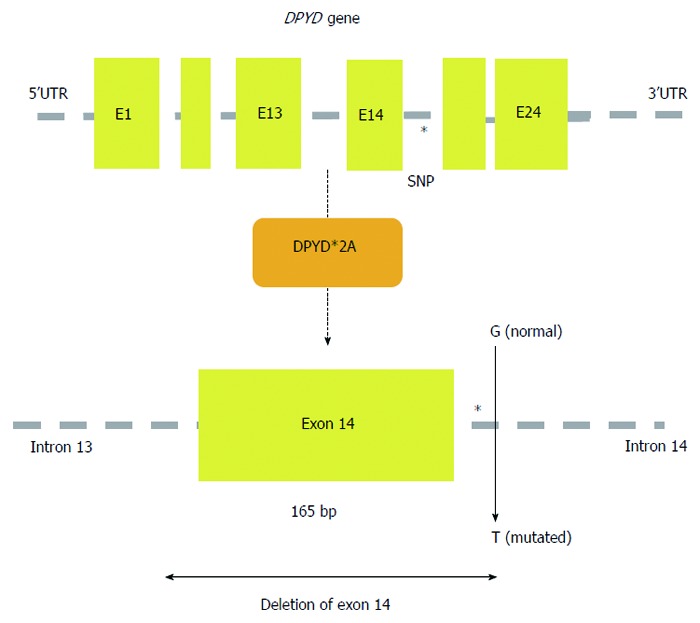
A schematic map of the human DPYD gene is shown with the location of SNP DPYD*2A (IVS14+1G>A); exon 14 is skipped as a result of the G>A translocation at intron 14.
Low DPD expression level should lead to reduced catabolism of 5-FU and therefore contribute to a more effective accumulation of the drug inside cells. On the other hand, high DPD activity in tumour tissue should be responsible for the development of drug resistance by reducing the cytotoxic effects of 5-FU. Also, genetic changes in the functioning of other genes encoding enzymatic proteins of the 5-FU metabolic pathway, such as DPYS (dihydropyrimidinase)[95] or UPB1 (β-ureidopropionase)[96] may contribute to a decrease in therapy effectiveness. Furthermore, it was proved that the patients with low expression of three genes, TYMS, DPYD and thymidine phosphorylase (TYMP) have a significantly longer survival time compared to patients with high expression of any of these genes[17]. A similar correlation between low expression of the DPYD gene determined using RT-PCR and better response to 5-FU based therapy was found in patients with advanced CRC treated with first-line capecitabine[97]. On the other hand, the results of recent studies in patients with metastatic CRC treated with fluoropyrimidine suggest that this correlation is weak or there is no evidence of an association between the expression of DPYD and effectiveness of chemotherapy[37,98,99]. The acquired uncertain evidence is derived mostly from retrospective clinical studies and suggests that low expression of the DPYD gene may be a sensitivity marker in tumour cells for fluoropyrimidines and thus allow us to predict the degree of response to treatment. However, currently little good quality clinical data have confirmed the predictive value of DPYD expression determination in order to predict the efficacy of 5-FU therapy in CRC patients[94].
TYMP
TYMP is the gene encoding thymidine phosphorylase (TP), an enzyme that catalyses phosphorylation of thymidine or deoxyuridine to thymine or uracil, and thus is essential for the nucleotide salvage pathway, that recovers pyrimidine nucleosides formed during RNA or DNA degradation[100]. Several studies suggest that TP is a promoter of tumour growth and metastasis by inhibiting apoptosis and induction of angiogenesis[100]. There is evidence that the level of TP expression is connected with angiogenesis, growth and progression of certain types of cancer[101]. An observed increase in TP expression in tumour tissues as compared to that occurring in normal tissues is visible inter alia in CRC[102]. In most of the analysed cases, high TP expression is related to aggressiveness of cancer and poor prognosis, although there are conflicting reports in this regard (Table 2)[100].
Table 2.
Gene/protein expression or metabolic enzyme activity in colorectal cancer cells and correlation with outcome of patients receiving fluoropyrimidine-based chemotherapy
| Treatment setting | Method | Patients (n) | Better response to chemotherapy | Form of the disease | Ref. |
| Thymidylate synthase (TYMS) [OMIM # 188350] | |||||
| 5-FU | RT-PCR | 29 | Low expression | mCRC | Iyevleva et al[24] |
| 5-FU | RT-PCR | 39 | Low expression | CRC | Ishida et al[25] |
| 5-FU | IHC | 57 | Low expression | mCRC | Hosokawa et al[26] |
| 5-FU | IHC | 62 | Low expression | aCRC | Ciaparrone et al[27] |
| 5-FU | RT-PCR | 92 | Low expression | CRC | Nakajima et al[28] |
| 5-FU | RT-PCR | 309 | Low expression | CRC | Kornmann et al[29] |
| 5-FU | IHC | 391 | Not significant | aCRC | Westra et al[438] |
| 5-FU | IHC | 945 | Not significant | CRC | Soong et al[107] |
| FUdR | IHC | 36 | Low expression | mCRC | Davies et al[31] |
| 5-FU/LV or 5-FU | RT-PCR | 29 | Low expression | mCRC | Kornmann et al[32] |
| 5-FU/LV | RT-PCR | 33 | Low expression | aCRC | Salonga et al[17] |
| 5-FU/LV | RT-PCR | 36 | Low expression | mCRC | Lenz et al[7] |
| 5-FU/LV | RT-PCR | 42 | Low expression | CRC | Leichman et al[19] |
| 5-FU/LV | RIA | 102 | Low expression | mCRC | Etienne et al[33] |
| 5-FU/OX | RT-PCR | 45 | Low expression | aCRC | Shirota et al[34] |
| 5-FU/MTX | IHC | 108 | Low expression | aCRC | Paradiso et al[35] |
| 5-FU or 5-FU/MTX or 5-FU/LV | IHC | 24 | Not significant | aCRC | Belvedere et al[439] |
| 5-FU or 5-FU/MTX or 5-FU/LV | IHC | 27 | Not significant | mCRC | Aschele et al[23] |
| 5-FU or 5-FU/MTX or 5-FU/LV | IHC | 48 | Low expression | mCRC | Aschele et al[36] |
| 5-FU/LV/CPT-11 | RT-PCR | 13 | Low expression | aCRC | Yanagisawa et al[37] |
| 5-FU/LV/CPT-11 | IHC | 54 | Low expression | aCRC | Bendardaf et al[38] |
| 5-FU/LV/CPT-11 | IHC | 57 | Not significant | aCRC | Paradiso et al[440] |
| UFT/LV | RT-PCR | 37 | Low expression | mCRC | Ichikawa et al[39] |
| Capecitabine | RT-PCR | 37 | Not significant | aCRC | Vallböhmer et al[97] |
| Capecitabine | IHC | 39 | Not significant | CRC | Lindebjerg et al[441] |
| Capecitabine/CPT-11 | IHC | 556 | Not significant | aCRC | Koopman et al[110] |
| 5-FU-based therapy | IHC | 681 | Not significant | CRC | Karlberg et al[442] |
| Dihydropyrimidine dehydrogenase (DPYD) (OMIM # 612779) | |||||
| 5-FU | RT-PCR | 29 | Not significant | mCRC | Iyevleva et al[24] |
| 5-FU | RT-PCR | 39 | Not significant | CRC | Ishida et al[25] |
| 5-FU | IHC | 62 | Low expression | aCRC | Ciaparrone et al[27] |
| 5-FU | IHC | 303 | Low expression | CRC | Jensen et al[443] |
| 5-FU | RT-PCR | 309 | Low expression | CRC | Kornmann et al[29] |
| 5-FU | IHC | 391 | Not significant | aCRC | Westra et al[438] |
| 5-FU | IHC | 945 | Not significant | CRC | Soong et al[107] |
| 5-FU/LV | RT-PCR | 33 | Low expression | aCRC | Salonga et al[17] |
| UFT/LV | RT-PCR | 37 | Low expression | mCRC | Ichikawa et al[39] |
| 5-FU/LV/CPT-11 | RT-PCR | 13 | Not significant | aCRC | Yanagisawa et al[37] |
| Capecitabine | RT-PCR | 37 | Low expression | aCRC | Vallböhmer et al[97] |
| Capecitabine/CPT-11 | RT-PCR | 67 | Not significant | aCRC | Meropol et al[98] |
| Capecitabine/CPT-11 | IHC | 556 | Low expression | aCRC | Koopman et al[110] |
| 5-FU-based therapy | ELISA | 64 | Low expression | aCRC | Oi et al[444] |
| 5-FU-based therapy | RT-PCR | 102 | Low expression | CRC | Lassman et al[445] |
| 5-FU-based therapy | RT-PCR | 144 | Low expression | aCRC | Gustavsson et al[446] |
| 5-FU-based therapy | IHC | 150 | Low expression | aCRC | Tokunaga et al[447] |
| Thymidine phosphorylase (TYMP) (OMIM # 131222) | |||||
| 5-FU | IHC | 62 | Not significant | aCRC | Ciaparrone et al[27] |
| 5-FU | IHC | 945 | Not significant | CRC | Soong et al[107] |
| 5-FU/LV | RT-PCR | 33 | Low expression | aCRC | Salonga et al[17] |
| 5-FU/LV/CPT-11 | RT-PCR | 13 | Not significant | aCRC | Yanagisawa et al[37] |
| Capecitabine | RT-PCR | 37 | Not significant | aCRC | Vallböhmer et al[97] |
| Capecitabine/OX | IHC | 41 | High expression | mCRC | Petrioli et al[448] |
| Capecitabine/CPT-11 | RT-PCR | 67 | High expression | aCRC | Meropol et al[98] |
| Capecitabine/CPT-11 | IHC | 556 | Not significant | aCRC | Koopman et al[110] |
| 5-FU-based therapy | RT-PCR | 144 | Low expression | aCRC | Gustavsson et al[446] |
| 5-FU-based therapy | IHC | 150 | Low expression | aCRC | Tokunaga et al[447] |
| Uridine monophosphate synthetase (UMPS) (OMIM #613891) | |||||
| 5-FU | RT-PCR | 38 | Not significant | mCRC | Sameshima et al[449] |
| 5-FU | RT-PCR | 39 | Not significant | CRC | Ishida et al[25] |
| 5-FU/LV/OX | RT-PCR | 58 | Not significant | CRC | Dong et al[450] |
| 5-FU/LV/cisplatin | RT-PCR | 22 | High expression | mCRC | Matsuyama et al[113] |
| UFT | RIA | 40 | High expression | CRC | Ichikawa et al[114] |
| UFT | RIA | 124 | High expression | CRC | Ochiai et al[115] |
| UFT | IHC | 150 | High expression | CRC | Tokunaga et al[116] |
| UFT | IHC | 160 | High expression | CRC | Tokunaga et al[117] |
| UFT/LV | RT-PCR | 37 | High expression | mCRC | Ichikawa et al[118] |
| UFT/LV | RT-PCR | 103 | High expression | CRC | Yamada et al[119] |
| 5-FU-based therapy | RT-PCR | 10 | Not significant | CRC | Ishibashi et al[451] |
| 5-FU-based therapy | RIA | 11 | Not significant | CRC | Yamada et al[452] |
| 5-FU-based therapy | RT-PCR | 52 | Not significant | CRC | Kinoshita et al[453] |
| 5-FU-based therapy | RIA | 54 | High expression | CRC | Fujii et al[120] |
| 5-FU-based therapy | RIA | 90 | High expression | CRC | Ochiai et al[121] |
5-FU: 5-fluorouracil; LV: Leucovorin; FUdR: 5-fluorodeoxyuridine; MTX: Methotrexate; OX: Oxaliplatin; UFT: Compound tegafur tablets; CPT-11: Irinotecan; CTX: Cetuximab; RT-PCR: Reverse trascriptase polymerase chain reaction; IHC: Immunohistochemistry; ELISA: Enzyme-linked immunosorbent ssay; RIA: Radioimmunoassay; CRC: Colorectal cancer; aCRC: Advanced colorectal cancer; mCRC: Metastatic colorectal cancer.
TP is involved in the metabolism of 5-FU, where catalysed by TP, 5-FU is converted to 5-fluoro-2’-deoxyuridine (5-FUDR) (Figure 1). This is the first stage of 5-FU activation in tumour cells consequently leading to inhibition of DNA synthesis by reducing the pool of available dTTP to the substrate of this reaction. Capecitabine, an oral form of 5-FU prodrug, is designed to reduce the gastrointestinal toxicity of 5’-deoxy-5-fluorouridine (5’DFUR) and to generate 5-FU preferentially at the tumour site[103]. 5’DFUR may be transformed in cancer cells in a reaction catalysed by TP or uridine phosphorylase[103,104]. Since TP expression is significantly higher in tumour cells, it allows targeted activation which minimizes the toxicity of such therapy[105]. In phase III clinical trials, metastatic CRC patients who were treated with capecitabine monotherapy had a significantly lower incidence of toxic effects in comparison to patients treated with 5FU/LV[106]. Moreover, since the enzymatic activity of TP is essential to obtain an adequate level of concentration of an active form of capecitabine, it may be a useful marker for predicting the effectiveness of chemotherapy using this drug[98].
Soong et al[107] published a study on the relationship between the expression level of TP (determined by microarrays and immunohistochemistry) and survival time of 945 CRC patients treated with 5-FU. The results of this study suggest that the low level of TP expression may be associated with the improved treatment outcomes observed, and may be a good predictive marker for response to 5-FU chemotherapy[107]. Also, the results presented by Salonga et al[17] confirm the link between low TP expression and a positive response to 5-FU. However, results different from the above were obtained by Meropol et al[98]. Patients with metastatic CRC treated with combination therapy using CPT-11 plus capecitabine (CAPIRI) were subjected to an assessment of TP protein expression in primary tumour tissues and metastases. Positive results for TP expression confirmed by IHC techniques were associated with a statistically significant longer time to progression (TTP) in comparison with those cases in which a low level of TP expression was found (8.7 mo vs 6.0 mo). Conversely, neither TS nor DPD, both enzymes that have been previously shown to correlate with resistance to 5-FU, were able to predict response to CAPIRI[98,108]. Presumably, the cells with higher expression of TP may exhibit an increased sensitivity to 5-FU, due to the increase in FdUMP concentration, which is the result of increased 5-FU activation. On the other hand, low TS expression may lead to serious DNA damage. Since cancer cells are characterized by a higher degree of proliferation compared to normal cells, low TS expression in tumour tissue may lead to a decrease in the dUMP substrate necessary for DNA synthesis, which would inhibit its replication and proliferation. Therefore, the low level of TS expression in tumour cells is associated with a less aggressive course of the disease and a more favourable prognosis in patients. In conclusion, a low level of TS expression may be prognostic rather than a predictor of fluoropyrimidines effectiveness[108,109]. However, the prognostic value of TS expression was not observed in one of the largest retrospective studies[110], which may give rise to questions as to whether further retrospective analysis can provide useful data to confirm the clinical significance of this marker. As highlighted in the meta-analysis by Popat et al[20], large methodological differences in individual primary studies make it difficult to come to decisive conclusions. The results of this analysis showed that patients whose tumour tissue had a high level of TS expression were observed to have worse OS compared to the group of patients with a low level of expression. However, as emphasized by the authors of the meta-analysis, the heterogeneity of the studies and possible publication bias do not allow a straightforward conclusion[20].
Uridine monophosphate synthetase
In mammalian cells, the last step of pyrimidine nucleotide synthesis involves the conversion of orotate to uridine monophosphate (UMP) and is catalysed by UMP synthase (UMPS). This bifunctional enzyme has 2 sequential activities, orotate phosphoribosyltransferase (OPRT) and orotidine-5-monophosphate decarboxylase (ODC)[111]. The protein product of the UMPS gene is the OPRT enzyme, which catalyses the conversion of 5-FU into FUMP, a common substrate for the production of 5-fluorouridine triphosphate and dUTP, two cytotoxic metabolites that target RNA and DNA, respectively. Muhale et al[112] showed that in the anabolic pathway of 5-FU, UMPS is the only gene that rounds out a manifestation of the phenotype of resistance to 5-FU. Furthermore, the high OPRT enzyme activity or increased expression of mRNA for UMPS gene is associated with longer survival times, suggesting that the UMPS may be a clinically useful marker for predicting the effectiveness of treatment with 5-FU[113-121]. In clinical in vitro studies carried out by Isshi et al[122], OPRT and DPD enzymatic activity was determined by radioassay in tumour tissues taken from patients diagnosed with CRC (n = 62) and fluorescein diacetate assay (FDA) or histoculture drug response assay (HDRA) were used to determine the chemosensitivity in relation to 5-FU. The chemosensitivity test proved positive in 60% of the specimens with ORPT activity of 0.413 (nmol/min per mg protein) or above and 50% of those with DPD activity of 30 (pmol/min per mg protein) or below. Of the patient specimens showing OPRT activity of 0.413 or above and DPD activity of 30 or below, 88.9% were positive for 5-FU sensitivity, suggesting the possibility that the combination of these two levels may be predictive of positive 5-FU sensitivity[122]. Tokunaga et al[116] indicated that high OPRT (IHC) expression in patients with CRC stage II-IV is associated with a longer OS, which was not confirmed in a study using RT-PCR in a smaller study group[37]. The prognostic value of UMPS/OPRT expression in both tumour and stromal cells, but each with an opposite effect on outcome, was an unexpected finding in a retrospective analysis of a large trial[110].
There are several described SNPs located in UMPS[123-126], including 286A>G (Arg96Gly), 1285G>C (Gly429Arg), 326T>G (Val109Gly), and 638G>C (Gly213Ala). Kitajima et al[123] analysed the effects of several SNPs gene UMPS (638G>C, 1050T>A, and 1336A>G) on the sensitivity to 5-FU in a group of 31 patients with CRC. They found no relationship between the effectiveness of treatment with 5-FU and frequency of any of the genetic variants among respondents[123]. In clinical in vitro trials it was shown that the functional polymorphism, Gly213Ala (638G>C) substitution, contributes to an increase in enzymatic OPRT activity[127]. With reference to the above results, in vivo studies showed that patients with substitution of 213Ala in the OPRT protein sequence, after exposure to 5-FU, experience much more severe symptoms of toxicity[124] such as grade 3 diarrhoea (P = 0.031) and grade 2-3 anorexia (P = 0.035)[125]. The probable mechanism of gastrointestinal toxicity is related to the incorporation of 5-FU into RNA (F-RNA), but not with inhibition of the biosynthesis of dTMP by conversion of 5-FU to FdUMP[128]. Therefore, 5-FU/LV administration at a higher OPRT enzymatic activity (especially with the homozygous genotype 638CC) significantly increases the level of F-RNA in enterocytes, which may increase the likelihood of severe diarrhoea[125].
There are still many unknown factors that may participate along with SNPs gene UMPS in chemosensitivity or mechanisms of resistance to 5-FU, which makes it necessary to analyse other regions of the gene including the promoter and regulatory region. A lack of confirmed reliable test data from in vivo studies on the correlation between the expression of UMPS/OPRT and the effectiveness of treatment with 5-FU, makes it now impossible to determine the potential clinical value of this marker.
Other potential factors
A total of 20 polymorphic variants and 20 haplotype systems of the CYP2A6 gene have been described, which encode P-450 cytochrome isoenzyme involved in the metabolic activation of tegafur (UFT). Based on the results obtained from genotype/haplotype-phenotype association tests, Wang et al[129] showed that the variant CYP2A6*4 is the main determinant contributing to the reduction of formed 5-FU with UFT, and the presence of the allele affects the level of decrease in CYP2A6 gene expression. A different correlation was observed in the case of 14 haplotype (a novel CYP2A6*1B alleles), which was associated with an increase in UTF microsomal activation to 5-FU, and the presence of the haplotype contributed to increased expression of CYP2A6. The authors speculate that the phenotype of increased metabolic activity of CYP2A6 may be the result of the sum of three different variants (22C>T, 1620T>C and a gene conversion in the 3’-UTR) included in this haplotype. Wang et al[129] conclude that variants CYP2A6*4 and CYP2A6*1B are major genetic factors responsible for inter-individual variation of UTF activation degree to 5-FU.
Microsatellite instability (MSI) is common in many types of tumours and is observed in 10%-14% of sporadic CRC. The MSI phenomenon is caused by mutations located in mismatch repair (MMR) genes, this group of genes are hMSH2, hMLH1 and hMSH6. Protein products of these genes are responsible for the repair of DNA damage caused during the replication process. It is believed that the MMR deficiency operation is one of the possible causes of resistance to fluoropyrimidines[130]. Meyers et al[131] showed that the restoration of a functional protein MLH1 in an MMR-deficient human colon cancer cell line contributes to increased sensitivity to 5-FU, which suggests that MMR deficiency in cells may be associated with resistance to 5-FU. It is likely that MMR deficiency in cancer cells contributes to increased tolerance to the presence of DNA damage occurring as a result of replication errors, instead of undergoing cell cycle arrest or death[132]. The results of several studies suggest that the presence of MMR deficit in tumour cells is associated with chemosensitivity to 5-FU based therapy[133]. Most of these studies found low sensitivity to 5-FU in the case of MMR deficiency, which was confirmed by a recent pooled reanalysis of randomized trials[134]. On the other hand, among patients with II and III stage CRC, prolonged survival time in cases with high MSI was detected[133,135,136]. In addition, when comparing the group of MSI patients with patients who were microsatellite stable it was found that MSI prolongs disease-free time, but is not beneficial in 5-FU adjuvant chemotherapy[137]. Furthermore, it was found that in most of these cases, where the tumours showed positive results for MSI, the expression was observed in wild-type p53[138] which is an important determinant of 5-FU sensitivity.
The tumour suppressor protein p53 plays a key role in the control of cell cycle progression and cell death[139]. It is estimated that in about 50% of cases with various types of tumours a number of mutations in P53 gene which encodes the p53 can be seen[140]. p53 is responsible for cell cycle arrest and directing cells to the apoptotic pathway in a situation where there is a risk of sustaining damage to the integrity of the genome preventing the transfer of damaged DNA into daughter cells. Longley et al[41] demonstrated that p53 and p53-target genes are activated in response to RNA-directed 5-FU cytotoxicity. Moreover, in vitro test results indicate that the loss of p53 functionality contributes to reducing chemosensitivity of cells to 5-FU[41,141]. Studies on expression have also shown that overexpression of p53 is correlated with resistance to 5-FU-based chemotherapy[136,142,143] although there is no conformity with the results obtained by other researchers[35]. The impact of the presence of specific mutations of P53 gene was also described, which may contribute to transformation and drug resistance[144]. Indeed, Pugacheva et al[145] suggested that certain p53 mutants may increase dUTPase expression, resulting in 5-FU resistance. Thus, 5-FU chemosensitivity may be dependent on the particular TP53 genotype.
IRINOTECAN
7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin (CPT-11) is a synthetic analogue of a naturally occurring alkaloid, camptothecin. CPT-11 was first approved for clinical use in Japan in 1994 for the treatment of small-cell lung cancer and hematologic malignancies, and then in 1995 in France for the treatment of advanced CRC. Finally, in 1996, CPT-11 was approved by the US Food and Drug Administration (FDA) and approved for use in the treatment of CRC in 1998. Currently, CPT-11 is mainly used in CRC diagnosed patients with metastases, with recorded relapse or progression after application of standard 5-FU-based therapy[146].
In preclinical screening tests using the HST-1 human squamous carcinoma cell line, SN-38, which is an active CPT-11 metabolite, exhibited the ability to increase the antitumour effect of such cytostatics as cisplatin, mitomycin C, 5-FU, and etoposide[147]. In in vitro tests using colon and hepatocellular carcinoma cell lines it was also observed that SN-38 had greater cytotoxic activity compared to cisplatin, mitomycin C, doxorubicin and 5-FU[148]. The in vivo tests showed that the positive response rate to CPT-11 monotherapy ranged from 17% to 27% of cases[149]. The effectiveness of CPT-11 based treatment was observed in both the group of patients for which this was the first application of treatment as well as in patients for whom 5-FU therapy was found to be ineffective[150]. The clinical application of the combination of CPT-11 with 5-FU/LV (FOLFIRI) resulted in a significant percentage increase in positive responses, prolonged time to tumour progression and survival. Efficacy was demonstrated both in chemotherapy-naive patients and those who progressed after 5-FU-based chemotherapy when compared with 5-FU/LV alone[151].
Tumour-specific somatic mutations and abnormal gene expression as well as germline genetic variations have been reported to be associated with CPT-11 therapeutic efficacy and toxicity. However, the available studies do not provide unequivocal confirmation that somatic mutations have a significant impact on the outcome of CPT-11 treatment, which prevents their usage as predictive markers. Generally, genetic variations may influence both the pharmacokinetics and pharmacodynamics of CPT-11[152-154]. Taking into account the results of previous preclinical and clinical tests, CPT-11 resistance phenotype may be associated with three different mechanisms: (1) insufficient intra-tumour accumulation of SN-38 (determined by pharmacokinetic factors); (2) a change in TOPI activity that decreases levels of the SN-38-Topo I-DNA complex (pharmacodynamic factors); and (3) alterations in the events downstream from the ternary complex, for example, apoptosis, cell cycle regulation, checkpoints, and DNA repair (pharmacodynamic factors)[155,156].
Carboxylesterase
Hydrolysis of the bulky dipiperidino moiety of CPT-11 produces the active metabolite SN-38. The enzymes responsible for these reactions have been identified as human carboxylesterases CES1, CES2 (Figure 5) and the recently described isoenzyme CES3. However, CES3 catalytic activity is low and therefore not likely to play a significant role in the metabolism of CPT-11. Several studies indicated that the CES2 isoenzyme plays a major role in CPT-11 and SN-38 hydrolysis[157].
Figure 5.
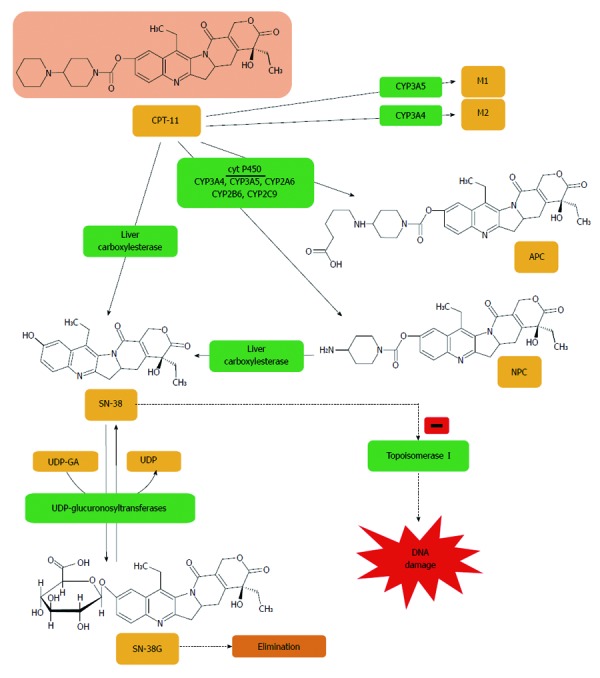
Irinotecan is metabolized to APC or NPC and potential other intermediate metabolites (M1, M2) via a cytochrome P450 mediated process. Neither 7-ethyl-10-[4-N-(5-aminopentanoic acid)-1-piperidino]carbonyloxycamptothecin (APC) nor 7-ethyl-10-(4-amino-1-piperidino)carbonyloxycamptothecin (NPC) contribute directly to irinotecan activity in vivo. NPC is further converted to 7-ethyl-10-hydroxy-camptothecin (SN-38) by carboxylesterase. All irinotecan metabolites are pH sensitive, thus are at risk of transforming from inactive to active products, and vice versa. SN-38 is subsequently conjugated predominantly by the enzyme UDP-glucuronosyltransferase 1A1 (UGT1A1) to form a glucuronide metabolite (SN-38G)[403].
Resequencing of CES1 and CES2 allowed the identification of SNPs and haplotype structure of these genes[158-163]. Numerous SNPs and haplotypes have been described in several populations: Europeans, Africans, and Asian-Americans[163]. Charasson et al[158] studied 115 cases (Caucasian population) for sequence analysis of all 12 exons of the CES2 gene and 5’ and 3’ untranslated regions, and identified 11 SNPs. One of these SNPs located at position 830 of gene (830C>G) was associated with a decrease in CES2 expression, which has been reported in 60 cases in the North American population[158]. The CPT-11 intra-tumour activation process is partially explained as some authors have provided experimental data indicating that the level of CES2 activity may be a predictor of CPT-11 toxicity[164], while others failed to detect CES2 activity in cultured cells[165].
Kubo et al[166] found 12 new SNPs located in the CES2 gene sequence including the nonsynonymous SNP 100C>T (Arg34Trp) and the SNP at the splice acceptor site of intron 8 (IVS8-2A>G). In vitro test results regarding functional characterization of these SNPs, as well as the additional nonsynonymous SNP 424G>A (Val142Met), suggest that the presence of 34Trp and 142Met variants is responsible for the loss of enzyme activity, and IVS8-2G allele is associated with a significant reduction in metabolic activity of CES2[166]. Kim et al[161], studying a Japanese population, based on linkage analysis of 21 polymorphisms of the CES2 gene, identified a panel comprising a number of haplotypes and found that some haplotypes were rare in the population, including nonsynonymous SNPs may contribute to the reduction of enzyme activity. Furthermore, Kim et al[161] found that patients who are carriers of nonsynonymous SNPs, 100C>T (Arg34Trp) or 1A>T (Met1Leu) have a significantly reduced ratio of (SN-38 + SN-38G)/CPT-11 area under the plasma concentration curve (AUC). In vitro test results regarding functional analysis of these SNPs allowed determination of their impact on the efficiency of translation and transcription of the CES2 gene. It has been shown that the presence of the 1A>T genetic variant does not affect the transcriptional activity of the gene, but it is important for the efficiency of the translation course[161]. These observations are the starting point for further research into CES2/CES2 pharmacogenetics, the results of which can be used in future to individualize dosing of CPT-11 and other prodrugs activated by carboxylesterases.
Carboxylesterase hydrolyzes CPT-11 to SN-38 primarily in the liver, but also in plasma and the gastrointestinal tract. It was found that the CES1 gene is highly expressed in the liver, which is the main organ responsible for the metabolic activation of CPT-11. It is likely that the genetic variants of CES1 can affect the concentration of CPT-11 metabolites in plasma. However, the clinical relevance of genetic determinants of CES1 on the pharmacokinetics/pharmacodynamics of CPT-11 is not fully understood. Functional human CES1 genes include CES1A1 and CES1A2 which are inversely located on chromosome 16q. In addition to structural variations of the CES1 gene family, several SNPs and small deletion/insertion variants were found. The influence of the -816C variant located in the CES1A2 promoter region on increased transcriptional activity of the CES1A2 gene was described. Furthermore, Tanimoto et al[167] showed that the mRNA expression level of the CES1A2 gene is related to the sensitivity of tumour cells to CPT-11. Besides, it was found that the polymorphism -816A>C is coupled to several other SNPs (-62T>C, -47G>C, -46G>T, -41C>G, -40A>G, -37G>C, -34del/G and -32G>T) located in the proximal promoter region, which is associated with increased transcription of CES1A2, as bound transcription factors such as Sp1 are found in this area[168]. The studies by Yoshimura et al[168] suggest that the genetic variant CES1A may affect the dose-dependent antitumour activity of CPT-11.
In conclusion, there are certain conditions relating to the impact of polymorphisms located in the CES1/CES2 genes on the metabolism of CPT-11, which, if they are confirmed in large clinical trials, in the future may allow the setting of individual regimens of CPT-11 in patients with cancer (Table 3).
Table 3.
Selected common polymorphisms of UGT1A1, UGT1A7, UGT1A9, CES2, CYP3A4, CYP3A5, MDR1, MRP1, MRP2, BCRP, OATP1B1 genes and their potential impact on functioning of proteins related to CPT-11 pharmacology
| dbSNP rs cluster ID | Type of polymorphism | Function | Ref. |
| UDP-glycosyltransferase 1A1 (UGT1A1) (OMIM # 191740) | |||
| rs8175347 | VNTR | [177,178,180,182,191,192,197,219,317,356,454-460] | |
| -53(TA)6>7 | UGT1A1*28, reduced activity | ||
| -53(TA)6>5 | UGT1A1*36, increased activity | ||
| -53(TA)6>8 | UGT1A1*37, reduced activity | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 8175347 | |||
| rs3755319 | SNP | [187] | |
| -3279T>G | UGT1A1*60, reduced activity | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 3755319 | |||
| rs10929302a | SNP | [192,404] | |
| -3156G>A | UGT1A1*93, reduced activity | ||
| rs887829b | -3140G>A | effect unknown | |
| ahttp://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 10929302 | |||
| bhttp://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 887829 | |||
| rs4148323 | SNP | [186,191,461] | |
| 211G>A | Gly71Arg, UGT1A1*6, reduced activity | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 4148323 | |||
| rs35350960 | SNP | [172,174,189] | |
| 686C>A | Pro229Gln, UGT1A1*27, reduced activity | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 35350960 | |||
| rs34993780 | SNP | [170,174,189] | |
| 1456T>G | Tyr486Asp, UGT1A1*7, reduced activity | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 34993780 | |||
| UDP-glycosyltransferase 1A7 (UGT1A7) (OMIM #606432) | |||
| rs17868323a | SNP | [188,189,197,237] | |
| 387T>G | Asn129Lys, UGT1A7*2 and *3, increased activity | ||
| rs17863778b | 391C>A | Arg131Lys, UGT1A7*2 and *3, increased activity | |
| rs11692021c | 622C>T | Trp208Arg, UGT1A7*3 and *4, reduced activity | |
| ahttp://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 17868323 | |||
| bhttp://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 17863778 | |||
| chttp://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 11692021 | |||
| UDP-glycosyltransferase 1A9 (UGT1A9) (OMIM #606434) | |||
| rs45625337 | VNTR | [190,197,462] | |
| –118(T)9>10 | UGT1A9*22, increased activity | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 45625337 | |||
| rs2741049 | SNP | [197,463] | |
| IVS1+399C>T | Effect unknown | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 2741049 | |||
| Carboxylesterase 2 (CES2) (OMIM #605278) | |||
| - | SNP | [159,161,166] | |
| 1A>T | Met1Leu, CES*5 | ||
| rs72547531a | 100C>T | Arg98Trp, CES*2 | |
| rs72547532b | 424G>A | Val206Met, CES*3 | |
| rs8192924c | 617G>A | Arg270His, CES*6 | |
| rs11075646d | 830C>G | Synonymous | |
| rs72547533e | IVS8-2A>G | Splicing defect, CES*4 | |
| a http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 72547531 | |||
| b http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 72547532 | |||
| c http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 8192924 | |||
| d http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 11075646 | |||
| e http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 72547533 | |||
| Cytochrome P450, subfamily IIIA, polypeptide 4 (CYP3A4) (OMIM #124010) | |||
| rs2740574a | SNP | [211,464,465] | |
| -392A>G | CYP3A4*1b, altered pharmacokinetics and toxicity | ||
| rs4986907b | 485G>A | CYP3A4*15, Arg162Gln | |
| rs4986908c | 520G>C | CYP3A4*10, Asp174His | |
| rs12721627d | 554C>G | CYP3A4*16, Thr185Ser | |
| rs4987161e | 566T>C | CYP3A4*17, Phe189Ser, altered pharmacokinetics | |
| rs55785340f | 664T>C | CYP3A4*2, Ser222Pro, altered pharmacokinetics and toxicity | |
| rs28371759g | 878T>C | CYP3A4*18, Leu293Pro, altered pharmacokinetics and toxicity | |
| ahttp://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 2740574 | |||
| bhttp://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 4986907 | |||
| chttp://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 4986908 | |||
| dhttp://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 12721627 | |||
| ehttp://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 4987161 | |||
| fhttp://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 55785340 | |||
| ghttp://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 28371759 | |||
| rs4986910 | SNP | [210,465] | |
| 1334T>C | CYP3A4*3, Met444Thr | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 4986910 | |||
| Cytochrome P450, subfamily IIIA, polypeptide 5 (CYP3A5) (OMIM #605325) | |||
| rs776746 | SNP | [179,464-467] | |
| 6986A>G | Synonymous | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 776746 | |||
| Multidrug resistance 1 (MDR1, ABCB1) (OMIM #171050) | |||
| rs1128503 | SNP | [210,211,217,460,467-469] | |
| 1236C>T | Synonymous, CTP-11 or SN-38 AUC ↑ | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 1128503 | |||
| rs2032582 | SNP | [217,468-470] | |
| 2677G>T/A | Ser893Ala or Ser893Thr | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 2032582 | |||
| rs1045642 | SNP | [179,217,468-475] | |
| 3435C>T | Synonymous, CTP-11 AUC ↑ | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 1045642 | |||
| rs10276036 | SNP | [207] | |
| IVS9-44A>G | Effect unknown | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 10276036 | |||
| Multidrug resistance-associated protein 1 (MRP1, ABCC1) (OMIM #158343) | |||
| rs35605 | SNP | [210,476] | |
| 1684T>C | Synonymous | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 35605 | |||
| rs17287570 | SNP | [237] | |
| c.1677+4951A>C | Effect unknown | ||
| rs3765129 | SNP | [207,210,476] | |
| IVS11-48C>T | Effect unknown | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 3765129 | |||
| rs2074087 | SNP | [476,477] | |
| IVC18-30C>G | Effect unknown | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 2074087 | |||
| Multidrug resistance-associated protein 2 (MRP2, ABCC2) (OMIM #601107) | |||
| rs1885301 | SNP | [477] | |
| -1549A>G | Effect unknown | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 1885301 | |||
| rs2804402 | SNP | [207] | |
| -1019A>G | Effect unknown | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 2804402 | |||
| rs717620 | SNP | [477-479] | |
| -24C>T | Effect unknown | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 717620 | |||
| rs2273697 | SNP | [467,479,480] | |
| 1249G>A | Val417Ile, effect unknown | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 2273697 | |||
| rs3740066 | SNP | [477,479,481] | |
| 3972C>T | Synonymous, CTP-11 or APC or SN-38G AUC ↑ | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 3740066 | |||
| Breast cancer resistance protein (BCRP, ABCG2) (OMIM #603756) | |||
| rs2622604a | SNP | [237] | |
| c.-19-17758A>G | Synonymous | ||
| rs3109823b | c.-19-3436G>A | Synonymous | |
| ahttp://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 2622604 | |||
| bhttp://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 3109823 | |||
| rs2231142 | SNP | [239-244,482] | |
| 421C>A | Gln141Lys, no significant changes in CPT-11 pharmacokinetics | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 2231142 | |||
| rs2231137 | SNP | [242,467,482] | |
| 34G>A | Val12Met, higher drug resistance in vitro (SN-38) | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 2231137 | |||
| rs1481012 | SNP | [483] | |
| c.841+179T>C | Synonymous | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 1481012 | |||
| Organic anion-transporting polypeptide 1B1 (OATP1B1, SLCO1B1) (OMIM #604843) | |||
| rs2306283 | SNP | [247-249,460,467,484] | |
| 388A>G | Asn130Asp, effect unknown | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 2306283 | |||
| rs4149056 | SNP | [247-249,460] | |
| 521T>C | Val174Ala, effect unknown | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 4149056 | |||
SNP: Single nucleotide polymorphism.
UDP-glycosyltransferase 1 family
SN-38 is glucuronidated, mainly in the liver, to SN-38 glucuronide (SN-38G) by the uridine diphosphate glucuronosyltransferase enzymes (UGTs), primarily the UDP-glycosyltransferase 1 family (UGT1As) isoenzyme. SN-38G metabolite is excreted into the bile and urine, where it can be removed from the body. However, rehydrolysis of SN-38G to SN-38, which can take place in the digestive tract under the influence of bacterial β-glucuronidase, can cause acute diarrhoea observed during treatment with CPT-11[169].
UGTs are one of the most important classes of enzyme proteins participating in the coupling reaction phase II of xenobiotic metabolism. Currently there are 17 human UGT isoenzymes described that have been assigned to one of two families identified as UGT1 and UGT2, which are further subdivided on the basis of amino acid sequence similarity into UGT1A, UGT2A and UGT2B subfamilies. Members of the UGT1 family are encoded by the UGT1A locus on chromosome 2q37, which contains 13 first exons, each having its own promoter and enhancer regions, which are spliced to identical exons 2-5 (Figure 6). UGT1A1 isoenzyme is responsible in humans for bilirubin conjugation with glucuronic acid, and some genetic variants located in the UGT1A1 gene are associated with the development of hyperbilirubinemic syndromes. These diseases, including Gilbert’s syndrome and Crigler-Najjar syndrome type I and II, are most often described in cases with no or low activity of UGT1A1 as a result of polymorphisms in the sequence of the promoter or coding region[170-172]. Two other isoenzymes, namely the liver UGT1A9 and extrahepatic UGT1A7 are considered important in the SN-38 enzymatic inactivation process. Several research groups have tested in vitro the impact of genetic variation in UGT1A1, UGT1A7 and UGT1A9 on the level of SN-38 glucuronidation[173,174]. Among the frequently occurring genetic variants in the UGT1A gene locus 100 SNPs were described, which are located both in the promoter region as well as the coding sequence of the UGT1A gene, many of these polymorphisms remain in linkage disequilibrium to the other alleles[175]. Determination of the possible clinical consequences of these functional changes is being studied, and has been fairly well documented for some of the identified alleles. A number of in vivo studies were aimed to determine the effect of different UGT1A genotypes on the pharmacokinetics and toxicity of CPT-11[176-183].
Figure 6.
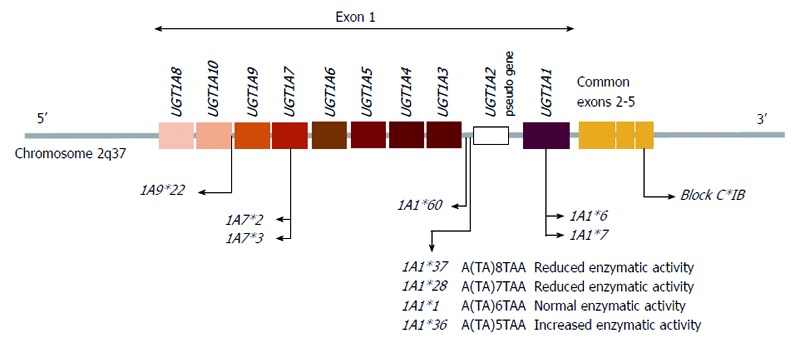
Graphic representation of the human UGT1A gene locus encoding the UGT1A enzymes and the major UGT1A1, 1A7 and 1A9 polymorphisms that are responsible for glucuronidation of SN-38. Individual first exons are positioned at the 5’ end of the chromosome and common exons 2-5 at the 3’ end. Individual exon 1 sequences are combined with exons 2-5 sequence, which is present in every UDP-glycosyltransferase 1A1 (UGT1A1) transcript, the intervening sequence of the primary transcript is eliminated by splicing[404]. The promoter variant, UGT1A1*28, *36 and *37 results from a TA insertion/deletion in the (TA)6TAA element of the UGT1A1 promoter region. This alteration leads to decreased/increased gene expression[184].
One of the best known UGT1A1 polymorphisms is VNTR concerning the number of repetitions of the dinucleotide part of TA [A(TA)nTAA, n = 5-8], which is located in the TATA sequence of the promoter region. The wild-type allele contains six repeats (TA)6 (UGT1A1*1), which are located between position -53 and -42 of the translational start codon. While (TA)7 (UGT1A1*28), an often quoted variant in Gilbert’s syndrome[172], in the in vitro study was responsible for a 63% reduction in translational activity compared to wild-type alleles[184]. Other variations such as (TA)5 (UGT1A1*36), and (TA)8 (UGT1A1*37), respectively, contribute to the growth and reduction of transcriptional activity, as observed in in vitro studies (Figure 6). Iyer et al[185] found that human hepatic tissue homozygous for the (TA)7/(TA)7 polymorphism and tissue heterozygous for the (TA)6/(TA)7 genotype had a significantly decreased rate of glucuronidation of SN-38 and bilirubin compared with tissue containing the reference sequence allele [(TA)6/(TA)6]. SN-38 glucuronidation decreased in the following manner: 6/6 > 6/7 > 7/7[185].
In addition, Han et al[186] investigated the genetic variation of the UGT1A gene. They showed that two SNPs UGT1A1*6 (211G>A, Gly71Arg) and UGT1A9*22 were important factors influencing the metabolism of CPT-11 and the toxicity of therapy[186]. Both studied polymorphisms affect the coupling efficiency of SN-38 with glucuronic acid which results in serious toxic effects[186]. The UGT1A1*60 allele is related to the presence of SNP -3279T>G, and is located in the distal enhancer region [phenobarbital-responsive enhancer module (PBREM)], and is another of the genetic variants of UGT1A1 which contributes to the reduction in gene transcription activity and an increase in bilirubin concentration in serum[187]. UGT1A1*27 (686C>A, Pro229Gln) is a rare nonsynonymous polymorphism in the population, in vitro studies have shown its relation with a reduced level of glucuronidation of SN-38, and it has been observed in patients with symptoms of Gilbert’s syndrome[174]. Another nonsynonymous variant is UGT1A1*7 (1456T>G, Tyr486Asp) recorded in an Asian population and is associated with Crigler-Najjar syndrome type II[170] for which a decrease in activity of the enzyme deactivation pathway of SN-38 was observed[174].
The frequently occurring functional SNPs of the UGT1A7 gene include: UGT1A7*2 [387T>G (Asn129Lys), 391C>A, (Arg131Lys)], UGT1A7*3 [387T>G (Asn129Lys), 391C>A, (Arg131Lys), 622C>T (Trp208Arg)], and UGT1A7*4 [622C>T (W208R)][188]. For these SNPs in clinical in vitro studies conditioned by UGT1A7*3 and UGT1A7*4, the phenotype shows a reduced rate of glucuronic acid conjugation with SN-38[189]. In contrast to these genetic variants, a common VNTR polymorphism -118(T)9>10 (UGT1A9*22), which is located in the promoter region of the UGT1A9 gene is associated with increased transcriptional activity, which has been confirmed in vitro[190].
First evidence from clinical trials on the role of UGT1A1*28 in the development of toxicity resulting from administration of CPT-11 was published by Ando et al[191]. They studied the relationship of the genetic variants of UGT1A1 with serious toxic effects (grade 4 leucopoenia and/or grade 3 or 4 diarrhoea) in a group of 118 Japanese patients undergoing CPT-11 therapy in a variety of regimens[191]. Also Innocenti et al[192] studying a group of 66 patients (including 50 Caucasians) treated with CPT-11 alone, demonstrated that the UGT1A1*28 allele is an important factor in the development of grade 4 neutropenia. In this study, it was observed that the incidence of severe neutropenia was much more common in patients with genotype (TA)7/(TA)7 (50%) compared to heterozygous (TA)6/(TA)7 (12%) and homozygous (TA)6/(TA)6 (0%). Moreover, another genetic variant, -3156G>A, is in strong linkage with UGT1A1*28 and was a better predictor of toxicity than the UGT1A1*28 polymorphism[192]. Also Marcuello et al[182] studied the effect of the UGT1A1*28 variant on the occurrence of severe toxic effects in a group of 95 cases with CRC (Caucasians) who were treated with CPT-11 containing regimens (5-FU or raltitrexed). In this study, the incidence of acute diarrhoea (grade 3 or 4) was significantly higher in patients who were carriers of UGT1A1*28 mutations [homozygous (50%) and heterozygous (33%)] in comparison to homozygotes of wild-type (17%). Also, symptoms of neutropenia were more frequently noted in the homozygotes group with the UGT1A1*28 allele, however, this relationship was not statistically significant[182]. The first systematic analysis of clinical studies on the impact of UGT1A1*28 on the effectiveness of CPT-11 therapy was published by Dias et al[193]. These results were generally supportive of the clinical utility of genotyping UGT1A1*28 prior to commencement of CPT-11 therapy in order to decrease the risk of severe neutropenia and diarrhoea through the pre-emptive dose reduction of CPT-11 for UGT1A1*28 homozygotes. The meta-analyses indicate that there is unlikely to be an important association between UGT1A1 genotype and ORR with CPT-11, however, this does not provide direct evidence that a dose reduction for UGT1A1*28 homozygotes will not lead to an important reduction in ORR[193]. Hu et al[194] published a meta-analysis of the relationship between the presence of UGT1A1*28 and the incidence of neutropenia induced by CPT-11. It has been shown that the presence of UGT1A1*28 is associated with an increased risk of developing neutropenia, not only in cases of medium or high CPT-11 dose applied, but also in patients treated with low doses of the drug. The dose-dependent manner of SN-38 glucuronidation explained why the association between UGT1A1*28 and neutropenia was dose dependent[194]. Also, Hu et al[195] published a meta-analysis of clinical studies on the relationship between the presence of the variant UGT1A1*28 and the risk of severe diarrhoea. Also in this case, in patients who are carriers of one or two mutant alleles [genotypes (TA)7/(TA)7 or (TA)6/(TA)7] there was an increased risk of severe diarrhoea induced by CPT-11. However, this increased risk was present only in the group of patients with high and medium drug dose[195]. This evidence supports the assessment of UGT1A1*28 in routine clinical practice. The FDA-approved diagnostic blood test (Invader®) is available specifically for testing the UGT1A1*1 (wild-type) and the UGT1A1*28 genotype. However, the proposed benefit of testing CRC patients for UGT1A1 genotype is that the risk for adverse drug-related side effects (e.g., severe neutropenia) among patients found to be homozygous for the *28 genotype can be reduced by lowering their initial and/or subsequent doses of CPT-11. The concomitant harm is that a reduction in CPT-11 dosage may also reduce the effectiveness of chemotherapy in tumour suppression and long-term survival[133,196].
In recent years, several studies were published on the effects of UGT1A polymorphisms on CPT-11 effectiveness in CRC cancer therapy. Marcuello et al[182] observed a trend in reduced OS in patients with genotype (TA)7/(TA)7 or (TA)6/(TA)7 in a study of 95 (Caucasians) cases with metastatic CRC who underwent therapy based on CPT-11. The probable reason for poor response to treatment, as concluded by the authors, was the need to reduce the dose of CPT-11 in patients with symptoms of severe diarrhoea, and who were carriers of the mutant allele UGT1A1*28. Toffoli et al[177] studying a group of 71 patients (Caucasian) with CRC and metastasis observed that in the homozygous group (TA)7/(TA)7 there was a higher percentage of positive responses to the treatment based on CPT-11 and longer survival time as compared to the homozygous group (TA)6/(TA)6. The authors suggested that toxicities in (TA)7/(TA)7 patients could be well-managed during the entire course of treatment without a reduction of CPT-11 dosage[177]. The impact of genetic variants of UGT1A7 on the effectiveness of therapy with capecitabine/CPT-11 was examined[197]. The analysis of 66 cases of CRC (including 55 Caucasians) demonstrated that the homozygous groups UGT1A7*2/*2 and UGT1A7*3/*3 showed low enzymatic activity and a lower incidence of severe diarrhoea (P = 0.003), but a higher percentage of positive responses to treatment (P = 0.013) compared with the other genotypes[197]. Also, considering the impact of another polymorphism located in the sequence UGT1A9 [-118 (T)9>10, UGT1A9*22], it was observed that the presence of genotype (T)9/(T)9 significantly reduced the toxicity (P = 0.002) and increased the degree of response to treatment (P = 0.047)[197]. These results suggest that the low activity phenotype of isoenzymes UGT1A7/1A9 conditioned by the presence of genetic variants is associated with a protective effect against toxicity such as severe diarrhoea. The authors explained that this observation may be due to reduced excretion of SN-38G to the intestine, where it is under the influence of bacterial β-glucuronidase hydrolysed to SN-38, responsible for toxic effects such as severe diarrhoea[197,198]. This finding also raised caution that higher intestinal levels of SN-38G can promote diarrhoea, while hepatic glucuronidation offers protection against neutropenia[197].
Cecchin et al[176] performed genotyping of (UGT1A1*28, UGT1A1*60, UGT1A1*93, UGT1A7*3 and UGT1A9*22) in a large group of 250 CRC patients with metastases treated with the FOLFIRI regimen. In addition, the study determined the relationship of these genetic variants with the incidence of severe hematologic and nonhematologic toxicities, the degree of response to therapy, and TTP and OS[176]. The results demonstrated that only the variant UGT1A7*3 may be a marker of severe hematologic toxicity after the application of the first cycle of therapy (P = 0.04). In addition, UGT1A1*28 allele and II haplotype (all the variant alleles, but not UGT1A9*22) were associated with a response indicator of the therapy (P = 0.01), and the UGT1A1*28 allele was also the only marker associated with TTP. The authors concluded that genetic variants near UGT1A1*28 may be predictors in CRC patients treated with FOLFIRI[176]. Liu et al[199] examined the impact of a polymorphic variant UGT1A1*28 on toxicity and the results of treatment in a group of 128 Chinese CRC patients with metastases undergoing therapy with FOLFIRI. It was found that, although the need to reduce the dose of CPT-11 was significantly higher in patients with genotype (TA)6/(TA)6 (P < 0.01), it had no significant effect on the rate of response to CPT-11 therapy, PFS and OS[199].
The above reports make it difficult to draw clear conclusions whether reduced UGT1A activity conditioned by the presence of genetic variants in the gene sequence only intensifies the anti-cancer activity of CPT-11, or results in a better response to treatment with the simultaneous increased frequency of severe toxic complications. It seems that the overall balance of the effectiveness/toxicity of the therapy depends primarily on the treatment regimen used. Moreover, the appearance of severe toxicities depends on the exposure levels of SN-38 in the tissues, however, the antitumour responses can be influenced by additional factors related to properties of target tumours, such as the tumour stage, acquisition of resistant factors, and sensitivity to other chemotherapeutic agents when combined.
CYP3A4 and CYP3A5
CYP3A4, which is highly expressed in the liver, is considered one of the major P-450 cytochrome isoenzymes involved in the metabolism of a large group of drugs. CYP3A4 and CYP3A5 are responsible for CPT-11 oxidation to the APC metabolite (7-Ethyl-10-(4-N-aminopentanoic acid)-1-piperidino)carbonyloxycamptothecin and inactive NPC (7-Ethyl-10-(4-amino-1-piperidino)carbonyloxycamptothecin), which can be hydrolysed to an active form of SN-38 (Figure 5). Inter-individual variation in CYP3A4 activity may contribute to changes in the pharmacokinetic parameters of CPT-11[200-202].
Several polymorphisms located in genes CYP3A4 and CYP3A5 have been described[203-206]. There are different SNPs for CYP3A4 and the frequencies of genotypes and alleles occurrence in different populations have been published. Relatively frequent SNPs are CYP3A4*2 (664T>C, Ser222Pro), CYP3A4*10 (520G>C, Asp174His), and CYP3A4*17 (566T>C, Phe189Ser) in Caucasians and Mexicans (2%-5%), CYP3A4*15 (485G>A, Arg162Gln) in African-Americans (2%-4%) and CYP3A4*16 (554C>G, Thr185Ser) and CYP3A4*18 (878T>C, Leu293Pro) in East Asians (1%-10%)[207]. Perhaps some of these genetic variants of CYP3A4 may have impact on the pharmacokinetics of CPT-11. An analysis of gene haplotypes of CYP3A4 conducted in a group of 416 cases from the Japanese population has allowed the identification of 25 haplotypes[208]. However, the influence of individual haplotypes on the pharmacokinetic parameters of CPT-11 was tested among 177 Japanese patients undergoing chemotherapy[209]. Haplotype *16B which consists of polymorphisms 554C>G (Thr185Ser) and IVS10+12G>A was present only in male patients, and in this group a significantly lower concentration ratio of APC/CPT-11 (in vivo tests of CYP3A4 activity) was observed compared with other patients. However, no relationship was observed between the genotypes and total clearance of CPT-11, and the frequency of toxicity symptoms in the study group[209]. Despite significant individual variability[206] and occurrence of more polymorphisms within genes CYP3A4 and CYP3A5, in the currently published studies there is no significant correlation between genotype CYP3A4/5 and the pharmacokinetics of CPT-11 or toxicity[210,211]. No significant correlation between genotypes CYP3A4/5 and the pharmacokinetic parameters of CPT-11 may be associated with the low frequency of alleles in most described genetic variants of CYP3A in the Caucasian population (e.g., CYP3A4*17, CYP3A4*18, and CYP3A5*1), or the presence of these variants does not result in measurable changes in enzyme activity in vivo (e.g., CYP3A4*1B)[157]. In conclusion, the current research findings do not support the clinical use of CYP3A4/5 genotyping in order to differentiate individual doses of CPT-11.
ABC and SLC transporters
In addition to the importance of the metabolism of CPT-11, the influence of the above-mentioned enzymes on the pharmacokinetics of the drug, and its own influence can also be demonstrated on different transporters, especially from the ABC (ATP-binding cassette transporter superfamily) group of transporters. ABC transporters play an important role in the pharmacology of CPT-11[157], and are one of the major causes of cancer cell resistance observed in vitro and in vivo[212]. A number of polymorphic variants of genes encoding proteins of ABC transporters and their potential impact on the transcription/expression and changes in transport activity have been described[213]. CPT-11, SN-38 and SN-38G are transported from cells to the extracellular environment via ABCB1 multidrug resistance (MDR1), ABCC1 multidrug resistance protein 1 (MRP1), ABCC2 multidrug resistance protein 2 (MRP2), ABCG2 breast cancer resistance protein (BCRP) and SLCO1B1 organic anion-transporting polypeptide 1B1 (OATP1B1) (Figure 7)[214]. Transport proteins which export CPT-11 and its metabolites to bile and urine were examined due to their potential impact on the effectiveness of anticancer therapy, and the occurrence of adverse reactions[215,216].
Figure 7.
UDP-glycosyltransferase 1 family. A: The active metabolite of irinotecan, SN-38, is a DNA topoisomerase I (TOP1) inhibitor which leads to cancer cell death. TOP1 is a nuclear enzyme required in replication, responsible for unwinding DNA and preventing lethal strand breaks. SN-38 is cytotoxic and destabilizes the TOP1-DNA covalent complex formed in colorectal cancer cells. SN-38 causes irreversible double strand breaks which lead to S phase arrest followed by cell death. To do so, SN-38 attaches to the complexes and blocks future replication forks preventing repair of double strand breaks[405]; B: Irinotecan uptake and transport into the liver is facilitated by: OATP1B1 (SLCO1B1), ABCB1, MRP1 (ABCC1), MRP2 (ABCC2), and MXR (ABCG2). Specifically, ABCB1 is present on the bile membrane and is responsible for the secretion of irinotecan and its metabolites into the liver[406]. Irinotecan is metabolized in the liver and converted to SN-38, the active metabolite and TOP1 inhibitor, by carboxylesterases (CE) mediated hydrolysis. SN-38 is then glucoronized to SN-38 glucuronic acid (SN-38G) and detoxified in the liver via conjugation by the UGT1A family, which releases SN-38G into the intestines for elimination[407]. Approximately 70% of SN-38 becomes SN-38G, which has 1/100 of the antitumour activity and is virtually inactive. In the intestinal lumen, bacterial β-glucuronidases can reverse the reaction and transform inactive SN-38G back into the active form SN-38. This factor contributes to varied toxicity, specifically dose limiting diarrhoea[198].
Studies regarding the influence of transport protein P-glycoprotein encoded by the gene ABCB1/MDR1 on CPT-11 pharmacology, have given ambiguous results. More than a dozen different polymorphisms have been identified in the sequence of the gene ABCB1. Research evaluating the impact of SNPs on the pharmacokinetics of CPT-11 typically focus on three well-known polymorphisms 1236C>T, 2677G>T/A and 3435C>T, which are in strong linkage disequilibrium[157]. Some studies have shown that both single genetic variants and haplotypes of ABCB1 can increase the bioavailability of CPT-11 and SN-38[210,217], while other studies have come to the opposite conclusion[216,218]. Furthermore, Korean studies found an association between the presence of wild-type ABCB1 and the occurrence of neutropenia[218], which was not confirmed by the results from American research[216]. Similarly, a lack of correlation between the occurrence of SNPs ABCB1 and toxicity of CTP-11 therapy was found in French studies[179]. On the other hand, studies by Glimelius et al[219] demonstrated that patients who are carriers of the mutated allele ABCB1 are less responsive to treatment with CPT-11. Carriers of at least one TT genotype of ABCB1 1236C>T, 2677G>T/A or 3435C>T were less likely to respond to treatment (OR = 0.32). A post hoc analysis showed that fewer patients with at least one ABCB1 1236T-2677T-3435T haplotype responded to treatment compared with others (43% vs 67%, P = 0.027)[219]. Given the conflicting results obtained in earlier research on the impact of genetic variants of ABCB1 on the effectiveness of CPT-11 therapy[179,210,216-218], the conclusions presented by Glimelius et al[219] need to be confirmed in in vivo studies in a larger population.
Several in vitro studies have shown that ABCC1/MRP1 is involved in the transport of CPT-11 and SN-38. The ABCC1 transporter is responsible for the efflux of SN-38 from the hepatocyte into the interstitial space[220]. Polymorphisms 462C>T, 1684T>C, 4002G>A, 14008G>A, 34215C>G, IVS9+8A>G, IVS30+18A>G, IVS11-48C>T and IVS18-30C>G in the ABCC1 gene have been identified[210,216]. Two SNPs of ABCC1, 1684T>C and IVS18-30C>G, are responsible for differentiated pharmacokinetic phenotypes of CPT-11 as measured by the AUC values for its metabolites: APC and SN-38G/SN-38. Polymorphism 1684T>C contributes to an increase in AUC value for SN-38, and SNP IVS11-48C>T causes a decrease in AUC for APC. The positive association between ABCC1 1684T>C and SN-38 AUC is consistent with increased transport of SN-38 from the hepatocyte into the plasma[216]. In comparison to the available data on the role of ABCB1 in drug resistance and bioavailability of CPT-11, the clinical significance of the genetic variation of ABCC1 is not sufficiently documented, and therefore further functional studies should be carried out to confirm these preliminary observations[216]. There are several rare variants of ABCC1, which may potentially affect the transport function, but the low frequency of occurrence of these alleles hinders unequivocal conclusions regarding their clinical significance in pharmacotherapy of CPT-11[221-224]. Similarly, there is insufficient evidence regarding the effect of the polymorphisms in the gene expression of ABCC1 measured by mRNA levels in lymphocytes or duodenal enterocytes[225].
In vivo tests on animals showed that the biliary excretion of CPT-11 carboxylate and SN-38 carboxylate, and both the lactone and carboxylate forms of SN-38G was lower in ABCC2-deficient rats[226]. Moreover, the impact of gene polymorphisms ABCC2/MRP2 on the bioavailability of CPT-11 has been described. Innocenti et al[192,227] examining a group of 64 cancer patients showed that the silent polymorphic variant 3972T>C was associated with the AUC value of CPT-11 (P = 0.02), for APC (P < 0.0001) and for the APC/CPT-11 ratio (P < 0.0001). Kitagawa et al[228] also studied the effects of gene SNPs of ABCC2 on the toxicity of CPT-11 therapy. However, in the 120 Japanese patients studied, there was no association between genetic variants 1249G>A, or -24C>T gene ABCC2 and the incidence of severe complications after treatment with CPT-11[228].
There are many studies confirming the important role of protein ABCG2/BCRP in the transport of CPT-11 and its metabolites. Scientific evidence supports the proposition that overexpression of ABCG2/ABCG2 leads to the development of drug resistance in tumour cells against drugs that are derivatives of camptothecin such as topotecan[229], CPT-11 and SN-38[230-233]. Several possible mechanisms were described which may contribute to drug resistance conditioned by the activity of gene ABCG2, such as: demethylation of CpG islets in the ABCG2 promoter resulting in increased gene transcription[234], gene amplification[235], and truncation at the 3’UTR of the ABCG2 mRNA, which is associated with a loss of the miRNA-159c binding site conferring higher mRNA stability[236]. Furthermore, it has recently been demonstrated that the ABCG2 mRNA content of liver metastatic tumour cells from CRC patients treated with CPT-11 is higher than that from CPT-11-naive patients[207]. Cha et al[237] suggested that the presence of introning SNP in gene sequence ABCG2 (rs2622604) may contribute to changes in transport protein activity which can effect the increase in CPT-11 concentration in cells. This may lead to an increased risk of severe myelosuppression (grades 3 and 4) in patients with this genetic variant[237]. The same research team also identified another SNP (rs3109823), which like the previous one had a strong association with severe myelosuppression[237]. Following this study, Poonkuzhali et al[238] showed that a polymorphic variant of rs2622604 was associated with decreased expression of ABCG2 measured by the level of mRNA. These results support the hypothesis that patients who are carriers of the rs2622604 negative variant, have in their livers, a low level of SN-38 excretion to the bile which leads to the growth of intracellular concentrations of SN-38 in hepatocytes. This, in turn, contributes to accumulation of CPT-11/SN-38 in the blood and an increased risk of severe myelosuppression. On the other hand, although described by Cha et al[237], another SNP rs3109823 showed a stronger association with myelosuppression than the variant rs2622604, and Poonkuzhali et al[238] did not prove it had an effect on the gene expression level of ABCG2.
Functional in vitro studies on the importance of amino acid substitution in the sequence of protein ABCG2 (Gln141Lys, 421C>A) have shown that it contributes to the reduction of transport activity substrates such as mitoxantrone, topotecan, SN-38[239,240], and therefore can contribute to an increase in cell chemosensitivity[241,242]. There were also several in vivo studies published on the effect of this polymorphism on the pharmacokinetics of CPT-11. de Jong et al[243] studied a group of 85 patients diagnosed with solid tumours who received chemotherapy based on CPT-11. They reported greater accumulation of SN-38 and SN-38 glucuronide in one of two homozygous carriers of the 421 variant alleles. However, the AUC of CPT-11 (P = 0.72) and its active metabolite SN-38 (P = 0.67) did not differ significantly between patients carrying the wild-type sequence and patients carrying at least one variant allele[243]. Also, the results of research published by Jada et al[244] confirmed the findings that there is no relationship between the presence of genetic variants 421C>A gene ABCG2, and the change in the pharmacokinetics of SN-38. Available results from this study suggest that the probable coexistence of SNPs other than 421C>A genetic variants [e.g., 34G>A (Val12Met) and 1322G>T (Ser441Asn)] of the gene ABCG2 may have some clinical implications for the pharmacology of CPT-11. Furthermore, additional in vitro and in vivo studies are needed to better clarify the role of the 34G>A polymorphism as this SNP is prevalent in many populations and there are many conflicting reports regarding the functional effects of this polymorphism[245]. Systematic prospective studies with well-chosen and less heterogeneous groups of patients should be conducted to provide more reliable evidence on the role of gene polymorphisms of ABCG2 on the pharmacokinetics of CPT-11.
Organic anion-transporting polypeptide 1B1 (OATP1B1, SLCO1B1), expressed on the basolateral membrane in hepatocytes, has been reported to contribute to the hepatic uptake of SN-38[246]. SLCO1B1 transports among others, CPT-11, SN-38 and SN-38G from blood to liver cells. Several polymorphic variants of the gene SLCO1B1, among them SLCO1B1*1b (388A>G) and SLCO1B1*5 (521T>C), have been described. In vitro research on the haplotype SLCO1B1*15, which is a combination of the SNPs, showed that it is responsible for a 50% reduction in the intracellular concentration of CPT-11, which may cause intra-individual variability in the toxicity of this drug[246,247]. Another pharmacokinetic study revealed that CPT-11 clearance was 3-fold reduced and systemic exposure to CPT-11 was enhanced in patients with the SLCO1B1*15 haplotype[248]. The literature also describes the case of a patient with severe toxic complications after CPT-11 treatment and the presence of the haplotype *15[249]. The effect of these SNPs and haplotype *15 on induction of CPT-11 toxicity should be confirmed by further in vivo studies. Other studies on the toxicity of CPT-11 and its effects on different genetic factors were carried by Takane et al[250]. By analysing three genetic variants of UGT1A1*6, UGT1A1*28 and SLCO1B1*15 a strong correlation was found between the presence of these alleles and excessive accumulation of SN-38, which resulted in severe toxic complications observed with the use of CPT-11.
In summary, it can be stated that frequent polymorphisms in genes encoding ABC and SLC transporters can have a significant impact on changes in the pharmacokinetics and pharmacodynamics of CPT-11. However, the practical application of previously published results will require additional study in vivo including CRC patients.
Topoisomerase I, DNA repair genes and cell cycle regulation
There is substantially less knowledge on CPT-11 pharmacodynamics, including DNA damage repair or cell death pathways, following the formation of camptothecin-TOPI-DNA complexes[251]. SN-38 is an inhibitor of topoisomerase I (TOPI) an enzyme that prevents the unfolding of DNA during transcription and replication. Scientists studying cancer cells which exhibited resistance to CPT-11, found that a possible cause of low sensitivity to the drug may be associated with the presence of mutations or low TOP1 gene expression[252,253]. The impact of the presence of different genetic variants of TOP1 gene expression was described, which may be a cause of primary drug resistance[254]. Genetic variation in the drug target of SN-38, as well as in cellular effectors responsible for DNA repair and apoptosis, are a potential source of clinically observed inter-individual variability in the efficacy and toxicity of treatment based on CPT-11[255]. Knowledge of the causes of drug resistance leading to CPT-11 treatment failure, provides the opportunity to better plan treatment and to predict the effects of therapy for an individual patient. The activity of numerous genes and proteins[155,255] and a mutual network of connections between various intracellular pathways are responsible for the phenotype of sensitivity to CPT-11. The molecular factors involved in CPT-11 pharmacodynamics may include: drug target-TOPI, cell cycle division 45-like protein (CDC45L), nuclear factor-κB (p50 subunit; NFκB1), poly (ADP-ribose) polymerase I (PARP1), tyrosyl DNA phosphodiesterase (TDP1), and X-ray cross complementation factor (XRCC1)[256-260].
XRCC1 plays a key role in base excision repair by forming a complex with DNA repair proteins including PARP1 and DNA polymerase β[261]. Hoskins et al[251] studied a group of 107 (European) patients with advanced CRC, treated with CPT-11. They conducted an analysis of the impact of genetic variant 1196G>A (Arg399Gln) of the gene XRCC1 on the efficacy of CPT-11 therapy. They found that patients who demonstrated a favourable response to treatment more commonly had the genotype 1196GG variant allele than 1196T (genotypes GA or AA) (46% vs 26%, P = 0.10). Patients homozygous for an XRCC1 haplotype (GGCC-G) were more likely to show an objective response to therapy than other patients (83% vs 30%, P = 0.02). This effect was also confirmed in a multivariate analysis (OR = 11.9, P = 0.04)[251]. A possible explanation for these findings is that the presence of the allele in the 1196G gene sequence XRCC1 conditioning the presence of arginine in the protein sequence XRCC1 (399ARG) leads to weaker DNA repair capacity, as compared with 1196A (399Gln). However, these findings, derived from in vivo studies, have not been confirmed in numerous in vitro studies, which unanimously showed that the presence of glutamine in codon 399 was associated with a reduced ability to repair DNA as assessed by the persistence of DNA adducts, elevated levels of sister chromatid exchanges, increased RBC glycophorin A, TP53 mutations, and prolonged cell cycle delay[262]. Hoskins et al[251] also investigated the effect of the gene variant IVS4+61 TOP1 on the frequency of severe neutropenia (grade 3/4). The cause of the differences observed in vivo in the toxicity of CPT-11 therapy and the frequency of different variants of the TOP1 gene, can be related to the stability of complexes SN-38-TOPI-DNA in bone marrow cells, which may lead to greater sensitivity and increased bone marrow toxicity. Furthermore, Hoskins et al[251] found that patients who are carriers of the homozygous CC gene haplotype PARP1 (with SNPs combination 852T>C-IVS19-297C>T) often suffer toxic effects due to CPT-11 treatment in comparison to patients with different arrangement of alleles in this haplotype. This observation suggests that the presence of the haplotype 852C-IVS19-297C is related to decreased DNA repair capacity by PARP1 protein, leading to increased loss of bone marrow cells and symptoms of neutropenia as a result of the cytotoxic effect of CPT-11[251].
In vitro research using colon/colorectal carcinoma cell lines, showed that there is a link between the presence of functional aberration in p53 and phenotype hypersensitivity to camptothecins[263-266], whereby some of the experimental test models showed only moderate cellular sensitivity[267]. Moreover, HT-29 colon carcinoma cells characterized by mutations in p53 had a much higher sensitivity to CPT-11 than control cells expressing wild-type p53[268]. Also, experiments with cell clones derived from tumour tissues with evidence of impaired activity of p53 showed that the apoptosis induction path is an important determinant of sensitivity to camptothecins. On the other hand, p53 is required for targeting apoptotic proteins in the sensitization of colon carcinoma to TNF-related apoptosis-inducing ligand (TRAIL) pathway therapy using CPT-11[269]. Most experimental data show that the initiation of apoptosis resulting from exposure to camptothecins is much weaker for cells with wild-type p53 compared with mutated p53. Tomicic et al[270] proposed that the phenotype conditioned by wild-type p53, formed in the presence of CPT-11 complexed with DNA and TOPI is degraded more easily, leading to the reduced DNA transcription/replication effect of camptothecins and contributes to the development of drug resistance. In cells lacking functional p53, TOP1-cc (TOP1-cleaved DNA 3’-phosphotyrosyl intermediates referred to as cleavable complexes) is not efficiently degraded upon transcription stalling, thus TOP1-linked single-strand breaks accumulate, which may interfere with DNA replication. p53 defective cells are, due to lack of p21 expression, only transiently arrested in G2, having no time to repair excessive camptothecin-induced replication-dependent double-strand breaks (DSB), thus undergoing mitotic cell death accompanied by apoptosis[270].
Malfunction of DSB repair mechanisms is essential for the survival of cancer cells and is one of the major reasons why these cells avoid the cytotoxic effects of camptothecin derivatives. Therefore, it seems reasonable to state that cells with a compromised DSB repair mechanism may have greater susceptibility to therapy based on camptothecins. The main paths of the DSB repair mechanisms include homologous recombination (HR) and non-homologous end-joining (NHEJ). Mutations in genes RAD51, XRCC2, BRCA2, RAD54 and MUS81 involved in HR contribute to the hypersensitivity of cells exposed to camptothecins because the protein products of these genes are essential for proper functioning of the HR pathway in S and G2 phases of the cell cycle[270]. The results indicate that DSB induced in cells by derivatives of camptothecin are repaired either by NHEJ or HR[270-272]. As HR requires replication it might even be the predominant route of defence against the killing effects of camptothecins that require replication to elicit cytotoxicity[270]. In conclusion, the decisive role in the creation of phenotype drug resistance to CPT-11 is the status of p53, the degree of degradation of the TOPI complex from DNA, DSB repair by HR on stalled replication forks, and downstream pro- and anti-apoptotic pathways, while the NHEJ pathway seems to be much less important[270].
OX
Within the last 40 years, a few thousand platinum derivatives have been synthesised and tested with regards to their anti-cancer activities. Among these compounds, the most interesting ones seem to be those discovered in the early 70s, such as derivatives of the 1, 2-diaminocyclohexane (DACH) carrier ligand that are non-cross-resistant with cisplatin. In the last two decades, many scientists searching for new and effective cytostatic medicines directed their research efforts towards this platinum derivative group. Interest in the DACH group compounds is associated with their beneficial properties in comparison with other platinum derivatives such as cisplatin or carboplatin. Not only do DACH compounds demonstrate less nephrotoxicity (as opposed to cisplatin) and myelosuppression (as opposed to carboplatin), but they also have higher efficacy in cancer which proved to be resistant to treatment with cisplatin. Research results in both cell lines and in vivo observations prove that the efficacy of DACH compounds, in comparison to cisplatin and carboplatin, may be related to breaking inner resistance to these cytostatics. The significant cytostatic activity of OX was proved during tests on several human cancer cell lines and is believed to be the most important platinum derivative from the DACH group[273,274].
Combination therapy with 5-FU/LV plus OX (FOLFOX) is currently a standard in treating gastric cancer and CRC with a 40% positive response ratio during first relapse therapy[275]. Despite the efficiency of combined therapy, a high percentage of patients show drug resistance to a higher or lower degree, which suggest that the therapeutic efficiency of FOLFOX is characterised by high variability. Since approval of the clinical application of OX in the treatment of patients with advanced CRC in 1999 in Europe and then in 2004 in the United States, access to data concerning OX pharmacology has grown significantly. In preclinical studies, OX showed activity towards colon cancer cell lines characterised by primary and acquired resistance to cisplatin[132]. Also, in many other experimental models with a phenotype of resistance to cisplatin it was shown that the sensitivity/drug resistance profiles of both platinum derivatives were different[276].
Resistance to platinum compounds, as is the case with other cytotoxic compounds, is multi-factorial and individual platinum derivatives have different degrees of cross-resistance. Generally, in the majority of studies of experimental cancers, carboplatin has cross-resistance with cisplatin, but not with OX. On the basis of numerous studies, six major cell drug resistance mechanisms towards platinum derivatives, have been identified[277,278]. Processes connected with transporting to and from cells could be included here, as they contribute to lower intracellular drug concentration. Also, an increase in drug detoxication may be of importance (e.g., increased concentration of sulphydril-containing molecules or activity of metabolic enzymes) or an increase in the quenching of DNA monoadducts. Lastly, in the cells with resistance to platinum compounds, a system of recognition and/or DNA damage repair may malfunction[279].
Intracellular drug accumulation
Membrane transporters and channels, collectively known as the transporters, are some of the best known factors determining chemosensitivity and drug resistance and the history of research into their significance in anticancer therapy dates back to the beginning of scientists’ interest in the causes of chemotherapy failure[280]. Only a small group of the known transporters have been recognised as relevant for intracellular accumulation of platinum derivatives. There is a broad review concerning membrane transporters and channels that can be found in the publications of Choi and Kim[281], Hall et al[282] and Liu et al[283].
Potential platinum uptake or influx transporters include copper transporter (CTR) proteins[284], organic cation transporters (OCTs) belonging to the SLC22 family[285] and an undefined cis-configuration specific platinum influx transporter[286]. In addition, some outward-directed drug transporters facilitating the active efflux of platinum compounds have been linked to decreased accumulation of platinum compounds and include adenosine triphosphate (ATP) binding cassette (ABC) multidrug transporters[287], and copper-transporting P-type adenosine triphosphatases (ATPases) (Figure 8). Insufficient intra-tumour concentration of platinum compounds is a critical factor determining both primary and secondary resistance. Lowered inflow and/or increased activity of outward-directed cellular transport is a frequent phenomenon in clones of chemoresistant cancer cells[280] exposed to cisplatin, OX[288] and carboplatin. However, currently, it is not quite clear whether and to what degree transporters help maintain therapeutic platinum concentrations in cancer cells, thus playing a crucial (clinically relevant) role in sensitivity and cell resistance to platinum derivatives[283]. During the last 15 years, a series of clinical studies have been designed to establish the connection between efficiency of chemotherapy based on OX and the level of expression of membrane transporters in both cancer cells and in healthy tissue. These studies of transporters including ATP7A, ATP7B, ABCC2, ABCG2, ABCB1, OCT2 and CTR1 are detailed below and summarized in Table 4.
Figure 8.
Intracellular drug accumulation. The free fraction of oxaliplatin is biotransformed non-enzymatically and subsequently forms complexes with chloride, glutathione (GSH), methionine (Met) and cysteine (Cys). Oxaliplatin undergoes non-enzymatic conversion in physiologic solutions to active derivatives via displacement of the labile oxalate ligand. Several transient reactive species are formed, including monoaquo DACH (1,2-diaminocyclohexane) platinum [Pt(H2O)Cl(DACH)]+ and diaquo DACH platinum [Pt(H2O)2(DACH)]2+, which covalently bind with macromolecules. There is no evidence of cytochrome P450-mediated metabolism in vitro. The major route of platinum elimination is renal excretion. The main mechanism of action is mediated through the formation of DNA adducts which is thought to be related to the anti-tumour effects of oxaliplatin. An important factor is the induction of apoptosis by the primary DNA-Pt lesions, which is possibly enhanced by the contribution of targets other than DNA. Several influx and efflux transporters such as organic cation transporters (OCTs) 1, 2 and 3 (SLC22A1, SLC22A2 and SLC22A3), copper efflux transporters (CTRs), P-type ATPases, ATP7A and ATP7B have been identified, which may play an important role in determining tumour sensitivity and/or resistance to oxaliplatin[408].
Table 4.
Selected common polymorphisms of MDR1, GSTP1, ERCC1, ERCC2, XRCC1 genes and their potential impact on functioning of proteins related to OX pharmacology
| dbSNP rs cluster ID | Type of polymorphism | Function | Ref. |
| Multidrug resistance 1 (MDR1, ABCB1) (OMIM #171050) | |||
| rs1128503 | SNP | [152,296,318,485] | |
| 1236C>T | Synonymous, effect unknown | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 1128503 | |||
| rs2032582 | SNP | [152,296] | |
| 2677G>T/A | Ser893Ala or Ser893Thr, the GG genotype carriers have the highest while the AT genotype carriers have the lowest levels of ABCB1 expression | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 2032582 | |||
| rs1045642 | SNP | [152,296,350,485] | |
| 3435C>T | Synonymous, TT genotype carriers have lower intestinal ABCB1 expression | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 1045642 | |||
| Glutathione S-transferase π (GSTP1) (OMIM #134660) | |||
| rs1138272 | SNP | [311,477] | |
| 341C>T | Ala114Val, altered enzyme kinetics, altered toxicity | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 1138272 | |||
| rs1695 | SNP | [51,180,311-329,467,477] | |
| 313A>G | Ile105Val, decreased enzymatic activity, altered toxicity | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 1695 | |||
| Excision repair cross-complementation group 1 (ERCC1) (OMIM #126380) | |||
| rs11615 | SNP | [51,313,344,345,357,486] | |
| 354T>C | Synonymous, decreased transcriptional activity of ERCC1 | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 11615 | |||
| rs3212948 | SNP | [487] | |
| 321+74C>G | Intronic SNP (intron 2), protective effect of the C allele to cancer risk | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 3212948 | |||
| Excision repair cross-complementation group 2 (ERCC2, XPD) (OMIM #126340) | |||
| rs13181 | SNP | [51,313,336,337,350,351,353,356,357,486] | |
| 2251A>C | Lys751Gln, the Gln allele is associated with a higher DNA adduct level or lower DNA repair capacity | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 13181 | |||
| rs1799793 | SNP | [313,336,337,353] | |
| 862G>A | Asp312Asn, lower DNA repair capacity for the Asn allele than the Asp allele | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 1799793 | |||
| X-ray cross complementation factor (XRCC1) (OMIM #194360) | |||
| rs25487 | SNP | [51,313,349,350,361-364,486] | |
| 1196A>G | Arg399Gln, reduced base excision repair function for Gln allele than the Arg allele | ||
| http://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?rs = 25487 | |||
SNP: Single nucleotide polymorphism.
The first clinical studies concerning the dependency between the results of treatment with platinum compounds in cancer chemotherapy and the expression of transporter concerned the P-type copper transporting ATPases ATP7A and ATP7B. In a study of 50 patients with an advanced stage of CRC and treated with 5-FU/LV/OX (FOLFOX) a correlation was observed between resistance and the level of expression of these transporters[289]. ATP7A and ATP7B involved in the sequestration and extrusion of copper from a compartment localized within the trans-Golgi network to the plasma membrane, have also been implicated in the efflux of platinum compounds[290]. While examining their CRC patients, Martinez-Balibrea et al[289] showed that low expression of the ATP7B gene measured by its level of mRNA was linked with significantly longer TTP (P = 0.0009) as opposed to the group of patients with a higher level of mRNA (12.14 mo vs 6.43 mo) who also had a greater risk of disease progression (HR = 3.56, P = 0.002). Furthermore, patients with both a low level of mRNA and ATP7B protein noted, had the longest TTP and benefitted from FOLFOX therapy most, as opposed to patients with a high level of mRNA and protein (14.64 mo vs 4.63 mo, respectively, P = 0.01)[289].
Various multidrug resistance-associated proteins (MRPs) belonging to the ABCC subfamily of ABC efflux transporters have been implicated in mediating resistance to platinum compounds[291]. Cancer cells resistant to platinum compounds are able to remove OX metabolites that are coupled with glutathione (GSH) into the intracellular environment via ATP transport dependent on hydrolysis through biological membranes[292]. On the basis of the above mechanism, it may be assumed that GHS accessibility and the effectiveness of conjunction with GHS are the key factors for the development of such resistance towards OX. Beretta et al[293] stated that some of the superfamily ABC transporters (ABCC1/MRP1 and ABCC4/MRP4) had significant expression in ovarian cancer cells with secondary OX resistance. Overexpression of ABCC1 or ABCC4 in cancer cell lines derived from ovarian cancer cells was connected with resistance to cisplatin and OX. The above results prove that the development of OX resistance is induced by the activity of MRP proteins, and it may be conducive to use cytostatics other than platinum derivatives that are not substrates of ABCC1 or ABCC4[293] in patients with relapsing cancer previously treated with OX. Furthermore, in other research it was observed that administering 5-FU inhibits the expression of ATP7B and human organic cation transporter 2 (OCT2) with a simultaneous 5.8-fold increase in the level of mRNA for the ABCC2 gene (MRP2) coding another transporter from ABCC[294]. Theile et al[294] proposed as one mechanism for FOLFOX synergism, the 5-FU mediated suppression of ATP7B, the overexpression of glutathione exporters such as MRP2 and the decrease in glutathione levels by the OX metabolite oxalate.
In studies of another transporter from the superfamily of ABC - ABCG2/BCRP it was found that overexpression may be a negative marker of OX therapy effectiveness[294]. Lin et al[295] tested the level of expression of protein ABCG2, measured by the IHC method, in a group of patients with CRC both in the primary and metastatic cancer tissue. They observed that lower expression of ABCG2 was noted more frequently in patients with better response to FOLFOX therapy than in patients with higher protein expression (63.6% vs 9.5%, respectively). Moreover, it was found that in the majority of cases the level of ABCG2 expression was higher in tissue derived from metastatic tissue than from primary tumours[295]. Therefore, Lin et al[295] concluded that ABCG2 expression is related to response to therapy based on FOLFOX among patients with metastatic CRC and that ABCG2 may be a selective marker in predicting the effectiveness of FOLFOX.
Wu et al[296] evaluated the influence of SNPs of ABCB1/MDR1 gene (1236C>T, 2677G>T/A and 3435C>T) on the outcome of treatment in CRC patients treated with OX-based therapy. Carriers of the 1236C>T variation of the ABCB1 gene had longer OS following post-operative OX therapy. Additionally, carriers of the 1236TT-2677TT-3435TT genotype combination had worse PFS (P = 0.043) and recurrence-free survival (P = 0.006)[296]. On the other hand, Yue et al[297] showed that SNPs of the ABCB1 gene were not pharmacogenetic factors which determined prognostics for chemosensitivity to OX-based therapy in CRC patients.
The SLC22 family of transporters includes several subgroups of proteins classified on the basis of position and transporting mechanisms. The subgroup of organic cation transporters (OCTs) consists of only three members: SLC22A1 (OCT1), SLC22A2 (OCT2) and SLC22A3 (OCT3)[285]. Currently, we have a limited range of accessible data concerning the connection between genetic variations and the level of OCT1 or OCT2 expression in tumour tissue and the results of treatment after administering therapy based on platinum derivatives. It is, however, postulated that these transporters may be of potential clinical importance as predictive markers. In an experimental model using transfected cells it was noted that the expression of the OCT1 gene significantly increased intracellular OX accumulation[298]. On the other hand, research results showed that OX is an excellent substrate for OCT2[298,299]. Zhang et al[298] showed that in transfected HEK293-hOCT2 cells, the amount of accumulated OX was 23.9-fold greater than that in control cells. Whereas, in the presence of cimetidine, which is an OCT2 inhibitor, the amount of accumulated OX was significantly lower. They also stated that in the transfected cells, the cytotoxic effect significantly increased following treatment with OX compared with control cells[298]. It is thought that OCT2 expression may modulate the sensitivity of CRC cells to OX. It is also postulated that the level of OCT2 expression may condition drug resistance in CRC patients treated with therapy based on a scheme including platinum[298]. However, the results of the above studies are not fully credible as while testing OCT2 expression in tissue, it was noted that a positive result was obtained in 11 of 20 tissue samples from patients with colon cancer, while a negative effect was obtained in 4 healthy tissue samples[300]. In contrast, all colon cancer cell lines investigated for transporter gene expression were found to lack OCT2 mRNA expression[298,300]. Therefore, it is worth stressing that if a significant role of OCT2 was proved to mediate transport of platinum derivatives in pre-clinical studies[298], the results of clinical studies do not confirm this observation.
The role and significance of copper influx and transporters efflux (CTRs) in cell accumulation of platinum compounds has been widely discussed in the literature[284,301,302]. CTR1 is an important transporting protein that is responsible for regulating copper concentrations, ensuring the biological balance of copper ion concentration. When the copper concentration is too low this leads to deactivation of enzymatic systems dependent on copper ions, whereas when the concentration is too high it causes cell toxicity[303]. Holzer et al[304] put forward a thesis that CTR1 plays an important role in OX accumulation only when exposed to a relatively low concentration (2 μmol/L) and does not have any relevance at higher OX concentrations. Furthermore, it is postulated that intracellular OX concentration is less dependent on the transporting activity of CTR1 than that of other platinum derivatives, e.g., cisplatin and carboplatin. Additionally, it was shown that similar to CTR1, CTR2 may also have analogical properties as a cisplatin and carboplatin concentration regulator and possibly OX as well[305]. Further in vivo research confirming the above hypotheses is necessary.
Clinical studies concerning transporters for platinum derivatives have concentrated on evaluation of the connection between intratumour expression of certain transporters and the results of treatment after chemotherapy based on platinum derivatives. The results of these studies are not completely certain due to many limitations. One of these limitations is the lack of functional research into transporting activity as accessible data focus on gene or protein expression using methods such as RT-PCR or IHC, respectively. Generally, correlations observed in the research were not supported by the analysis of pharmacokinetic variables in relation to accumulation of platinum derivatives in the tumour tissue, and the size of individual groups was small. Furthermore, it is necessary to conduct in vivo research into the meaning of genetic variability of membrane transporters and channels for gene expression and their influence on the pharmacokinetics and effectiveness of OX-based therapy.
Glutathione S-transferases
The phenotype of resistance to platinum derivatives may be dependent on the variable activity of detoxification channels. In the cytoplasm, platinating agents become acquated, which then enables them to react with thiol-containing molecules, including GSH and metallothioneins (Figure 8). In the cell, GSH plays the role of antioxidant which helps maintain a reductive intracellular environment by coupling oxidated particles with sulphydryl groups. It is assumed that high GSH concentration and/or metallothionein may cause deactivation of platinum compounds before they have a chance to interact with DNA in the nucleus (it is estimated that only 1% of the dosage that enters the cell stands a chance of bonding with nuclear DNA[306]) to quench Pt-DNA monoadducts before conversion to more lethal diadducts, or the efflux of Pt-glutathione conjugates[307,308]. There is ample evidence to show that glutathione S-transferers (GSTs) belonging to the superfamily of dimeric enzymes of the second metabolism phase are responsible for a differential sensitivity profile towards anticancer drugs, including platinum derivatives[309]. GSTs are coded by genes belonging to at least five main groups: α (GSTA1), μ (GSTM1), π (GSTP1), σ (GSTS1) and θ (GSTT1). Many of these genes have genetic polymorphisms that influence their transcription and/or enzymatic activity of the proteins coded by them[310]. One of the isoenzymes from the GSTs family - GSTP1, has high expression in CRC tissues and partakes in detoxication processes of platinum derivatives, therefore, it may be a source of drug resistance in some patients treated with therapy based on cytostatics that are platinum analogues. The published research suggests a connection between some of the polymorphic variables of GSTP1 gene and the increase in effectiveness of anticancer therapy[51].
Two major polymorphisms in GSTP1 - 313A>G (Ile105Val) and 341C>T (Ala114Val) - induce amino acid changes in the electrophile-binding active site of the enzyme[311]. SNP 313A>G, responsible for substitution of isoleucine through valine in codon 105 (Ile105Val) causes lowered enzymatic activity of GSTP1[312]. There are a few clinical studies available which refer to the influence of this polymorphism on the frequency of occurrence of toxic effects due to FOLFOX or IROX therapy (CPT-11/OX) in patients with metastatic CRC[180,313,314]. McLeod et al[180] state that in a group of patients treated with FOLFOX, who were homozygous for the 105Val variation, treatment discontinuation was more frequent due to symptoms of neurotoxicity (P = 0.01). However, the necessity to discontinue therapy was not dependent on the frequency of occurrence of individual genotypes in groups treated with other combinations (IROX or capecitabine/OX). Most probably, the presence of the 313GG genotype is connected with significant lowering of the catabolic activity of GSTP1 than it is the case of allele 313A carriers (genotypes 313AG or 313AA), which leads to increased OX accumulation and thus a greater risk of 3rd degree neurotoxicity[313,314]. On the other hand, Inada et al[315], while examining CRC patients, demonstrated that genotype 313AA carriers were more likely to develop early OX-induced grade 1 peripheral neurotoxicity than patients with 313G alleles (313AG or 313GG), but they did not observe a connection between the frequency of these genetic variations and the risk of grade ≥ 2 neurotoxicity. In addition, the results of other research did not confirm the existence of SNP 313A>G dependence and neurotoxicity of OX therapy[316-321].
As replacing isoleucine with valine (Ile105Val) leads to a lowering of the cell’s ability to protect itself against cytotoxic factors, this polymorphism may contribute to an increase in chemosensitivity to OX[312]. A few clinical studies showed that patients with the 313GG genotype benefitted more from combined therapy including OX than patients with the 313A allele[51,322-324]. However, three recently published studies on the efficiency of FOLFOX in patients with advanced CRC, on the basis of genotyping GSTP1 gene for SNP 313A>G, showed no connection between the presence of the allele and PFS[313,321,325]. Ye et al[326] performed a systematic analysis of five clinical studies[314,325,327-329] involving 415 CRC patients treated with OX. In this analysis, no dependence between the 313A>G polymorphism and the level of response to OX-based therapy (P = 0.13) was confirmed[326]. In order to put forward any definite conclusions concerning the predictive significance of SNP 313A>G, it is necessary to carry out clinical research on a large group of patients.
Among the available clinical data, studies on copy number variations (CNV) of GSTT1 and its potential influence on the toxicity of OX-based therapy have been observed. While investigating CNV of GSTT1, Goekkurt et al[330] found no statistically relevant dependence between genetic variables of this gene and the frequency of toxic effects due to therapy in patients with gastric cancer, although there was a trend showing that patients with the null variant were less likely to develop hematologic toxicity. Two other clinical studies of patients with metastatic CRC treated with OX did not confirm the hypothesis of the potential influence of CNV of GSTT1 on therapy toxicity[316,317]. It is necessary to conduct further research which would clearly resolve the role of genetic GSTs variability in the development of toxicity in CRC patients undergoing treatment which includes OX.
Nucleotide excision repair pathway (ERCC1, ERCC2, XRCC1)
Blocking the process of DNA replication using platinum derivatives by creating adducts with nuclear nucleic acid leads to the induction of apoptosis and the death of cancer cells[331,332]. The observed inter-individual variability in the ability to recognise and repair such DNA damage through the nucleotide excision repair (NER) pathway is one of the factors that may influence the success of OX-based therapy. DNA strands are separated and a DNA residue containing the adducts is removed (Figure 9). The mechanism of recognition and repair of the damaged DNA fragments itself is dependent on several factors. Lowered efficiency of the DNA repair system may, in consequence, lead to the increased sensitivity of cancer cells to therapy which includes platinum compounds[333]. excision repair cross-complementation group 1 (ERCC1) and ERCC2 protein [otherwise known as xeroderma pigmentosum group D (XPD)] are the two main compounds of the NER group that play a crucial role in regulation of the activity of other elements that are part of the NER pathway. Together with xeroderma pigmentosum group F (XPF) protein, ERCC1 is responsible for recognising these places in the DNA strand where adducts are located, whereas ERCC2 is a subunit of human transcriptional initiation factor TFIIH with ATP-dependent helicase activity[334]. Considering the above, it may be assumed that functional SNPs in ERCC1 and ERCC2 genes may directly contribute to the phenotype sensitivity to platinum compounds, such as OX, through conditioning congenital suboptimal activity of the NER pathway. For genes ERCC1 and ERCC2, there are several frequent and probably functional SNPs described, among them are 354C>T and 8092C>A in the ERCC1 gene, which contribute to the changes in activity measured by the level of mRNA[335] and ERCC2 SNPs 312G>A gene (Asp312Asn), and 2251T>G (Lys751Gln) are recognised as determinants of suboptimal activity of the DNA repair system[336,337]. Study results suggest that ERCC1 is a potential predictive marker of response to therapy based on platinum compounds due to the fact that low ERCC1 expression is connected to cancer cells’ sensitivity to chemotherapy with those drugs[34,338-340].
Figure 9.
Nucleotide excision repair pathway. (1) DNA damage formed by platinum agents leads to DNA double helix distortion. Several distinct complexes are involved in sequential steps than can be summarized as DNA damage recognition (XPCHR23B), damage demarcation, and verification (TFIIH), assembly of a preincision complex (RPA and XPA) and helix unwinding (XPB and XPD); (2) Endonuclease recruitment with dual incision of the damaged strand on the 5’ side (ERCC1-XPF heterodimers) and 3’ side (XPG) followed by the removal of the excised oligomer; (3) DNA repair synthesis to fill in the resulting gap; and (4) ligation. ERCC1: Excision repair cross-complementation group 1; Pol σ/ε : Polymerase σ/ε; RFC: Replication factor C; TFIIH: Transcription factor II H; XP (A,B,C,D,F,G): Xeroderma pigmentosum complementation group (A,B,C,D,F,G)[340].
Shirota et al[34] were the first research group to study the influence of ERCC1 gene expression on the results of treatment in 50 patients with advanced stage CRC and the phenotype of resistance in those treated with 5-FU/OX. They stated that patients with high intra-tumour ERCC1 expression measured by mRNA level had shorter survival time than patients with a lower level of expression (P = 0.008)[34]. Uchida et al[341], while examining 91 patients treated with a combination of capecitabine/OX stated that a high mRNA level for the ERCC1 gene was associated with shorter time to treatment failure compared to patients with lower expression (P = 0.046). In another study, low expression of the ERCC1 gene was also associated with better response to both primary (P = 0.047) and secondary chemotherapy, although in the latter case this association was on the verge of statistical relevance (P = 0.054). Furthermore, high expression of the ERCC1 gene was related to shorter OS in primary therapy (P = 0.014)[342]. The above results from clinical studies support the hypothesis put forward at the beginning regarding the influence of ERCC1 gene expression on the results of treatment with platinum derivatives, whereas a high level of mRNA may be the cause of clinical resistance to OX.
The literature also describes polymorphisms located in the ERCC1 gene sequence, one of them being a silent SNP 354C>T (Arg118Arg). Although the mechanism through which this SNP influences the change in ERCC1 activity is not fully known, it is postulated that AAC codon exchange on a rarely occurring AAT influences the effectiveness of the translation process, however, for 354T allele, there is a decrease in protein expression of about 50%[343]. In two clinical studies of patients with advanced CRC, it was observed that carriers of the 354TT genotype had higher response rates to OX treatment[344] and longer PFS[345]. However, in five other studies, the survival time of patients with CRC was longer in genotype 354CC carriers[51,313,314,339,346]. While examining 168 patients, Chang et al[346] showed that in a group with genotypes which included allele 354T (354CT or 354TT), poorer treatment results were noted in comparison with those of patients with genotype 354CC [in terms of response (P = 0.01), PFS (P = 0.01) and OS (P = 0.01)]. Additionally, while evaluating the association between genetic variants 354C>T and protein expression determined by IHC, it was shown that a higher level of expression was related to the presence of allele 354T[346]. In addition, Chen et al[314], while examining 166 patients, pointed out that carriers of genotypes with at least one 354T allele were characterised by poor response (P = 0.01) and shorter OS (P = 0.01). Park et al[339] also found a significant correlation between polymorphic variants in codon 118 and treatment outcome in 106 patients with advanced refractory CRC receiving 5-FU/OX. For patients with genotype 354CC, median survival time was 15.3 mo, while in a group of allele 354T (354CT and 354TT genotypes) carriers it was only 11.1 mo.
Partially different from fluoropyrimidine genes previously described, the frequency of these polymorphisms varied with race and may account for reduced response rates in Black patients compared with Caucasian patients, as expressed by Goldberg et al[347] and confirmed in more recent studies, as in the subgroup of patients in the CAIRO study[110]. It is postulated that the differences in the observed associations and the strength of the correlations may be connected with inter-population differences in the frequency of occurrence of alleles and genotypes. For instance, the frequency of occurrence of SNP 354C>T (Arg118Arg) in an East Asian population was much lower than that in other ethnic groups[340].
The presence of allele 354T in the ERCC1 gene is connected with the change in the expression of gene/protein[339], while allele 2251G which is a variation of the ERCC2 gene was described as having influence on a low number of X-ray induced chromatic aberrations[336]. Carriers of genotype 2251TT had a 7-fold greater risk of suboptimal repair of DNA damage compared to carriers of allele 2251G (genotypes 2251GG or 2251GT)[336]. It is postulated that patients who have both allele 354T (ERCC1) and 2251G (ERCC2) that are connected with a highly efficient detection system and DNA damage repair, may have resistance to OX, thus contributing to a worse prognosis. However, the results of clinical studies do not confirm the above hypothesis. The 2251T>G (Lys751Gln) polymorphism did not show any relation with survival time compared with the frequency of genotype dispersion in patients with gastro-oesophageal cancer[348,349] and CRC[350,351] who underwent treatment based on various platinum derivatives. Whereas, studies of the synonymous SNP Arg156Arg (C>A) ERCC2 gene carried out in patients with gastric cancer treated with OX showed that carriers of A allele (genotypes CA or AA) were characterised by a higher response rate and longer TTP compared to patients with genotype CC[352]. A similar trend was observed in the studies by Park et al[353], who examined patients with metastatic CRC, and noted that the presence of A allele contributed to better treatment response and longer median survival compared to patients with different variants of the ERCC2 gene. Functional studies confirmed the SNPs influence of the ERCC1 (354C>T) and ERCC2 (2251T>G) genes on the phenotype of NER pathway efficiency[335,354,355]. In a study of 73 patients treated with 5-FU/OX it was observed that in patients with the genotype 2251TT (751Lys/Lys) median survival time was 17.4 mo, while for carriers of genotypes with the 2251G allele it was 12.8 mo (751Lys/Gln) and 3.3 mo (751Gln/Gln) (P = 0.02)[353]. The influence on genetic variants of the genes ERCC1 and ERCC2 was also studied in a group of 166 metastatic CRC patients who were treated with a combination of 5-FU/LV/OX (FOLFOX4)[356]. In the analysis of associations between SNPs and the results of treatment it was shown that the occurrence of each of the genotypes ERCC1-354TT, ERCC2-2251AC and ERCC2-2251CC, independently of each other, was related to shorter PFS. The median PFS was 11.2 mo for patients without any of the three genotypes, 9.8 mo for those with one of the high-risk genotypes, and 8 mo for those with both the ERCC1-354TT and either ERCC2-2251AC or -2251CC genotypes (P = 0.002)[356]. In the meta-analysis published by Yin et al[357] it was shown that SNPs 354C>T (ERCC1) and 2251T>G (ERCC2) may be clinically useful in the evaluation of treatment results in patients with gastric cancer and CRC who underwent treatment which included OX (FOLFOX or XELOX). However, as the authors of this analysis emphasise, it is necessary to carry out wide and well-planned prospective clinical studies to clearly show the utility of these markers in clinical practise[357].
Apart from studies which focused on the analysis of individual determinants of therapy efficiency such as SNPs, a joined analysis of a few potential predictive factors in forecasting the effects of chemotherapy in CRC patients was also carried out. Kim et al[358] assessed the expression of proteins ERCC1, TS and GSTP1 using IHC for potential application in predicting the effects of therapy in 70 patients with advanced stage CRC who underwent treatment with 5-FU/OX. They observed that positive expression occurred in 55.7% (ERCC1), 68.6% (TS) and 71.4% (GSTP1) of the analysed cases. It was confirmed that a low level of TS expression was related to better chemotherapy outcome (P = 0.009), however, in the case of ERCC1 and GSTP1 proteins there was no statistically relevant association between the level of expression and efficiency of treatment (P = 0.768, P = 0.589, respectively). The median OS was significantly longer in patients with negative ERCC1 protein expression (P = 0.0474). Additionally, patients with positive expression of both ERCC1 and TS had poorer OS (P = 0.0017). Also, multi-variant analysis confirmed that positive expression of ERCC1 and TS significantly influenced OS (HR = 1.72, P = 0.023), which justifies simultaneous clinical application of the two markers for predicting the efficiency of 5-FU/OX therapy[358].
Apart from the NER pathway, the base pair excision repair pathway (BER) may also influence the efficiency of therapy based on platinum derivatives. XRCC1 plays a key role in the BER pathway and it has been demonstrated that the Arg399Gln (1196A>G) substitution in the XRCC1 gene is associated with increased levels of DNA damage markers[359]. This relatively frequently occurring polymorphism probably contributes to the change in XRCC1 protein conformation in the domain binding other elements of the BER complex, which may lead to a decrease in the efficiency of the DNA repair system. A deficiency in DNA repair pathways has been shown to confer resistance to several drugs, including platinum compounds[360]. It was shown that the presence of allele 399Arg (1196A) is associated with better survival time in patients with gastric[349] and lung cancer[361] undergoing chemotherapy with platinum derivatives. Also, Suh et al[362] observed that better treatment outcomes in patients with metastatic CRC treated with FOLFOX occurred in those where the presence of allele 399Arg (1196A) was noted. However, the results of other clinical studies published in patients with advanced CRC and gastric cancer treated with OX, did not confirm the above observations[51,313,350]. Liang et al[363] attempted to analyse the influence of both polymorphisms on genes engaged in DNA repair processes: ERCC1 (354C>T) and XRCC1 (1196A>G). They studied a group of 113 patients diagnosed with metastatic CRC who underwent chemotherapy that included OX. The analysis of individual SNPs showed no significant influence of these polymorphisms on prediction of disease control rates (DCR) or OS (P = 0.662 and 0.631, respectively). However, while evaluating the influence of the combination of both SNPs, a significant correlation between genetic variations of ERCC1 (354C>T) and XRCC1 (1196A>G), DCR (P = 0.01) and OS (P = 0.001), were independently observed. This was the first study to prove the importance of the clinical application of genetic determinants located in ERCC1 and XRCC1 genes in the selection of patients with metastatic CRC who were expected to benefit most from OX-based therapy[363]. Subsequent results obtained by Stoehlmacher et al[364], who studied the influence of Arg399Gln (1196A>G) polymorphism on the efficiency of treatment with 5-FU/OX in 61 patients with metastatic CRC, confirmed the significance of this SNP as a predictive marker. Seventy-three percent of patients with the favourable 399Arg/Arg (1196AA) genotype responded to treatment, and patients who possessed at least one 399Gln (1196A) allelic polymorphism in XRCC1 were 5.2-fold more likely to fail 5-FU/OX chemotherapy[364].
Among the available data, one clinical study conducted a multivariate analysis of a few of the predictive factors described above in patients with refractory CRC who underwent treatment with the 5-FU/OX combination. Analysis of multiple gene polymorphisms proved that the efficiency of such therapy may be dependent on the presence of two or more unfavourable variants for genes ERCC1, ERCC2, TYMS and GSTP1 as the carriers of these SNPs were characterised by a significantly shorter OS[51]. In summary, for the successful prediction of the effectiveness of a particular therapy, a few predictive markers need to be applied where several cytostatic drugs are used in a combination therapy.
MMR and apoptosis regulation
The cytotoxic effects caused by OX are stronger than those caused by cisplatin due to the result of a stronger reduction in DNA damage[365]. Resistance to cytostatic platinum derivatives is probably the result of variable functionality of the proteins responsible for recognising damage resulting from Pt-DNA adducts[366]. MMR is a highly conserved, strand-specific repair pathway which is a multi-stage process initiated when DNA damage is recognised by specific proteins[367]. In many types of cancer, various defects in activity of these proteins are noted, particularly three proteins: MSH2, MSH6 and MLH1[368]. In a situation when MMR shows a deficit in activity, this results in the accumulation of numerous types of DNA damage in the genome, which leads to MSI[369]. Experimental data have shown that MMR deficits are associated with resistance to the cytotoxic activity of alkylating agents[370]. Studies of DNA repair mechanisms after exposure to cisplatin showed that Pt-DNA adducts are recognised by the complex of MMR proteins[371]. The MMR pathway is one of the factors influencing cisplatin activity, which was proved by pre-clinical studies where cells with deficient activity of proteins MLH1, MSH2 and MSH6 had the phenotype of moderate resistance to cisplatin, but remained sensitive to the cytotoxic activity of OX[276,372]. Interestingly, Pt-DNA adducts are recognised by MSH1 protein only when damage occurs after cells are exposed to cisplatin, but not when Pt-DNA adducts are created due to the influence of OX[371,372]. Therefore, even though the MMR pathway is a key element in the mechanism of DNA repair, this system seems not to recognise Pt-DNA adducts created following exposure to OX. Generally, it is assumed that if attempts to repair Pt-induced DNA damage fail, this eventually leads to initiation of apoptosis[373,374]. Adducts induced by OX do not activate JNK (JNK-c-Jun NH2-terminal kinase, also known as stress activated protein kinase) and c-Abl (a nuclear protein)[375], which allow OX to maintain its cytotoxic activity in both MMR-proficient and -deficient cells[372,375]. Cisplatin depends on an intact MMR system for maximal cytotoxicity and for signalling apoptosis via the JNK-mediated pathway[371,375,376]. The binding of the MMR complex to Pt-DNA adducts appears to increase the cytotoxicity of the adducts[377], either by activating downstream signalling pathways that lead to apoptosis[375] or by causing “futile cycling” during translation synthesis past Pt-DNA adducts[372]. Therefore, cisplatin and OX have a different ability for activating signal paths to induce apoptosis in response to Pt-DNA adducts, which may be the basis of the observed differences in the profile of drug resistance in these platinum derivatives[378].
Protein p53 mediates the transduction of a signal induced by DNA damage following exposure to cisplatin[379]. p53 interacts with several significant elements that are part of the NER pathway, such as xeroderma pigmentosum, complementation group C (XPC), TFIIH and replication protein A (RPA), which points to its role in supervising the DNA repair process[380]. While testing 60 different cell lines, Vekris et al[381] showed that the expression of p53 was positively correlated with cell sensitivity to four different platinum derivatives: cisplatin, carboplatin, OX and tetraplatin. As p53 takes part in apoptosis induction and participates in the process of removing Pt-DNA adducts created by platinum derivatives, this protein may contribute to both chemosensitivity and drug resistance[382]. A systematic analysis of cellular sensitivity to OX in relation to p53 status in pairs of cisplatin-sensitive and -resistant cells showed that OX is less potent than cisplatin in cisplatin-sensitive cell lines, whereas it was capable of overcoming cisplatin resistance in the majority of sublines. Cell sensitivity to OX seems also dependent on the occurrence of genetic variants in gene TP53. While studying the cell line A431 which is characterised by a mutation in codon 273 of p53, it was observed that it has high resistance to OX[276].
Clinical application of the above in vitro studies to test a various panel of factors influencing the phenotype of chemosensitivity or drug resistance will require a series of in vivo studies with the participation of well selected groups of patients. Currently available data from pre-clinical studies show the potential significance of some molecular factors connected with the DNA repair processes and those participating in control of the cell cycle and apoptosis, which could serve as predictive markers in forecasting the efficiency of OX therapy in CRC patients.
FUTURE PERSPECTIVES IN PERSONALIZED MEDICINE FOR THE TREATMENT OF COLORECTAL CANCER
The last few decades have resulted in huge progress in understanding the complex processes regulating the growth and development of tumours. However, the major challenge in basic and clinical research is to solve the problem of primary and secondary drug resistance, which in many cases significantly reduces the antitumour efficacy of therapy. Early research on the development of new chemotherapeutic agents with significant antitumour potency, led to the introduction in oncology practice of few effective drugs, including those currently used in the treatment of CRC. Although they strongly induce apoptosis in intensively dividing cells, their strongest drawback is that they have the same effect on both cancer cells and healthy tissue. Therefore, to maintain the effectiveness of cancer treatment, it is necessary to use a maximum dose that provides a strong cytotoxic effect against tumour tissue, while minimizing toxicity to a patient. On the other hand, the intensive development of molecular tests in the last two decades initiated the development of “targeted” drugs and new treatment strategies such as targeted therapy. These new techniques have increased the hope of achieving substantial benefits in patients for whom the use of cytostatics proved not to be very effective. The main advantage of targeted therapy is the ability to avoid toxic effects of the drug with little impact on healthy cells. However, soon after the first research reports on targeted therapy and its high potential in clinical applications, drug resistance still remains a problem even with these ‘‘smart drugs’’. Similar to conventional cytostatics, resistance to a new class of drugs is an important issue in oncology[383]. It should be noted that drug resistance remains the most critical factor in the success of therapy. Currently, the main problem for researchers working on the effectiveness of cancer treatment is how to produce a rational treatment plan based on the classic cytostatic drugs and targeted drugs. Overcoming resistance in many cases is only possible by selecting an appropriate drug combination and optimal dosing during the treatment cycle. Due to the fact that many of the drug-resistance mechanisms are determined by individual patient characteristics, the key to successful therapy may be personalized cancer medicine. However, in recent years most scientists conducting research in the field of molecular mechanisms of drug resistance have focused on individual processes associated with metabolism, biodistribution, and anticancer drug mechanisms. Such research does not include the wider context and different body processes that constitute the effectiveness of a therapeutic strategy[384].
In the current paradigm accepted by scientists, it is considered that individual differences in response are the results of individual patient features that can be identified at a molecular level. These features are subject to genetic variation and the environmental pattern which are specific for each patient. It can be assumed that understanding the molecular mechanisms of inter-individual differences in the effectiveness of cancer treatment will allow the optimization of cancer therapy. Therefore, in the past two decades there has been a significant research effort to acquire information on the mechanisms responsible for the effectiveness of therapies. The approach that underlies individualized medicine is based on the assumption that by using molecular profiling and a set of biomarkers we can improve treatment efficacy in a patient, prolonging survival time and/or reduce the risk of serious complications[385].
Is it possible to apply these concepts in the individualization of treatment of CRC patients in the near future? In the previous chapters a variety of prognostic and predictive markers were described, which in recent years have been subject to various test procedures in order to determine their potential clinical value in the treatment of CRC. A technological breakthrough in molecular studies, as observed in recent years [single-nucleotide polymorphism arrays, complementary DNA microarrays, DNA methylation and microRNA (miRNA) profiling as well as next-generation sequencing] also made it possible to create individual molecular profiling for patients which is more profitable in economic terms. The data obtained using these high-throughput methods give hope for the practical application of various biomarkers to predict the effectiveness of treatment in individual patients with CRC.
Of the main variables affecting the therapeutic efficacy of cytostatics, the level of DNA synthesis and/or the intensity of cell division are important, and in the case of targeted drugs, the expression level of molecules in a signalling pathway in which the drug is targeted. As in the case of cytostatic drugs, the predominant mechanism of drug resistance is a wide panel of pharmacokinetic factors, and for targeted therapy it is mainly processes related to pharmacodynamics (genetic alteration/mutation of the target itself, persistent activation of downstream signalling pathways, and bypass mechanisms). Such a clear distinction does not describe the complexity of drug resistance mechanisms. Given the holistic nature of personalized medicine, it is necessary to develop and validate a wider panel of biomarkers which would reflect the multifactor mechanisms of resistance. In addition, when using predictors in clinical practice, we must take into account different therapeutic objectives which are set for specific subgroups of patients. From the point of view of drug resistance in cancer therapy, at least two main objectives require to be met in personalized medicine: (1) risk minimization of inducing resistance; and (2) breaking existing primary or secondary resistance. Finding the optimum combination of drugs and dosage regimen can, in many cases, lead to better efficiency in first-line treatment, and prevent cancer relapse. Furthermore, an equally important problem is the selection of resistant clones during the first treatment cycle, which in the case of relapse can significantly reduce the therapeutic efficacy of new combinations of drugs. Use of dynamic-response markers in clinical practice that would allow monitoring of the course of treatment is a promising line of research in personalized medicine. Changing the level of expression of marker genes or activity of posttranslational protein modification during the course of therapy has been assesssed in several studies. Analysis of molecular changes taking place during treatment may provide information regarding the development of resistance resulting from drug exposure, which is particularly important in the context of the existence of secondary drug resistance mechanisms. In such cases, a change in treatment regimen may be important for the future of a patient.
There are several main obstacles which currently prevent the full application of personalized medicine in clinical practice, despite significant progress in the study of causes of drug resistance in the treatment of CRC. Inter-individual differences in the response to treatment in patients with CRC may be subject to genetic and epigenetic features which can be classified as genomic aberrations [e.g., MSI[386,387], chromosomal instability (CIN)[388,389] and CpG island methylator phenotype (CIMP)[390-393]] as well as polymorphic variation (e.g., SNP or VNTR). This multifactor substrate conditioning efficacy in CRC makes it difficult to plan reliable research on predicting markers. In addition, the available clinical data indicate that CRCs are a molecularly heterogeneous group of neoplasms, which is why it is important to plan future studies taking into account this heterogeneity. Only this type of approach will provide a link between specific molecular features and effectiveness of the treatment. Another of the existing barriers for development of personalized medicine is the need for invasive biological sampling. A large part of the results of clinical trials on CRC drug resistance is based on the analysis of biological material derived from tumour biopsy. The possibility of using blood serum may be a way of solving this problem[394]. Another barrier that prevents truly individualized treatment of CRC patients is the small amount of research data that could connect mutation analysis and gene expression during the course of translation and activity of specific marker proteins. The main research stream based on transcriptome analysis does not provide information on protein expression, and mRNA level does not allow the determination of protein activity. It was not until recently when proteome analysis (proteomics) was developed, including important protein-protein interactions, that a number of new drugs for targeted therapy, such as inhibitors of kinases and their substrates were developed. Analysis of the activity of individual proteins involved in intracellular signal transduction is a very important aspect of research on tumour biology, and as shown by Pierobon et al[395], the level of protein expression and the level of protein activation (e.g., phosphorylation) do not always correlate, suggesting that the latter could be a better predictive biomarker for patient stratification. In conclusion, due to the heterogeneity of CRC and the complexity of drug resistance, prediction of the effectiveness of treatment in individual patients should be based on prediction biomarkers derived from the genome and proteome. Analysis of multiple markers is also justified as most modern standards of CRC treatment use a combination of several anticancer drugs. Combination therapy is based on the inhibition of tumour cells on several molecular levels. In order to rationally combine different therapies that would presumably be more effective than monotherapy, it is therefore necessary to use an integrated approach for the analysis of multiple pathways simultaneously. In this way, it will be possible to highlight pathway alterations that can be targeted by different agents.
The most recent data in the field of biomarker research show that only the interdisciplinary research approach, using combined analysis of genome and proteome, makes it possible to recognise prognostic and predictive factors which will help select patients in terms of relevant clinical features for individualized therapy[396]. Among a number of potential predictive markers described in the preceding sections of this review, only a small number were found to be clinically useful. In many cases, the analysis of the same marker provided contradictory data sometimes leading to opposing conclusions. There may be several reasons for these discrepancies, including the following: (1) methodological differences (prevalence of retrospective studies); (2) use of different and non-standardized research techniques; (3) use of inappropriate statistical analysis for a given type of data; and (4) diverse and/or insufficiently large groups of patients[385]. Therefore, to increase the credibility of preclinical and clinical prediction, it is necessary when planning research to take into account all variables which can affect the outcome of the analysis. Adoption of uniform research standards in the form of guidelines, such as reporting recommendations for tumour MARKer prognostic studies[397], provide an opportunity to obtain reliable data. Moreover, the current retrospective analysis, the results of which suggest a correlation should only be used as a source of hypotheses to be verified during the course of later well-designed studies.
In summary, from a clinical point of view, there is a need for innovative patient stratification methods which, based on validated biomarkers, will help clinicians to make correct therapeutic decisions. The effectiveness of anticancer drugs such as classical cytostatics and targeted drugs should be carefully reviewed in properly selected groups of patients whose common molecular profile will determine susceptibility or resistance to treatment[398]. The implementation of new technologies has led to the accumulation of huge amounts of genomic and proteomic data and the identification and validation of predictive biomarkers for existing and new targeted therapies, and will likely improve patient outcomes in the future[399]. Although the initial costs of cancer management and personalized medicine may be high[400], in the future they should result in significant benefits from both a clinical and economical perspective.
Footnotes
P- Reviewer: Fang BL, Lakatos PL, Nishida T S- Editor: Gou SX L- Editor: Webster JR E- Editor: Wang CH
References
- 1.Porteous M. Insights from next generation sequencing of the cancer genome. J R Coll Physicians Edinb. 2011;41:323. doi: 10.4997/JRCPE.2011.408. [DOI] [PubMed] [Google Scholar]
- 2.Russell C, Rahman A, Mohammed AR. Application of genomics, proteomics and metabolomics in drug discovery, development and clinic. Ther Deliv. 2013;4:395–413. doi: 10.4155/tde.13.4. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez de Castro D, Clarke PA, Al-Lazikani B, Workman P. Personalized cancer medicine: molecular diagnostics, predictive biomarkers, and drug resistance. Clin Pharmacol Ther. 2013;93:252–259. doi: 10.1038/clpt.2012.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cascorbi I, Bruhn O, Werk AN. Challenges in pharmacogenetics. Eur J Clin Pharmacol. 2013;69 Suppl 1:17–23. doi: 10.1007/s00228-013-1492-x. [DOI] [PubMed] [Google Scholar]
- 5.Abrahams E, Silver M. The case for personalized medicine. J Diabetes Sci Technol. 2009;3:680–684. doi: 10.1177/193229680900300411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spławiński J, Kuźniar J. Clinical trials: active control vs placebo--what is ethical? Sci Eng Ethics. 2004;10:73–79. doi: 10.1007/s11948-004-0065-x. [DOI] [PubMed] [Google Scholar]
- 7.Lenz HJ, Hayashi K, Salonga D, Danenberg KD, Danenberg PV, Metzger R, Banerjee D, Bertino JR, Groshen S, Leichman LP, et al. p53 point mutations and thymidylate synthase messenger RNA levels in disseminated colorectal cancer: an analysis of response and survival. Clin Cancer Res. 1998;4:1243–1250. [PubMed] [Google Scholar]
- 8.Loupakis F, Schirripa M, Zhang W, Falcone A, Lenz H-J. Pharmacogenetic Concerns in Metastatic Colorectal Cancer Therapy. Curr Colorectal Cancer Rep. 2012;8:263–271. [Google Scholar]
- 9.Stoehlmacher J. Prediction of efficacy and side effects of chemotherapy in colorectal cancer. Recent Results Cancer Res. 2007;176:81–88. doi: 10.1007/978-3-540-46091-6_8. [DOI] [PubMed] [Google Scholar]
- 10.Heidelberger C, Chaudhuri nk, Danneberg P, Mooren D, Griesbach L, Duschinsky R, Schnitzer RJ, Pleven E, Scheiner J. Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature. 1957;179:663–666. doi: 10.1038/179663a0. [DOI] [PubMed] [Google Scholar]
- 11.Chaudhuri NK, Montag BJ, Heidelberger C. Studies on fluorinated pyrimidines. III. The metabolism of 5-fluorouracil-2-C14 and 5-fluoroorotic-2-C14 acid in vivo. Cancer Res. 1958;18:318–328. [PubMed] [Google Scholar]
- 12.Cassidy J, Saltz L, Twelves C, Van Cutsem E, Hoff P, Kang Y, Saini JP, Gilberg F, Cunningham D. Efficacy of capecitabine versus 5-fluorouracil in colorectal and gastric cancers: a meta-analysis of individual data from 6171 patients. Ann Oncol. 2011;22:2604–2609. doi: 10.1093/annonc/mdr031. [DOI] [PubMed] [Google Scholar]
- 13.Grogan L, Sotos GA, Allegra CJ. Leucovorin modulation of fluorouracil. Oncology (Williston Park) 1993;7:63–72; discussion 75-76. [PubMed] [Google Scholar]
- 14.Thirion P, Michiels S, Pignon JP, Buyse M, Braud AC, Carlson RW, O’Connell M, Sargent P, Piedbois P. Modulation of fluorouracil by leucovorin in patients with advanced colorectal cancer: an updated meta-analysis. J Clin Oncol. 2004;22:3766–3775. doi: 10.1200/JCO.2004.03.104. [DOI] [PubMed] [Google Scholar]
- 15.Zhou JY, Shi R, Yu HL, Zeng Y, Zheng WL, Ma WL. The association between two polymorphisms in the TS gene and risk of cancer: a systematic review and pooled analysis. Int J Cancer. 2012;131:2103–2116. doi: 10.1002/ijc.27465. [DOI] [PubMed] [Google Scholar]
- 16.Berger SH, Jenh CH, Johnson LF, Berger FG. Thymidylate synthase overproduction and gene amplification in fluorodeoxyuridine-resistant human cells. Mol Pharmacol. 1985;28:461–467. [PubMed] [Google Scholar]
- 17.Salonga D, Danenberg KD, Johnson M, Metzger R, Groshen S, Tsao-Wei DD, Lenz HJ, Leichman CG, Leichman L, Diasio RB, et al. Colorectal tumors responding to 5-fluorouracil have low gene expression levels of dihydropyrimidine dehydrogenase, thymidylate synthase, and thymidine phosphorylase. Clin Cancer Res. 2000;6:1322–1327. [PubMed] [Google Scholar]
- 18.Leichman L, Lenz HJ, Leichman CG, Groshen S, Danenberg K, Baranda J, Spears CP, Boswell W, Silberman H, Ortega A. Quantitation of intratumoral thymidylate synthase expression predicts for resistance to protracted infusion of 5-fluorouracil and weekly leucovorin in disseminated colorectal cancers: preliminary report from an ongoing trial. Eur J Cancer. 1995;31A:1306–1310. doi: 10.1016/0959-8049(95)00326-e. [DOI] [PubMed] [Google Scholar]
- 19.Leichman CG, Lenz HJ, Leichman L, Danenberg K, Baranda J, Groshen S, Boswell W, Metzger R, Tan M, Danenberg PV. Quantitation of intratumoral thymidylate synthase expression predicts for disseminated colorectal cancer response and resistance to protracted-infusion fluorouracil and weekly leucovorin. J Clin Oncol. 1997;15:3223–3229. doi: 10.1200/JCO.1997.15.10.3223. [DOI] [PubMed] [Google Scholar]
- 20.Popat S, Matakidou A, Houlston RS. Thymidylate synthase expression and prognosis in colorectal cancer: a systematic review and meta-analysis. J Clin Oncol. 2004;22:529–536. doi: 10.1200/JCO.2004.05.064. [DOI] [PubMed] [Google Scholar]
- 21.Qiu LX, Tang QY, Bai JL, Qian XP, Li RT, Liu BR, Zheng MH. Predictive value of thymidylate synthase expression in advanced colorectal cancer patients receiving fluoropyrimidine-based chemotherapy: evidence from 24 studies. Int J Cancer. 2008;123:2384–2389. doi: 10.1002/ijc.23822. [DOI] [PubMed] [Google Scholar]
- 22.Marsh S, McKay JA, Curran S, Murray GI, Cassidy J, McLeod HL. Primary colorectal tumour is not an accurate predictor of thymidylate synthase in lymph node metastasis. Oncol Rep. 2002;9:231–234. [PubMed] [Google Scholar]
- 23.Aschele C, Debernardis D, Tunesi G, Maley F, Sobrero A. Thymidylate synthase protein expression in primary colorectal cancer compared with the corresponding distant metastases and relationship with the clinical response to 5-fluorouracil. Clin Cancer Res. 2000;6:4797–4802. [PubMed] [Google Scholar]
- 24.Iyevleva AG, Buslov KG, Togo AV, Matsko DE, Filimonenko VP, Moiseyenko VM, Imyanitov EN. Measurement of DPD and TS transcripts aimed to predict clinical benefit from fluoropyrimidines: confirmation of the trend in Russian colorectal cancer series and caution regarding the gene referees. Onkologie. 2007;30:295–300. doi: 10.1159/000102046. [DOI] [PubMed] [Google Scholar]
- 25.Ishida H, Shirakawa K, Ohsawa T, Sobajima J, Hayashi Y, Nakada H, Yokoyama M, Hashimoto D. [Expression of mRNA levels of thymidylate synthase, dihydropyrimidine dehydrogenase, and orotate phosphoribosyltransferase of colorectal cancer--relationships among mRNA levels in association with response to 5-FU based treatment] Gan To Kagaku Ryoho. 2005;32:1929–1934. [PubMed] [Google Scholar]
- 26.Hosokawa A, Yamada Y, Shimada Y, Muro K, Hamaguchi T, Morita H, Araake M, Orita H, Shirao K. Prognostic significance of thymidylate synthase in patients with metastatic colorectal cancer who receive protracted venous infusions of 5-fluorouracil. Int J Clin Oncol. 2004;9:388–392. doi: 10.1007/s10147-004-0425-1. [DOI] [PubMed] [Google Scholar]
- 27.Ciaparrone M, Quirino M, Schinzari G, Zannoni G, Corsi DC, Vecchio FM, Cassano A, La Torre G, Barone C. Predictive role of thymidylate synthase, dihydropyrimidine dehydrogenase and thymidine phosphorylase expression in colorectal cancer patients receiving adjuvant 5-fluorouracil. Oncology. 2006;70:366–377. doi: 10.1159/000098110. [DOI] [PubMed] [Google Scholar]
- 28.Nakajima TE, Yamada Y, Shimoda T, Matsubara J, Kato K, Hamaguchi T, Shimada Y, Okayama Y, Oka T, Shirao K. Combination of O6-methylguanine-DNA methyltransferase and thymidylate synthase for the prediction of fluoropyrimidine efficacy. Eur J Cancer. 2008;44:400–407. doi: 10.1016/j.ejca.2007.11.010. [DOI] [PubMed] [Google Scholar]
- 29.Kornmann M, Schwabe W, Sander S, Kron M, Sträter J, Polat S, Kettner E, Weiser HF, Baumann W, Schramm H, et al. Thymidylate synthase and dihydropyrimidine dehydrogenase mRNA expression levels: predictors for survival in colorectal cancer patients receiving adjuvant 5-fluorouracil. Clin Cancer Res. 2003;9:4116–4124. [PubMed] [Google Scholar]
- 30.Link KH, Kornmann M, Butzer U, Leder G, Sunelaitis E, Pillasch J, Salonga D, Danenberg KD, Danenberg PV, Beger HG. Thymidylate synthase quantitation and in vitro chemosensitivity testing predicts responses and survival of patients with isolated nonresectable liver tumors receiving hepatic arterial infusion chemotherapy. Cancer. 2000;89:288–296. [PubMed] [Google Scholar]
- 31.Davies MM, Johnston PG, Kaur S, Allen-Mersh TG. Colorectal liver metastasis thymidylate synthase staining correlates with response to hepatic arterial floxuridine. Clin Cancer Res. 1999;5:325–328. [PubMed] [Google Scholar]
- 32.Kornmann M, Link KH, Lenz HJ, Pillasch J, Metzger R, Butzer U, Leder GH, Weindel M, Safi F, Danenberg KD, et al. Thymidylate synthase is a predictor for response and resistance in hepatic artery infusion chemotherapy. Cancer Lett. 1997;118:29–35. doi: 10.1016/s0304-3835(97)00220-6. [DOI] [PubMed] [Google Scholar]
- 33.Etienne MC, Chazal M, Laurent-Puig P, Magné N, Rosty C, Formento JL, Francoual M, Formento P, Renée N, Chamorey E, et al. Prognostic value of tumoral thymidylate synthase and p53 in metastatic colorectal cancer patients receiving fluorouracil-based chemotherapy: phenotypic and genotypic analyses. J Clin Oncol. 2002;20:2832–2843. doi: 10.1200/JCO.2002.09.091. [DOI] [PubMed] [Google Scholar]
- 34.Shirota Y, Stoehlmacher J, Brabender J, Xiong YP, Uetake H, Danenberg KD, Groshen S, Tsao-Wei DD, Danenberg PV, Lenz HJ. ERCC1 and thymidylate synthase mRNA levels predict survival for colorectal cancer patients receiving combination oxaliplatin and fluorouracil chemotherapy. J Clin Oncol. 2001;19:4298–4304. doi: 10.1200/JCO.2001.19.23.4298. [DOI] [PubMed] [Google Scholar]
- 35.Paradiso A, Simone G, Petroni S, Leone B, Vallejo C, Lacava J, Romero A, Machiavelli M, De Lena M, Allegra CJ, et al. Thymidilate synthase and p53 primary tumour expression as predictive factors for advanced colorectal cancer patients. Br J Cancer. 2000;82:560–567. doi: 10.1054/bjoc.1999.0964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Aschele C, Debernardis D, Bandelloni R, Cascinu S, Catalano V, Giordani P, Barni S, Turci D, Drudi G, Lonardi S, et al. Thymidylate synthase protein expression in colorectal cancer metastases predicts for clinical outcome to leucovorin-modulated bolus or infusional 5-fluorouracil but not methotrexate-modulated bolus 5-fluorouracil. Ann Oncol. 2002;13:1882–1892. doi: 10.1093/annonc/mdf327. [DOI] [PubMed] [Google Scholar]
- 37.Yanagisawa Y, Maruta F, Iinuma N, Ishizone S, Koide N, Nakayama J, Miyagawa S. Modified Irinotecan/5FU/Leucovorin therapy in advanced colorectal cancer and predicting therapeutic efficacy by expression of tumor-related enzymes. Scand J Gastroenterol. 2007;42:477–484. doi: 10.1080/00365520600994418. [DOI] [PubMed] [Google Scholar]
- 38.Bendardaf R, Lamlum H, Elzagheid A, Ristamäki R, Pyrhönen S. Thymidylate synthase expression levels: a prognostic and predictive role in advanced colorectal cancer. Oncol Rep. 2005;14:657–662. doi: 10.3892/or.14.3.657. [DOI] [PubMed] [Google Scholar]
- 39.Ichikawa W, Uetake H, Shirota Y, Yamada H, Nishi N, Nihei Z, Sugihara K, Hirayama R. Combination of dihydropyrimidine dehydrogenase and thymidylate synthase gene expressions in primary tumors as predictive parameters for the efficacy of fluoropyrimidine-based chemotherapy for metastatic colorectal cancer. Clin Cancer Res. 2003;9:786–791. [PubMed] [Google Scholar]
- 40.Chu E, Koeller DM, Johnston PG, Zinn S, Allegra CJ. Regulation of thymidylate synthase in human colon cancer cells treated with 5-fluorouracil and interferon-gamma. Mol Pharmacol. 1993;43:527–533. [PubMed] [Google Scholar]
- 41.Longley DB, Boyer J, Allen WL, Latif T, Ferguson PR, Maxwell PJ, McDermott U, Lynch M, Harkin DP, Johnston PG. The role of thymidylate synthase induction in modulating p53-regulated gene expression in response to 5-fluorouracil and antifolates. Cancer Res. 2002;62:2644–2649. [PubMed] [Google Scholar]
- 42.Chu E, Voeller DM, Jones KL, Takechi T, Maley GF, Maley F, Segal S, Allegra CJ. Identification of a thymidylate synthase ribonucleoprotein complex in human colon cancer cells. Mol Cell Biol. 1994;14:207–213. doi: 10.1128/mcb.14.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suh KW, Kim JH, Kim YB, Kim J, Jeong S. Thymidylate synthase gene polymorphism as a prognostic factor for colon cancer. J Gastrointest Surg. 2005;9:336–342. doi: 10.1016/j.gassur.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 44.Gosens MJ, Moerland E, Lemmens VP, Rutten HT, Tan-Go I, van den Brule AJ. Thymidylate synthase genotyping is more predictive for therapy response than immunohistochemistry in patients with colon cancer. Int J Cancer. 2008;123:1941–1949. doi: 10.1002/ijc.23740. [DOI] [PubMed] [Google Scholar]
- 45.Fernández-Contreras ME, Sánchez-Prudencio S, Sánchez-Hernández JJ, García de Paredes ML, Gisbert JP, Roda-Navarro P, Gamallo C. Thymidylate synthase expression pattern, expression level and single nucleotide polymorphism are predictors for disease-free survival in patients of colorectal cancer treated with 5-fluorouracil. Int J Oncol. 2006;28:1303–1310. [PubMed] [Google Scholar]
- 46.Gusella M, Frigo AC, Bolzonella C, Marinelli R, Barile C, Bononi A, Crepaldi G, Menon D, Stievano L, Toso S, et al. Predictors of survival and toxicity in patients on adjuvant therapy with 5-fluorouracil for colorectal cancer. Br J Cancer. 2009;100:1549–1557. doi: 10.1038/sj.bjc.6605052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Páez D, Paré L, Altés A, Sancho-Poch FJ, Petriz L, Garriga J, Monill JM, Salazar J, del Rio E, Barnadas A, et al. Thymidylate synthase germline polymorphisms in rectal cancer patients treated with neoadjuvant chemoradiotherapy based on 5-fluorouracil. J Cancer Res Clin Oncol. 2010;136:1681–1689. doi: 10.1007/s00432-010-0826-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Villafranca E, Okruzhnov Y, Dominguez MA, García-Foncillas J, Azinovic I, Martínez E, Illarramendi JJ, Arias F, Martínez Monge R, Salgado E, et al. Polymorphisms of the repeated sequences in the enhancer region of the thymidylate synthase gene promoter may predict downstaging after preoperative chemoradiation in rectal cancer. J Clin Oncol. 2001;19:1779–1786. doi: 10.1200/JCO.2001.19.6.1779. [DOI] [PubMed] [Google Scholar]
- 49.Graziano F, Ruzzo A, Loupakis F, Santini D, Catalano V, Canestrari E, Maltese P, Bisonni R, Fornaro L, Baldi G, et al. Liver-only metastatic colorectal cancer patients and thymidylate synthase polymorphisms for predicting response to 5-fluorouracil-based chemotherapy. Br J Cancer. 2008;99:716–721. doi: 10.1038/sj.bjc.6604555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marcuello E, Altés A, del Rio E, César A, Menoyo A, Baiget M. Single nucleotide polymorphism in the 5’ tandem repeat sequences of thymidylate synthase gene predicts for response to fluorouracil-based chemotherapy in advanced colorectal cancer patients. Int J Cancer. 2004;112:733–737. doi: 10.1002/ijc.20487. [DOI] [PubMed] [Google Scholar]
- 51.Stoehlmacher J, Park DJ, Zhang W, Yang D, Groshen S, Zahedy S, Lenz HJ. A multivariate analysis of genomic polymorphisms: prediction of clinical outcome to 5-FU/oxaliplatin combination chemotherapy in refractory colorectal cancer. Br J Cancer. 2004;91:344–354. doi: 10.1038/sj.bjc.6601975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lurje G, Manegold PC, Ning Y, Pohl A, Zhang W, Lenz HJ. Thymidylate synthase gene variations: predictive and prognostic markers. Mol Cancer Ther. 2009;8:1000–1007. doi: 10.1158/1535-7163.MCT-08-0219. [DOI] [PubMed] [Google Scholar]
- 53.Horie N, Aiba H, Oguro K, Hojo H, Takeishi K. Functional analysis and DNA polymorphism of the tandemly repeated sequences in the 5’-terminal regulatory region of the human gene for thymidylate synthase. Cell Struct Funct. 1995;20:191–197. doi: 10.1247/csf.20.191. [DOI] [PubMed] [Google Scholar]
- 54.Kawakami K, Watanabe G. Identification and functional analysis of single nucleotide polymorphism in the tandem repeat sequence of thymidylate synthase gene. Cancer Res. 2003;63:6004–6007. [PubMed] [Google Scholar]
- 55.Pullarkat ST, Stoehlmacher J, Ghaderi V, Xiong YP, Ingles SA, Sherrod A, Warren R, Tsao-Wei D, Groshen S, Lenz HJ. Thymidylate synthase gene polymorphism determines response and toxicity of 5-FU chemotherapy. Pharmacogenomics J. 2001;1:65–70. doi: 10.1038/sj.tpj.6500012. [DOI] [PubMed] [Google Scholar]
- 56.Mandola MV, Stoehlmacher J, Muller-Weeks S, Cesarone G, Yu MC, Lenz HJ, Ladner RD. A novel single nucleotide polymorphism within the 5’ tandem repeat polymorphism of the thymidylate synthase gene abolishes USF-1 binding and alters transcriptional activity. Cancer Res. 2003;63:2898–2904. [PubMed] [Google Scholar]
- 57.Mandola MV, Stoehlmacher J, Zhang W, Groshen S, Yu MC, Iqbal S, Lenz HJ, Ladner RD. A 6 bp polymorphism in the thymidylate synthase gene causes message instability and is associated with decreased intratumoral TS mRNA levels. Pharmacogenetics. 2004;14:319–327. doi: 10.1097/00008571-200405000-00007. [DOI] [PubMed] [Google Scholar]
- 58.Ulrich CM, Bigler J, Velicer CM, Greene EA, Farin FM, Potter JD. Searching expressed sequence tag databases: discovery and confirmation of a common polymorphism in the thymidylate synthase gene. Cancer Epidemiol Biomarkers Prev. 2000;9:1381–1385. [PubMed] [Google Scholar]
- 59.Schaaf MJ, Cidlowski JA. AUUUA motifs in the 3’UTR of human glucocorticoid receptor alpha and beta mRNA destabilize mRNA and decrease receptor protein expression. Steroids. 2002;67:627–636. doi: 10.1016/s0039-128x(02)00015-6. [DOI] [PubMed] [Google Scholar]
- 60.Lu JW, Gao CM, Wu JZ, Cao HX, Tajima K, Feng JF. Polymorphism in the 3’-untranslated region of the thymidylate synthase gene and sensitivity of stomach cancer to fluoropyrimidine-based chemotherapy. J Hum Genet. 2006;51:155–160. doi: 10.1007/s10038-005-0339-4. [DOI] [PubMed] [Google Scholar]
- 61.Pinedo HM, Peters GF. Fluorouracil: biochemistry and pharmacology. J Clin Oncol. 1988;6:1653–1664. doi: 10.1200/JCO.1988.6.10.1653. [DOI] [PubMed] [Google Scholar]
- 62.Frosst P, Blom HJ, Milos R, Goyette P, Sheppard CA, Matthews RG, Boers GJ, den Heijer M, Kluijtmans LA, van den Heuvel LP. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat Genet. 1995;10:111–113. doi: 10.1038/ng0595-111. [DOI] [PubMed] [Google Scholar]
- 63.Bagley PJ, Selhub J. A common mutation in the methylenetetrahydrofolate reductase gene is associated with an accumulation of formylated tetrahydrofolates in red blood cells. Proc Natl Acad Sci USA. 1998;95:13217–13220. doi: 10.1073/pnas.95.22.13217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16:966–975. doi: 10.1038/cdd.2009.33. [DOI] [PubMed] [Google Scholar]
- 65.Sohn KJ, Croxford R, Yates Z, Lucock M, Kim YI. Effect of the methylenetetrahydrofolate reductase C677T polymorphism on chemosensitivity of colon and breast cancer cells to 5-fluorouracil and methotrexate. J Natl Cancer Inst. 2004;96:134–144. doi: 10.1093/jnci/djh015. [DOI] [PubMed] [Google Scholar]
- 66.Cohen V, Panet-Raymond V, Sabbaghian N, Morin I, Batist G, Rozen R. Methylenetetrahydrofolate reductase polymorphism in advanced colorectal cancer: a novel genomic predictor of clinical response to fluoropyrimidine-based chemotherapy. Clin Cancer Res. 2003;9:1611–1615. [PubMed] [Google Scholar]
- 67.Etienne MC, Formento JL, Chazal M, Francoual M, Magné N, Formento P, Bourgeon A, Seitz JF, Delpero JR, Letoublon C, et al. Methylenetetrahydrofolate reductase gene polymorphisms and response to fluorouracil-based treatment in advanced colorectal cancer patients. Pharmacogenetics. 2004;14:785–792. doi: 10.1097/00008571-200412000-00001. [DOI] [PubMed] [Google Scholar]
- 68.Jakobsen A, Nielsen JN, Gyldenkerne N, Lindeberg J. Thymidylate synthase and methylenetetrahydrofolate reductase gene polymorphism in normal tissue as predictors of fluorouracil sensitivity. J Clin Oncol. 2005;23:1365–1369. doi: 10.1200/JCO.2005.06.219. [DOI] [PubMed] [Google Scholar]
- 69.Marcuello E, Altés A, Menoyo A, Rio ED, Baiget M. Methylenetetrahydrofolate reductase gene polymorphisms: genomic predictors of clinical response to fluoropyrimidine-based chemotherapy? Cancer Chemother Pharmacol. 2006;57:835–840. doi: 10.1007/s00280-005-0089-1. [DOI] [PubMed] [Google Scholar]
- 70.van der Put NM, Gabreëls F, Stevens EM, Smeitink JA, Trijbels FJ, Eskes TK, van den Heuvel LP, Blom HJ. A second common mutation in the methylenetetrahydrofolate reductase gene: an additional risk factor for neural-tube defects? Am J Hum Genet. 1998;62:1044–1051. doi: 10.1086/301825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weisberg I, Tran P, Christensen B, Sibani S, Rozen R. A second genetic polymorphism in methylenetetrahydrofolate reductase (MTHFR) associated with decreased enzyme activity. Mol Genet Metab. 1998;64:169–172. doi: 10.1006/mgme.1998.2714. [DOI] [PubMed] [Google Scholar]
- 72.Sharma R, Hoskins JM, Rivory LP, Zucknick M, London R, Liddle C, Clarke SJ. Thymidylate synthase and methylenetetrahydrofolate reductase gene polymorphisms and toxicity to capecitabine in advanced colorectal cancer patients. Clin Cancer Res. 2008;14:817–825. doi: 10.1158/1078-0432.CCR-07-0425. [DOI] [PubMed] [Google Scholar]
- 73.Zhang W, Press OA, Haiman CA, Yang DY, Gordon MA, Fazzone W, El-Khoueiry A, Iqbal S, Sherrod AE, Lurje G, et al. Association of methylenetetrahydrofolate reductase gene polymorphisms and sex-specific survival in patients with metastatic colon cancer. J Clin Oncol. 2007;25:3726–3731. doi: 10.1200/JCO.2007.11.4710. [DOI] [PubMed] [Google Scholar]
- 74.Zintzaras E, Ziogas DC, Kitsios GD, Papathanasiou AA, Lau J, Raman G. MTHFR gene polymorphisms and response to chemotherapy in colorectal cancer: a meta-analysis. Pharmacogenomics. 2009;10:1285–1294. doi: 10.2217/pgs.09.59. [DOI] [PubMed] [Google Scholar]
- 75.Afzal S, Gusella M, Vainer B, Vogel UB, Andersen JT, Broedbaek K, Petersen M, Jimenez-Solem E, Bertolaso L, Barile C, et al. Combinations of polymorphisms in genes involved in the 5-Fluorouracil metabolism pathway are associated with gastrointestinal toxicity in chemotherapy-treated colorectal cancer patients. Clin Cancer Res. 2011;17:3822–3829. doi: 10.1158/1078-0432.CCR-11-0304. [DOI] [PubMed] [Google Scholar]
- 76.Diasio RB, Harris BE. Clinical pharmacology of 5-fluorouracil. Clin Pharmacokinet. 1989;16:215–237. doi: 10.2165/00003088-198916040-00002. [DOI] [PubMed] [Google Scholar]
- 77.Ezzeldin H, Diasio R. Dihydropyrimidine dehydrogenase deficiency, a pharmacogenetic syndrome associated with potentially life-threatening toxicity following 5-fluorouracil administration. Clin Colorectal Cancer. 2004;4:181–189. doi: 10.3816/ccc.2004.n.018. [DOI] [PubMed] [Google Scholar]
- 78.Mattison LK, Soong R, Diasio RB. Implications of dihydropyrimidine dehydrogenase on 5-fluorouracil pharmacogenetics and pharmacogenomics. Pharmacogenomics. 2002;3:485–492. doi: 10.1517/14622416.3.4.485. [DOI] [PubMed] [Google Scholar]
- 79.Etienne MC, Lagrange JL, Dassonville O, Fleming R, Thyss A, Renée N, Schneider M, Demard F, Milano G. Population study of dihydropyrimidine dehydrogenase in cancer patients. J Clin Oncol. 1994;12:2248–2253. doi: 10.1200/JCO.1994.12.11.2248. [DOI] [PubMed] [Google Scholar]
- 80.Ridge SA, Sludden J, Brown O, Robertson L, Wei X, Sapone A, Fernandez-Salguero PM, Gonzalez FJ, Vreken P, van Kuilenburg AB, et al. Dihydropyrimidine dehydrogenase pharmacogenetics in Caucasian subjects. Br J Clin Pharmacol. 1998;46:151–156. doi: 10.1046/j.1365-2125.1998.00751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ofverholm A, Arkblad E, Skrtic S, Albertsson P, Shubbar E, Enerbäck C. Two cases of 5-fluorouracil toxicity linked with gene variants in the DPYD gene. Clin Biochem. 2010;43:331–334. doi: 10.1016/j.clinbiochem.2009.09.024. [DOI] [PubMed] [Google Scholar]
- 82.Johnson MR, Wang K, Tillmanns S, Albin N, Diasio RB. Structural organization of the human dihydropyrimidine dehydrogenase gene. Cancer Res. 1997;57:1660–1663. [PubMed] [Google Scholar]
- 83.Yu J, McLeod HL, Ezzeldin HH, Diasio RB. Methylation of the DPYD promoter and dihydropyrimidine dehydrogenase deficiency. Clin Cancer Res. 2006;12:3864; author reply 3864. doi: 10.1158/1078-0432.CCR-06-0549. [DOI] [PubMed] [Google Scholar]
- 84.Bakkeren JA, De Abreu RA, Sengers RC, Gabreëls FJ, Maas JM, Renier WO. Elevated urine, blood and cerebrospinal fluid levels of uracil and thymine in a child with dihydrothymine dehydrogenase deficiency. Clin Chim Acta. 1984;140:247–256. doi: 10.1016/0009-8981(84)90206-7. [DOI] [PubMed] [Google Scholar]
- 85.Van Kuilenburg AB, Vreken P, Abeling NG, Bakker HD, Meinsma R, Van Lenthe H, De Abreu RA, Smeitink JA, Kayserili H, Apak MY, et al. Genotype and phenotype in patients with dihydropyrimidine dehydrogenase deficiency. Hum Genet. 1999;104:1–9. doi: 10.1007/pl00008711. [DOI] [PubMed] [Google Scholar]
- 86.van Kuilenburg AB, Muller EW, Haasjes J, Meinsma R, Zoetekouw L, Waterham HR, Baas F, Richel DJ, van Gennip AH. Lethal outcome of a patient with a complete dihydropyrimidine dehydrogenase (DPD) deficiency after administration of 5-fluorouracil: frequency of the common IVS14+1G& gt; A mutation causing DPD deficiency. Clin Cancer Res. 2001;7:1149–1153. [PubMed] [Google Scholar]
- 87.Van Kuilenburg AB, Meinsma R, Zoetekouw L, Van Gennip AH. High prevalence of the IVS14 + 1G& gt; A mutation in the dihydropyrimidine dehydrogenase gene of patients with severe 5-fluorouracil-associated toxicity. Pharmacogenetics. 2002;12:555–558. doi: 10.1097/00008571-200210000-00007. [DOI] [PubMed] [Google Scholar]
- 88.Ciccolini J, Mercier C, Evrard A, Dahan L, Boyer JC, Duffaud F, Richard K, Blanquicett C, Milano G, Blesius A, et al. A rapid and inexpensive method for anticipating severe toxicity to fluorouracil and fluorouracil-based chemotherapy. Ther Drug Monit. 2006;28:678–685. doi: 10.1097/01.ftd.0000245771.82720.c7. [DOI] [PubMed] [Google Scholar]
- 89.Meinsma R, Fernandez-Salguero P, Van Kuilenburg AB, Van Gennip AH, Gonzalez FJ. Human polymorphism in drug metabolism: mutation in the dihydropyrimidine dehydrogenase gene results in exon skipping and thymine uracilurea. DNA Cell Biol. 1995;14:1–6. doi: 10.1089/dna.1995.14.1. [DOI] [PubMed] [Google Scholar]
- 90.Wei X, McLeod HL, McMurrough J, Gonzalez FJ, Fernandez-Salguero P. Molecular basis of the human dihydropyrimidine dehydrogenase deficiency and 5-fluorouracil toxicity. J Clin Invest. 1996;98:610–615. doi: 10.1172/JCI118830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.van Kuilenburg AB, Haasjes J, Richel DJ, Zoetekouw L, Van Lenthe H, De Abreu RA, Maring JG, Vreken P, van Gennip AH. Clinical implications of dihydropyrimidine dehydrogenase (DPD) deficiency in patients with severe 5-fluorouracil-associated toxicity: identification of new mutations in the DPD gene. Clin Cancer Res. 2000;6:4705–4712. [PubMed] [Google Scholar]
- 92.Amstutz U, Farese S, Aebi S, Largiadèr CR. Dihydropyrimidine dehydrogenase gene variation and severe 5-fluorouracil toxicity: a haplotype assessment. Pharmacogenomics. 2009;10:931–944. doi: 10.2217/pgs.09.28. [DOI] [PubMed] [Google Scholar]
- 93.Amstutz U, Farese S, Aebi S, Largiadèr CR. Hypermethylation of the DPYD promoter region is not a major predictor of severe toxicity in 5-fluorouracil based chemotherapy. J Exp Clin Cancer Res. 2008;27:54. doi: 10.1186/1756-9966-27-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Scartozzi M, Maccaroni E, Giampieri R, Pistelli M, Bittoni A, Del Prete M, Berardi R, Cascinu S. 5-Fluorouracil pharmacogenomics: still rocking after all these years? Pharmacogenomics. 2011;12:251–265. doi: 10.2217/pgs.10.167. [DOI] [PubMed] [Google Scholar]
- 95.van Kuilenburg AB, Meinsma R, Zonnenberg BA, Zoetekouw L, Baas F, Matsuda K, Tamaki N, van Gennip AH. Dihydropyrimidinase deficiency and severe 5-fluorouracil toxicity. Clin Cancer Res. 2003;9:4363–4367. [PubMed] [Google Scholar]
- 96.Thomas HR, Ezzeldin HH, Guarcello V, Mattison LK, Fridley BL, Diasio RB. Genetic regulation of beta-ureidopropionase and its possible implication in altered uracil catabolism. Pharmacogenet Genomics. 2008;18:25–35. doi: 10.1097/FPC.0b013e3282f2f134. [DOI] [PubMed] [Google Scholar]
- 97.Vallböhmer D, Yang DY, Kuramochi H, Shimizu D, Danenberg KD, Lindebjerg J, Nielsen JN, Jakobsen A, Danenberg PV. DPD is a molecular determinant of capecitabine efficacy in colorectal cancer. Int J Oncol. 2007;31:413–418. [PubMed] [Google Scholar]
- 98.Meropol NJ, Gold PJ, Diasio RB, Andria M, Dhami M, Godfrey T, Kovatich AJ, Lund KA, Mitchell E, Schwarting R. Thymidine phosphorylase expression is associated with response to capecitabine plus irinotecan in patients with metastatic colorectal cancer. J Clin Oncol. 2006;24:4069–4077. doi: 10.1200/JCO.2005.05.2084. [DOI] [PubMed] [Google Scholar]
- 99.Vallböhmer D, Kuramochi H, Shimizu D, Danenberg KD, Lindebjerg J, Nielsen JN, Jakobsen A, Danenberg PV. Molecular factors of 5-fluorouracil metabolism in colorectal cancer: analysis of primary tumor and lymph node metastasis. Int J Oncol. 2006;28:527–533. doi: 10.3892/ijo.28.2.527. [DOI] [PubMed] [Google Scholar]
- 100.Bronckaers A, Gago F, Balzarini J, Liekens S. The dual role of thymidine phosphorylase in cancer development and chemotherapy. Med Res Rev. 2009;29:903–953. doi: 10.1002/med.20159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Miyadera K, Sumizawa T, Haraguchi M, Yoshida H, Konstanty W, Yamada Y, Akiyama S. Role of thymidine phosphorylase activity in the angiogenic effect of platelet derived endothelial cell growth factor/thymidine phosphorylase. Cancer Res. 1995;55:1687–1690. [PubMed] [Google Scholar]
- 102.Takebayashi Y, Yamada K, Miyadera K, Sumizawa T, Furukawa T, Kinoshita F, Aoki D, Okumura H, Yamada Y, Akiyama S, et al. The activity and expression of thymidine phosphorylase in human solid tumours. Eur J Cancer. 1996;32A:1227–1232. doi: 10.1016/0959-8049(96)00061-5. [DOI] [PubMed] [Google Scholar]
- 103.Walko CM, Lindley C. Capecitabine: a review. Clin Ther. 2005;27:23–44. doi: 10.1016/j.clinthera.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 104.Temmink OH, de Bruin M, Turksma AW, Cricca S, Laan AC, Peters GJ. Activity and substrate specificity of pyrimidine phosphorylases and their role in fluoropyrimidine sensitivity in colon cancer cell lines. Int J Biochem Cell Biol. 2007;39:565–575. doi: 10.1016/j.biocel.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 105.Schüller J, Cassidy J, Dumont E, Roos B, Durston S, Banken L, Utoh M, Mori K, Weidekamm E, Reigner B. Preferential activation of capecitabine in tumor following oral administration to colorectal cancer patients. Cancer Chemother Pharmacol. 2000;45:291–297. doi: 10.1007/s002800050043. [DOI] [PubMed] [Google Scholar]
- 106.Lamberti C, Sauerbruch T, Glasmacher A. Adjuvant capecitabine is at least as effective as fluorouracil plus leucovorin for survival in people with resected stage III colon cancer. Cancer Treat Rev. 2005;31:648–652. doi: 10.1016/j.ctrv.2005.09.009. [DOI] [PubMed] [Google Scholar]
- 107.Soong R, Shah N, Salto-Tellez M, Tai BC, Soo RA, Han HC, Ng SS, Tan WL, Zeps N, Joseph D, et al. Prognostic significance of thymidylate synthase, dihydropyrimidine dehydrogenase and thymidine phosphorylase protein expression in colorectal cancer patients treated with or without 5-fluorouracil-based chemotherapy. Ann Oncol. 2008;19:915–919. doi: 10.1093/annonc/mdm599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Aprile G, Mazzer M, Moroso S, Puglisi F. Pharmacology and therapeutic efficacy of capecitabine: focus on breast and colorectal cancer. Anticancer Drugs. 2009;20:217–229. doi: 10.1097/CAD.0b013e3283293fd4. [DOI] [PubMed] [Google Scholar]
- 109.Allegra CJ, Paik S, Colangelo LH, Parr AL, Kirsch I, Kim G, Klein P, Johnston PG, Wolmark N, Wieand HS. Prognostic value of thymidylate synthase, Ki-67, and p53 in patients with Dukes’ B and C colon cancer: a National Cancer Institute-National Surgical Adjuvant Breast and Bowel Project collaborative study. J Clin Oncol. 2003;21:241–250. doi: 10.1200/JCO.2003.05.044. [DOI] [PubMed] [Google Scholar]
- 110.Koopman M, Venderbosch S, van Tinteren H, Ligtenberg MJ, Nagtegaal I, Van Krieken JH, Punt CJ. Predictive and prognostic markers for the outcome of chemotherapy in advanced colorectal cancer, a retrospective analysis of the phase III randomised CAIRO study. Eur J Cancer. 2009;45:1999–2006. doi: 10.1016/j.ejca.2009.04.017. [DOI] [PubMed] [Google Scholar]
- 111.Evans DR, Guy HI. Mammalian pyrimidine biosynthesis: fresh insights into an ancient pathway. J Biol Chem. 2004;279:33035–33038. doi: 10.1074/jbc.R400007200. [DOI] [PubMed] [Google Scholar]
- 112.Muhale FA, Wetmore BA, Thomas RS, McLeod HL. Systems pharmacology assessment of the 5-fluorouracil pathway. Pharmacogenomics. 2011;12:341–350. doi: 10.2217/pgs.10.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Matsuyama R, Togo S, Shimizu D, Momiyama N, Ishikawa T, Ichikawa Y, Endo I, Kunisaki C, Suzuki H, Hayasizaki Y, et al. Predicting 5-fluorouracil chemosensitivity of liver metastases from colorectal cancer using primary tumor specimens: three-gene expression model predicts clinical response. Int J Cancer. 2006;119:406–413. doi: 10.1002/ijc.21843. [DOI] [PubMed] [Google Scholar]
- 114.Ishikawa M, Miyauchi T, Kashiwagi Y. Clinical implications of thymidylate synthetase, dihydropyrimidine dehydrogenase and orotate phosphoribosyl transferase activity levels in colorectal carcinoma following radical resection and administration of adjuvant 5-FU chemotherapy. BMC Cancer. 2008;8:188. doi: 10.1186/1471-2407-8-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ochiai T, Nishimura K, Noguchi H, Kitajima M, Tsuruoka Y, Takahashi Y, Tsukada A, Watanabe E, Nagaoka I, Futagawa S. Prognostic impact of orotate phosphoribosyl transferase activity in resectable colorectal cancers treated by 5-fluorouracil-based adjuvant chemotherapy. J Surg Oncol. 2006;94:45–50. doi: 10.1002/jso.20553. [DOI] [PubMed] [Google Scholar]
- 116.Tokunaga Y, Sasaki H, Saito T. Clinical role of orotate phosphoribosyl transferase and dihydropyrimidine dehydrogenase in colorectal cancer treated with postoperative fluoropyrimidine. Surgery. 2007;141:346–353. doi: 10.1016/j.surg.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 117.Tokunaga Y, Ohnishi T, Sasaki H. [Investigation of chemotherapy based on enzyme expression and drug sensitivity test in colorectal cancer] Gan To Kagaku Ryoho. 2011;38:69–73. [PubMed] [Google Scholar]
- 118.Ichikawa W, Uetake H, Shirota Y, Yamada H, Takahashi T, Nihei Z, Sugihara K, Sasaki Y, Hirayama R. Both gene expression for orotate phosphoribosyltransferase and its ratio to dihydropyrimidine dehydrogenase influence outcome following fluoropyrimidine-based chemotherapy for metastatic colorectal cancer. Br J Cancer. 2003;89:1486–1492. doi: 10.1038/sj.bjc.6601335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yamada H, Iinuma H, Watanabe T. Prognostic value of 5-fluorouracil metabolic enzyme genes in Dukes’ stage B and C colorectal cancer patients treated with oral 5-fluorouracil-based adjuvant chemotherapy. Oncol Rep. 2008;19:729–735. [PubMed] [Google Scholar]
- 120.Fujii R, Seshimo A, Kameoka S. Relationships between the expression of thymidylate synthase, dihydropyrimidine dehydrogenase, and orotate phosphoribosyltransferase and cell proliferative activity and 5-fluorouracil sensitivity in colorectal carcinoma. Int J Clin Oncol. 2003;8:72–78. doi: 10.1007/s101470300013. [DOI] [PubMed] [Google Scholar]
- 121.Ochiai T, Nishimura K, Noguchi H, Kitajima M, Tsukada A, Watanabe E, Nagaoka I, Futagawa S. Prognostic impact of orotate phosphoribosyl transferase among 5-fluorouracil metabolic enzymes in resectable colorectal cancers treated by oral 5-fluorouracil-based adjuvant chemotherapy. Int J Cancer. 2006;118:3084–3088. doi: 10.1002/ijc.21779. [DOI] [PubMed] [Google Scholar]
- 122.Isshi K, Sakuyama T, Gen T, Nakamura Y, Kuroda T, Katuyama T, Maekawa Y. Predicting 5-FU sensitivity using human colorectal cancer specimens: comparison of tumor dihydropyrimidine dehydrogenase and orotate phosphoribosyl transferase activities with in vitro chemosensitivity to 5-FU. Int J Clin Oncol. 2002;7:335–342. doi: 10.1007/s101470200051. [DOI] [PubMed] [Google Scholar]
- 123.Kitajima M, Takita N, Hata M, Maeda T, Sakamoto K, Kamano T, Ochiai T. The relationship between 5-fluorouracil sensitivity and single nucleotide polymorphisms of the orotate phosphoribosyl transferase gene in colorectal cancer. Oncol Rep. 2006;15:161–165. [PubMed] [Google Scholar]
- 124.Ichikawa W, Takahashi T, Suto K, Sasaki Y, Hirayama R. Orotate phosphoribosyltransferase gene polymorphism predicts toxicity in patients treated with bolus 5-fluorouracil regimen. Clin Cancer Res. 2006;12:3928–3934. doi: 10.1158/1078-0432.CCR-05-2665. [DOI] [PubMed] [Google Scholar]
- 125.Tsunoda A, Nakao K, Watanabe M, Matsui N, Ooyama A, Kusano M. Associations of various gene polymorphisms with toxicity in colorectal cancer patients receiving oral uracil and tegafur plus leucovorin: a prospective study. Ann Oncol. 2011;22:355–361. doi: 10.1093/annonc/mdq358. [DOI] [PubMed] [Google Scholar]
- 126.Gusella M, Bertolaso L, Bolzonella C, Pasini F, Padrini R. Frequency of uridine monophosphate synthase Gly(213)Ala polymorphism in Caucasian gastrointestinal cancer patients and healthy subjects, investigated by means of new, rapid genotyping assays. Genet Test Mol Biomarkers. 2011;15:691–695. doi: 10.1089/gtmb.2011.0021. [DOI] [PubMed] [Google Scholar]
- 127.Suchi M, Mizuno H, Kawai Y, Tsuboi T, Sumi S, Okajima K, Hodgson ME, Ogawa H, Wada Y. Molecular cloning of the human UMP synthase gene and characterization of point mutations in two hereditary orotic aciduria families. Am J Hum Genet. 1997;60:525–539. [PMC free article] [PubMed] [Google Scholar]
- 128.Houghton JA, Houghton PJ, Wooten RS. Mechanism of induction of gastrointestinal toxicity in the mouse by 5-fluorouracil, 5-fluorouridine, and 5-fluoro-2’-deoxyuridine. Cancer Res. 1979;39:2406–2413. [PubMed] [Google Scholar]
- 129.Wang H, Bian T, Liu D, Jin T, Chen Y, Lin A, Chen C. Association analysis of CYP2A6 genotypes and haplotypes with 5-fluorouracil formation from tegafur in human liver microsomes. Pharmacogenomics. 2011;12:481–492. doi: 10.2217/pgs.10.202. [DOI] [PubMed] [Google Scholar]
- 130.Carethers JM, Chauhan DP, Fink D, Nebel S, Bresalier RS, Howell SB, Boland CR. Mismatch repair proficiency and in vitro response to 5-fluorouracil. Gastroenterology. 1999;117:123–131. doi: 10.1016/s0016-5085(99)70558-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Meyers M, Wagner MW, Hwang HS, Kinsella TJ, Boothman DA. Role of the hMLH1 DNA mismatch repair protein in fluoropyrimidine-mediated cell death and cell cycle responses. Cancer Res. 2001;61:5193–5201. [PubMed] [Google Scholar]
- 132.Raymond E, Chaney SG, Taamma A, Cvitkovic E. Oxaliplatin: a review of preclinical and clinical studies. Ann Oncol. 1998;9:1053–1071. doi: 10.1023/a:1008213732429. [DOI] [PubMed] [Google Scholar]
- 133.Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005;23:609–618. doi: 10.1200/JCO.2005.01.086. [DOI] [PubMed] [Google Scholar]
- 134.Sargent DJ, Marsoni S, Thibodeau SN, Labianca R, Hamilton SR, Torri V, Monges G, Ribic C, Grothey A, Gallinger S. Confirmation of deficient mismatch repair (dMMR) as a predictive marker for lack of benefit from 5-FU based chemotherapy in stage II and III colon cancer (CC): A pooled molecular reanalysis of randomized chemotherapy trials. J Clin Oncol. 2008;26:4008. [Google Scholar]
- 135.Gryfe R, Kim H, Hsieh ET, Aronson MD, Holowaty EJ, Bull SB, Redston M, Gallinger S. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N Engl J Med. 2000;342:69–77. doi: 10.1056/NEJM200001133420201. [DOI] [PubMed] [Google Scholar]
- 136.Elsaleh H, Powell B, McCaul K, Grieu F, Grant R, Joseph D, Iacopetta B. P53 alteration and microsatellite instability have predictive value for survival benefit from chemotherapy in stage III colorectal carcinoma. Clin Cancer Res. 2001;7:1343–1349. [PubMed] [Google Scholar]
- 137.Lim SB, Jeong SY, Lee MR, Ku JL, Shin YK, Kim WH, Park JG. Prognostic significance of microsatellite instability in sporadic colorectal cancer. Int J Colorectal Dis. 2004;19:533–537. doi: 10.1007/s00384-004-0596-2. [DOI] [PubMed] [Google Scholar]
- 138.Mori S, Ogata Y, Shirouzu K. Biological features of sporadic colorectal carcinoma with high-frequency microsatellite instability: special reference to tumor proliferation and apoptosis. Int J Clin Oncol. 2004;9:322–329. doi: 10.1007/s10147-004-0406-4. [DOI] [PubMed] [Google Scholar]
- 139.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408:307–310. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 140.Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 141.Bunz F, Hwang PM, Torrance C, Waldman T, Zhang Y, Dillehay L, Williams J, Lengauer C, Kinzler KW, Vogelstein B. Disruption of p53 in human cancer cells alters the responses to therapeutic agents. J Clin Invest. 1999;104:263–269. doi: 10.1172/JCI6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Liang JT, Huang KC, Cheng YM, Hsu HC, Cheng AL, Hsu CH, Yeh KH, Wang SM, Chang KJ. P53 overexpression predicts poor chemosensitivity to high-dose 5-fluorouracil plus leucovorin chemotherapy for stage IV colorectal cancers after palliative bowel resection. Int J Cancer. 2002;97:451–457. doi: 10.1002/ijc.1637. [DOI] [PubMed] [Google Scholar]
- 143.Garrity MM, Burgart LJ, Mahoney MR, Windschitl HE, Salim M, Wiesenfeld M, Krook JE, Michalak JC, Goldberg RM, O’Connell MJ, et al. Prognostic value of proliferation, apoptosis, defective DNA mismatch repair, and p53 overexpression in patients with resected Dukes’ B2 or C colon cancer: a North Central Cancer Treatment Group Study. J Clin Oncol. 2004;22:1572–1582. doi: 10.1200/JCO.2004.10.042. [DOI] [PubMed] [Google Scholar]
- 144.van Oijen MG, Slootweg PJ. Gain-of-function mutations in the tumor suppressor gene p53. Clin Cancer Res. 2000;6:2138–2145. [PubMed] [Google Scholar]
- 145.Pugacheva EN, Ivanov AV, Kravchenko JE, Kopnin BP, Levine AJ, Chumakov PM. Novel gain of function activity of p53 mutants: activation of the dUTPase gene expression leading to resistance to 5-fluorouracil. Oncogene. 2002;21:4595–4600. doi: 10.1038/sj.onc.1205704. [DOI] [PubMed] [Google Scholar]
- 146.Wu AHB, Yeo KTJ. Pharmacogenomic testing in current clinical practice: implementation in the clinical laboratory. New York: Humana Press; 2011. [Google Scholar]
- 147.Masumoto N, Nakano S, Esaki T, Tatsumoto T, Fujishima H, Baba E, Nakamura M, Niho Y. Sequence-dependent modulation of anticancer drug activities by 7-ethyl-10-hydroxycamptothecin in an HST-1 human squamous carcinoma cell line. Anticancer Res. 1995;15:405–409. [PubMed] [Google Scholar]
- 148.Matsuoka H, Yano K, Seo Y, Saito T, Tomoda H, Takiguchi S, Kono A. Cytotoxicity of CPT-11 for gastrointestinal cancer cells cultured on fixed-contact-sensitive plates. Anticancer Drugs. 1995;6:413–418. doi: 10.1097/00001813-199506000-00008. [DOI] [PubMed] [Google Scholar]
- 149.Shimada Y, Rougier P, Pitot H. Efficacy of CPT-11 (irinotecan) as a single agent in metastatic colorectal cancer. Eur J Cancer. 1996;32A Suppl 3:S13–S17. doi: 10.1016/0959-8049(96)00292-4. [DOI] [PubMed] [Google Scholar]
- 150.Rougier P, Bugat R, Douillard JY, Culine S, Suc E, Brunet P, Becouarn Y, Ychou M, Marty M, Extra JM, et al. Phase II study of irinotecan in the treatment of advanced colorectal cancer in chemotherapy-naive patients and patients pretreated with fluorouracil-based chemotherapy. J Clin Oncol. 1997;15:251–260. doi: 10.1200/JCO.1997.15.1.251. [DOI] [PubMed] [Google Scholar]
- 151.Clarke SJ, Yip S, Brown C, van Hazel GA, Ransom DT, Goldstein D, Jeffrey GM, Tebbutt NC, Buck M, Lowenthal RM, et al. Single-agent irinotecan or FOLFIRI as second-line chemotherapy for advanced colorectal cancer; results of a randomised phase II study (DaVINCI) and meta-analysis [corrected] Eur J Cancer. 2011;47:1826–1836. doi: 10.1016/j.ejca.2011.04.024. [DOI] [PubMed] [Google Scholar]
- 152.Cortejoso L, López-Fernández LA. Pharmacogenetic markers of toxicity for chemotherapy in colorectal cancer patients. Pharmacogenomics. 2012;13:1173–1191. doi: 10.2217/pgs.12.95. [DOI] [PubMed] [Google Scholar]
- 153.Freyer G, Duret A, Milano G, Chatelut E, Rebischung C, Delord JP, Merrouche Y, Lledo G, Etienne MC, Falandry C. Pharmacogenetic tailoring of irinotecan-based first-line chemotherapy in metastatic colorectal cancer: results of a pilot study. Anticancer Res. 2011;31:359–366. [PubMed] [Google Scholar]
- 154.Shimoyama S. Pharmacogenetics of irinotecan: An ethnicity-based prediction of irinotecan adverse events. World J Gastrointest Surg. 2010;2:14–21. doi: 10.4240/wjgs.v2.i1.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Rasheed ZA, Rubin EH. Mechanisms of resistance to topoisomerase I-targeting drugs. Oncogene. 2003;22:7296–7304. doi: 10.1038/sj.onc.1206935. [DOI] [PubMed] [Google Scholar]
- 156.Pommier Y, Pourquier P, Urasaki Y, Wu J, Laco GS. Topoisomerase I inhibitors: selectivity and cellular resistance. Drug Resist Updat. 1999;2:307–318. doi: 10.1054/drup.1999.0102. [DOI] [PubMed] [Google Scholar]
- 157.Smith NF, Figg WD, Sparreboom A. Pharmacogenetics of irinotecan metabolism and transport: an update. Toxicol In Vitro. 2006;20:163–175. doi: 10.1016/j.tiv.2005.06.045. [DOI] [PubMed] [Google Scholar]
- 158.Charasson V, Bellott R, Meynard D, Longy M, Gorry P, Robert J. Pharmacogenetics of human carboxylesterase 2, an enzyme involved in the activation of irinotecan into SN-38. Clin Pharmacol Ther. 2004;76:528–535. doi: 10.1016/j.clpt.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 159.Bellott R, Le Morvan V, Charasson V, Laurand A, Colotte M, Zanger UM, Klein K, Smith D, Bonnet J, Robert J. Functional study of the 830C& gt; G polymorphism of the human carboxylesterase 2 gene. Cancer Chemother Pharmacol. 2008;61:481–488. doi: 10.1007/s00280-007-0493-9. [DOI] [PubMed] [Google Scholar]
- 160.Wu MH, Chen P, Wu X, Liu W, Strom S, Das S, Cook EH, Rosner GL, Dolan ME. Determination and analysis of single nucleotide polymorphisms and haplotype structure of the human carboxylesterase 2 gene. Pharmacogenetics. 2004;14:595–605. doi: 10.1097/00008571-200409000-00004. [DOI] [PubMed] [Google Scholar]
- 161.Kim SR, Sai K, Tanaka-Kagawa T, Jinno H, Ozawa S, Kaniwa N, Saito Y, Akasawa A, Matsumoto K, Saito H, et al. Haplotypes and a novel defective allele of CES2 found in a Japanese population. Drug Metab Dispos. 2007;35:1865–1872. doi: 10.1124/dmd.107.015339. [DOI] [PubMed] [Google Scholar]
- 162.Sanghani SP, Sanghani PC, Schiel MA, Bosron WF. Human carboxylesterases: an update on CES1, CES2 and CES3. Protein Pept Lett. 2009;16:1207–1214. doi: 10.2174/092986609789071324. [DOI] [PubMed] [Google Scholar]
- 163.Marsh S, Xiao M, Yu J, Ahluwalia R, Minton M, Freimuth RR, Kwok PY, McLeod HL. Pharmacogenomic assessment of carboxylesterases 1 and 2. Genomics. 2004;84:661–668. doi: 10.1016/j.ygeno.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 164.van Ark-Otte J, Kedde MA, van der Vijgh WJ, Dingemans AM, Jansen WJ, Pinedo HM, Boven E, Giaccone G. Determinants of CPT-11 and SN-38 activities in human lung cancer cells. Br J Cancer. 1998;77:2171–2176. doi: 10.1038/bjc.1998.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Pavillard V, Agostini C, Richard S, Charasson V, Montaudon D, Robert J. Determinants of the cytotoxicity of irinotecan in two human colorectal tumor cell lines. Cancer Chemother Pharmacol. 2002;49:329–335. doi: 10.1007/s00280-001-0416-0. [DOI] [PubMed] [Google Scholar]
- 166.Kubo T, Kim SR, Sai K, Saito Y, Nakajima T, Matsumoto K, Saito H, Shirao K, Yamamoto N, Minami H, et al. Functional characterization of three naturally occurring single nucleotide polymorphisms in the CES2 gene encoding carboxylesterase 2 (HCE-2) Drug Metab Dispos. 2005;33:1482–1487. doi: 10.1124/dmd.105.005587. [DOI] [PubMed] [Google Scholar]
- 167.Tanimoto K, Kaneyasu M, Shimokuni T, Hiyama K, Nishiyama M. Human carboxylesterase 1A2 expressed from carboxylesterase 1A1 and 1A2 genes is a potent predictor of CPT-11 cytotoxicity in vitro. Pharmacogenet Genomics. 2007;17:1–10. doi: 10.1097/01.fpc.0000230110.18957.50. [DOI] [PubMed] [Google Scholar]
- 168.Yoshimura M, Kimura T, Ishii M, Ishii K, Matsuura T, Geshi E, Hosokawa M, Muramatsu M. Functional polymorphisms in carboxylesterase1A2 (CES1A2) gene involves specific protein 1 (Sp1) binding sites. Biochem Biophys Res Commun. 2008;369:939–942. doi: 10.1016/j.bbrc.2008.02.120. [DOI] [PubMed] [Google Scholar]
- 169.Ando Y, Hasegawa Y. Clinical pharmacogenetics of irinotecan (CPT-11) Drug Metab Rev. 2005;37:565–574. doi: 10.1080/03602530500316254. [DOI] [PubMed] [Google Scholar]
- 170.Aono S, Yamada Y, Keino H, Hanada N, Nakagawa T, Sasaoka Y, Yazawa T, Sato H, Koiwai O. Identification of defect in the genes for bilirubin UDP-glucuronosyl-transferase in a patient with Crigler-Najjar syndrome type II. Biochem Biophys Res Commun. 1993;197:1239–1244. doi: 10.1006/bbrc.1993.2610. [DOI] [PubMed] [Google Scholar]
- 171.Aono S, Yamada Y, Keino H, Sasaoka Y, Nakagawa T, Onishi S, Mimura S, Koiwai O, Sato H. A new type of defect in the gene for bilirubin uridine 5’-diphosphate-glucuronosyltransferase in a patient with Crigler-Najjar syndrome type I. Pediatr Res. 1994;35:629–632. doi: 10.1203/00006450-199406000-00002. [DOI] [PubMed] [Google Scholar]
- 172.Aono S, Adachi Y, Uyama E, Yamada Y, Keino H, Nanno T, Koiwai O, Sato H. Analysis of genes for bilirubin UDP-glucuronosyltransferase in Gilbert’s syndrome. Lancet. 1995;345:958–959. doi: 10.1016/s0140-6736(95)90702-5. [DOI] [PubMed] [Google Scholar]
- 173.Villeneuve L, Girard H, Fortier LC, Gagné JF, Guillemette C. Novel functional polymorphisms in the UGT1A7 and UGT1A9 glucuronidating enzymes in Caucasian and African-American subjects and their impact on the metabolism of 7-ethyl-10-hydroxycamptothecin and flavopiridol anticancer drugs. J Pharmacol Exp Ther. 2003;307:117–128. doi: 10.1124/jpet.103.054072. [DOI] [PubMed] [Google Scholar]
- 174.Jinno H, Tanaka-Kagawa T, Hanioka N, Saeki M, Ishida S, Nishimura T, Ando M, Saito Y, Ozawa S, Sawada J. Glucuronidation of 7-ethyl-10-hydroxycamptothecin (SN-38), an active metabolite of irinotecan (CPT-11), by human UGT1A1 variants, G71R, P229Q, and Y486D. Drug Metab Dispos. 2003;31:108–113. doi: 10.1124/dmd.31.1.108. [DOI] [PubMed] [Google Scholar]
- 175.Strassburg CP, Kalthoff S, Ehmer U. Variability and function of family 1 uridine-5’-diphosphate glucuronosyltransferases (UGT1A) Crit Rev Clin Lab Sci. 2008;45:485–530. doi: 10.1080/10408360802374624. [DOI] [PubMed] [Google Scholar]
- 176.Cecchin E, Innocenti F, D’Andrea M, Corona G, De Mattia E, Biason P, Buonadonna A, Toffoli G. Predictive role of the UGT1A1, UGT1A7, and UGT1A9 genetic variants and their haplotypes on the outcome of metastatic colorectal cancer patients treated with fluorouracil, leucovorin, and irinotecan. J Clin Oncol. 2009;27:2457–2465. doi: 10.1200/JCO.2008.19.0314. [DOI] [PubMed] [Google Scholar]
- 177.Toffoli G, Cecchin E, Corona G, Russo A, Buonadonna A, D’Andrea M, Pasetto LM, Pessa S, Errante D, De Pangher V, et al. The role of UGT1A1*28 polymorphism in the pharmacodynamics and pharmacokinetics of irinotecan in patients with metastatic colorectal cancer. J Clin Oncol. 2006;24:3061–3068. doi: 10.1200/JCO.2005.05.5400. [DOI] [PubMed] [Google Scholar]
- 178.Rouits E, Charasson V, Pétain A, Boisdron-Celle M, Delord JP, Fonck M, Laurand A, Poirier AL, Morel A, Chatelut E, et al. Pharmacokinetic and pharmacogenetic determinants of the activity and toxicity of irinotecan in metastatic colorectal cancer patients. Br J Cancer. 2008;99:1239–1245. doi: 10.1038/sj.bjc.6604673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Côté JF, Kirzin S, Kramar A, Mosnier JF, Diebold MD, Soubeyran I, Thirouard AS, Selves J, Laurent-Puig P, Ychou M. UGT1A1 polymorphism can predict hematologic toxicity in patients treated with irinotecan. Clin Cancer Res. 2007;13:3269–3275. doi: 10.1158/1078-0432.CCR-06-2290. [DOI] [PubMed] [Google Scholar]
- 180.McLeod HL, Sargent DJ, Marsh S, Green EM, King CR, Fuchs CS, Ramanathan RK, Williamson SK, Findlay BP, Thibodeau SN, et al. Pharmacogenetic predictors of adverse events and response to chemotherapy in metastatic colorectal cancer: results from North American Gastrointestinal Intergroup Trial N9741. J Clin Oncol. 2010;28:3227–3233. doi: 10.1200/JCO.2009.21.7943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Massacesi C, Terrazzino S, Marcucci F, Rocchi MB, Lippe P, Bisonni R, Lombardo M, Pilone A, Mattioli R, Leon A. Uridine diphosphate glucuronosyl transferase 1A1 promoter polymorphism predicts the risk of gastrointestinal toxicity and fatigue induced by irinotecan-based chemotherapy. Cancer. 2006;106:1007–1016. doi: 10.1002/cncr.21722. [DOI] [PubMed] [Google Scholar]
- 182.Marcuello E, Altés A, Menoyo A, Del Rio E, Gómez-Pardo M, Baiget M. UGT1A1 gene variations and irinotecan treatment in patients with metastatic colorectal cancer. Br J Cancer. 2004;91:678–682. doi: 10.1038/sj.bjc.6602042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Rouits E, Boisdron-Celle M, Dumont A, Guérin O, Morel A, Gamelin E. Relevance of different UGT1A1 polymorphisms in irinotecan-induced toxicity: a molecular and clinical study of 75 patients. Clin Cancer Res. 2004;10:5151–5159. doi: 10.1158/1078-0432.CCR-03-0548. [DOI] [PubMed] [Google Scholar]
- 184.Beutler E, Gelbart T, Demina A. Racial variability in the UDP-glucuronosyltransferase 1 (UGT1A1) promoter: a balanced polymorphism for regulation of bilirubin metabolism? Proc Natl Acad Sci USA. 1998;95:8170–8174. doi: 10.1073/pnas.95.14.8170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Iyer L, Hall D, Das S, Mortell MA, Ramírez J, Kim S, Di Rienzo A, Ratain MJ. Phenotype-genotype correlation of in vitro SN-38 (active metabolite of irinotecan) and bilirubin glucuronidation in human liver tissue with UGT1A1 promoter polymorphism. Clin Pharmacol Ther. 1999;65:576–582. doi: 10.1016/S0009-9236(99)70078-0. [DOI] [PubMed] [Google Scholar]
- 186.Han JY, Lim HS, Shin ES, Yoo YK, Park YH, Lee JE, Jang IJ, Lee DH, Lee JS. Comprehensive analysis of UGT1A polymorphisms predictive for pharmacokinetics and treatment outcome in patients with non-small-cell lung cancer treated with irinotecan and cisplatin. J Clin Oncol. 2006;24:2237–2244. doi: 10.1200/JCO.2005.03.0239. [DOI] [PubMed] [Google Scholar]
- 187.Sugatani J, Yamakawa K, Yoshinari K, Machida T, Takagi H, Mori M, Kakizaki S, Sueyoshi T, Negishi M, Miwa M. Identification of a defect in the UGT1A1 gene promoter and its association with hyperbilirubinemia. Biochem Biophys Res Commun. 2002;292:492–497. doi: 10.1006/bbrc.2002.6683. [DOI] [PubMed] [Google Scholar]
- 188.Guillemette C, Ritter JK, Auyeung DJ, Kessler FK, Housman DE. Structural heterogeneity at the UDP-glucuronosyltransferase 1 locus: functional consequences of three novel missense mutations in the human UGT1A7 gene. Pharmacogenetics. 2000;10:629–644. doi: 10.1097/00008571-200010000-00006. [DOI] [PubMed] [Google Scholar]
- 189.Gagné JF, Montminy V, Belanger P, Journault K, Gaucher G, Guillemette C. Common human UGT1A polymorphisms and the altered metabolism of irinotecan active metabolite 7-ethyl-10-hydroxycamptothecin (SN-38) Mol Pharmacol. 2002;62:608–617. doi: 10.1124/mol.62.3.608. [DOI] [PubMed] [Google Scholar]
- 190.Yamanaka H, Nakajima M, Katoh M, Hara Y, Tachibana O, Yamashita J, McLeod HL, Yokoi T. A novel polymorphism in the promoter region of human UGT1A9 gene (UGT1A9*22) and its effects on the transcriptional activity. Pharmacogenetics. 2004;14:329–332. doi: 10.1097/00008571-200405000-00008. [DOI] [PubMed] [Google Scholar]
- 191.Ando Y, Saka H, Ando M, Sawa T, Muro K, Ueoka H, Yokoyama A, Saitoh S, Shimokata K, Hasegawa Y. Polymorphisms of UDP-glucuronosyltransferase gene and irinotecan toxicity: a pharmacogenetic analysis. Cancer Res. 2000;60:6921–6926. [PubMed] [Google Scholar]
- 192.Innocenti F, Undevia SD, Iyer L, Chen PX, Das S, Kocherginsky M, Karrison T, Janisch L, Ramírez J, Rudin CM, et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol. 2004;22:1382–1388. doi: 10.1200/JCO.2004.07.173. [DOI] [PubMed] [Google Scholar]
- 193.Dias MM, McKinnon RA, Sorich MJ. Impact of the UGT1A1*28 allele on response to irinotecan: a systematic review and meta-analysis. Pharmacogenomics. 2012;13:889–899. doi: 10.2217/pgs.12.68. [DOI] [PubMed] [Google Scholar]
- 194.Hu ZY, Yu Q, Pei Q, Guo C. Dose-dependent association between UGT1A1*28 genotype and irinotecan-induced neutropenia: low doses also increase risk. Clin Cancer Res. 2010;16:3832–3842. doi: 10.1158/1078-0432.CCR-10-1122. [DOI] [PubMed] [Google Scholar]
- 195.Hu ZY, Yu Q, Zhao YS. Dose-dependent association between UGT1A1*28 polymorphism and irinotecan-induced diarrhoea: a meta-analysis. Eur J Cancer. 2010;46:1856–1865. doi: 10.1016/j.ejca.2010.02.049. [DOI] [PubMed] [Google Scholar]
- 196.Winder T, Lenz HJ. Molecular predictive and prognostic markers in colon cancer. Cancer Treat Rev. 2010;36:550–556. doi: 10.1016/j.ctrv.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 197.Carlini LE, Meropol NJ, Bever J, Andria ML, Hill T, Gold P, Rogatko A, Wang H, Blanchard RL. UGT1A7 and UGT1A9 polymorphisms predict response and toxicity in colorectal cancer patients treated with capecitabine/irinotecan. Clin Cancer Res. 2005;11:1226–1236. [PubMed] [Google Scholar]
- 198.Brandi G, de Rosa F, Biasco G. Irinotecan toxicity: genes or intestinal microflora? Br J Cancer. 2009;100:1017. doi: 10.1038/sj.bjc.6604957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Liu CY, Chen PM, Chiou TJ, Liu JH, Lin JK, Lin TC, Chen WS, Jiang JK, Wang HS, Wang WS. UGT1A1*28 polymorphism predicts irinotecan-induced severe toxicities without affecting treatment outcome and survival in patients with metastatic colorectal carcinoma. Cancer. 2008;112:1932–1940. doi: 10.1002/cncr.23370. [DOI] [PubMed] [Google Scholar]
- 200.Rivory LP, Haaz MC, Canal P, Lokiec F, Armand JP, Robert J. Pharmacokinetic interrelationships of irinotecan (CPT-11) and its three major plasma metabolites in patients enrolled in phase I/II trials. Clin Cancer Res. 1997;3:1261–1266. [PubMed] [Google Scholar]
- 201.Haaz MC, Riché C, Rivory LP, Robert J. Biosynthesis of an aminopiperidino metabolite of irinotecan [7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecine] by human hepatic microsomes. Drug Metab Dispos. 1998;26:769–774. [PubMed] [Google Scholar]
- 202.Haaz MC, Rivory L, Riché C, Vernillet L, Robert J. Metabolism of irinotecan (CPT-11) by human hepatic microsomes: participation of cytochrome P-450 3A and drug interactions. Cancer Res. 1998;58:468–472. [PubMed] [Google Scholar]
- 203.Rebbeck TR, Jaffe JM, Walker AH, Wein AJ, Malkowicz SB. Modification of clinical presentation of prostate tumors by a novel genetic variant in CYP3A4. J Natl Cancer Inst. 1998;90:1225–1229. doi: 10.1093/jnci/90.16.1225. [DOI] [PubMed] [Google Scholar]
- 204.Agundez JA. Cytochrome P450 gene polymorphism and cancer. Curr Drug Metab. 2004;5:211–224. doi: 10.2174/1389200043335621. [DOI] [PubMed] [Google Scholar]
- 205.Bozina N, Bradamante V, Lovrić M. Genetic polymorphism of metabolic enzymes P450 (CYP) as a susceptibility factor for drug response, toxicity, and cancer risk. Arh Hig Rada Toksikol. 2009;60:217–242. doi: 10.2478/10004-1254-60-2009-1885. [DOI] [PubMed] [Google Scholar]
- 206.Xie HG, Wood AJ, Kim RB, Stein CM, Wilkinson GR. Genetic variability in CYP3A5 and its possible consequences. Pharmacogenomics. 2004;5:243–272. doi: 10.1517/phgs.5.3.243.29833. [DOI] [PubMed] [Google Scholar]
- 207.Fujiwara Y, Minami H. An overview of the recent progress in irinotecan pharmacogenetics. Pharmacogenomics. 2010;11:391–406. doi: 10.2217/pgs.10.19. [DOI] [PubMed] [Google Scholar]
- 208.Fukushima-Uesaka H, Saito Y, Watanabe H, Shiseki K, Saeki M, Nakamura T, Kurose K, Sai K, Komamura K, Ueno K, et al. Haplotypes of CYP3A4 and their close linkage with CYP3A5 haplotypes in a Japanese population. Hum Mutat. 2004;23:100. doi: 10.1002/humu.9210. [DOI] [PubMed] [Google Scholar]
- 209.Sai K, Saito Y, Fukushima-Uesaka H, Kurose K, Kaniwa N, Kamatani N, Shirao K, Yamamoto N, Hamaguchi T, Kunitoh H, et al. Impact of CYP3A4 haplotypes on irinotecan pharmacokinetics in Japanese cancer patients. Cancer Chemother Pharmacol. 2008;62:529–537. doi: 10.1007/s00280-007-0634-1. [DOI] [PubMed] [Google Scholar]
- 210.Mathijssen RH, Marsh S, Karlsson MO, Xie R, Baker SD, Verweij J, Sparreboom A, McLeod HL. Irinotecan pathway genotype analysis to predict pharmacokinetics. Clin Cancer Res. 2003;9:3246–3253. [PubMed] [Google Scholar]
- 211.Mathijssen RH, de Jong FA, van Schaik RH, Lepper ER, Friberg LE, Rietveld T, de Bruijn P, Graveland WJ, Figg WD, Verweij J, et al. Prediction of irinotecan pharmacokinetics by use of cytochrome P450 3A4 phenotyping probes. J Natl Cancer Inst. 2004;96:1585–1592. doi: 10.1093/jnci/djh298. [DOI] [PubMed] [Google Scholar]
- 212.Sodani K, Patel A, Kathawala RJ, Chen ZS. Multidrug resistance associated proteins in multidrug resistance. Chin J Cancer. 2012;31:58–72. doi: 10.5732/cjc.011.10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Fromm MF, Kim RB. Drug transporters. Heidelberg: Springer; 2011. [Google Scholar]
- 214.Ishikawa T, Kim RB, König Jr. Pharmacogenomics of human drug transporters: clinical impacts. Hoboken, NJ: John Wiley & Sons; 2013. [Google Scholar]
- 215.Kim TW, Innocenti F. Insights, challenges, and future directions in irinogenetics. Ther Drug Monit. 2007;29:265–270. doi: 10.1097/FTD.0b013e318068623b. [DOI] [PubMed] [Google Scholar]
- 216.Innocenti F, Kroetz DL, Schuetz E, Dolan ME, Ramírez J, Relling M, Chen P, Das S, Rosner GL, Ratain MJ. Comprehensive pharmacogenetic analysis of irinotecan neutropenia and pharmacokinetics. J Clin Oncol. 2009;27:2604–2614. doi: 10.1200/JCO.2008.20.6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Sai K, Kaniwa N, Itoda M, Saito Y, Hasegawa R, Komamura K, Ueno K, Kamakura S, Kitakaze M, Shirao K, et al. Haplotype analysis of ABCB1/MDR1 blocks in a Japanese population reveals genotype-dependent renal clearance of irinotecan. Pharmacogenetics. 2003;13:741–757. doi: 10.1097/00008571-200312000-00005. [DOI] [PubMed] [Google Scholar]
- 218.Han JY, Lim HS, Yoo YK, Shin ES, Park YH, Lee SY, Lee JE, Lee DH, Kim HT, Lee JS. Associations of ABCB1, ABCC2, and ABCG2 polymorphisms with irinotecan-pharmacokinetics and clinical outcome in patients with advanced non-small cell lung cancer. Cancer. 2007;110:138–147. doi: 10.1002/cncr.22760. [DOI] [PubMed] [Google Scholar]
- 219.Glimelius B, Garmo H, Berglund A, Fredriksson LA, Berglund M, Kohnke H, Byström P, Sørbye H, Wadelius M. Prediction of irinotecan and 5-fluorouracil toxicity and response in patients with advanced colorectal cancer. Pharmacogenomics J. 2011;11:61–71. doi: 10.1038/tpj.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Chen ZS, Furukawa T, Sumizawa T, Ono K, Ueda K, Seto K, Akiyama SI. ATP-Dependent efflux of CPT-11 and SN-38 by the multidrug resistance protein (MRP) and its inhibition by PAK-104P. Mol Pharmacol. 1999;55:921–928. [PubMed] [Google Scholar]
- 221.Conrad S, Kauffmann HM, Ito K, Deeley RG, Cole SP, Schrenk D. Identification of human multidrug resistance protein 1 (MRP1) mutations and characterization of a G671V substitution. J Hum Genet. 2001;46:656–663. doi: 10.1007/s100380170017. [DOI] [PubMed] [Google Scholar]
- 222.Conrad S, Kauffmann HM, Ito K, Leslie EM, Deeley RG, Schrenk D, Cole SP. A naturally occurring mutation in MRP1 results in a selective decrease in organic anion transport and in increased doxorubicin resistance. Pharmacogenetics. 2002;12:321–330. doi: 10.1097/00008571-200206000-00008. [DOI] [PubMed] [Google Scholar]
- 223.Leslie EM, Létourneau IJ, Deeley RG, Cole SP. Functional and structural consequences of cysteine substitutions in the NH2 proximal region of the human multidrug resistance protein 1 (MRP1/ABCC1) Biochemistry. 2003;42:5214–5224. doi: 10.1021/bi027076n. [DOI] [PubMed] [Google Scholar]
- 224.Létourneau IJ, Deeley RG, Cole SP. Functional characterization of non-synonymous single nucleotide polymorphisms in the gene encoding human multidrug resistance protein 1 (MRP1/ABCC1) Pharmacogenet Genomics. 2005;15:647–657. doi: 10.1097/01.fpc.0000173484.51807.48. [DOI] [PubMed] [Google Scholar]
- 225.Moriya Y, Nakamura T, Horinouchi M, Sakaeda T, Tamura T, Aoyama N, Shirakawa T, Gotoh A, Fujimoto S, Matsuo M, et al. Effects of polymorphisms of MDR1, MRP1, and MRP2 genes on their mRNA expression levels in duodenal enterocytes of healthy Japanese subjects. Biol Pharm Bull. 2002;25:1356–1359. doi: 10.1248/bpb.25.1356. [DOI] [PubMed] [Google Scholar]
- 226.Chu XY, Kato Y, Niinuma K, Sudo KI, Hakusui H, Sugiyama Y. Multispecific organic anion transporter is responsible for the biliary excretion of the camptothecin derivative irinotecan and its metabolites in rats. J Pharmacol Exp Ther. 1997;281:304–314. [PubMed] [Google Scholar]
- 227.Innocenti F, Undevia SD, Chen PX, Das S, Ramirez J, Dolan ME, Relling MV, Kroetz DL, Ratain MJ. Pharmacogenetic analysis of interindividual irinotecan (CPT-11) pharmacokinetic (PK) variability: Evidence for a functional variant of ABCC2. J Clin Oncol. 2004;22:2010. [Google Scholar]
- 228.Kitagawa C, Ando M, Ando Y, Sekido Y, Usui M, Takahashi K, Shimokata K, Hasegawa Y. Genetic polymorphisms of the multidrug resistance-associated protein 2 gene (ABCC2) and Irinotecan toxicity. J Clin Oncol. 2004;22:2009. [Google Scholar]
- 229.Maliepaard M, van Gastelen MA, de Jong LA, Pluim D, van Waardenburg RC, Ruevekamp-Helmers MC, Floot BG, Schellens JH. Overexpression of the BCRP/MXR/ABCP gene in a topotecan-selected ovarian tumor cell line. Cancer Res. 1999;59:4559–4563. [PubMed] [Google Scholar]
- 230.Schellens JH, Maliepaard M, Scheper RJ, Scheffer GL, Jonker JW, Smit JW, Beijnen JH, Schinkel AH. Transport of topoisomerase I inhibitors by the breast cancer resistance protein. Potential clinical implications. Ann N Y Acad Sci. 2000;922:188–194. doi: 10.1111/j.1749-6632.2000.tb07037.x. [DOI] [PubMed] [Google Scholar]
- 231.Kawabata S, Oka M, Soda H, Shiozawa K, Nakatomi K, Tsurutani J, Nakamura Y, Doi S, Kitazaki T, Sugahara K, et al. Expression and functional analyses of breast cancer resistance protein in lung cancer. Clin Cancer Res. 2003;9:3052–3057. [PubMed] [Google Scholar]
- 232.Candeil L, Gourdier I, Peyron D, Vezzio N, Copois V, Bibeau F, Orsetti B, Scheffer GL, Ychou M, Khan QA, et al. ABCG2 overexpression in colon cancer cells resistant to SN38 and in irinotecan-treated metastases. Int J Cancer. 2004;109:848–854. doi: 10.1002/ijc.20032. [DOI] [PubMed] [Google Scholar]
- 233.Yoshikawa M, Ikegami Y, Sano K, Yoshida H, Mitomo H, Sawada S, Ishikawa T. Transport of SN-38 by the wild type of human ABC transporter ABCG2 and its inhibition by quercetin, a natural flavonoid. J Exp Ther Oncol. 2004;4:25–35. [PubMed] [Google Scholar]
- 234.Bram EE, Stark M, Raz S, Assaraf YG. Chemotherapeutic drug-induced ABCG2 promoter demethylation as a novel mechanism of acquired multidrug resistance. Neoplasia. 2009;11:1359–1370. doi: 10.1593/neo.91314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Bram EE, Ifergan I, Grimberg M, Lemke K, Skladanowski A, Assaraf YG. C421 allele-specific ABCG2 gene amplification confers resistance to the antitumor triazoloacridone C-1305 in human lung cancer cells. Biochem Pharmacol. 2007;74:41–53. doi: 10.1016/j.bcp.2007.03.028. [DOI] [PubMed] [Google Scholar]
- 236.To KK, Robey RW, Knutsen T, Zhan Z, Ried T, Bates SE. Escape from hsa-miR-519c enables drug-resistant cells to maintain high expression of ABCG2. Mol Cancer Ther. 2009;8:2959–2968. doi: 10.1158/1535-7163.MCT-09-0292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Cha PC, Mushiroda T, Zembutsu H, Harada H, Shinoda N, Kawamoto S, Shimoyama R, Nishidate T, Furuhata T, Sasaki K, et al. Single nucleotide polymorphism in ABCG2 is associated with irinotecan-induced severe myelosuppression. J Hum Genet. 2009;54:572–580. doi: 10.1038/jhg.2009.80. [DOI] [PubMed] [Google Scholar]
- 238.Poonkuzhali B, Lamba J, Strom S, Sparreboom A, Thummel K, Watkins P, Schuetz E. Association of breast cancer resistance protein/ABCG2 phenotypes and novel promoter and intron 1 single nucleotide polymorphisms. Drug Metab Dispos. 2008;36:780–795. doi: 10.1124/dmd.107.018366. [DOI] [PubMed] [Google Scholar]
- 239.Morisaki K, Robey RW, Ozvegy-Laczka C, Honjo Y, Polgar O, Steadman K, Sarkadi B, Bates SE. Single nucleotide polymorphisms modify the transporter activity of ABCG2. Cancer Chemother Pharmacol. 2005;56:161–172. doi: 10.1007/s00280-004-0931-x. [DOI] [PubMed] [Google Scholar]
- 240.Sparreboom A, Loos WJ, Burger H, Sissung TM, Verweij J, Figg WD, Nooter K, Gelderblom H. Effect of ABCG2 genotype on the oral bioavailability of topotecan. Cancer Biol Ther. 2005;4:650–658. doi: 10.4161/cbt.4.6.1731. [DOI] [PubMed] [Google Scholar]
- 241.Mizuarai S, Aozasa N, Kotani H. Single nucleotide polymorphisms result in impaired membrane localization and reduced atpase activity in multidrug transporter ABCG2. Int J Cancer. 2004;109:238–246. doi: 10.1002/ijc.11669. [DOI] [PubMed] [Google Scholar]
- 242.Tamura A, Wakabayashi K, Onishi Y, Takeda M, Ikegami Y, Sawada S, Tsuji M, Matsuda Y, Ishikawa T. Re-evaluation and functional classification of non-synonymous single nucleotide polymorphisms of the human ATP-binding cassette transporter ABCG2. Cancer Sci. 2007;98:231–239. doi: 10.1111/j.1349-7006.2006.00371.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.de Jong FA, Marsh S, Mathijssen RH, King C, Verweij J, Sparreboom A, McLeod HL. ABCG2 pharmacogenetics: ethnic differences in allele frequency and assessment of influence on irinotecan disposition. Clin Cancer Res. 2004;10:5889–5894. doi: 10.1158/1078-0432.CCR-04-0144. [DOI] [PubMed] [Google Scholar]
- 244.Jada SR, Lim R, Wong CI, Shu X, Lee SC, Zhou Q, Goh BC, Chowbay B. Role of UGT1A1*6, UGT1A1*28 and ABCG2 c.421C& gt; A polymorphisms in irinotecan-induced neutropenia in Asian cancer patients. Cancer Sci. 2007;98:1461–1467. doi: 10.1111/j.1349-7006.2007.00541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Gradhand U, Kim RB. Pharmacogenomics of MRP transporters (ABCC1-5) and BCRP (ABCG2) Drug Metab Rev. 2008;40:317–354. doi: 10.1080/03602530801952617. [DOI] [PubMed] [Google Scholar]
- 246.Nozawa T, Minami H, Sugiura S, Tsuji A, Tamai I. Role of organic anion transporter OATP1B1 (OATP-C) in hepatic uptake of irinotecan and its active metabolite, 7-ethyl-10-hydroxycamptothecin: in vitro evidence and effect of single nucleotide polymorphisms. Drug Metab Dispos. 2005;33:434–439. doi: 10.1124/dmd.104.001909. [DOI] [PubMed] [Google Scholar]
- 247.Han JY, Lim HS, Shin ES, Yoo YK, Park YH, Lee JE, Kim HT, Lee JS. Influence of the organic anion-transporting polypeptide 1B1 (OATP1B1) polymorphisms on irinotecan-pharmacokinetics and clinical outcome of patients with advanced non-small cell lung cancer. Lung Cancer. 2008;59:69–75. doi: 10.1016/j.lungcan.2007.07.019. [DOI] [PubMed] [Google Scholar]
- 248.Xiang X, Jada SR, Li HH, Fan L, Tham LS, Wong CI, Lee SC, Lim R, Zhou QY, Goh BC, et al. Pharmacogenetics of SLCO1B1 gene and the impact of *1b and *15 haplotypes on irinotecan disposition in Asian cancer patients. Pharmacogenet Genomics. 2006;16:683–691. doi: 10.1097/01.fpc.0000230420.05221.71. [DOI] [PubMed] [Google Scholar]
- 249.Takane H, Miyata M, Burioka N, Kurai J, Fukuoka Y, Suyama H, Shigeoka Y, Otsubo K, Ieiri I, Shimizu E. Severe toxicities after irinotecan-based chemotherapy in a patient with lung cancer: a homozygote for the SLCO1B1*15 allele. Ther Drug Monit. 2007;29:666–668. doi: 10.1097/FTD.0b013e3181357364. [DOI] [PubMed] [Google Scholar]
- 250.Takane H, Kawamoto K, Sasaki T, Moriki K, Moriki K, Kitano H, Higuchi S, Otsubo K, Ieiri I. Life-threatening toxicities in a patient with UGT1A1*6/*28 and SLCO1B1*15/*15 genotypes after irinotecan-based chemotherapy. Cancer Chemother Pharmacol. 2009;63:1165–1169. doi: 10.1007/s00280-008-0864-x. [DOI] [PubMed] [Google Scholar]
- 251.Hoskins JM, Marcuello E, Altes A, Marsh S, Maxwell T, Van Booven DJ, Paré L, Culverhouse R, McLeod HL, Baiget M. Irinotecan pharmacogenetics: influence of pharmacodynamic genes. Clin Cancer Res. 2008;14:1788–1796. doi: 10.1158/1078-0432.CCR-07-1472. [DOI] [PubMed] [Google Scholar]
- 252.Takatani H, Oka M, Fukuda M, Narasaki F, Nakano R, Ikeda K, Terashi K, Kinoshita A, Soda H, Kanda T, et al. Gene mutation analysis and quantitation of DNA topoisomerase I in previously untreated non-small cell lung carcinomas. Jpn J Cancer Res. 1997;88:160–165. doi: 10.1111/j.1349-7006.1997.tb00361.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Tsurutani J, Nitta T, Hirashima T, Komiya T, Uejima H, Tada H, Syunichi N, Tohda A, Fukuoka M, Nakagawa K. Point mutations in the topoisomerase I gene in patients with non-small cell lung cancer treated with irinotecan. Lung Cancer. 2002;35:299–304. doi: 10.1016/s0169-5002(01)00425-1. [DOI] [PubMed] [Google Scholar]
- 254.McLeod HL, Keith WN. Variation in topoisomerase I gene copy number as a mechanism for intrinsic drug sensitivity. Br J Cancer. 1996;74:508–512. doi: 10.1038/bjc.1996.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Pommier Y. Camptothecins and topoisomerase I: a foot in the door. Targeting the genome beyond topoisomerase I with camptothecins and novel anticancer drugs: importance of DNA replication, repair and cell cycle checkpoints. Curr Med Chem Anticancer Agents. 2004;4:429–434. doi: 10.2174/1568011043352777. [DOI] [PubMed] [Google Scholar]
- 256.Reid RJ, Fiorani P, Sugawara M, Bjornsti MA. CDC45 and DPB11 are required for processive DNA replication and resistance to DNA topoisomerase I-mediated DNA damage. Proc Natl Acad Sci USA. 1999;96:11440–11445. doi: 10.1073/pnas.96.20.11440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Malanga M, Althaus FR. Poly(ADP-ribose) reactivates stalled DNA topoisomerase I and Induces DNA strand break resealing. J Biol Chem. 2004;279:5244–5248. doi: 10.1074/jbc.C300437200. [DOI] [PubMed] [Google Scholar]
- 258.Barthelmes HU, Habermeyer M, Christensen MO, Mielke C, Interthal H, Pouliot JJ, Boege F, Marko D. TDP1 overexpression in human cells counteracts DNA damage mediated by topoisomerases I and II. J Biol Chem. 2004;279:55618–55625. doi: 10.1074/jbc.M405042200. [DOI] [PubMed] [Google Scholar]
- 259.Park SY, Lam W, Cheng YC. X-ray repair cross-complementing gene I protein plays an important role in camptothecin resistance. Cancer Res. 2002;62:459–465. [PubMed] [Google Scholar]
- 260.Cusack JC, Liu R, Houston M, Abendroth K, Elliott PJ, Adams J, Baldwin AS. Enhanced chemosensitivity to CPT-11 with proteasome inhibitor PS-341: implications for systemic nuclear factor-kappaB inhibition. Cancer Res. 2001;61:3535–3540. [PubMed] [Google Scholar]
- 261.El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003;31:5526–5533. doi: 10.1093/nar/gkg761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Hu Z, Ma H, Chen F, Wei Q, Shen H. XRCC1 polymorphisms and cancer risk: a meta-analysis of 38 case-control studies. Cancer Epidemiol Biomarkers Prev. 2005;14:1810–1818. doi: 10.1158/1055-9965.EPI-04-0793. [DOI] [PubMed] [Google Scholar]
- 263.Magrini R, Bhonde MR, Hanski ML, Notter M, Scherübl H, Boland CR, Zeitz M, Hanski C. Cellular effects of CPT-11 on colon carcinoma cells: dependence on p53 and hMLH1 status. Int J Cancer. 2002;101:23–31. doi: 10.1002/ijc.10565. [DOI] [PubMed] [Google Scholar]
- 264.Bhonde MR, Hanski ML, Notter M, Gillissen BF, Daniel PT, Zeitz M, Hanski C. Equivalent effect of DNA damage-induced apoptotic cell death or long-term cell cycle arrest on colon carcinoma cell proliferation and tumour growth. Oncogene. 2006;25:165–175. doi: 10.1038/sj.onc.1209017. [DOI] [PubMed] [Google Scholar]
- 265.Gupta M, Fan S, Zhan Q, Kohn KW, O’Connor PM, Pommier Y. Inactivation of p53 increases the cytotoxicity of camptothecin in human colon HCT116 and breast MCF-7 cancer cells. Clin Cancer Res. 1997;3:1653–1660. [PubMed] [Google Scholar]
- 266.te Poele RH, Joel SP. Schedule-dependent cytotoxicity of SN-38 in p53 wild-type and mutant colon adenocarcinoma cell lines. Br J Cancer. 1999;81:1285–1293. doi: 10.1038/sj.bjc.6694370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Goldwasser F, Shimizu T, Jackman J, Hoki Y, O’Connor PM, Kohn KW, Pommier Y. Correlations between S and G2 arrest and the cytotoxicity of camptothecin in human colon carcinoma cells. Cancer Res. 1996;56:4430–4437. [PubMed] [Google Scholar]
- 268.Abal M, Bras-Goncalves R, Judde JG, Fsihi H, De Cremoux P, Louvard D, Magdelenat H, Robine S, Poupon MF. Enhanced sensitivity to irinotecan by Cdk1 inhibition in the p53-deficient HT29 human colon cancer cell line. Oncogene. 2004;23:1737–1744. doi: 10.1038/sj.onc.1207299. [DOI] [PubMed] [Google Scholar]
- 269.Wang S, El-Deiry WS. Requirement of p53 targets in chemosensitization of colonic carcinoma to death ligand therapy. Proc Natl Acad Sci USA. 2003;100:15095–15100. doi: 10.1073/pnas.2435285100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Tomicic MT, Kaina B. Topoisomerase degradation, DSB repair, p53 and IAPs in cancer cell resistance to camptothecin-like topoisomerase I inhibitors. Biochim Biophys Acta. 2013;1835:11–27. doi: 10.1016/j.bbcan.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 271.Adachi N, So S, Koyama H. Loss of nonhomologous end joining confers camptothecin resistance in DT40 cells. Implications for the repair of topoisomerase I-mediated DNA damage. J Biol Chem. 2004;279:37343–37348. doi: 10.1074/jbc.M313910200. [DOI] [PubMed] [Google Scholar]
- 272.Otsuki M, Seki M, Kawabe Y, Inoue E, Dong YP, Abe T, Kato G, Yoshimura A, Tada S, Enomoto T. WRN counteracts the NHEJ pathway upon camptothecin exposure. Biochem Biophys Res Commun. 2007;355:477–482. doi: 10.1016/j.bbrc.2007.01.175. [DOI] [PubMed] [Google Scholar]
- 273.Tashiro T, Kawada Y, Sakurai Y, Kidani Y. Antitumor activity of a new platinum complex, oxalato (trans-l-1,2-diaminocyclohexane)platinum (II): new experimental data. Biomed Pharmacother. 1989;43:251–260. doi: 10.1016/0753-3322(89)90004-8. [DOI] [PubMed] [Google Scholar]
- 274.Rixe O, Ortuzar W, Alvarez M, Parker R, Reed E, Paull K, Fojo T. Oxaliplatin, tetraplatin, cisplatin, and carboplatin: spectrum of activity in drug-resistant cell lines and in the cell lines of the National Cancer Institute’s Anticancer Drug Screen panel. Biochem Pharmacol. 1996;52:1855–1865. doi: 10.1016/s0006-2952(97)81490-6. [DOI] [PubMed] [Google Scholar]
- 275.Tournigand C, André T, Achille E, Lledo G, Flesh M, Mery-Mignard D, Quinaux E, Couteau C, Buyse M, Ganem G, et al. FOLFIRI followed by FOLFOX6 or the reverse sequence in advanced colorectal cancer: a randomized GERCOR study. J Clin Oncol. 2004;22:229–237. doi: 10.1200/JCO.2004.05.113. [DOI] [PubMed] [Google Scholar]
- 276.Manic S, Gatti L, Carenini N, Fumagalli G, Zunino F, Perego P. Mechanisms controlling sensitivity to platinum complexes: role of p53 and DNA mismatch repair. Curr Cancer Drug Targets. 2003;3:21–29. doi: 10.2174/1568009033333727. [DOI] [PubMed] [Google Scholar]
- 277.Gately DP, Howell SB. Cellular accumulation of the anticancer agent cisplatin: a review. Br J Cancer. 1993;67:1171–1176. doi: 10.1038/bjc.1993.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Perez RP. Cellular and molecular determinants of cisplatin resistance. Eur J Cancer. 1998;34:1535–1542. doi: 10.1016/s0959-8049(98)00227-5. [DOI] [PubMed] [Google Scholar]
- 279.Meijer C, Mulder NH, Hospers GA, Uges DR, de Vries EG. The role of glutathione in resistance to cisplatin in a human small cell lung cancer cell line. Br J Cancer. 1990;62:72–77. doi: 10.1038/bjc.1990.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Wernyj RP, Morin PJ. Molecular mechanisms of platinum resistance: still searching for the Achilles’ heel. Drug Resist Updat. 2004;7:227–232. doi: 10.1016/j.drup.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 281.Choi MK, Kim DD. Platinum transporters and drug resistance. Arch Pharm Res. 2006;29:1067–1073. doi: 10.1007/BF02969293. [DOI] [PubMed] [Google Scholar]
- 282.Hall MD, Okabe M, Shen DW, Liang XJ, Gottesman MM. The role of cellular accumulation in determining sensitivity to platinum-based chemotherapy. Annu Rev Pharmacol Toxicol. 2008;48:495–535. doi: 10.1146/annurev.pharmtox.48.080907.180426. [DOI] [PubMed] [Google Scholar]
- 283.Liu JJ, Lu J, McKeage MJ. Membrane transporters as determinants of the pharmacology of platinum anticancer drugs. Curr Cancer Drug Targets. 2012;12:962–986. doi: 10.2174/156800912803251199. [DOI] [PubMed] [Google Scholar]
- 284.Howell SB, Safaei R, Larson CA, Sailor MJ. Copper transporters and the cellular pharmacology of the platinum-containing cancer drugs. Mol Pharmacol. 2010;77:887–894. doi: 10.1124/mol.109.063172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Koepsell H, Endou H. The SLC22 drug transporter family. Pflugers Arch. 2004;447:666–676. doi: 10.1007/s00424-003-1089-9. [DOI] [PubMed] [Google Scholar]
- 286.Helleman J, Burger H, Hamelers IH, Boersma AW, de Kroon AI, Stoter G, Nooter K. Impaired cisplatin influx in an A2780 mutant cell line: evidence for a putative, cis-configuration-specific, platinum influx transporter. Cancer Biol Ther. 2006;5:943–949. doi: 10.4161/cbt.5.8.2876. [DOI] [PubMed] [Google Scholar]
- 287.Surowiak P, Materna V, Kaplenko I, Spaczynski M, Dolinska-Krajewska B, Gebarowska E, Dietel M, Zabel M, Lage H. ABCC2 (MRP2, cMOAT) can be localized in the nuclear membrane of ovarian carcinomas and correlates with resistance to cisplatin and clinical outcome. Clin Cancer Res. 2006;12:7149–7158. doi: 10.1158/1078-0432.CCR-06-0564. [DOI] [PubMed] [Google Scholar]
- 288.Hector S, Bolanowska-Higdon W, Zdanowicz J, Hitt S, Pendyala L. In vitro studies on the mechanisms of oxaliplatin resistance. Cancer Chemother Pharmacol. 2001;48:398–406. doi: 10.1007/s002800100363. [DOI] [PubMed] [Google Scholar]
- 289.Martinez-Balibrea E, Martínez-Cardús A, Musulén E, Ginés A, Manzano JL, Aranda E, Plasencia C, Neamati N, Abad A. Increased levels of copper efflux transporter ATP7B are associated with poor outcome in colorectal cancer patients receiving oxaliplatin-based chemotherapy. Int J Cancer. 2009;124:2905–2910. doi: 10.1002/ijc.24273. [DOI] [PubMed] [Google Scholar]
- 290.Samimi G, Katano K, Holzer AK, Safaei R, Howell SB. Modulation of the cellular pharmacology of cisplatin and its analogs by the copper exporters ATP7A and ATP7B. Mol Pharmacol. 2004;66:25–32. doi: 10.1124/mol.66.1.25. [DOI] [PubMed] [Google Scholar]
- 291.Zhou SF, Wang LL, Di YM, Xue CC, Duan W, Li CG, Li Y. Substrates and inhibitors of human multidrug resistance associated proteins and the implications in drug development. Curr Med Chem. 2008;15:1981–2039. doi: 10.2174/092986708785132870. [DOI] [PubMed] [Google Scholar]
- 292.Suzuki T, Nishio K, Tanabe S. The MRP family and anticancer drug metabolism. Curr Drug Metab. 2001;2:367–377. doi: 10.2174/1389200013338289. [DOI] [PubMed] [Google Scholar]
- 293.Beretta GL, Benedetti V, Cossa G, Assaraf YG, Bram E, Gatti L, Corna E, Carenini N, Colangelo D, Howell SB, et al. Increased levels and defective glycosylation of MRPs in ovarian carcinoma cells resistant to oxaliplatin. Biochem Pharmacol. 2010;79:1108–1117. doi: 10.1016/j.bcp.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 294.Theile D, Grebhardt S, Haefeli WE, Weiss J. Involvement of drug transporters in the synergistic action of FOLFOX combination chemotherapy. Biochem Pharmacol. 2009;78:1366–1373. doi: 10.1016/j.bcp.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 295.Lin PC, Lin HH, Lin JK, Lin CC, Yang SH, Li AF, Chen WS, Chang SC. Expression of ABCG2 associated with tumor response in metastatic colorectal cancer patients receiving first-line FOLFOX therapy--preliminary evidence. Int J Biol Markers. 2013;28:182–186. doi: 10.5301/jbm.5000004. [DOI] [PubMed] [Google Scholar]
- 296.Wu H, Kang H, Liu Y, Xiao Q, Zhang Y, Sun M, Liu D, Wang Z, Zhao H, Yao W, et al. Association of ABCB1 genetic polymorphisms with susceptibility to colorectal cancer and therapeutic prognosis. Pharmacogenomics. 2013;14:897–911. doi: 10.2217/pgs.13.78. [DOI] [PubMed] [Google Scholar]
- 297.Yue AM, Xie ZB, Zhao HF, Guo SP, Shen YH, Wang HP. Associations of ABCB1 and XPC genetic polymorphisms with susceptibility to colorectal cancer and therapeutic prognosis in a Chinese population. Asian Pac J Cancer Prev. 2013;14:3085–3091. doi: 10.7314/apjcp.2013.14.5.3085. [DOI] [PubMed] [Google Scholar]
- 298.Zhang S, Lovejoy KS, Shima JE, Lagpacan LL, Shu Y, Lapuk A, Chen Y, Komori T, Gray JW, Chen X, et al. Organic cation transporters are determinants of oxaliplatin cytotoxicity. Cancer Res. 2006;66:8847–8857. doi: 10.1158/0008-5472.CAN-06-0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Yokoo S, Yonezawa A, Masuda S, Fukatsu A, Katsura T, Inui K. Differential contribution of organic cation transporters, OCT2 and MATE1, in platinum agent-induced nephrotoxicity. Biochem Pharmacol. 2007;74:477–487. doi: 10.1016/j.bcp.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 300.Burger H, Zoumaro-Djayoon A, Boersma AW, Helleman J, Berns EM, Mathijssen RH, Loos WJ, Wiemer EA. Differential transport of platinum compounds by the human organic cation transporter hOCT2 (hSLC22A2) Br J Pharmacol. 2010;159:898–908. doi: 10.1111/j.1476-5381.2009.00569.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Larson CA, Blair BG, Safaei R, Howell SB. The role of the mammalian copper transporter 1 in the cellular accumulation of platinum-based drugs. Mol Pharmacol. 2009;75:324–330. doi: 10.1124/mol.108.052381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Rabik CA, Maryon EB, Kasza K, Shafer JT, Bartnik CM, Dolan ME. Role of copper transporters in resistance to platinating agents. Cancer Chemother Pharmacol. 2009;64:133–142. doi: 10.1007/s00280-008-0860-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Safaei R. Role of copper transporters in the uptake and efflux of platinum containing drugs. Cancer Lett. 2006;234:34–39. doi: 10.1016/j.canlet.2005.07.046. [DOI] [PubMed] [Google Scholar]
- 304.Holzer AK, Manorek GH, Howell SB. Contribution of the major copper influx transporter CTR1 to the cellular accumulation of cisplatin, carboplatin, and oxaliplatin. Mol Pharmacol. 2006;70:1390–1394. doi: 10.1124/mol.106.022624. [DOI] [PubMed] [Google Scholar]
- 305.Blair BG, Larson CA, Safaei R, Howell SB. Copper transporter 2 regulates the cellular accumulation and cytotoxicity of Cisplatin and Carboplatin. Clin Cancer Res. 2009;15:4312–4321. doi: 10.1158/1078-0432.CCR-09-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Verstraete S, Heudi O, Cailleux A, Allain P. Comparison of the reactivity of oxaliplatin, pt(diaminocyclohexane)Cl2 and pt(diaminocyclohexane1)(OH2)2(2+) with guanosine and L-methionine. J Inorg Biochem. 2001;84:129–135. doi: 10.1016/s0162-0134(00)00208-7. [DOI] [PubMed] [Google Scholar]
- 307.Kelley SL, Basu A, Teicher BA, Hacker MP, Hamer DH, Lazo JS. Overexpression of metallothionein confers resistance to anticancer drugs. Science. 1988;241:1813–1815. doi: 10.1126/science.3175622. [DOI] [PubMed] [Google Scholar]
- 308.Vescio RA, Connors KM, Bordin GM, Robb JA, Youngkin T, Umbreit JN, Hoffman RM. The distinction of small cell and non-small cell lung cancer by growth in native-state histoculture. Cancer Res. 1990;50:6095–6099. [PubMed] [Google Scholar]
- 309.Townsend DM, Tew KD. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene. 2003;22:7369–7375. doi: 10.1038/sj.onc.1206940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Lo HW, Ali-Osman F. Genetic polymorphism and function of glutathione S-transferases in tumor drug resistance. Curr Opin Pharmacol. 2007;7:367–374. doi: 10.1016/j.coph.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 311.Ali-Osman F, Akande O, Antoun G, Mao JX, Buolamwini J. Molecular cloning, characterization, and expression in Escherichia coli of full-length cDNAs of three human glutathione S-transferase Pi gene variants. Evidence for differential catalytic activity of the encoded proteins. J Biol Chem. 1997;272:10004–10012. doi: 10.1074/jbc.272.15.10004. [DOI] [PubMed] [Google Scholar]
- 312.Watson MA, Stewart RK, Smith GB, Massey TE, Bell DA. Human glutathione S-transferase P1 polymorphisms: relationship to lung tissue enzyme activity and population frequency distribution. Carcinogenesis. 1998;19:275–280. doi: 10.1093/carcin/19.2.275. [DOI] [PubMed] [Google Scholar]
- 313.Ruzzo A, Graziano F, Loupakis F, Rulli E, Canestrari E, Santini D, Catalano V, Ficarelli R, Maltese P, Bisonni R, et al. Pharmacogenetic profiling in patients with advanced colorectal cancer treated with first-line FOLFOX-4 chemotherapy. J Clin Oncol. 2007;25:1247–1254. doi: 10.1200/JCO.2006.08.1844. [DOI] [PubMed] [Google Scholar]
- 314.Chen YC, Tzeng CH, Chen PM, Lin JK, Lin TC, Chen WS, Jiang JK, Wang HS, Wang WS. Influence of GSTP1 I105V polymorphism on cumulative neuropathy and outcome of FOLFOX-4 treatment in Asian patients with colorectal carcinoma. Cancer Sci. 2010;101:530–535. doi: 10.1111/j.1349-7006.2009.01418.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Inada M, Sato M, Morita S, Kitagawa K, Kawada K, Mitsuma A, Sawaki M, Fujita K, Ando Y. Associations between oxaliplatin-induced peripheral neuropathy and polymorphisms of the ERCC1 and GSTP1 genes. Int J Clin Pharmacol Ther. 2010;48:729–734. doi: 10.5414/cpp48729. [DOI] [PubMed] [Google Scholar]
- 316.Etienne-Grimaldi MC, Milano G, Maindrault-Goebel F, Chibaudel B, Formento JL, Francoual M, Lledo G, André T, Mabro M, Mineur L, et al. Methylenetetrahydrofolate reductase (MTHFR) gene polymorphisms and FOLFOX response in colorectal cancer patients. Br J Clin Pharmacol. 2010;69:58–66. doi: 10.1111/j.1365-2125.2009.03556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Boige V, Mendiboure J, Pignon JP, Loriot MA, Castaing M, Barrois M, Malka D, Trégouët DA, Bouché O, Le Corre D, et al. Pharmacogenetic assessment of toxicity and outcome in patients with metastatic colorectal cancer treated with LV5FU2, FOLFOX, and FOLFIRI: FFCD 2000-05. J Clin Oncol. 2010;28:2556–2564. doi: 10.1200/JCO.2009.25.2106. [DOI] [PubMed] [Google Scholar]
- 318.Braun MS, Richman SD, Thompson L, Daly CL, Meade AM, Adlard JW, Allan JM, Parmar MK, Quirke P, Seymour MT. Association of molecular markers with toxicity outcomes in a randomized trial of chemotherapy for advanced colorectal cancer: the FOCUS trial. J Clin Oncol. 2009;27:5519–5528. doi: 10.1200/JCO.2008.21.6283. [DOI] [PubMed] [Google Scholar]
- 319.Paré L, Marcuello E, Altés A, del Río E, Sedano L, Salazar J, Cortés A, Barnadas A, Baiget M. Pharmacogenetic prediction of clinical outcome in advanced colorectal cancer patients receiving oxaliplatin/5-fluorouracil as first-line chemotherapy. Br J Cancer. 2008;99:1050–1055. doi: 10.1038/sj.bjc.6604671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Kweekel DM, Koopman M, Antonini NF, Van der Straaten T, Nortier JW, Gelderblom H, Punt CJ, Guchelaar HJ. GSTP1 Ile105Val polymorphism correlates with progression-free survival in MCRC patients treated with or without irinotecan: a study of the Dutch Colorectal Cancer Group. Br J Cancer. 2008;99:1316–1321. doi: 10.1038/sj.bjc.6604654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Kweekel DM, Gelderblom H, Antonini NF, Van der Straaten T, Nortier JW, Punt CJ, Guchelaar HJ. Glutathione-S-transferase pi (GSTP1) codon 105 polymorphism is not associated with oxaliplatin efficacy or toxicity in advanced colorectal cancer patients. Eur J Cancer. 2009;45:572–578. doi: 10.1016/j.ejca.2008.10.015. [DOI] [PubMed] [Google Scholar]
- 322.Sweeney C, McClure GY, Fares MY, Stone A, Coles BF, Thompson PA, Korourian S, Hutchins LF, Kadlubar FF, Ambrosone CB. Association between survival after treatment for breast cancer and glutathione S-transferase P1 Ile105Val polymorphism. Cancer Res. 2000;60:5621–5624. [PubMed] [Google Scholar]
- 323.Allan JM, Wild CP, Rollinson S, Willett EV, Moorman AV, Dovey GJ, Roddam PL, Roman E, Cartwright RA, Morgan GJ. Polymorphism in glutathione S-transferase P1 is associated with susceptibility to chemotherapy-induced leukemia. Proc Natl Acad Sci USA. 2001;98:11592–11597. doi: 10.1073/pnas.191211198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Stoehlmacher J, Park DJ, Zhang W, Groshen S, Tsao-Wei DD, Yu MC, Lenz HJ. Association between glutathione S-transferase P1, T1, and M1 genetic polymorphism and survival of patients with metastatic colorectal cancer. J Natl Cancer Inst. 2002;94:936–942. doi: 10.1093/jnci/94.12.936. [DOI] [PubMed] [Google Scholar]
- 325.Le Morvan V, Smith D, Laurand A, Brouste V, Bellott R, Soubeyran I, Mathoulin-Pelissier S, Robert J. Determination of ERCC2 Lys751Gln and GSTP1 Ile105Val gene polymorphisms in colorectal cancer patients: relationships with treatment outcome. Pharmacogenomics. 2007;8:1693–1703. doi: 10.2217/14622416.8.12.1693. [DOI] [PubMed] [Google Scholar]
- 326.Ye F, Liu Z, Tan A, Liao M, Mo Z, Yang X. XRCC1 and GSTP1 polymorphisms and prognosis of oxaliplatin-based chemotherapy in colorectal cancer: a meta-analysis. Cancer Chemother Pharmacol. 2013;71:733–740. doi: 10.1007/s00280-012-2067-8. [DOI] [PubMed] [Google Scholar]
- 327.Chai H, Pan J, Zhang X, Zhang X, Shen X, Li H, Zhang K, Yang C, Sheng H, Gao H. ERCC1 C118T associates with response to FOLFOX4 chemotherapy in colorectal cancer patients in Han Chinese. Int J Clin Exp Med. 2012;5:186–194. [PMC free article] [PubMed] [Google Scholar]
- 328.Hong J, Han SW, Ham HS, Kim TY, Choi IS, Kim BS, Oh DY, Im SA, Kang GH, Bang YJ, et al. Phase II study of biweekly S-1 and oxaliplatin combination chemotherapy in metastatic colorectal cancer and pharmacogenetic analysis. Cancer Chemother Pharmacol. 2011;67:1323–1331. doi: 10.1007/s00280-010-1425-7. [DOI] [PubMed] [Google Scholar]
- 329.Lamas MJ, Duran G, Balboa E, Bernardez B, Touris M, Vidal Y, Gallardo E, Lopez R, Carracedo A, Barros F. Use of a comprehensive panel of biomarkers to predict response to a fluorouracil-oxaliplatin regimen in patients with metastatic colorectal cancer. Pharmacogenomics. 2011;12:433–442. doi: 10.2217/pgs.10.196. [DOI] [PubMed] [Google Scholar]
- 330.Goekkurt E, Al-Batran SE, Hartmann JT, Mogck U, Schuch G, Kramer M, Jaeger E, Bokemeyer C, Ehninger G, Stoehlmacher J. Pharmacogenetic analyses of a phase III trial in metastatic gastroesophageal adenocarcinoma with fluorouracil and leucovorin plus either oxaliplatin or cisplatin: a study of the arbeitsgemeinschaft internistische onkologie. J Clin Oncol. 2009;27:2863–2873. doi: 10.1200/JCO.2008.19.1718. [DOI] [PubMed] [Google Scholar]
- 331.Faivre S, Chan D, Salinas R, Woynarowska B, Woynarowski JM. DNA strand breaks and apoptosis induced by oxaliplatin in cancer cells. Biochem Pharmacol. 2003;66:225–237. doi: 10.1016/s0006-2952(03)00260-0. [DOI] [PubMed] [Google Scholar]
- 332.Reed E. ERCC1 and clinical resistance to platinum-based therapy. Clin Cancer Res. 2005;11:6100–6102. doi: 10.1158/1078-0432.CCR-05-1083. [DOI] [PubMed] [Google Scholar]
- 333.Kweekel DM, Gelderblom H, Guchelaar HJ. Pharmacology of oxaliplatin and the use of pharmacogenomics to individualize therapy. Cancer Treat Rev. 2005;31:90–105. doi: 10.1016/j.ctrv.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 334.Reardon JT, Vaisman A, Chaney SG, Sancar A. Efficient nucleotide excision repair of cisplatin, oxaliplatin, and Bis-aceto-ammine-dichloro-cyclohexylamine-platinum(IV) (JM216) platinum intrastrand DNA diadducts. Cancer Res. 1999;59:3968–3971. [PubMed] [Google Scholar]
- 335.Yu JJ, Lee KB, Mu C, Li Q, Abernathy TV, Bostick-Bruton F, Reed E. Comparison of two human ovarian carcinoma cell lines (A2780/CP70 and MCAS) that are equally resistant to platinum, but differ at codon 118 of the ERCC1 gene. Int J Oncol. 2000;16:555–560. doi: 10.3892/ijo.16.3.555. [DOI] [PubMed] [Google Scholar]
- 336.Lunn RM, Helzlsouer KJ, Parshad R, Umbach DM, Harris EL, Sanford KK, Bell DA. XPD polymorphisms: effects on DNA repair proficiency. Carcinogenesis. 2000;21:551–555. doi: 10.1093/carcin/21.4.551. [DOI] [PubMed] [Google Scholar]
- 337.Duell EJ, Wiencke JK, Cheng TJ, Varkonyi A, Zuo ZF, Ashok TD, Mark EJ, Wain JC, Christiani DC, Kelsey KT. Polymorphisms in the DNA repair genes XRCC1 and ERCC2 and biomarkers of DNA damage in human blood mononuclear cells. Carcinogenesis. 2000;21:965–971. doi: 10.1093/carcin/21.5.965. [DOI] [PubMed] [Google Scholar]
- 338.Vilmar A, Sørensen JB. Excision repair cross-complementation group 1 (ERCC1) in platinum-based treatment of non-small cell lung cancer with special emphasis on carboplatin: a review of current literature. Lung Cancer. 2009;64:131–139. doi: 10.1016/j.lungcan.2008.08.006. [DOI] [PubMed] [Google Scholar]
- 339.Park DJ, Zhang W, Stoehlmacher J, Tsao-Wei D, Groshen S, Gil J, Yun J, Sones E, Mallik N, Lenz HJ. ERCC1 gene polymorphism as a predictor for clinical outcome in advanced colorectal cancer patients treated with platinum-based chemotherapy. Clin Adv Hematol Oncol. 2003;1:162–166. [PubMed] [Google Scholar]
- 340.Bohanes P, Labonte MJ, Lenz HJ. A review of excision repair cross-complementation group 1 in colorectal cancer. Clin Colorectal Cancer. 2011;10:157–164. doi: 10.1016/j.clcc.2011.03.024. [DOI] [PubMed] [Google Scholar]
- 341.Uchida K, Danenberg PV, Danenberg KD, Grem JL. Thymidylate synthase, dihydropyrimidine dehydrogenase, ERCC1, and thymidine phosphorylase gene expression in primary and metastatic gastrointestinal adenocarcinoma tissue in patients treated on a phase I trial of oxaliplatin and capecitabine. BMC Cancer. 2008;8:386. doi: 10.1186/1471-2407-8-386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Lenz HJ, Zhang W, Shi MM, Jacques C, Barrett JC, Danenberg KD, Hoffmann AC, Trarbach T, Folprecht G, Meinhardt G, et al. ERCC-1 gene expression levels and outcome to FOLFOX chemotherapy in patients enrolled in CONFIRM1 and CONFIRM2. J Clin Oncol. 2008;26:4131. [Google Scholar]
- 343.Yu JJ, Mu C, Lee KB, Okamoto A, Reed EL, Bostick-Bruton F, Mitchell KC, Reed E. A nucleotide polymorphism in ERCC1 in human ovarian cancer cell lines and tumor tissues. Mutat Res. 1997;382:13–20. doi: 10.1016/s1383-5726(97)00004-6. [DOI] [PubMed] [Google Scholar]
- 344.Viguier J, Boige V, Miquel C, Pocard M, Giraudeau B, Sabourin JC, Ducreux M, Sarasin A, Praz F. ERCC1 codon 118 polymorphism is a predictive factor for the tumor response to oxaliplatin/5-fluorouracil combination chemotherapy in patients with advanced colorectal cancer. Clin Cancer Res. 2005;11:6212–6217. doi: 10.1158/1078-0432.CCR-04-2216. [DOI] [PubMed] [Google Scholar]
- 345.Martinez-Balibrea E, Abad A, Aranda E, Sastre J, Manzano JL, Díaz-Rubio E, Gómez-España A, Aparicio J, García T, Maestu I, et al. Pharmacogenetic approach for capecitabine or 5-fluorouracil selection to be combined with oxaliplatin as first-line chemotherapy in advanced colorectal cancer. Eur J Cancer. 2008;44:1229–1237. doi: 10.1016/j.ejca.2008.03.025. [DOI] [PubMed] [Google Scholar]
- 346.Chang PM, Tzeng CH, Chen PM, Lin JK, Lin TC, Chen WS, Jiang JK, Wang HS, Wang WS. ERCC1 codon 118 C→T polymorphism associated with ERCC1 expression and outcome of FOLFOX-4 treatment in Asian patients with metastatic colorectal carcinoma. Cancer Sci. 2009;100:278–283. doi: 10.1111/j.1349-7006.2008.01031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Goldberg RM, McLeod HL, Sargent DJ, Morton RF, Green EM, Fuchs C, Ramanathan RK, Williamson SK, Findlay BP, Pitot HC, et al. Genetic polymorphisms, toxicity, and response rate in African Americans (AA) with metastatic colorectal cancer (MCRC) compared to Caucasians (C) when treated with IFL, FOLFOX or IROX in Intergroup N9741. J Clin Oncol. 2006;24:3503. [Google Scholar]
- 348.Ruzzo A, Graziano F, Kawakami K, Watanabe G, Santini D, Catalano V, Bisonni R, Canestrari E, Ficarelli R, Menichetti ET, et al. Pharmacogenetic profiling and clinical outcome of patients with advanced gastric cancer treated with palliative chemotherapy. J Clin Oncol. 2006;24:1883–1891. doi: 10.1200/JCO.2005.04.8322. [DOI] [PubMed] [Google Scholar]
- 349.Liu B, Wei J, Zou Z, Qian X, Nakamura T, Zhang W, Ding Y, Feng J, Yu L. Polymorphism of XRCC1 predicts overall survival of gastric cancer patients receiving oxaliplatin-based chemotherapy in Chinese population. Eur J Hum Genet. 2007;15:1049–1053. doi: 10.1038/sj.ejhg.5201884. [DOI] [PubMed] [Google Scholar]
- 350.Braun MS, Richman SD, Quirke P, Daly C, Adlard JW, Elliott F, Barrett JH, Selby P, Meade AM, Stephens RJ, et al. Predictive biomarkers of chemotherapy efficacy in colorectal cancer: results from the UK MRC FOCUS trial. J Clin Oncol. 2008;26:2690–2698. doi: 10.1200/JCO.2007.15.5580. [DOI] [PubMed] [Google Scholar]
- 351.Monzo M, Moreno I, Navarro A, Ibeas R, Artells R, Gel B, Martinez F, Moreno J, Hernandez R, Navarro-Vigo M. Single nucleotide polymorphisms in nucleotide excision repair genes XPA, XPD, XPG and ERCC1 in advanced colorectal cancer patients treated with first-line oxaliplatin/fluoropyrimidine. Oncology. 2007;72:364–370. doi: 10.1159/000113534. [DOI] [PubMed] [Google Scholar]
- 352.Keam B, Im SA, Han SW, Ham HS, Kim MA, Oh DY, Lee SH, Kim JH, Kim DW, Kim TY, et al. Modified FOLFOX-6 chemotherapy in advanced gastric cancer: Results of phase II study and comprehensive analysis of polymorphisms as a predictive and prognostic marker. BMC Cancer. 2008;8:148. doi: 10.1186/1471-2407-8-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Park DJ, Stoehlmacher J, Zhang W, Tsao-Wei DD, Groshen S, Lenz HJ. A Xeroderma pigmentosum group D gene polymorphism predicts clinical outcome to platinum-based chemotherapy in patients with advanced colorectal cancer. Cancer Res. 2001;61:8654–8658. [PubMed] [Google Scholar]
- 354.Shi Q, Wang LE, Bondy ML, Brewster A, Singletary SE, Wei Q. Reduced DNA repair of benzo[a]pyrene diol epoxide-induced adducts and common XPD polymorphisms in breast cancer patients. Carcinogenesis. 2004;25:1695–1700. doi: 10.1093/carcin/bgh167. [DOI] [PubMed] [Google Scholar]
- 355.Qiao Y, Spitz MR, Shen H, Guo Z, Shete S, Hedayati M, Grossman L, Mohrenweiser H, Wei Q. Modulation of repair of ultraviolet damage in the host-cell reactivation assay by polymorphic XPC and XPD/ERCC2 genotypes. Carcinogenesis. 2002;23:295–299. doi: 10.1093/carcin/23.2.295. [DOI] [PubMed] [Google Scholar]
- 356.Ruzzo A, Graziano F, Loupakis F, Santini D, Catalano V, Bisonni R, Ficarelli R, Fontana A, Andreoni F, Falcone A, et al. Pharmacogenetic profiling in patients with advanced colorectal cancer treated with first-line FOLFIRI chemotherapy. Pharmacogenomics J. 2008;8:278–288. doi: 10.1038/sj.tpj.6500463. [DOI] [PubMed] [Google Scholar]
- 357.Yin M, Yan J, Martinez-Balibrea E, Graziano F, Lenz HJ, Kim HJ, Robert J, Im SA, Wang WS, Etienne-Grimaldi MC, et al. ERCC1 and ERCC2 polymorphisms predict clinical outcomes of oxaliplatin-based chemotherapies in gastric and colorectal cancer: a systemic review and meta-analysis. Clin Cancer Res. 2011;17:1632–1640. doi: 10.1158/1078-0432.CCR-10-2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Kim SH, Kwon HC, Oh SY, Lee DM, Lee S, Lee JH, Roh MS, Kim DC, Park KJ, Choi HJ, et al. Prognostic value of ERCC1, thymidylate synthase, and glutathione S-transferase pi for 5-FU/oxaliplatin chemotherapy in advanced colorectal cancer. Am J Clin Oncol. 2009;32:38–43. doi: 10.1097/COC.0b013e31817be58e. [DOI] [PubMed] [Google Scholar]
- 359.Lunn RM, Langlois RG, Hsieh LL, Thompson CL, Bell DA. XRCC1 polymorphisms: effects on aflatoxin B1-DNA adducts and glycophorin A variant frequency. Cancer Res. 1999;59:2557–2561. [PubMed] [Google Scholar]
- 360.Monaco R, Rosal R, Dolan MA, Pincus MR, Brandt-Rauf PW. Conformational effects of a common codon 399 polymorphism on the BRCT1 domain of the XRCC1 protein. Protein J. 2007;26:541–546. doi: 10.1007/s10930-007-9095-y. [DOI] [PubMed] [Google Scholar]
- 361.Gurubhagavatula S, Liu G, Park S, Zhou W, Su L, Wain JC, Lynch TJ, Neuberg DS, Christiani DC. XPD and XRCC1 genetic polymorphisms are prognostic factors in advanced non-small-cell lung cancer patients treated with platinum chemotherapy. J Clin Oncol. 2004;22:2594–2601. doi: 10.1200/JCO.2004.08.067. [DOI] [PubMed] [Google Scholar]
- 362.Suh KW, Kim JH, Kim do Y, Kim YB, Lee C, Choi S. Which gene is a dominant predictor of response during FOLFOX chemotherapy for the treatment of metastatic colorectal cancer, the MTHFR or XRCC1 gene? Ann Surg Oncol. 2006;13:1379–1385. doi: 10.1245/s10434-006-9112-y. [DOI] [PubMed] [Google Scholar]
- 363.Liang J, Jiang T, Yao RY, Liu ZM, Lv HY, Qi WW. The combination of ERCC1 and XRCC1 gene polymorphisms better predicts clinical outcome to oxaliplatin-based chemotherapy in metastatic colorectal cancer. Cancer Chemother Pharmacol. 2010;66:493–500. doi: 10.1007/s00280-009-1186-3. [DOI] [PubMed] [Google Scholar]
- 364.Stoehlmacher J, Ghaderi V, Iobal S, Groshen S, Tsao-Wei D, Park D, Lenz HJ. A polymorphism of the XRCC1 gene predicts for response to platinum based treatment in advanced colorectal cancer. Anticancer Res. 2001;21:3075–3079. [PubMed] [Google Scholar]
- 365.Raymond E, Faivre S, Woynarowski JM, Chaney SG. Oxaliplatin: mechanism of action and antineoplastic activity. Semin Oncol. 1998;25:4–12. [PubMed] [Google Scholar]
- 366.Chaney SG, Campbell SL, Bassett E, Wu Y. Recognition and processing of cisplatin- and oxaliplatin-DNA adducts. Crit Rev Oncol Hematol. 2005;53:3–11. doi: 10.1016/j.critrevonc.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 367.Jascur T, Boland CR. Structure and function of the components of the human DNA mismatch repair system. Int J Cancer. 2006;119:2030–2035. doi: 10.1002/ijc.22023. [DOI] [PubMed] [Google Scholar]
- 368.Wheeler JM, Beck NE, Kim HC, Tomlinson IP, Mortensen NJ, Bodmer WF. Mechanisms of inactivation of mismatch repair genes in human colorectal cancer cell lines: the predominant role of hMLH1. Proc Natl Acad Sci USA. 1999;96:10296–10301. doi: 10.1073/pnas.96.18.10296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Peltomäki P. Role of DNA mismatch repair defects in the pathogenesis of human cancer. J Clin Oncol. 2003;21:1174–1179. doi: 10.1200/JCO.2003.04.060. [DOI] [PubMed] [Google Scholar]
- 370.Plumb JA, Strathdee G, Sludden J, Kaye SB, Brown R. Reversal of drug resistance in human tumor xenografts by 2’-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer Res. 2000;60:6039–6044. [PubMed] [Google Scholar]
- 371.Fink D, Nebel S, Aebi S, Zheng H, Cenni B, Nehmé A, Christen RD, Howell SB. The role of DNA mismatch repair in platinum drug resistance. Cancer Res. 1996;56:4881–4886. [PubMed] [Google Scholar]
- 372.Vaisman A, Varchenko M, Umar A, Kunkel TA, Risinger JI, Barrett JC, Hamilton TC, Chaney SG. The role of hMLH1, hMSH3, and hMSH6 defects in cisplatin and oxaliplatin resistance: correlation with replicative bypass of platinum-DNA adducts. Cancer Res. 1998;58:3579–3585. [PubMed] [Google Scholar]
- 373.Barry MA, Behnke CA, Eastman A. Activation of programmed cell death (apoptosis) by cisplatin, other anticancer drugs, toxins and hyperthermia. Biochem Pharmacol. 1990;40:2353–2362. doi: 10.1016/0006-2952(90)90733-2. [DOI] [PubMed] [Google Scholar]
- 374.Jung Y, Lippard SJ. Direct cellular responses to platinum-induced DNA damage. Chem Rev. 2007;107:1387–1407. doi: 10.1021/cr068207j. [DOI] [PubMed] [Google Scholar]
- 375.Nehmé A, Baskaran R, Nebel S, Fink D, Howell SB, Wang JY, Christen RD. Induction of JNK and c-Abl signalling by cisplatin and oxaliplatin in mismatch repair-proficient and -deficient cells. Br J Cancer. 1999;79:1104–1110. doi: 10.1038/sj.bjc.6690176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 376.Raymond E, Faivre S, Chaney S, Woynarowski J, Cvitkovic E. Cellular and molecular pharmacology of oxaliplatin. Mol Cancer Ther. 2002;1:227–235. [PubMed] [Google Scholar]
- 377.Fink D, Zheng H, Nebel S, Norris PS, Aebi S, Lin TP, Nehmé A, Christen RD, Haas M, MacLeod CL, et al. In vitro and in vivo resistance to cisplatin in cells that have lost DNA mismatch repair. Cancer Res. 1997;57:1841–1845. [PubMed] [Google Scholar]
- 378.Ahmad S. Platinum-DNA interactions and subsequent cellular processes controlling sensitivity to anticancer platinum complexes. Chem Biodivers. 2010;7:543–566. doi: 10.1002/cbdv.200800340. [DOI] [PubMed] [Google Scholar]
- 379.Wang D, Lippard SJ. Cellular processing of platinum anticancer drugs. Nat Rev Drug Discov. 2005;4:307–320. doi: 10.1038/nrd1691. [DOI] [PubMed] [Google Scholar]
- 380.Adimoolam S, Ford JM. p53 and regulation of DNA damage recognition during nucleotide excision repair. DNA Repair (Amst) 2003;2:947–954. doi: 10.1016/s1568-7864(03)00087-9. [DOI] [PubMed] [Google Scholar]
- 381.Vekris A, Meynard D, Haaz MC, Bayssas M, Bonnet J, Robert J. Molecular determinants of the cytotoxicity of platinum compounds: the contribution of in silico research. Cancer Res. 2004;64:356–362. doi: 10.1158/0008-5472.can-03-2258. [DOI] [PubMed] [Google Scholar]
- 382.Fojta M, Pivonkova H, Brazdova M, Kovarova L, Palecek E, Pospisilova S, Vojtesek B, Kasparkova J, Brabec V. Recognition of DNA modified by antitumor cisplatin by “latent” and “active” protein p53. Biochem Pharmacol. 2003;65:1305–1316. doi: 10.1016/s0006-2952(03)00078-9. [DOI] [PubMed] [Google Scholar]
- 383.Masui K, Gini B, Wykosky J, Zanca C, Mischel PS, Furnari FB, Cavenee WK. A tale of two approaches: complementary mechanisms of cytotoxic and targeted therapy resistance may inform next-generation cancer treatments. Carcinogenesis. 2013;34:725–738. doi: 10.1093/carcin/bgt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Casado E, De Castro J, Belda-Iniesta C, Cejas P, Feliu J, Sereno M, González-Barón M. Molecular markers in colorectal cancer: genetic bases for a customised treatment. Clin Transl Oncol. 2007;9:549–554. doi: 10.1007/s12094-007-0102-8. [DOI] [PubMed] [Google Scholar]
- 385.Silvestri A, Pin E, Huijbers A, Pellicani R, Parasido EM, Pierobon M, Petricoin E, Liotta L, Belluco C. Individualized therapy for metastatic colorectal cancer. J Intern Med. 2013;274:1–24. doi: 10.1111/joim.12070. [DOI] [PubMed] [Google Scholar]
- 386.Kim ST, Lee J, Park SH, Park JO, Lim HY, Kang WK, Kim JY, Kim YH, Chang DK, Rhee PL, et al. Clinical impact of microsatellite instability in colon cancer following adjuvant FOLFOX therapy. Cancer Chemother Pharmacol. 2010;66:659–667. doi: 10.1007/s00280-009-1206-3. [DOI] [PubMed] [Google Scholar]
- 387.Zaanan A, Fléjou JF, Emile JF, Des GG, Cuilliere-Dartigues P, Malka D, Lecaille C, Validire P, Louvet C, Rougier P, et al. Defective mismatch repair status as a prognostic biomarker of disease-free survival in stage III colon cancer patients treated with adjuvant FOLFOX chemotherapy. Clin Cancer Res. 2011;17:7470–7478. doi: 10.1158/1078-0432.CCR-11-1048. [DOI] [PubMed] [Google Scholar]
- 388.Postma C, Koopman M, Buffart TE, Eijk PP, Carvalho B, Peters GJ, Ylstra B, van Krieken JH, Punt CJ, Meijer GA. DNA copy number profiles of primary tumors as predictors of response to chemotherapy in advanced colorectal cancer. Ann Oncol. 2009;20:1048–1056. doi: 10.1093/annonc/mdn738. [DOI] [PubMed] [Google Scholar]
- 389.Lee AJ, Endesfelder D, Rowan AJ, Walther A, Birkbak NJ, Futreal PA, Downward J, Szallasi Z, Tomlinson IP, Howell M, et al. Chromosomal instability confers intrinsic multidrug resistance. Cancer Res. 2011;71:1858–1870. doi: 10.1158/0008-5472.CAN-10-3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Jover R, Nguyen TP, Pérez-Carbonell L, Zapater P, Payá A, Alenda C, Rojas E, Cubiella J, Balaguer F, Morillas JD, et al. 5-Fluorouracil adjuvant chemotherapy does not increase survival in patients with CpG island methylator phenotype colorectal cancer. Gastroenterology. 2011;140:1174–1181. doi: 10.1053/j.gastro.2010.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 391.Iacopetta B, Kawakami K, Watanabe T. Predicting clinical outcome of 5-fluorouracil-based chemotherapy for colon cancer patients: is the CpG island methylator phenotype the 5-fluorouracil-responsive subgroup? Int J Clin Oncol. 2008;13:498–503. doi: 10.1007/s10147-008-0854-3. [DOI] [PubMed] [Google Scholar]
- 392.Shen L, Catalano PJ, Benson AB, O’Dwyer P, Hamilton SR, Issa JP. Association between DNA methylation and shortened survival in patients with advanced colorectal cancer treated with 5-fluorouracil based chemotherapy. Clin Cancer Res. 2007;13:6093–6098. doi: 10.1158/1078-0432.CCR-07-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Miyaki Y, Suzuki K, Koizumi K, Kato T, Saito M, Kamiyama H, Maeda T, Shibata K, Shiya N, Konishi F. Identification of a potent epigenetic biomarker for resistance to camptothecin and poor outcome to irinotecan-based chemotherapy in colon cancer. Int J Oncol. 2012;40:217–226. doi: 10.3892/ijo.2011.1189. [DOI] [PubMed] [Google Scholar]
- 394.Kasahara K, Arao T, Sakai K, Matsumoto K, Sakai A, Kimura H, Sone T, Horiike A, Nishio M, Ohira T, et al. Impact of serum hepatocyte growth factor on treatment response to epidermal growth factor receptor tyrosine kinase inhibitors in patients with non-small cell lung adenocarcinoma. Clin Cancer Res. 2010;16:4616–4624. doi: 10.1158/1078-0432.CCR-10-0383. [DOI] [PubMed] [Google Scholar]
- 395.Pierobon M, Calvert V, Belluco C, Garaci E, Deng J, Lise M, Nitti D, Mammano E, De Marchi F, Liotta L, et al. Multiplexed cell signaling analysis of metastatic and nonmetastatic colorectal cancer reveals COX2-EGFR signaling activation as a potential prognostic pathway biomarker. Clin Colorectal Cancer. 2009;8:110–117. doi: 10.3816/CCC.2009.n.018. [DOI] [PubMed] [Google Scholar]
- 396.de Wit M, Fijneman RJ, Verheul HM, Meijer GA, Jimenez CR. Proteomics in colorectal cancer translational research: biomarker discovery for clinical applications. Clin Biochem. 2013;46:466–479. doi: 10.1016/j.clinbiochem.2012.10.039. [DOI] [PubMed] [Google Scholar]
- 397.McShane LM, Altman DG, Sauerbrei W, Taube SE, Gion M, Clark GM. REporting recommendations for tumor MARKer prognostic studies (REMARK) Nat Clin Pract Urol. 2005;2:416–422. [PubMed] [Google Scholar]
- 398.Walther A, Johnstone E, Swanton C, Midgley R, Tomlinson I, Kerr D. Genetic prognostic and predictive markers in colorectal cancer. Nat Rev Cancer. 2009;9:489–499. doi: 10.1038/nrc2645. [DOI] [PubMed] [Google Scholar]
- 399.Heinemann V, Douillard JY, Ducreux M, Peeters M. Targeted therapy in metastatic colorectal cancer -- an example of personalised medicine in action. Cancer Treat Rev. 2013;39:592–601. doi: 10.1016/j.ctrv.2012.12.011. [DOI] [PubMed] [Google Scholar]
- 400.Vijayaraghavan A, Efrusy MB, Göke B, Kirchner T, Santas CC, Goldberg RM. Cost-effectiveness of KRAS testing in metastatic colorectal cancer patients in the United States and Germany. Int J Cancer. 2012;131:438–445. doi: 10.1002/ijc.26400. [DOI] [PubMed] [Google Scholar]
- 401.Longley DB, Allen WL, Johnston PG. Drug resistance, predictive markers and pharmacogenomics in colorectal cancer. Biochim Biophys Acta. 2006;1766:184–196. doi: 10.1016/j.bbcan.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 402.Sommer H, Santi DV. Purification and amino acid analysis of an active site peptide from thymidylate synthetase containing covalently bound 5-fluoro-2’-deoxyuridylate and methylenetetrahydrofolate. Biochem Biophys Res Commun. 1974;57:689–695. doi: 10.1016/0006-291x(74)90601-9. [DOI] [PubMed] [Google Scholar]
- 403.Santos A, Zanetta S, Cresteil T, Deroussent A, Pein F, Raymond E, Vernillet L, Risse ML, Boige V, Gouyette A, et al. Metabolism of irinotecan (CPT-11) by CYP3A4 and CYP3A5 in humans. Clin Cancer Res. 2000;6:2012–2020. [PubMed] [Google Scholar]
- 404.Strassburg CP, Lankisch TO, Manns MP, Ehmer U. Family 1 uridine-5’-diphosphate glucuronosyltransferases (UGT1A): from Gilbert’s syndrome to genetic organization and variability. Arch Toxicol. 2008;82:415–433. doi: 10.1007/s00204-008-0314-x. [DOI] [PubMed] [Google Scholar]
- 405.Paillas S, Causse A, Marzi L, de Medina P, Poirot M, Denis V, Vezzio-Vie N, Espert L, Arzouk H, Coquelle A, et al. MAPK14/p38α confers irinotecan resistance to TP53-defective cells by inducing survival autophagy. Autophagy. 2012;8:1098–1112. doi: 10.4161/auto.20268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Kroetz DL. Role for drug transporters beyond tumor resistance: hepatic functional imaging and genotyping of multidrug resistance transporters for the prediction of irinotecan toxicity. J Clin Oncol. 2006;24:4225–4227. doi: 10.1200/JCO.2006.07.2355. [DOI] [PubMed] [Google Scholar]
- 407.Han JY, Lim HS, Park YH, Lee SY, Lee JS. Integrated pharmacogenetic prediction of irinotecan pharmacokinetics and toxicity in patients with advanced non-small cell lung cancer. Lung Cancer. 2009;63:115–120. doi: 10.1016/j.lungcan.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 408.Rabik CA, Dolan ME. Molecular mechanisms of resistance and toxicity associated with platinating agents. Cancer Treat Rev. 2007;33:9–23. doi: 10.1016/j.ctrv.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Salgado J, Zabalegui N, Gil C, Monreal I, Rodríguez J, García-Foncillas J. Polymorphisms in the thymidylate synthase and dihydropyrimidine dehydrogenase genes predict response and toxicity to capecitabine-raltitrexed in colorectal cancer. Oncol Rep. 2007;17:325–328. [PubMed] [Google Scholar]
- 410.Park DJ, Stoehlmacher J, Zhang W, Tsao-Wei D, Groshen S, Lenz HJ. Thymidylate synthase gene polymorphism predicts response to capecitabine in advanced colorectal cancer. Int J Colorectal Dis. 2002;17:46–49. doi: 10.1007/s003840100358. [DOI] [PubMed] [Google Scholar]
- 411.Marsh S, McKay JA, Cassidy J, McLeod HL. Polymorphism in the thymidylate synthase promoter enhancer region in colorectal cancer. Int J Oncol. 2001;19:383–386. doi: 10.3892/ijo.19.2.383. [DOI] [PubMed] [Google Scholar]
- 412.Schwab M, Zanger UM, Marx C, Schaeffeler E, Klein K, Dippon J, Kerb R, Blievernicht J, Fischer J, Hofmann U, et al. Role of genetic and nongenetic factors for fluorouracil treatment-related severe toxicity: a prospective clinical trial by the German 5-FU Toxicity Study Group. J Clin Oncol. 2008;26:2131–2138. doi: 10.1200/JCO.2006.10.4182. [DOI] [PubMed] [Google Scholar]
- 413.Hitre E, Budai B, Adleff V, Czeglédi F, Horváth Z, Gyergyay F, Lövey J, Kovács T, Orosz Z, Láng I, et al. Influence of thymidylate synthase gene polymorphisms on the survival of colorectal cancer patients receiving adjuvant 5-fluorouracil. Pharmacogenet Genomics. 2005;15:723–730. doi: 10.1097/01.fpc.0000175598.42141.59. [DOI] [PubMed] [Google Scholar]
- 414.Lecomte T, Ferraz JM, Zinzindohoué F, Loriot MA, Tregouet DA, Landi B, Berger A, Cugnenc PH, Jian R, Beaune P, et al. Thymidylate synthase gene polymorphism predicts toxicity in colorectal cancer patients receiving 5-fluorouracil-based chemotherapy. Clin Cancer Res. 2004;10:5880–5888. doi: 10.1158/1078-0432.CCR-04-0169. [DOI] [PubMed] [Google Scholar]
- 415.Dotor E, Cuatrecases M, Martínez-Iniesta M, Navarro M, Vilardell F, Guinó E, Pareja L, Figueras A, Molleví DG, Serrano T, et al. Tumor thymidylate synthase 1494del6 genotype as a prognostic factor in colorectal cancer patients receiving fluorouracil-based adjuvant treatment. J Clin Oncol. 2006;24:1603–1611. doi: 10.1200/JCO.2005.03.5253. [DOI] [PubMed] [Google Scholar]
- 416.Robien K, Ulrich CM. 5,10-Methylenetetrahydrofolate reductase polymorphisms and leukemia risk: a HuGE minireview. Am J Epidemiol. 2003;157:571–582. doi: 10.1093/aje/kwg024. [DOI] [PubMed] [Google Scholar]
- 417.Deenen MJ, Tol J, Burylo AM, Doodeman VD, de Boer A, Vincent A, Guchelaar HJ, Smits PH, Beijnen JH, Punt CJ, et al. Relationship between single nucleotide polymorphisms and haplotypes in DPYD and toxicity and efficacy of capecitabine in advanced colorectal cancer. Clin Cancer Res. 2011;17:3455–3468. doi: 10.1158/1078-0432.CCR-10-2209. [DOI] [PubMed] [Google Scholar]
- 418.Magné N, Etienne-Grimaldi MC, Cals L, Renée N, Formento JL, Francoual M, Milano G. Dihydropyrimidine dehydrogenase activity and the IVS14+1G& gt; A mutation in patients developing 5FU-related toxicity. Br J Clin Pharmacol. 2007;64:237–240. doi: 10.1111/j.1365-2125.2007.02869.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.van Kuilenburg AB, Meijer J, Mul AN, Meinsma R, Schmid V, Dobritzsch D, Hennekam RC, Mannens MM, Kiechle M, Etienne-Grimaldi MC, et al. Intragenic deletions and a deep intronic mutation affecting pre-mRNA splicing in the dihydropyrimidine dehydrogenase gene as novel mechanisms causing 5-fluorouracil toxicity. Hum Genet. 2010;128:529–538. doi: 10.1007/s00439-010-0879-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Johnson MR, Wang K, Diasio RB. Profound dihydropyrimidine dehydrogenase deficiency resulting from a novel compound heterozygote genotype. Clin Cancer Res. 2002;8:768–774. [PubMed] [Google Scholar]
- 421.Gross E, Ullrich T, Seck K, Mueller V, de Wit M, von Schilling C, Meindl A, Schmitt M, Kiechle M. Detailed analysis of five mutations in dihydropyrimidine dehydrogenase detected in cancer patients with 5-fluorouracil-related side effects. Hum Mutat. 2003;22:498. doi: 10.1002/humu.9201. [DOI] [PubMed] [Google Scholar]
- 422.Morel A, Boisdron-Celle M, Fey L, Soulie P, Craipeau MC, Traore S, Gamelin E. Clinical relevance of different dihydropyrimidine dehydrogenase gene single nucleotide polymorphisms on 5-fluorouracil tolerance. Mol Cancer Ther. 2006;5:2895–2904. doi: 10.1158/1535-7163.MCT-06-0327. [DOI] [PubMed] [Google Scholar]
- 423.Tanaka D, Hishida A, Matsuo K, Iwata H, Shinoda M, Yamamura Y, Kato T, Hatooka S, Mitsudomi T, Kagami Y, et al. Polymorphism of dihydropyrimidine dehydrogenase (DPYD) Cys29Arg and risk of six malignancies in Japanese. Nagoya J Med Sci. 2005;67:117–124. [PubMed] [Google Scholar]
- 424.Kleibl Z, Fidlerova J, Kleiblova P, Kormunda S, Bilek M, Bouskova K, Sevcik J, Novotny J. Influence of dihydropyrimidine dehydrogenase gene (DPYD) coding sequence variants on the development of fluoropyrimidine-related toxicity in patients with high-grade toxicity and patients with excellent tolerance of fluoropyrimidine-based chemotherapy. Neoplasma. 2009;56:303–316. doi: 10.4149/neo_2009_04_303. [DOI] [PubMed] [Google Scholar]
- 425.Gross E, Busse B, Riemenschneider M, Neubauer S, Seck K, Klein HG, Kiechle M, Lordick F, Meindl A. Strong association of a common dihydropyrimidine dehydrogenase gene polymorphism with fluoropyrimidine-related toxicity in cancer patients. PLoS One. 2008;3:e4003. doi: 10.1371/journal.pone.0004003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Sistonen J, Smith C, Fu YK, Largiadèr CR. A new DPYD genotyping assay for improving the safety of 5-fluorouracil therapy. Clin Chim Acta. 2012;414:109–111. doi: 10.1016/j.cca.2012.08.015. [DOI] [PubMed] [Google Scholar]
- 427.Mueller F, Büchel B, Köberle D, Schürch S, Pfister B, Krähenbühl S, Froehlich TK, Largiader CR, Joerger M. Gender-specific elimination of continuous-infusional 5-fluorouracil in patients with gastrointestinal malignancies: results from a prospective population pharmacokinetic study. Cancer Chemother Pharmacol. 2013;71:361–370. doi: 10.1007/s00280-012-2018-4. [DOI] [PubMed] [Google Scholar]
- 428.Wei X, Elizondo G, Sapone A, McLeod HL, Raunio H, Fernandez-Salguero P, Gonzalez FJ. Characterization of the human dihydropyrimidine dehydrogenase gene. Genomics. 1998;51:391–400. doi: 10.1006/geno.1998.5379. [DOI] [PubMed] [Google Scholar]
- 429.He YF, Wei W, Zhang X, Li YH, Li S, Wang FH, Lin XB, Li ZM, Zhang DS, Huang HQ, et al. Analysis of the DPYD gene implicated in 5-fluorouracil catabolism in Chinese cancer patients. J Clin Pharm Ther. 2008;33:307–314. doi: 10.1111/j.1365-2710.2008.00898.x. [DOI] [PubMed] [Google Scholar]
- 430.Zhang XP, Bai ZB, Chen BA, Feng JF, Yan F, Jiang Z, Zhong YJ, Wu JZ, Chen L, Lu ZH, et al. Polymorphisms of dihydropyrimidine dehydrogenase gene and clinical outcomes of gastric cancer patients treated with fluorouracil-based adjuvant chemotherapy in Chinese population. Chin Med J (Engl) 2012;125:741–746. [PubMed] [Google Scholar]
- 431.van Kuilenburg AB, Dobritzsch D, Meinsma R, Haasjes J, Waterham HR, Nowaczyk MJ, Maropoulos GD, Hein G, Kalhoff H, Kirk JM, et al. Novel disease-causing mutations in the dihydropyrimidine dehydrogenase gene interpreted by analysis of the three-dimensional protein structure. Biochem J. 2002;364:157–163. doi: 10.1042/bj3640157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 432.Loganayagam A, Arenas-Hernandez M, Fairbanks L, Ross P, Sanderson JD, Marinaki AM. The contribution of deleterious DPYD gene sequence variants to fluoropyrimidine toxicity in British cancer patients. Cancer Chemother Pharmacol. 2010;65:403–406. doi: 10.1007/s00280-009-1147-x. [DOI] [PubMed] [Google Scholar]
- 433.Collie-Duguid ES, Etienne MC, Milano G, McLeod HL. Known variant DPYD alleles do not explain DPD deficiency in cancer patients. Pharmacogenetics. 2000;10:217–223. doi: 10.1097/00008571-200004000-00002. [DOI] [PubMed] [Google Scholar]
- 434.Ezzeldin HH, Lee AM, Mattison LK, Diasio RB. Methylation of the DPYD promoter: an alternative mechanism for dihydropyrimidine dehydrogenase deficiency in cancer patients. Clin Cancer Res. 2005;11:8699–8705. doi: 10.1158/1078-0432.CCR-05-1520. [DOI] [PubMed] [Google Scholar]
- 435.Seck K, Riemer S, Kates R, Ullrich T, Lutz V, Harbeck N, Schmitt M, Kiechle M, Diasio R, Gross E. Analysis of the DPYD gene implicated in 5-fluorouracil catabolism in a cohort of Caucasian individuals. Clin Cancer Res. 2005;11:5886–5892. doi: 10.1158/1078-0432.CCR-04-1784. [DOI] [PubMed] [Google Scholar]
- 436.Loganayagam A, Arenas Hernandez M, Corrigan A, Fairbanks L, Lewis CM, Harper P, Maisey N, Ross P, Sanderson JD, Marinaki AM. Pharmacogenetic variants in the DPYD, TYMS, CDA and MTHFR genes are clinically significant predictors of fluoropyrimidine toxicity. Br J Cancer. 2013;108:2505–2515. doi: 10.1038/bjc.2013.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Boisdron-Celle M, Remaud G, Traore S, Poirier AL, Gamelin L, Morel A, Gamelin E. 5-Fluorouracil-related severe toxicity: a comparison of different methods for the pretherapeutic detection of dihydropyrimidine dehydrogenase deficiency. Cancer Lett. 2007;249:271–282. doi: 10.1016/j.canlet.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 438.Westra JL, Hollema H, Schaapveld M, Platteel I, Oien KA, Keith WN, Mauritz R, Peters GJ, Buys CH, Hofstra RM, et al. Predictive value of thymidylate synthase and dihydropyrimidine dehydrogenase protein expression on survival in adjuvantly treated stage III colon cancer patients. Ann Oncol. 2005;16:1646–1653. doi: 10.1093/annonc/mdi316. [DOI] [PubMed] [Google Scholar]
- 439.Belvedere O, Puglisi F, Di Loreto C, Cataldi P, Guglielmi A, Aschele C, Sobrero A. Lack of correlation between immunohistochemical expression of E2F-1, thymidylate synthase expression and clinical response to 5-fluorouracil in advanced colorectal cancer. Ann Oncol. 2004;15:55–58. doi: 10.1093/annonc/mdh018. [DOI] [PubMed] [Google Scholar]
- 440.Paradiso A, Xu J, Mangia A, Chiriatti A, Simone G, Zito A, Montemurro S, Giuliani F, Maiello E, Colucci G. Topoisomerase-I, thymidylate synthase primary tumour expression and clinical efficacy of 5-FU/CPT-11 chemotherapy in advanced colorectal cancer patients. Int J Cancer. 2004;111:252–258. doi: 10.1002/ijc.20208. [DOI] [PubMed] [Google Scholar]
- 441.Lindebjerg J, Nielsen JN, Hoeffding LD, Jakobsen A. Immunohistochemical expression of thymidylate synthase as predictor of response to capecitabine in patients with advanced colorectal adenocarcinoma. APMIS. 2005;113:600–602. doi: 10.1111/j.1600-0463.2005.apm_201.x. [DOI] [PubMed] [Google Scholar]
- 442.Karlberg M, Ohrling K, Edler D, Hallström M, Ullén H, Ragnhammar P. Prognostic and predictive value of thymidylate synthase expression in primary colorectal cancer. Anticancer Res. 2010;30:645–651. [PubMed] [Google Scholar]
- 443.Jensen SA, Vainer B, Sørensen JB. The prognostic significance of thymidylate synthase and dihydropyrimidine dehydrogenase in colorectal cancer of 303 patients adjuvantly treated with 5-fluorouracil. Int J Cancer. 2007;120:694–701. doi: 10.1002/ijc.22318. [DOI] [PubMed] [Google Scholar]
- 444.Oi K, Makino M, Ozaki M, Takemoto H, Yamane N, Nakamura S, Ikeguchi M, Kaibara N. Immunohistochemical dihydropyrimidine dehydrogenase expression is a good prognostic indicator for patients with Dukes’ C colorectal cancer. Anticancer Res. 2004;24:273–279. [PubMed] [Google Scholar]
- 445.Lassmann S, Hennig M, Rosenberg R, Nährig J, Schreglmann J, Krause F, Poignee-Heger M, Nekarda H, Höfler H, Werner M. Thymidine phosphorylase, dihydropyrimidine dehydrogenase and thymidylate synthase mRNA expression in primary colorectal tumors-correlation to tumor histopathology and clinical follow-up. Int J Colorectal Dis. 2006;21:238–247. doi: 10.1007/s00384-005-0767-9. [DOI] [PubMed] [Google Scholar]
- 446.Gustavsson B, Kaiser C, Carlsson G, Wettergren Y, Odin E, Lindskog EB, Niyikiza C, Ma D. Molecular determinants of efficacy for 5-FU-based treatments in advanced colorectal cancer: mRNA expression for 18 chemotherapy-related genes. Int J Cancer. 2009;124:1220–1226. doi: 10.1002/ijc.23852. [DOI] [PubMed] [Google Scholar]
- 447.Tokunaga Y, Takahashi K, Saito T. Clinical role of thymidine phosphorylase and dihydropyrimidine dehydrogenase in colorectal cancer treated with postoperative fluoropyrimidine. Hepatogastroenterology. 2005;52:1715–1721. [PubMed] [Google Scholar]
- 448.Petrioli R, Bargagli G, Lazzi S, Pascucci A, Francini E, Bellan C, Conca R, Martellucci I, Fiaschi AI, Lorenzi B, et al. Thymidine phosphorylase expression in metastatic sites is predictive for response in patients with colorectal cancer treated with continuous oral capecitabine and biweekly oxaliplatin. Anticancer Drugs. 2010;21:313–319. doi: 10.1097/CAD.0b013e328334d88a. [DOI] [PubMed] [Google Scholar]
- 449.Sameshima S, Tomozawa S, Horikoshi H, Motegi K, Hirayama I, Koketsu S, Okada T, Kojima M, Kon Y, Sawada T. 5-Fluorouracil-related gene expression in hepatic artery infusion-treated patients with hepatic metastases from colorectal carcinomas. Anticancer Res. 2008;28:389–393. [PubMed] [Google Scholar]
- 450.Dong Q, Huang S, Li Y, Liu J. [Expressions of orotate phosphoribosyltransferase in colorectal carcinoma and its correlations with toxicities of chemotherapy] Nanfang Yike Daxue Xuebao. 2012;32:1179–1181. [PubMed] [Google Scholar]
- 451.Ishibashi K, Sobajima J, Ohsawa T, Yokoyama M, Miyazaki T, Nakada H, Gonda T, Ishida H. [Expression of mRNA levels of thymidylate synthase, thymidine phosphorylase, dihydropyrimidine dehydrogenase and orotate phosphoribosyltransferase in diffusely infiltrating colorectal cancer] Gan To Kagaku Ryoho. 2007;34:1073–1077. [PubMed] [Google Scholar]
- 452.Yamada T, Tanaka N, Yokoi K, Ishikawa N, Seya T, Horiba K, Kanazawa Y, Shirakawa T, Ohkawa K, Kudoh H, et al. [Prediction of sensitivity to 5-fluorouracil (5-fu) by metabolic and target enzyme activities in colon cancer] Gan To Kagaku Ryoho. 2006;33:1603–1609. [PubMed] [Google Scholar]
- 453.Kinoshita M, Kodera Y, Hibi K, Nakayama G, Inoue T, Ohashi N, Ito Y, Koike M, Fujiwara M, Nakao A. Gene expression profile of 5-fluorouracil metabolic enzymes in primary colorectal cancer: potential as predictive parameters for response to fluorouracil-based chemotherapy. Anticancer Res. 2007;27:851–856. [PubMed] [Google Scholar]
- 454.de Jong FA, Kehrer DF, Mathijssen RH, Creemers GJ, de Bruijn P, van Schaik RH, Planting AS, van der Gaast A, Eskens FA, Janssen JT, et al. Prophylaxis of irinotecan-induced diarrhea with neomycin and potential role for UGT1A1*28 genotype screening: a double-blind, randomized, placebo-controlled study. Oncologist. 2006;11:944–954. doi: 10.1634/theoncologist.11-8-944. [DOI] [PubMed] [Google Scholar]
- 455.Kweekel DM, Gelderblom H, Van der Straaten T, Antonini NF, Punt CJ, Guchelaar HJ. UGT1A1*28 genotype and irinotecan dosage in patients with metastatic colorectal cancer: a Dutch Colorectal Cancer Group study. Br J Cancer. 2008;99:275–282. doi: 10.1038/sj.bjc.6604461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456.Minami H, Sai K, Saeki M, Saito Y, Ozawa S, Suzuki K, Kaniwa N, Sawada J, Hamaguchi T, Yamamoto N, et al. Irinotecan pharmacokinetics/pharmacodynamics and UGT1A genetic polymorphisms in Japanese: roles of UGT1A1*6 and *28. Pharmacogenet Genomics. 2007;17:497–504. doi: 10.1097/FPC.0b013e328014341f. [DOI] [PubMed] [Google Scholar]
- 457.Lamas MJ, Duran G, Balboa E, Bernardez B, Candamio S, Vidal Y, Mosquera A, Giraldez JM, Lopez R, Carracedo A, et al. The value of genetic polymorphisms to predict toxicity in metastatic colorectal patients with irinotecan-based regimens. Cancer Chemother Pharmacol. 2012;69:1591–1599. doi: 10.1007/s00280-012-1866-2. [DOI] [PubMed] [Google Scholar]
- 458.Shulman K, Cohen I, Barnett-Griness O, Kuten A, Gruber SB, Lejbkowicz F, Rennert G. Clinical implications of UGT1A1*28 genotype testing in colorectal cancer patients. Cancer. 2011;117:3156–3162. doi: 10.1002/cncr.25735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Martinez-Balibrea E, Abad A, Martínez-Cardús A, Ginés A, Valladares M, Navarro M, Aranda E, Marcuello E, Benavides M, Massutí B, et al. UGT1A and TYMS genetic variants predict toxicity and response of colorectal cancer patients treated with first-line irinotecan and fluorouracil combination therapy. Br J Cancer. 2010;103:581–589. doi: 10.1038/sj.bjc.6605776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Rhodes KE, Zhang W, Yang D, Press OA, Gordon M, Vallböhmer D, Schultheis AM, Lurje G, Ladner RD, Fazzone W, et al. ABCB1, SLCO1B1 and UGT1A1 gene polymorphisms are associated with toxicity in metastatic colorectal cancer patients treated with first-line irinotecan. Drug Metab Lett. 2007;1:23–30. doi: 10.2174/187231207779814328. [DOI] [PubMed] [Google Scholar]
- 461.Ando Y, Saka H, Asai G, Sugiura S, Shimokata K, Kamataki T. UGT1A1 genotypes and glucuronidation of SN-38, the active metabolite of irinotecan. Ann Oncol. 1998;9:845–847. doi: 10.1023/a:1008438109725. [DOI] [PubMed] [Google Scholar]
- 462.Innocenti F, Liu W, Chen P, Desai AA, Das S, Ratain MJ. Haplotypes of variants in the UDP-glucuronosyltransferase1A9 and 1A1 genes. Pharmacogenet Genomics. 2005;15:295–301. doi: 10.1097/01213011-200505000-00004. [DOI] [PubMed] [Google Scholar]
- 463.Ramírez J, Liu W, Mirkov S, Desai AA, Chen P, Das S, Innocenti F, Ratain MJ. Lack of association between common polymorphisms in UGT1A9 and gene expression and activity. Drug Metab Dispos. 2007;35:2149–2153. doi: 10.1124/dmd.107.015446. [DOI] [PubMed] [Google Scholar]
- 464.Roco A, Quiñones L, Agúndez JA, García-Martín E, Squicciarini V, Miranda C, Garay J, Farfán N, Saavedra I, Cáceres D, et al. Frequencies of 23 functionally significant variant alleles related with metabolism of antineoplastic drugs in the chilean population: comparison with caucasian and asian populations. Front Genet. 2012;3:229. doi: 10.3389/fgene.2012.00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Bethke L, Webb E, Sellick G, Rudd M, Penegar S, Withey L, Qureshi M, Houlston R. Polymorphisms in the cytochrome P450 genes CYP1A2, CYP1B1, CYP3A4, CYP3A5, CYP11A1, CYP17A1, CYP19A1 and colorectal cancer risk. BMC Cancer. 2007;7:123. doi: 10.1186/1471-2407-7-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Yang X, Zhang B, Molony C, Chudin E, Hao K, Zhu J, Gaedigk A, Suver C, Zhong H, Leeder JS, et al. Systematic genetic and genomic analysis of cytochrome P450 enzyme activities in human liver. Genome Res. 2010;20:1020–1036. doi: 10.1101/gr.103341.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Dai Z, Papp AC, Wang D, Hampel H, Sadee W. Genotyping panel for assessing response to cancer chemotherapy. BMC Med Genomics. 2008;1:24. doi: 10.1186/1755-8794-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Balcerczak E, Panczyk M, Piaskowski S, Pasz-Walczak G, Sałagacka A, Mirowski M. ABCB1/MDR1 gene polymorphisms as a prognostic factor in colorectal cancer. Int J Colorectal Dis. 2010;25:1167–1176. doi: 10.1007/s00384-010-0961-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Panczyk M, Balcerczak E, Piaskowski S, Jamroziak K, Pasz-Walczak G, Mirowski M. ABCB1 gene polymorphisms and haplotype analysis in colorectal cancer. Int J Colorectal Dis. 2009;24:895–905. doi: 10.1007/s00384-009-0724-0. [DOI] [PubMed] [Google Scholar]
- 470.He T, Mo A, Zhang K, Liu L. ABCB1/MDR1 gene polymorphism and colorectal cancer risk: a meta-analysis of case-control studies. Colorectal Dis. 2013;15:12–18. doi: 10.1111/j.1463-1318.2012.02919.x. [DOI] [PubMed] [Google Scholar]
- 471.Zhao L, Li K, Li W, Yang Z. Association between the C3435T polymorphism of ABCB1/MDR1 gene (rs1045642) and colorectal cancer susceptibility: a meta-analysis based on 11,339 subjects. Tumour Biol. 2013;34:1949–1957. doi: 10.1007/s13277-013-0740-0. [DOI] [PubMed] [Google Scholar]
- 472.Campa D, Sainz J, Pardini B, Vodickova L, Naccarati A, Rudolph A, Novotny J, Försti A, Buch S, von Schönfels W, et al. A comprehensive investigation on common polymorphisms in the MDR1/ABCB1 transporter gene and susceptibility to colorectal cancer. PLoS One. 2012;7:e32784. doi: 10.1371/journal.pone.0032784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.He T, Mo A, Zhang K, Liu L. ABCB1/MDR1 gene polymorphism and colorectal cancer risk: a meta-analysis of case-control studies. Colorectal Dis. 2013;15:12–18. doi: 10.1111/j.1463-1318.2012.02919.x. [DOI] [PubMed] [Google Scholar]
- 474.Lara PN, Natale R, Crowley J, Lenz HJ, Redman MW, Carleton JE, Jett J, Langer CJ, Kuebler JP, Dakhil SR, et al. Phase III trial of irinotecan/cisplatin compared with etoposide/cisplatin in extensive-stage small-cell lung cancer: clinical and pharmacogenomic results from SWOG S0124. J Clin Oncol. 2009;27:2530–2535. doi: 10.1200/JCO.2008.20.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Zhou Q, Sparreboom A, Tan EH, Cheung YB, Lee A, Poon D, Lee EJ, Chowbay B. Pharmacogenetic profiling across the irinotecan pathway in Asian patients with cancer. Br J Clin Pharmacol. 2005;59:415–424. doi: 10.1111/j.1365-2125.2004.02330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Wang Z, Sew PH, Ambrose H, Ryan S, Chong SS, Lee EJ, Lee CG. Nucleotide sequence analyses of the MRP1 gene in four populations suggest negative selection on its coding region. BMC Genomics. 2006;7:111. doi: 10.1186/1471-2164-7-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Cecchin E, D’Andrea M, Lonardi S, Zanusso C, Pella N, Errante D, De Mattia E, Polesel J, Innocenti F, Toffoli G. A prospective validation pharmacogenomic study in the adjuvant setting of colorectal cancer patients treated with the 5-fluorouracil/leucovorin/oxaliplatin (FOLFOX4) regimen. Pharmacogenomics J. 2013;13:403–409. doi: 10.1038/tpj.2012.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Campa D, Müller P, Edler L, Knoefel L, Barale R, Heussel CP, Thomas M, Canzian F, Risch A. A comprehensive study of polymorphisms in ABCB1, ABCC2 and ABCG2 and lung cancer chemotherapy response and prognosis. Int J Cancer. 2012;131:2920–2928. doi: 10.1002/ijc.27567. [DOI] [PubMed] [Google Scholar]
- 479.Sun N, Sun X, Chen B, Cheng H, Feng J, Cheng L, Lu Z. MRP2 and GSTP1 polymorphisms and chemotherapy response in advanced non-small cell lung cancer. Cancer Chemother Pharmacol. 2010;65:437–446. doi: 10.1007/s00280-009-1046-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Tian C, Ambrosone CB, Darcy KM, Krivak TC, Armstrong DK, Bookman MA, Davis W, Zhao H, Moysich K, Gallion H, et al. Common variants in ABCB1, ABCC2 and ABCG2 genes and clinical outcomes among women with advanced stage ovarian cancer treated with platinum and taxane-based chemotherapy: a Gynecologic Oncology Group study. Gynecol Oncol. 2012;124:575–581. doi: 10.1016/j.ygyno.2011.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Akaba K, Kimura T, Sasaki A, Tanabe S, Ikegami T, Hashimoto M, Umeda H, Yoshida H, Umetsu K, Chiba H, et al. Neonatal hyperbilirubinemia and mutation of the bilirubin uridine diphosphate-glucuronosyltransferase gene: a common missense mutation among Japanese, Koreans and Chinese. Biochem Mol Biol Int. 1998;46:21–26. doi: 10.1080/15216549800203512. [DOI] [PubMed] [Google Scholar]
- 482.Tamura A, Watanabe M, Saito H, Nakagawa H, Kamachi T, Okura I, Ishikawa T. Functional validation of the genetic polymorphisms of human ATP-binding cassette (ABC) transporter ABCG2: identification of alleles that are defective in porphyrin transport. Mol Pharmacol. 2006;70:287–296. doi: 10.1124/mol.106.023556. [DOI] [PubMed] [Google Scholar]
- 483.Campa D, Pardini B, Naccarati A, Vodickova L, Novotny J, Försti A, Hemminki K, Barale R, Vodicka P, Canzian F. A gene-wide investigation on polymorphisms in the ABCG2/BRCP transporter and susceptibility to colorectal cancer. Mutat Res. 2008;645:56–60. doi: 10.1016/j.mrfmmm.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 484.Di Martino MT, Arbitrio M, Leone E, Guzzi PH, Rotundo MS, Ciliberto D, Tomaino V, Fabiani F, Talarico D, Sperlongano P, et al. Single nucleotide polymorphisms of ABCC5 and ABCG1 transporter genes correlate to irinotecan-associated gastrointestinal toxicity in colorectal cancer patients: a DMET microarray profiling study. Cancer Biol Ther. 2011;12:780–787. doi: 10.4161/cbt.12.9.17781. [DOI] [PubMed] [Google Scholar]
- 485.Cortejoso L, García MI, García-Alfonso P, González-Haba E, Escolar F, Sanjurjo M, López-Fernández LA. Differential toxicity biomarkers for irinotecan- and oxaliplatin-containing chemotherapy in colorectal cancer. Cancer Chemother Pharmacol. 2013;71:1463–1472. doi: 10.1007/s00280-013-2145-6. [DOI] [PubMed] [Google Scholar]
- 486.Kweekel DM, Antonini NF, Nortier JW, Punt CJ, Gelderblom H, Guchelaar HJ. Explorative study to identify novel candidate genes related to oxaliplatin efficacy and toxicity using a DNA repair array. Br J Cancer. 2009;101:357–362. doi: 10.1038/sj.bjc.6605134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 487.Ma H, Xu L, Yuan J, Shao M, Hu Z, Wang F, Wang Y, Yuan W, Qian J, Wang Y, et al. Tagging single nucleotide polymorphisms in excision repair cross-complementing group 1 (ERCC1) and risk of primary lung cancer in a Chinese population. Pharmacogenet Genomics. 2007;17:417–423. doi: 10.1097/01.fpc.0000239975.77088.17. [DOI] [PubMed] [Google Scholar]