

Abstract
In this review, we discuss the findings and concepts underlying the “persistence mechanisms” of Helicobacter pylori (H. pylori), a spiral-shaped, Gram-negative rod bacterium that was discovered as a gastric pathogen by Marshall and Warren in 1984. H. pylori colonizes the gastric mucosa of nearly half of the human population. Infections appear in early childhood and, if not treated, persist for life. The presence or absence of symptoms and their severity depend on multiple bacterial components, host susceptibility and environmental factors, which allow H. pylori to switch between pathogenicity and commensalism. Many studies have shown that H. pylori components may facilitate the colonization process and the immune response of the host during the course of H. pylori infection. These H. pylori-driven interactions might result from positive or negative modulation. Among the negative immunomodulators, a prominent position is occupied by a vacuolating toxin A (VacA) and cytotoxin-associated gene A (CagA) protein. However, in light of the recent studies that are presented in this review, it is necessary to enrich this panel with H. pylori lipopolysaccharide (LPS). Together with CagA and VacA, LPS suppresses the elimination of H. pylori bacteria from the gastric mucosa by interfering with the activity of innate and adaptive immune cells, diminishing the inflammatory response, and affecting the adaptive T lymphocyte response, thus facilitating the development of chronic infections. The complex strategy of H. pylori bacteria for survival in the gastric mucosa of the host involves both structural modifications of LPS lipid A to diminish its endotoxic properties and the expression and variation of Lewis determinants, arranged in O-specific chains of H. pylori LPS. By mimicking host components, this phenomenon leaves these bacteria “invisible” to immune cells. Together, these mechanisms allow H. pylori to survive and live for many years within their hosts.
Keywords: Helicobacter pylori, Lipopolysaccharide, Immune response, Adaptation, Inflammation
Core tip: Helicobacter pylori (H. pylori), a Gram-negative bacterium, colonizes the gastric mucosa of at least half of the human population and possesses a unique lipopolysaccharide (LPS) structure. According to recent studies, this structure contributes to the immunomodulatory properties of LPS. The structural rearrangements of H. pylori LPS, especially in relation to the lipid A and O side chains, result in a unique pattern of interactions between the bacterium and the host. In this review, we report and discuss the actual findings underlying the LPS-driven “persistence mechanisms” of H. pylori, explaining how structural modifications may allow these bacteria to “live in peace” within a human host.
INTRODUCTION
In this review, we discuss the findings and concepts underlying the “persistence mechanisms” of Helicobacter pylori (H. pylori), a human pathogen colonizing the gastric mucosa. We focus on aspects concerning the structure, bioactivity, and immunomodulatory properties of H. pylori lipopolysaccharide (LPS) and its contribution to the development of chronic gastritis. Today, we know that LPS is the main outer membrane component of Gram-negative bacteria and that LPS has strong immunostimulatory and inflammatory capacities. Recent years have provided new insights into the H. pylori LPS structure and the receptors of the innate immune system that are involved in its recognition and signal transduction pathways. These new data allow a more precise definition of the harmful and beneficial effects of H. pylori LPS. There is a growing number of theories suggesting that H. pylori LPS has evolved during the co-existence of these bacteria with human hosts since the migrations out of Africa 60000 years ago, enabling these bacteria “to live in peace”[1].
HELICOBACTER PYLORI: BETWEEN PATHOGENICITY AND COMMENSALISM
H. pylori is a spiral-shaped, Gram-negative rod bacteria that was discovered as a gastric pathogen by Marshall et al[2]. H. pylori colonizes the gastric mucosa of nearly half of the human population. Infections appear in early childhood and, if not treated, persist for life[3]. Disease development depends on multiple bacterial virulence components, host susceptibility, and environmental factors[4]. A characteristic symptom of H. pylori infection is an excessive inflammatory response, which results in the development of pathological processes in the gastric epithelium, such as erosions, ulcers, changes in cell phenotype, excessive cell proliferation, and the secretion of proinflammatory cytokines[5-7]. The balance of bacterial factors and the host inflammatory response to H. pylori infection determines the outcome of the disease[8]. Most H. pylori-infected individuals (80% to 90%) remain clinically asymptomatic throughout their lifespan. In most cases, H. pylori colonizes the stomach for many decades before symptoms appear, which distinguishes this bacterium from bacterial pathogens that cause acute infections. However, even in asymptomatic subjects, H. pylori induces histological gastritis, due to the infiltration of the gastric mucosa by immune cells[9]. Approximately 10% of infected subjects develop symptomatic gastritis, peptic ulcer, or even gastric cancer. Despite the high prevalence of H. pylori infection in Africa and South Asia, the incidence of gastric cancer in those areas is much lower than in other regions. Such geographic differences in pathology can be explained in part by the presence of different types of H. pylori virulence factors[4,10-13]. Many studies have been performed to define what makes H. pylori a pathogen and, at the same time, why so many people do not suffer due to infection. Generally, penetration of H. pylori through the gastric mucosa results in the recruitment of host immune defenses to the site of infection and the development of acute inflammation. If the course of events is favorable to the host, the bacteria are eliminated, and healing begins. However, when acute inflammation fails to fight the infection, a chronic inflammatory response occurs. Among several mediators engaged in propagating gastric inflammation after H. pylori infection, cyclooxygenase-2[14], together with reactive oxygen species (ROS) and reactive nitrogen species (RNS)[15] might be the principal ones. Excessive ROS/RNS production correlates well with histopathological mucosal damage and with bacterial load[14,15]. The permanent recruitment of inflammatory cells to infectious foci results in progressive tissue damage. Asymptomatic H. pylori-infected individuals likely develop very weak gastric inflammation in response to H. pylori. In such conditions, the bacteria are not eliminated. A type of balance is created between the microbes and the host, which limits the recruitment of immune cells and prevents the development of gastric pathologies.
It is known that H. pylori is a genetically diverse species[5,7,8,16]. H. pylori strains isolated from different hosts possess various virulence capacities, and their genetic diversity may also appear within the gastric niche of a single human host. Most H. pylori-related diseases depend on the expression of specific protein virulence factors, such as cytotoxin-associated gene A (CagA) and vacuolating cytotoxin A (VacA)[7]. A recent study on H. pylori-host protein interactions confirmed that the type IV secretion system (TIVSS) can translocate CagA to gastric epithelial cells using human β1 integrin as a receptor[17]. H. pylori strains that are positive for the Cag TIVSS more successfully cause chronic infections in humans. In contrast, it has been observed that the activity of the TIVSS system may limit H. pylori colonization. The study in a mouse model has shown that an exchange of DNA between genetically heterogeneous H. pylori strains may support chronic colonization[18,19]. Other well-characterized H. pylori virulence factors include urease[20,21] and several types of outer membrane proteins, such as Hop proteins (Hsp70/Hsp90-organizing Hsp)[22] and blood antigen-binding adhesins[23]. However, the panel of H. pylori proteins is very complex and, as shown in a proteomics study, includes over 1200 compounds[24,25]. The role of the majority of these proteins in the course of H. pylori-related diseases needs to be clarified. It is interesting that different H. pylori proteins may interact with various host molecules and be involved in numerous pathogenic signaling pathways.
Key findings
H. pylori demonstrates high affinity to the gastric mucosa and is well adapted to live in the acidic environment of the stomach. In general, the interactions of H. pylori with host cells resemble a lifelong homeostasis, but in certain patients, these microbes cause pathological changes, such as ulcers or gastric cancers. It is not clear why certain patients remain asymptomatic, whereas others develop illness. Recently, both bacterial virulence factors and host susceptibility have been recognized as contributors to H. pylori pathogenicity.
UNIQUE STRUCTURAL AND BIOLOGICAL FEATURES OF H. PYLORI LPS - A KEY PLAYER IN THE PERSISTENCE STRATEGY
Although H. pylori, as a gastric pathogen, expresses several unquestionable virulence factors that are related to the cytotoxicity of this bacterium, its most potent feature is the ability to persist for years within the gastric epithelium of the host. This unique, adaptive property occurs due to a complex mechanism of H. pylori colonization and persistence, which is mediated by proteins, glycoconjugates, and lipids exposed on the surface of this bacterium. Well-characterized adhesins in H. pylori include blood antigen-binding adhesin (BabA), which interacts with the host’s Lewisb (Leb), and sialic acid-binding adhesin, which binds sialylated LewisX (LeX). There are many other H. pylori adhesion molecules, such as hemagglutinin, which interacts with sialic acid exposed on human tissues, and less-characterized adhesion molecules that interact with host extracellular matrix (ECM) molecules[12,23,26,27]. Among H. pylori virulence factors, LPS plays a special role due to its unique structural and biological properties, which differ from those of the LPSs typical of other Gram-negative bacteria. In general, these unique features concern modifications of lipid A, resulting in a reduction in the endotoxic and proinflammatory properties of H. pylori LPS[28,29] and the expression of O chains. These chains are structurally related to Lewis blood-group antigens, which are also expressed by human cells. This phenomenon, called molecular mimicry, most likely makes these pathogens less sensitive to recognition by the immune cells of the host[30]. The direct cytotoxic effect of H. pylori LPS on immune cells and epithelial as well as endothelial barriers and its immunomodulatory activity, mediated through different cytokines, are being investigated.
General structure of bacterial LPSs: A summary
Bacterial LPSs are described as heat-stable, non-protein, endotoxic cell wall components of Gram-negative bacteria that consist of conserved and highly variable regions[31,32]. Conserved parts of LPS are shared between various bacterial species and are critical for cell survival. In contrast, the variable regions of LPS are not essential for the existence of bacteria but promote evolutionary variations, which are crucial for the adaptation of microbes to the host or environment. In general, the structures of LPSs from various bacterial species and serotypes of Gram-negative bacteria are similar. These amphiphilic molecules contain two parts: a hydrophilic polysaccharide and lipid A, which constitutes a covalently bound hydrophobic lipid component. The polysaccharide moiety contains a core region and an O-specific chain, with a sequence of repeating units of identical polysaccharides, which are composed of several sugar monomers. Due to the great variety of monosaccharides occurring in the O-specific chain, this region of LPS is unique to each endotoxin and characterizes the serotype. The core region is less variable and is subdivided into outer and inner cores. The outer core may contain different sugar moieties: D-glucose, D-galactose, 2-amino-2-deoxy-D-galactose, and 2-amino-2-deoxy-D-galactose. The inner core contains 2-keto-3-deoxy-octulosonic acid (Kdo) and heptose residues, which are phosphorylated. Lipid A is known as the bioactive center of LPS and is composed of 1,6-linked D-glucosamine disaccharide, which is phosphorylated and possesses up to six acyl residues. There are differences in the length, position, and number of fatty acids. Lipid A is responsible for the endotoxic properties of LPS[32], and its bioactivity is defined as cytokine-inducing capacity. The minimal requirements for this activity include the presence of a molecule composed of two gluco-configurated hexosamine residues, two phosphate groups, and six fatty acids, as in lipid A of Escherichia coli (E. coli), which has been established as standard LPS (Figure 1)[33]. Modifications in the chemical structure of LPS, such as different phosphorylation or acylation patterns of hexosamine disaccharide, result in a varied ability to induce monokine production by immune cells. Endotoxicity is usually caused by monomeric LPS molecules, although at a higher concentration, the physical structure of LPS is also crucial for its bioactivity[34].
Figure 1.
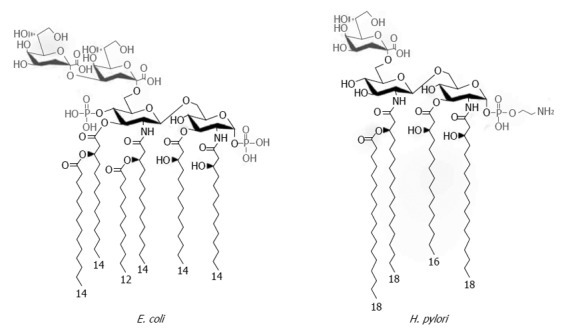
Chemical structures of Helicobacter pylori and Escherichia coli Kdo-lipid A domains. The majority of Escherichia coli (E. coli) species express lipid A composed of two gluco-configurated hexosamine residues, two phosphate groups at the 1- and 4’- positions, and six C12-C14 long fatty acids. In contrast, the low immunogenic lipid A of Helicobacter pylori (H. pylori) is a tetraacylated molecule with the long acyl chains C16-C18 that is lacking the 4’-phosphate group. Modified from Cullen et al[34].
Lipid A of H. pylori LPS: A weak bioactive center
Similarly to other Gram-negative bacteria, H. pylori contains a cell envelope composed of poly- or oligosaccharides that are covalently linked to lipid A of LPS. Fresh clinical isolates produce a high-molecular-weight, smooth LPS with an O-antigen, which, in an in vitro culture, may convert into a rough form without the O side chain[35,36]. Lipid A of H. pylori LPS has a unique structure and lower biological activity compared with lipid A from other bacteria[28,29]. Probably it evolved due to adaptation to long-term infections in the gastric milieu. Structural analysis of H. pylori lipid A of the rough and smooth types has indicated that these lipids contain fatty acids that are longer than those present in lipid A of enterobacterial species. The predominant form is tetraacyl lipid A, with long acyl chains of 16 to 18 carbons and underphosphorylation, which are unusual properties for H. pylori lipid A (Figure 1)[34,37]. Studies on reference lipid A from Salmonella minnesota and E. coli have shown that specific phosphorylation patterns and fatty acid compositions and a particular degree of acylation are necessary for LPS to express its full biological activity[31]. The underphosphorylation and underacylation of H. pylori lipid A moieties promote the lipid’s low endotoxicity and biological activity[26]. H. pylori LPSs, interpreted in the context of their lipid A, possess a significantly lower ability to induce the production of cytokines, nitric oxide, and prostaglandin E2; show diminished potential to activate natural killer (NK) cells; barely induce the expression of E-selectin; and are unable to impair the activity of suppressor T lymphocytes[38-42]. These suggest that this lipid should be treated not as a typical endotoxin, but rather as a type of matrix that enables H. pylori to interact with and modulate the host environment[37].
O side chains of H. pylori LPS: The idea of molecular mimicry
Considering the role of H. pylori LPS in the course of gastroduodenal infections, several studies have analyzed the relevance of O side-chain repeating units. In contrast to other pathogens, H. pylori LPS O antigen is strongly conserved, which suggests that its role in the pathogenesis of H. pylori is not related to weak endotoxic activity. The O side chain of H. pylori can be fucosylated and thus mimics human Lewis molecules and other related blood-group antigens: LeX; LeY; and, in certain strains, Lea, Leb, Lec, sialyl-LeX, and H-1 antigens. The side chain can also mimic the antigens of blood groups A and B[29,42,43]. Inactivation of the H. pylori LeX and LeY determinants results in the unsuccessful colonization of mice in vivo[44-46]. The biosynthesis of H. pylori LPS follows a pathway that is different from all established synthesis cascades. The translocation of the O chain is accomplished by Wzk translocase, which is essential for the cell surface expression of Le antigens[47]. During persistent infection, the diversity of H. pylori LPS tends to increase[48], especially in relation to poly (C) tracts in the gene encoding α-1,3-fucosyltransferases as well as a poly (C) tract and poly (TAA) repeats in the gene encoding α-1,2-fucosyltransferase[49,50]. The phenomenon of LPS phase variation results in increased heterogeneity of the bacterial population which enables H. pylori to more precisely adapt to the changing environment of the gastric mucosa[36,51]. Considering the similarities between LeX and Lea or LeY and Leb determinants, it has been suggested that the host Le phenotype may preferentially select for the expression of Le determinants by H. pylori[52]. Thus, the host milieu promotes a selection of bacterial strains with particular characteristics that facilitate adaptation and survival in the gastric mucosa of the individual host and shape the bacterial community structure[47]. An investigation of the genetic basis of H. pylori Le biosynthesis showed that despite functional similarity, low sequence homology occurs between bacterial and mammalian α-1,3/4- and α-1,2-fucosyltransferases. Thus, the expression of Le antigens in H. pylori and these antigens’ role in the mimicry phenomenon could be influenced by several factors: the regulation of fucosyltransferase genes, the activity and expression levels of functional enzymes, the preferences of the expressed enzyme for distinctive acceptor molecules, and the availability of activated sugar intermediates[44].
The poorly endotoxic properties of H. pylori LPS and the expression of LeX or LeY determinants in O-specific chains suggest that antigenic mimicry induced by H. pylori LPS may allow the bacterium to avoid the host immune response via the induction of potentially autoreactive anti-LeX or anti-LeY antibodies, as well as by strengthening the adhesion process. The functional role of H. pylori Lewis antigens in the adhesion of bacteria to the human gastric mucosa has been reevaluated by studying the in situ adhesion of the strains with or without the expression of BabA in patients with various gastric pathologies. It was concluded that BabA is a major H. pylori adhesin that mediates H. pylori binding to gastric epithelial cells via a mucosal Leb blood-group antigen. In contrast, it was reported that H. pylori LPS LeX determinants play a distinct but minor role in adhesion to the human gastric epithelium[53] and that related strains are not able to colonize C3H/HeJ mice[54]. However, more recent studies showed that the polymeric LeX structure in the O antigen of H. pylori LPS is specifically recognized by β-galactoside-binding lectin (galectin-3), which has been identified as a gastric receptor[55]. Previously, it was shown that the core region of LPS may bind laminin (a molecule of the host ECM) and thus contribute to the colonization process[56].
Key findings
Among H. pylori virulence factors, a special role is played by LPS. This molecule’s unique structure and low biological activity promote the survival of the bacterium in the gastric mucosa, despite the existence of various immune barriers. Underphosphorylation and underacylation of lipid A result in the low endotoxicity and biological activity of H. pylori LPS. Fucosylation of the H. pylori O antigen results in the phenomenon of antigenic mimicry, in which bacterial fucose residues imitate human Le blood-group antigens. This imitation allows the bacterium to avoid host immune responses and contributes to prolonged infection.
HOST CELLULAR IMMUNE BARRIERS VS H. PYLORI LPS
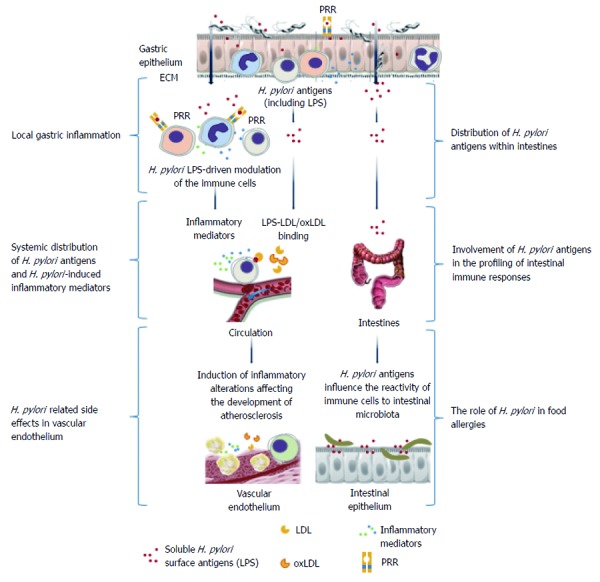
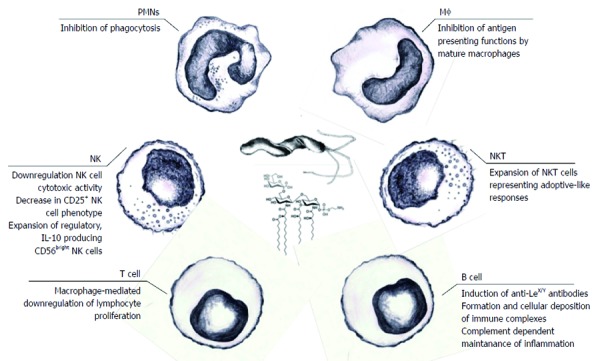
Pathogens may modulate host immune responses by interfering with host recognition and signal transduction mechanisms[57]. During the co-evolution of H. pylori and its host, the bacterium evolved complex strategies to impair immune defenses while maintaining a limited, balanced inflammatory response[58]. Several H. pylori compounds reduce the inflammatory response and the recognition of bacterial antigens by the host immune system (Figure 2). The immunomodulatory activities of VacA, CagA, γ-glutamyl transpeptidase, arginase, and several adhesins have been shown[3,58-64]. The release of H. pylori LPS into an infectious niche may play an important role in the induction of both local and systemic inflammatory responses. This induction may be a consequence of direct LPS-cell interactions or cell-to-cell cytokine-mediated crosstalk. Changes occurring in the gastric epithelium colonized by H. pylori lead to the penetration of bacterial antigens through the basal membrane, where they bind to the ECM and, via pathogen recognition receptors (PRRs), interact with immune cells infiltrating the lamina propria and induce the production of cytokines. Moreover, the soluble antigens of H. pylori may enter the circulation and induce systemic effects[65,66] (Figure 3).
Figure 2.
Helicobacter pylori lipopolysaccharide-mediated immunomodulation. The effector functions of both natural and adaptive immune cells could be downregulated by various lipopolysaccharide (LPS)-dependent mechanisms, making the persistence of Helicobacter pylori (H. pylori) infection possible. These probable mechanisms might include LPS-dependent escape of phagocytosis due to apoptosis of polymorphonuclear cells, downregulation of the cytotoxic activity of natural killer (NK) cells and expansion of NK cells producing regulatory interleukin (IL)-10, expansion of natural killer T cells, inhibition of antigen presentation by mature macrophages, and downregulation of the lymphocyte proliferation indirectly caused by LPS-driven macrophage arrest. Stimulation of B lymphocytes in response to H. pylori LPS may result in the production of anti-Lewis antibodies, which, together with host Lewis antigens, form immune complexes which may induce complement (C)-dependent inflammation. ECM: Extracellular matrix; PRRs: Pathogen recognition receptors; oxLDL: Oxidized low density lipoprotein (LDL).
Figure 3.
Local and systemic consequences of Helicobacter pylori-driven immunomodulation. Helicobacter pylori (H. pylori) lipopolysaccharide (LPS) may facilitate colonization of the gastric mucosa through extracellular matrix proteins and induction of local inflammation. Increased permeability of the gastric epithelium due to the activity of H. pylori proteins such as urease and vacuolating cytotoxin, which contribute to the degradation of intercellular tight junctions, may result in broad interactions of soluble H. pylori LPS with host cells. This LPS can bind to various immune cells infiltrating the mucosal basal lamina or even enter the circulation, where the LPS may contribute to the inflammatory response via a direct influence on immune cells or through the formation of complexes with low density lipoprotein (LDL)s, and possibly oxidized LDL. Released H. pylori LPS can also move along the gastrointestinal tract and, together with the intestinal microbiota, may induce complex immunoregulatory mechanisms. These interactions can result in an increased expression of adhesins, pathogen recognition receptors (PRRs), and proinflammatory cytokines. Different PRRs can mediate the interactions of H. pylori LPSs with the immune cells, including Toll-like receptor 4 or 2, dendritic cell-specific intracellular adhesion molecule-3-grabbing non-integrin, and triggering receptor expressed on myeloid cells-like receptors. The signaling pathways triggered by these receptors may cause cytokine secretion. NKT: Natural killer T lymphocytes; PMNs: Polymorphonuclear cells.
H. pylori LPS: An atypical PAMP
H. pylori, similarly to many intestinal microorganisms, has developed mechanisms to vary its LPS chemistry to subvert recognition by innate immune receptors[67]. Bacterial LPSs are pathogen-associated molecular patterns (PAMPs), or so-called “alarmins”, which allow the recognition of bacterial invasion and trigger the innate immune response of the host[68]. LPSs and other PAMPs, including lipoproteins, peptidoglycan, flagellin, double-stranded RNA, CpG-DNA, and zymosan, are recognized by PRRs on the cells of the first line of immune defense: macrophages and dendritic cells (DCs). Toll-like receptors (TLRs) and nucleotide-binding domain, leucine-rich repeat-containing receptors (NLRs) are well-characterized PRRs that display a high degree of PAMP specificity. In contrast to the adaptive response, innate immunity is not specific. Each type of PRR can differentiate microbial components from similar structures, i.e., patterns or molecular signals, that are unique to host cells, allowing distinction between self and non-self[68,69]. The recognition of PAMPs induces the production of proinflammatory cytokines and the maturation of DCs, which in turn result in the activation of a specific immune response[70,71].
In the context of infections caused by Gram-negative bacteria, LPS constitutes the most dominant and suitable PAMP[72,73]. The structure of lipid A, the expression of Le antigens, and the fucosylation patterns in H. pylori LPS may have multiple effects on immune responses. The interactions of lipid A with host cells are mediated by cellular and soluble receptors: (1) serum LPS-binding protein (LBP); (2) the membrane CD14 receptor; (3) sCD14 protein, a soluble form of the CD14 receptor; and (4) TLR4 bound to MD2 molecule, that is used for signaling via recruitment of the TIR domain containing MyD88 and TIRAP adaptor proteins or, alternatively, of the TRAM and TRIF proteins, leading to the activation of nuclear factor κB or IRF3/7 transcription factors. This events result in the production of proinflammatory cytokines and type I interferons (IFNs)[73,74]. Studies conducted on two types of cellular systems, 70Z/3 cells expressing CD14 and an epithelial cell lineage with CD14-dependent activation, confirmed that H. pylori LPS, although much less active than E. coli LPS, stimulates cells to secrete interleukin (IL)-8 via CD14[75]. Lower efficiency in LBP binding may be one of the explanations for the low biological activity of H. pylori LPS, which is especially observed in the activation of human monocytes. Although H. pylori LPS binds LBP less efficiently than does E. coli LPS[76], the level of circulating LBP significantly rises in H. pylori-infected patients with gastritis and is even more elevated in H. pylori-infected patients with coronary heart disease (CHD), most likely as a consequence of the chronic inflammatory response and endothelial dysfunction[77]. Through LBP, LPS is detoxified in the circulation, mainly by its incorporation into low-density lipoproteins (LDLs)[78]. However, considering LPS/LBP/LDL interactions in CHD patients may serve as a mechanism that enables the deposition of H. pylori LPS in the vascular endothelium, thus enhancing the TLR4-dependent inflammatory response.
The variation in bacterial lipid A structures might be a successful strategy for escaping recognition by the innate immune system and blocking TLR4 signaling. In vitro studies showed that moieties of lipid A from H. pylori LPS are poorly recognized by human TLR4[79]. This phenomenon is due to the presence of fewer but longer (16- to 18-carbon) acyl chains. The potential of these pathogens to cause severe chronic disease in humans is attributed, at least in part, to their relatively low ability to activate TLR4 signaling[67]. However, the advantage of this mechanism for the persistence of H. pylori is limited in the gastric milieu because human gastric epithelial cells do not express functional TLR4, which is present on the surface of monocytes and macrophages[80]. H. pylori lipid A, which is unable to trigger a signal through TLR4, can act through TLR2, expressed on both epithelial cells and macrophages, which may compensate for a lack of sufficient TLR4 signaling[81]. It has been shown that LPSs from certain H. pylori strains can antagonize TLR4. The antagonistic activity of H. pylori LPS and its binding to TLR2 might give the bacterium an advantage over the host, and this phenomenon might be associated with the clinical outcome of H. pylori infection[79]. It was shown that the presence of cag pathogenicity island (PAI) genes may be crucial for the synthesis of bioactive lipid A molecules that stimulate TLR4-mediated expression of mitogen oxidase 1 (Mox1) in gastric pit cells, which could be involved in the initiation of mucosal cell inflammatory and immune responses against H. pylori infection[82,83]. However, the genomic and molecular bases for the linkage between cagPAI genes and the synthesis of bioactive lipid A remain unsolved. Until recently, lipid A was considered the only component of LPS, determining its biological activity. However, since the discovery of LeX in the LPS of H. pylori, several different biological functions have been attributed to the presence of Le antigens. Several proposed functions include enhancement of colonization, escape from host recognition, modulation of the activity of immune cells, and induction of gastric autoimmunity[44].
It has been shown that due to LPS phase variation, H. pylori evades binding to surfactant protein D (SP-D) and may persist in the gastric mucosa[44,58]. This phenomenon caused by changes in the fucosylation of the O chain, which is concomitant with slipped-strand mispairing in a poly (C) tract of the fucosyltransferase A (fut T1) gene[83]. Recently, two antigenic types of H. pylori LPS have been proposed: LPS with a highly antigenic epitope (HA-LPS) and a weakly antigenic epitope (WA-LPS), which is expressed after the removal of a β-N-acetyl-D-glucosamine residue, a highly antigenic determinant. This antigenic conversion, resulting with the loss or lack of only one β-linked D-GlCNAc residue, may serve as an escape mechanism for H. pylori, enabling the bacterium to evade the host antibody-mediated immune response. In the presence of SP-D, WA-LPS possesses a stronger ability to upregulate TLR4 expression, proliferation of epithelial cells, inflammation and tumorigenesis[84].
Whether the LeX and LeY determinants are involved in the pathways of cytokine signaling is unclear. It has been shown that H. pylori LPSs, with or without LeXY determinants, differ in their ability to induce the production of inflammatory cytokines by peripheral blood mononuclear leukocytes (PBMLs). The lack of Lewis determinants in H. pylori LPS results in a significant reduction in its ability to induce TNF-α secretion compared with H. pylori LPS containing LeXY antigenic moieties or standard E. coli LPS[85]. Little is known about the recognition of bacterial Le determinants by the immune cells of the host. Successful pathogens may manipulate signaling through bacterial structures that modify TLR signaling. For instance, Mycobacterium tuberculosis, due to the interaction of mannose-capped lipoarabinomannan (ManLAM) with the mannose receptor, inhibits LPS-induced IL-12 production by macrophages[86]. Additional receptors such as DC-specific intercellular adhesion molecule (ICAM)-3-grabbing non-integrin (DC-SIGN) on DCs, work together with the mannose receptor to inhibit antimicrobial and anti-inflammatory responses by modifying the effects of subsequent TLR activation[87]. It has been shown that whole H. pylori bacteria and their LPSs bind to DC-SIGN not only via Le carbohydrates but also through galactose moieties[88,89]. It was suggested that differences in H. pylori binding affinity for DC-SIGN are determined by the phase variation[90]. The ability of H. pylori to bind DC-SIGN might allow the bacterium to modulate the T helper cell 1 (Th1)/T helper cell 2 (Th2) balance and could be crucial for the control of H. pylori infections. Activated lymphocytes more effectively control bacterial growth and might diminish inflammation in the gastric mucosa by releasing cytokines, most likely of the Th2 type[90]. Beyond TLRs and DC-SIGN receptors, other molecules, such as triggering receptor expressed on myeloid cells (TREM)-like and nucleotide-binding oligomerization domain 1 (NOD1) receptors, are also engaged in H. pylori-driven immune responses[91]. As shown in recent studies, the stimulation of gastric epithelial cells by H. pylori-derived NOD1 ligands leads to a production of host defense factors, such as IFN-β, IFN-γ, and IFN-γ-induced protein 10 (IP-10), all of which regulate bacterial growth. The molecular mechanisms of NOD1-mediated mucosal host defense against H. pylori are not completely understood but are at least partially dependent on the presence of a functional TIVSS. Interestingly, TLR-dependent proinflammatory cytokine responses are remarkably enhanced in the presence of NOD1 ligands. Thus, synergetic TLR-NOD1 activation in the gastric epithelium should be considered[92].
ESCAPING PHAGOCYTOSIS
Host recognition of microbial invasion induce the recruitment of neutrophils and mononuclear cells into the lamina propria of the gastric mucosa which depends on the density and avidity of adhesion-mediated molecules expressed on leukocytes and endothelial cells. The majority of adhesins, such as ICAM-1 and vascular adhesin molecule-1, are upregulated in the antral mucosa in patients with H. pylori-related gastritis. A higher level of E-selectin was shown in the gastric mucosa of individuals infected with H. pylori strains positive for the cagPAI. These findings indicate that E-selectin and its ligands preferentially mediate the extravasation of mononuclear cells and their migration into inflamed tissues[93]. In this context, a question arises: why are the phagocytes, not able to effectively destroy and eliminate H. pylori? It has been shown that several mechanisms allow H. pylori to alter phagocyte functions, especially by evading opsonization, actively retarding phagocytosis, affecting membrane trafficking, and inhibiting phagosome maturation[94]. The anti-phagocytic properties of H. pylori depend on the presence of the cag PAI[95], sialic acid-specific and heparan sulfate-binding proteins[96] and on the expression of various ECM proteins[26]. H. pylori LPS may also have various effects on phagocytes. In an in vitro study, H. pylori LPS diminished the ingestion of H. pylori by human peripheral blood polymorphonuclear leukocytes (PMNs)[97]. This anti-phagocytic effect of H. pylori LPS was dose dependent and neutralized by recombinant LPS-binding protein (rLBP). LBP is thought to primarily function in facilitating LPS-stimulated cytokine synthesis by monocytes and macrophages. However, LBP also enhances the later stages, affecting bactericidal/permeability-increasing protein (BPI) activity and opsonizing Gram-negative bacteria and thus playing a more direct role in host defense[98]. It is possible that LBP reduces the concentration of BPI required to kill bacteria[99]. The LPS-mediated attachment of H. pylori to laminin may affect the effectiveness of the engulfment process[100]. This mechanism may limit the availability of bacteria to phagocytes. Another anti-phagocytic mechanism of H. pylori LPS could be associated with LPS-driven apoptotic changes in phagocytes[97]. Previous findings implicated caspase-3 involvement in gastric mucosal inflammatory responses to H. pylori LPS and suggested the participation of nitric oxide synthase 2 in the amplification of the cell death signaling cascade[101]. In vivo, H. pylori LPS may diminish bacteria elimination and, despite abundant granulocyte infiltration in the gastric mucosa, promote persistence of the infection.
MODULATION OF NK CELL ACTIVITY
There is a growing set of data concerning the interplay of H. pylori antigens with NK cells and its consequences in the course of infection. The main function of NK cells, which are large granular lymphocytes representing the innate immune system, is to promote major histocompatibility complex (MHC)-unrestricted killing of both target cells infected by viruses or bacteria and tumor cells[102]. NK cell-dependent responses are especially important in defense against pathogens that induce extensive cell damage, apoptosis, or cancerogenic changes. H. pylori bacteria fulfill all of these criteria. The predominance, heterogeneity, and distribution of NK cells at different sites within the gastric mucosa reflect a potential functional role for these cells during H. pylori infections[103]. CD8-CD16-CD56+bright NK cells in the gastric mucosa of H. pylori-infected subjects have likely adapted to respond to such bacterial infections. NK cells may respond by robust IFN-γ secretion for both direct H. pylori stimulation and indirect antigen presenting cell (APC)-dependent cytokine activation[104-106]. Regarding the anti-phagocytic properties of H. pylori LPS, it is unclear how this LPS influences NK cell activity compared with the effects of other surface H. pylori antigens and standard E. coli LPS. It was found that H. pylori LPS, but not E. coli LPS or H. pylori glycine-acid extract (GE) of surface antigens, downregulated the natural cytotoxic activity of peripheral blood lymphocytes, which also correlated with a diminished ability of PBMLs to produce IFN-γ and IL-2 and a retained but very low secretion of IL-10[107]. H. pylori LPS-driven downregulation was more intense in H. pylori-infected individuals. The activity of H. pylori LPS, but not GE or E. coli LPS, was related to a decrease in the cytotoxic properties and propagation of NK cells (CD3-CD56+). The H. pylori LPS-driven downregulation of lymphocyte cytotoxicity was also accompanied by the expansion of natural killer T (NKT) (CD3+CD56+) cells exclusively in H. pylori-infected donors. This might be explained by the previous in vivo stimulation of immune cells with H. pylori antigens, including LPS, and the adaptive-like properties of NKT cells[108]. The abundant population of NK cells in H. pylori-infected individuals expresses the CD56bright phenotype, exhibiting regulatory rather than cytotoxic activity[109]. Lindgren et al[104] suggested that CD56bright NK cells present in the gastric mucosa respond to bacterial infections by cytokine production, which influences the course of innate defense. Thus, the response of NK cells to H. pylori most likely depends on the type of antigenic challenge. H. pylori antigens may differ in their ability to induce a different cytokine network, which in turn may positively (by GE antigens) or negatively (by LPS) modulate NK cell cytotoxic and secretory activities[107]. The H. pylori LPS-mediated decrease in lymphocyte cytotoxicity is accompanied by a lack of CD3-CD56+CD25+ NK cell propagation, inhibition of proinflammatory cytokine expression, and intense expansion of IL-10-producing NK cells. These results are particularly interesting in the context of the results obtained for E. coli LPS, which was found to stimulate the propagation of human CD56+CD3- NK cells, followed by a substantial increase in cytolytic activity and IFN-γ production[110]. This expansion might be driven by monocyte-derived DCs[111]. Using a guinea pig model, it was confirmed that H. pylori GE antigens stimulated the cytotoxic activity of lymph node lymphocytes in vitro, whereas H. pylori LPS did not possess such potential and thus might have promoted the development of chronic infection[112].
T LYMPHOCYTES AND ADAPTIVE IMMUNE RESPONSES
LPSs have various effects, mainly on myeloid cell lineages, including induction of acute phase responses, leukocytosis, moderate fever, infiltration of defense cells into the site of infection, and activation of antimicrobial mechanisms. If not excessive, this response is crucial for the elimination of pathogens. The role of LPS in the development of an antimicrobial humoral response was evaluated in a mouse model where LPS induced the polyclonal activation of B lymphocytes, and secretion of antibodies with different specificities was observed[33,113]. The mechanisms of LPS involvement in adaptive cellular antimicrobial immunity are not yet known. In several studies, LPS has been shown to stimulate human T lymphocytes to proliferate and secrete the Th1-type cytokines IFN-γ, IL-12, and TNF-α. However, T cell activation depends on accessory myeloid cells that provide costimulatory signals[114-116]. Although all of these findings support the hypothesis that LPS could be involved in cellular adaptive antimicrobial immunity, it has been shown that humans may be divided into LPS responders and non-responders[115]. The type of LPS-driven cell response may have implications for the outcomes of infections with Gram-negative bacteria, including H. pylori. It has been shown that H. pylori LPS itself possesses a very weak, if any, capacity to stimulate the proliferation of PBMLs from H. pylori-infected dyspeptic patients[117] or the lymphocytes of guinea pigs with induced H. pylori infection[112]. However, in the presence of IL-2, a lymphocyte growth factor, H. pylori lysates or LPS can effectively stimulate human lymphocytes. The role of the interaction between IL-2 and the macrophage CD14 surface antigen, serving as the LPS-binding site, in the activation of human monocytes was reported by Bosco et al[118]. In another study, it was shown that downregulation of H. pylori LPS-driven PBML proliferation was determined by the susceptibility of host cells to bacterial LPS and could have been a result of the direct influence of H. pylori LPS on non-adherent lymphocytes or a consequence of an indirect effect of the LPS preparations on monocyte-derived macrophages, known to be the main targets of bacterial LPS[119]. These results revealed that in the milieu of H. pylori LPS, but not E. coli LPS, only mature macrophages from LPS responders expressed inhibitory activity toward lymphocytes which could result from changes in cytokine production; the expression of surface co-stimulatory molecules; the production of non-protein mediators, such as oxygen free radicals; and changes in TLR4 expression on macrophages[120]. H. pylori LPS not only activates phagocytes to release oxygen free radicals but also increased nicotinamide adenine dinucleotide phosphate oxidase and TLR4 expression on gastric epithelial cells leading to elevation of oxidative stress[15]. It has been shown that elimination of regulatory lymphocytes from the population of H. pylori-specific memory T cells increased their proliferation in response to H. pylori antigens and abolished the effect of anergy[121]. It could be speculated that H. pylori LPS affects the macrophage-lymphocyte interactions contributing to prolonged H. pylori infection. Immunosuppression induced by H. pylori protein components, such as VacA and CagA antigen, has also been suggested to be involved in the chronic nature of H. pylori infections[59-64,122].
These bacteria may alter the Th1/Th2 balance to evade the immune response of the host. Th1 polarization of the gastric immune response may be induced by different H. pylori components, including LPSs, enhancing IFN-γ and IL-12 secretion and inhibiting IL-2 production and cell proliferation, which may be necessary for Th2 responses[123,124]. It has also been shown that H. pylori modulates the Th1/Th2 cell balance through a phase-variable interaction between LPS and DC-SIGN. Lewis-positive H. pylori variants were shown to bind DC-SIGN C-type lectin present on gastric DCs, and this interaction blocked Th1 cell development, whereas Lewis-negative H. pylori variants that were not involved in binding to DC-SIGN induced a strong Th1 response[90].
A growing number of reports suggest that the Th17 cells identified by their production of IL-17 are involved in H. pylori induced inflammation[125]. H. pylori colonization and H. pylori-induced inflammation were more intense in wild-type mice than in IL-17-deficient mice, indicating that IL-17 may not contribute to protection, instead playing a role in the development of inflammation[126]. In another study, anti-IL-17 treatment resulted in a reduced H. pylori burden and lower inflammation in the stomach, suggesting that IL-17 neutralization contributes to bacterial clearance and prevents inflammatory processes[127]. It has been also shown that H. pylori specific tumor infiltrating lymphocytes (TILs) of gastric mucosa produce IL-17[128]. The results obtained in animal models are in line with findings based on real-time PCR and Western blotting analyses of human gastric biopsies, in which the upregulation of IL-17 occurred at both the RNA and the protein levels in H. pylori-infected biopsies compared with uninfected biopsies[129,130]. The direct implication of LPS or any other H. pylori antigen in IL-17 induction has not been clarified yet. However, regardless of the presence or absence of Th1 or Th17 cells, the results described here support the suggestion that gastritis due to H. pylori is the result of complex interactions between at least several different T cell subsets.
Key findings
What distinguishes H. pylori from other gastric pathogens is its ability to avoid and modulate host immune mechanisms to maintain balanced inflammation in the mucus layer of the stomach. These mechanisms include the manipulation or downregulation of TLR signaling pathways, SP-D binding, and modulation of the network of cytokines secreted by immune cells. All of these mechanisms are determined by the unique structure of H. pylori LPS, and mainly the low acylation of lipid A and phase variation in relation to the O antigen. The diminished elimination of H. pylori due to the interference of its LPS in the process of phagocytosis and its negative modulation of NK cell cytotoxic and cytokine activities are also being considered. Although the involvement of H. pylori LPS in the mechanisms of adaptive immune responses is not yet known, the H. pylori LPS-driven alterations in lymphocyte proliferation and macrophage-lymphocyte interactions may contribute to the lifelong persistence of H. pylori infection.
WHAT ABOUT ANTI-H. PYLORI LPS ANTIBODIES AND AUTOREACTIVE RESPONSES?
It has been hypothesized that H. pylori infections induce antibodies that are potentially autoreactive[131-133]. Half of H. pylori-infected individuals are positive for the production of serum autoantibodies potentially crossreacting with gastric parietal cells. The correlation between the eradication of H. pylori and a subsequent decrease in serum anti-H. pylori IgG levels, resulting in the regression of autoimmune gastritis, seems to confirm this suggestion. Candidates for the antigens responsible for the molecular mimicry that causes autoreactivity include H. pylori Hsp60, Le antigens and H+,K+-ATPase[131-133]. Opinions on a possible role for antigenic mimicry in the interaction between H. pylori Hsp60 and the human gastric mucosa are varied because this antigen was not found to be involved in the autoreactive response observed in individuals infected with H. pylori[134]. However, in the sera of H. pylori-infected patients, elevated titers of anti-H. pylori LPS antibodies have been detected[131]. The epitope specificity of human anti-LPS antibodies has been widely discussed. Although controversial, certain data suggest that long-term H. pylori infections could induce autoreactive anti-Lewis antibodies that cross-react with the gastric mucosa, contributing to the development of gastric pathologies[44,132]. However, sera from H. pylori-infected patients containing anti-canalicular autoantibodies, despite intense binding to LeX- or LeY-positive H. pylori strains, still express antigastric autoreactivity. It cannot be excluded that anti-LeX and anti-LeY antibodies occur naturally under physiological conditions because these antibodies are detected in the majority of H. pylori-uninfected individuals. However, the elevated levels of these antibodies have been detected more frequently in the sera of H. pylori-infected than uninfected individuals, which suggests that infections with these bacteria may promote specific anti-Le antibody production[135]. Anti-LeX antibodies, depending on their isotype, may play a different role during H. pylori infections. The readiness of the host to respond to Le determinants using IgG, but not IgM, could be a risk factor for H. pylori-related pathologies. Anti-LeX IgM was found to facilitate the phagocytosis of LeX-positive, but not LeX-negative, strains, suggesting a protective role. In contrast, increased levels of anti-LeX and anti-LeXY antibodies of the IgG class, either soluble or bound in immune complexes (ICs), might contribute to the pathogenesis of chronic H. pylori infections[136,137]. It is worth mentioning that ICs containing H. pylori antigens have been detected in the glomeruli of patients with membranous nephropathy and coexisting H. pylori infection[138]. Le-anti-Le ICs are also suspected to play a role in the development or the maintenance of the inflammatory response in H. pylori-infected subjects with CHD[75]. It has been shown on a murine model that immunization of animals with H. pylori induces anti-LeXY antibodies that cross-react with the gastric epithelium, and particularly with the gastric H+,K+-ATPase, the proton pump localized to the parietal cell canaliculi[30]. However, the cross-reactive antibodies are specifically directed toward peptide, and not sugar, epitopes. In another study using a mouse model, it was shown that chronic H. pylori infection stimulated the production of antibodies interacting with the parietal cell canaliculi that were coupled with synthetic Le antigens[139]. Recent studies have confirmed that anti-Le antibodies are present in most patients with H. pylori infection, including those with gastric cancer. The intensity of the anti-Le response is independent of the host Le phenotype but related to the bacterial Lewis phenotype[140]. The role of anti-Le antibodies during H. pylori infections remains unclear. It has been suggested that H. pylori anti-LeXY antibodies are locally produced and rapidly bind to gastric mucosal epitopes. Thus, these antibodies do not appear in serum. In healthy humans, cellular barriers, innate immunity, and an efficient physiological clearance system work together to neutralize gastrointestinal LPS. For instance, nutritional lipids guarantee the maintenance of relatively low systemic levels of LPS[141]. However, evidence of increased permeability of the epithelial and endothelial barriers in the milieu of H. pylori products, such as urease and VacA, may suggest the need for a change in the approach to H. pylori LPS immunogenicity[66]. In this case, the strong binding of cholesterol by H. pylori surface components should also be taken into account[142,143].
Key findings
The role of autoimmune responses in the pathologies associated with H. pylori infection must also be taken into consideration. The molecular mimicry involved in autoreactivity might be directly induced by H. pylori surface antigens, of which LPS is the most prominent cross-reactive candidate. Gastric pathologies can be induced by anti-Le antibodies that cross-react with Le determinants exposed on the gastric epithelium. Le-anti-Le ICs are also suspected to be associated with the maintenance of H. pylori-related inflammatory responses.
CONCLUSION
The data summarized in this review support the suggestion that H. pylori LPS evolved during the co-existence of these bacteria with their host, enabling the bacteria to “live in peace”. This phenomenon at least partially depends on the immunomodulatory properties of H. pylori LPS and its influence on host cell barriers and the condition and responsiveness of immune cells. Many studies have shown that H. pylori components may facilitate the colonization process, and especially the immune response of the host, during the course of H. pylori infection. This H. pylori-driven interaction might result from positive or negative modulation. Among the negative immunomodulators, a prominent position is occupied by a vacuolating toxin and CagA protein. However, in light of the recent studies presented in this review, it is necessary to enrich this panel with H. pylori LPS. Together with CagA and VacA, this LPS suppresses the elimination of H. pylori bacteria from the gastric mucosa by interfering with the activity of innate and adaptive immune cells. In particular, the LPS diminishes the inflammatory response and affects the adaptive T lymphocyte response, thus facilitating the development of chronic infections. The complex strategy of H. pylori bacteria for surviving in the gastric mucosa of the host involves both structural modifications of LPS lipid A to diminish its endotoxic properties and the expression and variation of Lewis determinants, arranged in O-specific chains of H. pylori LPS. By mimicking host components, this phenomenon leaves these bacteria “invisible” to immune cells. Taken together, these mechanisms allow H. pylori to survive and live for many years within their hosts.
Footnotes
Supported by Certain results presented in this review derived from studies that were supported by the Polish Ministry of Science and Higher Education grants, N401 021 31/0379, N N401 015 136, N N303 451 738 and UMO-2013/09/N/NZ6/ 00805
P- Reviewer: Actis GC, Codoner-Franch P, D'Elios MM, Handa O, Watanabe T S- Editor: Gou SX L- Editor: A E- Editor: Wang CH
References
- 1.Moodley Y, Linz B, Bond RP, Nieuwoudt M, Soodyall H, Schlebusch CM, Bernhöft S, Hale J, Suerbaum S, Mugisha L, et al. Age of the association between Helicobacter pylori and man. PLoS Pathog. 2012;8:e1002693. doi: 10.1371/journal.ppat.1002693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet. 1984;1:1311–1315. doi: 10.1016/s0140-6736(84)91816-6. [DOI] [PubMed] [Google Scholar]
- 3.Mahdavi J, Sondén B, Hurtig M, Olfat FO, Forsberg L, Roche N, Angstrom J, Larsson T, Teneberg S, Karlsson KA, et al. Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science. 2002;297:573–578. doi: 10.1126/science.1069076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suzuki R, Shiota S, Yamaoka Y. Molecular epidemiology, population genetics, and pathogenic role of Helicobacter pylori. Infect Genet Evol. 2012;12:203–213. doi: 10.1016/j.meegid.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Israel DA, Salama N, Arnold CN, Moss SF, Ando T, Wirth HP, Tham KT, Camorlinga M, Blaser MJ, Falkow S, et al. Helicobacter pylori strain-specific differences in genetic content, identified by microarray, influence host inflammatory responses. J Clin Invest. 2001;107:611–620. doi: 10.1172/JCI11450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Versalovic J. Helicobacter pylori. Pathology and diagnostic strategies. Am J Clin Pathol. 2003;119:403–412. [PubMed] [Google Scholar]
- 7.Chmiela M, Michetti P. Inflammation, immunity, vaccines for Helicobacter infection. Helicobacter. 2006;11 Suppl 1:21–26. doi: 10.1111/j.1478-405X.2006.00422.x. [DOI] [PubMed] [Google Scholar]
- 8.Matteo MJ, Armitano RI, Granados G, Wonaga AD, Sánches C, Olmos M, Catalano M. Helicobacter pylori oipA, vacA and dupA genetic diversity in individual hosts. J Med Microbiol. 2010;59:89–95. doi: 10.1099/jmm.0.011684-0. [DOI] [PubMed] [Google Scholar]
- 9.Portal-Celhay C, Perez-Perez GI. Immune responses to Helicobacter pylori colonization: mechanisms and clinical outcomes. Clin Sci (Lond) 2006;110:305–314. doi: 10.1042/CS20050232. [DOI] [PubMed] [Google Scholar]
- 10.Hunt RH. The role of Helicobacter pylori in pathogenesis: the spectrum of clinical outcomes. Scand J Gastroenterol Suppl. 1996;220:3–9. [PubMed] [Google Scholar]
- 11.Peek RM, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat Rev Cancer. 2002;2:28–37. doi: 10.1038/nrc703. [DOI] [PubMed] [Google Scholar]
- 12.de Jonge R, Pot RG, Loffeld RJ, van Vliet AH, Kuipers EJ, Kusters JG. The functional status of the Helicobacter pylori sabB adhesin gene as a putative marker for disease outcome. Helicobacter. 2004;9:158–164. doi: 10.1111/j.1083-4389.2004.00213.x. [DOI] [PubMed] [Google Scholar]
- 13.Kim KK, Kim HB. Protein interaction network related to Helicobacter pylori infection response. World J Gastroenterol. 2009;15:4518–4528. doi: 10.3748/wjg.15.4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JS, Cho JY, Song H, Kim EH, Hahm KB. Revaprazan, a novel acid pump antagonist, exerts anti-inflammatory action against Helicobacter pylori-induced COX-2 expression by inactivating Akt signaling. J Clin Biochem Nutr. 2012;51:77–83. doi: 10.3164/jcbn.11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Handa O, Naito Y, Yoshikawa T. Redox biology and gastric carcinogenesis: the role of Helicobacter pylori. Redox Rep. 2011;16:1–7. doi: 10.1179/174329211X12968219310756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blaser MJ, Berg DE. Helicobacter pylori genetic diversity and risk of human disease. J Clin Invest. 2001;107:767–773. doi: 10.1172/JCI12672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schuelein R, Everingham P, Kwok T. Integrin-mediated type IV secretion by Helicobacter: what makes it tick? Trends Microbiol. 2011;19:211–216. doi: 10.1016/j.tim.2011.01.010. [DOI] [PubMed] [Google Scholar]
- 18.Jungblut PR, Schiele F, Zimny-Arndt U, Ackermann R, Schmid M, Lange S, Stein R, Pleissner KP. Helicobacter pylori proteomics by 2-DE/MS, 1-DE-LC/MS and functional data mining. Proteomics. 2010;10:182–193. doi: 10.1002/pmic.200900361. [DOI] [PubMed] [Google Scholar]
- 19.Dorer MS, Cohen IE, Sessler TH, Fero J, Salama NR. Natural competence promotes Helicobacter pylori chronic infection. Infect Immun. 2013;81:209–215. doi: 10.1128/IAI.01042-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunn BE, Campbell GP, Perez-Perez GI, Blaser MJ. Purification and characterization of urease from Helicobacter pylori. J Biol Chem. 1990;265:9464–9469. [PubMed] [Google Scholar]
- 21.Labigne A, Cussac V, Courcoux P. Shuttle cloning and nucleotide sequences of Helicobacter pylori genes responsible for urease activity. J Bacteriol. 1991;173:1920–1931. doi: 10.1128/jb.173.6.1920-1931.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Evans DJ, Evans DG. Helicobacter pylori adhesins: review and perspectives. Helicobacter. 2000;5:183–195. doi: 10.1046/j.1523-5378.2000.00029.x. [DOI] [PubMed] [Google Scholar]
- 23.Borén T, Falk P, Roth KA, Larson G, Normark S. Attachment of Helicobacter pylori to human gastric epithelium mediated by blood group antigens. Science. 1993;262:1892–1895. doi: 10.1126/science.8018146. [DOI] [PubMed] [Google Scholar]
- 24.Rain JC, Selig L, De Reuse H, Battaglia V, Reverdy C, Simon S, Lenzen G, Petel F, Wojcik J, Schächter V, et al. The protein-protein interaction map of Helicobacter pylori. Nature. 2001;409:211–215. doi: 10.1038/35051615. [DOI] [PubMed] [Google Scholar]
- 25.Du X. On coupling analysis of complex network and pathogenesis dynamics of gastroduodenal disease. Inf Technol J. 2013;12:429–433. [Google Scholar]
- 26.Ljungh A, Moran AP, Wadström T. Interactions of bacterial adhesins with extracellular matrix and plasma proteins: pathogenic implications and therapeutic possibilities. FEMS Immunol Med Microbiol. 1996;16:117–126. doi: 10.1111/j.1574-695X.1996.tb00128.x. [DOI] [PubMed] [Google Scholar]
- 27.Olfat FO, Zheng Q, Oleastro M, Voland P, Borén T, Karttunen R, Engstrand L, Rad R, Prinz C, Gerhard M. Correlation of the Helicobacter pylori adherence factor BabA with duodenal ulcer disease in four European countries. FEMS Immunol Med Microbiol. 2005;44:151–156. doi: 10.1016/j.femsim.2004.10.010. [DOI] [PubMed] [Google Scholar]
- 28.Muotiala A, Helander IM, Pyhälä L, Kosunen TU, Moran AP. Low biological activity of Helicobacter pylori lipopolysaccharide. Infect Immun. 1992;60:1714–1716. doi: 10.1128/iai.60.4.1714-1716.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moran AP, Aspinall GO. Unique structural and biological features of Helicobacter pylori lipopolysaccharides. Prog Clin Biol Res. 1998;397:37–49. [PubMed] [Google Scholar]
- 30.Appelmelk BJ, Negrini R, Moran AP, Kuipers EJ. Molecular mimicry between Helicobacter pylori and the host. Trends Microbiol. 1997;5:70–73. doi: 10.1016/S0966-842X(96)10084-6. [DOI] [PubMed] [Google Scholar]
- 31.Rietschel ET, Kirikae T, Schade FU, Mamat U, Schmidt G, Loppnow H, Ulmer AJ, Zähringer U, Seydel U, Di Padova F. Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB J. 1994;8:217–225. doi: 10.1096/fasebj.8.2.8119492. [DOI] [PubMed] [Google Scholar]
- 32.Rietschel ET, Brade H, Holst O, Brade L, Müller-Loennies S, Mamat U, Zähringer U, Beckmann F, Seydel U, Brandenburg K, et al. Bacterial endotoxin: Chemical constitution, biological recognition, host response, and immunological detoxification. Curr Top Microbiol Immunol. 1996;216:39–81. doi: 10.1007/978-3-642-80186-0_3. [DOI] [PubMed] [Google Scholar]
- 33.Heine H, Rietschel ET, Ulmer AJ. The biology of endotoxin. Mol Biotechnol. 2001;19:279–296. doi: 10.1385/MB:19:3:279. [DOI] [PubMed] [Google Scholar]
- 34.Cullen TW, Giles DK, Wolf LN, Ecobichon C, Boneca IG, Trent MS. Helicobacter pylori versus the host: remodeling of the bacterial outer membrane is required for survival in the gastric mucosa. PLoS Pathog. 2011;7:e1002454. doi: 10.1371/journal.ppat.1002454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walsh EJ, Moran AP. Influence of medium composition on the growth and antigen expression of Helicobacter pylori. J Appl Microbiol. 1997;83:67–75. doi: 10.1046/j.1365-2672.1997.00164.x. [DOI] [PubMed] [Google Scholar]
- 36.Moran AP, Knirel YA, Senchenkova SN, Widmalm G, Hynes SO, Jansson PE. Phenotypic variation in molecular mimicry between Helicobacter pylori lipopolysaccharides and human gastric epithelial cell surface glycoforms. Acid-induced phase variation in Lewis(x) and Lewis(y) expression by H. Pylori lipopolysaccharides. J Biol Chem. 2002;277:5785–5795. doi: 10.1074/jbc.M108574200. [DOI] [PubMed] [Google Scholar]
- 37.Moran AP, Lindner B, Walsh EJ. Structural characterization of the lipid A component of Helicobacter pylori rough- and smooth-form lipopolysaccharides. J Bacteriol. 1997;179:6453–6463. doi: 10.1128/jb.179.20.6453-6463.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baker PJ, Hraba T, Taylor CE, Stashak PW, Fauntleroy MB, Zähringer U, Takayama K, Sievert TR, Hronowski X, Cotter RJ. Molecular structures that influence the immunomodulatory properties of the lipid A and inner core region oligosaccharides of bacterial lipopolysaccharides. Infect Immun. 1994;62:2257–2269. doi: 10.1128/iai.62.6.2257-2269.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Darveau RP, Cunningham MD, Bailey T, Seachord C, Ratcliffe K, Bainbridge B, Dietsch M, Page RC, Aruffo A. Ability of bacteria associated with chronic inflammatory disease to stimulate E-selectin expression and promote neutrophil adhesion. Infect Immun. 1995;63:1311–1317. doi: 10.1128/iai.63.4.1311-1317.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mattsby-Baltzer I, Mielniczuk Z, Larsson L, Lindgren K, Goodwin S. Lipid A in Helicobacter pylori. Infect Immun. 1992;60:4383–4387. doi: 10.1128/iai.60.10.4383-4387.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pérez-Pérez GI, Shepherd VL, Morrow JD, Blaser MJ. Activation of human THP-1 cells and rat bone marrow-derived macrophages by Helicobacter pylori lipopolysaccharide. Infect Immun. 1995;63:1183–1187. doi: 10.1128/iai.63.4.1183-1187.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Semeraro N, Montemurro P, Piccoli C, Muolo V, Colucci M, Giuliani G, Fumarola D, Pece S, Moran AP. Effect of Helicobacter pylori lipopolysaccharide (LPS) and LPS derivatives on the production of tissue factor and plasminogen activator inhibitor type 2 by human blood mononuclear cells. J Infect Dis. 1996;174:1255–1260. doi: 10.1093/infdis/174.6.1255. [DOI] [PubMed] [Google Scholar]
- 43.Monteiro MA, Chan KH, Rasko DA, Taylor DE, Zheng PY, Appelmelk BJ, Wirth HP, Yang M, Blaser MJ, Hynes SO, et al. Simultaneous expression of type 1 and type 2 Lewis blood group antigens by Helicobacter pylori lipopolysaccharides. Molecular mimicry between h. pylori lipopolysaccharides and human gastric epithelial cell surface glycoforms. J Biol Chem. 1998;273:11533–11543. doi: 10.1074/jbc.273.19.11533. [DOI] [PubMed] [Google Scholar]
- 44.Moran AP. Relevance of fucosylation and Lewis antigen expression in the bacterial gastroduodenal pathogen Helicobacter pylori. Carbohydr Res. 2008;343:1952–1965. doi: 10.1016/j.carres.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 45.Logan SM, Conlan JW, Monteiro MA, Wakarchuk WW, Altman E. Functional genomics of Helicobacter pylori: identification of a beta-1,4 galactosyltransferase and generation of mutants with altered lipopolysaccharide. Mol Microbiol. 2000;35:1156–1167. doi: 10.1046/j.1365-2958.2000.01784.x. [DOI] [PubMed] [Google Scholar]
- 46.Appelmelk BJ, Vandenbroucke-Grauls CM. H pylori and Lewis antigens. Gut. 2000;47:10–11. doi: 10.1136/gut.47.1.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hug I, Couturier MR, Rooker MM, Taylor DE, Stein M, Feldman MF. Helicobacter pylori lipopolysaccharide is synthesized via a novel pathway with an evolutionary connection to protein N-glycosylation. PLoS Pathog. 2010;6:e1000819. doi: 10.1371/journal.ppat.1000819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nilsson C, Skoglund A, Moran AP, Annuk H, Engstrand L, Normark S. Lipopolysaccharide diversity evolving in Helicobacter pylori communities through genetic modifications in fucosyltransferases. PLoS One. 2008;3:e3811. doi: 10.1371/journal.pone.0003811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Appelmelk BJ, Martin SL, Monteiro MA, Clayton CA, McColm AA, Zheng P, Verboom T, Maaskant JJ, van den Eijnden DH, Hokke CH, et al. Phase variation in Helicobacter pylori lipopolysaccharide due to changes in the lengths of poly(C) tracts in alpha3-fucosyltransferase genes. Infect Immun. 1999;67:5361–5366. doi: 10.1128/iai.67.10.5361-5366.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang G, Rasko DA, Sherburne R, Taylor DE. Molecular genetic basis for the variable expression of Lewis Y antigen in Helicobacter pylori: analysis of the alpha (1,2) fucosyltransferase gene. Mol Microbiol. 1999;31:1265–1274. doi: 10.1046/j.1365-2958.1999.01268.x. [DOI] [PubMed] [Google Scholar]
- 51.Skoglund A, Bäckhed HK, Nilsson C, Björkholm B, Normark S, Engstrand L. A changing gastric environment leads to adaptation of lipopolysaccharide variants in Helicobacter pylori populations during colonization. PLoS One. 2009;4:e5885. doi: 10.1371/journal.pone.0005885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blaser MJ. Helicobacter-pylori - Balance and Imbalance. USA: Lippincott Williams & WilkinsPhiladelphia; 1997. pp. S15–S18. [Google Scholar]
- 53.Mahdavi J, Borén T, Vandenbroucke-Grauls C, Appelmelk BJ. Limited role of lipopolysaccharide Lewis antigens in adherence of Helicobacter pylori to the human gastric epithelium. Infect Immun. 2003;71:2876–2880. doi: 10.1128/IAI.71.5.2876-2880.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takata T, El-Omar E, Camorlinga M, Thompson SA, Minohara Y, Ernst PB, Blaser MJ. Helicobacter pylori does not require Lewis X or Lewis Y expression to colonize C3H/HeJ mice. Infect Immun. 2002;70:3073–3079. doi: 10.1128/IAI.70.6.3073-3079.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fowler M, Thomas RJ, Atherton J, Roberts IS, High NJ. Galectin-3 binds to Helicobacter pylori O-antigen: it is upregulated and rapidly secreted by gastric epithelial cells in response to H. pylori adhesion. Cell Microbiol. 2006;8:44–54. doi: 10.1111/j.1462-5822.2005.00599.x. [DOI] [PubMed] [Google Scholar]
- 56.Valkonen KH, Wadström T, Moran AP. Interaction of lipopolysaccharides of Helicobacter pylori with basement membrane protein laminin. Infect Immun. 1994;62:3640–3648. doi: 10.1128/iai.62.9.3640-3648.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Portnoy DA. Manipulation of innate immunity by bacterial pathogens. Curr Opin Immunol. 2005;17:25–28. doi: 10.1016/j.coi.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 58.Khamri W, Moran AP, Worku ML, Karim QN, Walker MM, Annuk H, Ferris JA, Appelmelk BJ, Eggleton P, Reid KB, et al. Variations in Helicobacter pylori lipopolysaccharide to evade the innate immune component surfactant protein D. Infect Immun. 2005;73:7677–7686. doi: 10.1128/IAI.73.11.7677-7686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Paziak-Domańska B, Chmiela M, Jarosińska A, Rudnicka W. Potential role of CagA in the inhibition of T cell reactivity in Helicobacter pylori infections. Cell Immunol. 2000;202:136–139. doi: 10.1006/cimm.2000.1654. [DOI] [PubMed] [Google Scholar]
- 60.Gebert B, Fischer W, Weiss E, Hoffmann R, Haas R. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science. 2003;301:1099–1102. doi: 10.1126/science.1086871. [DOI] [PubMed] [Google Scholar]
- 61.Zabaleta J, McGee DJ, Zea AH, Hernández CP, Rodriguez PC, Sierra RA, Correa P, Ochoa AC. Helicobacter pylori arginase inhibits T cell proliferation and reduces the expression of the TCR zeta-chain (CD3zeta) J Immunol. 2004;173:586–593. doi: 10.4049/jimmunol.173.1.586. [DOI] [PubMed] [Google Scholar]
- 62.Gerhard M, Schmees C, Voland P, Endres N, Sander M, Reindl W, Rad R, Oelsner M, Decker T, Mempel M, et al. A secreted low-molecular-weight protein from Helicobacter pylori induces cell-cycle arrest of T cells. Gastroenterology. 2005;128:1327–1339. doi: 10.1053/j.gastro.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 63.Torres VJ, VanCompernolle SE, Sundrud MS, Unutmaz D, Cover TL. Helicobacter pylori vacuolating cytotoxin inhibits activation-induced proliferation of human T and B lymphocyte subsets. J Immunol. 2007;179:5433–5440. doi: 10.4049/jimmunol.179.8.5433. [DOI] [PubMed] [Google Scholar]
- 64.Schmees C, Prinz C, Treptau T, Rad R, Hengst L, Voland P, Bauer S, Brenner L, Schmid RM, Gerhard M. Inhibition of T-cell proliferation by Helicobacter pylori gamma-glutamyl transpeptidase. Gastroenterology. 2007;132:1820–1833. doi: 10.1053/j.gastro.2007.02.031. [DOI] [PubMed] [Google Scholar]
- 65.Dubreuil JD, Giudice GD, Rappuoli R. Helicobacter pylori interactions with host serum and extracellular matrix proteins: potential role in the infectious process. Microbiol Mol Biol Rev. 2002;66:617–629, table of contents. doi: 10.1128/MMBR.66.4.617-629.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wessler S, Backert S. Molecular mechanisms of epithelial-barrier disruption by Helicobacter pylori. Trends Microbiol. 2008;16:397–405. doi: 10.1016/j.tim.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 67.Miller SI, Ernst RK, Bader MW. LPS, TLR4 and infectious disease diversity. Nat Rev Microbiol. 2005;3:36–46. doi: 10.1038/nrmicro1068. [DOI] [PubMed] [Google Scholar]
- 68.Janeway CA, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 69.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 70.Hoebe K, Janssen E, Beutler B. The interface between innate and adaptive immunity. Nat Immunol. 2004;5:971–974. doi: 10.1038/ni1004-971. [DOI] [PubMed] [Google Scholar]
- 71.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 72.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 73.Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. Int Rev Immunol. 2011;30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- 74.Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 75.Kirkland T, Viriyakosol S, Perez-Perez GI, Blaser MJ. Helicobacter pylori lipopolysaccharide can activate 70Z/3 cells via CD14. Infect Immun. 1997;65:604–608. doi: 10.1128/iai.65.2.604-608.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dixon DR, Darveau RP. Lipopolysaccharide heterogeneity: innate host responses to bacterial modification of lipid a structure. J Dent Res. 2005;84:584–595. doi: 10.1177/154405910508400702. [DOI] [PubMed] [Google Scholar]
- 77.Grebowska A, Rechciński T, Bak-Romaniszyn L, Czkwianianc E, Moran A, Druszczyńska M, Kowalewicz-Kulbat M, Owczarek A, Dziuba M, Krzemińska-Pakuła M, et al. Potential role of LPS in the outcome of Helicobacter pylori related diseases. Pol J Microbiol. 2006;55:25–30. [PubMed] [Google Scholar]
- 78.Wurfel MM, Kunitake ST, Lichenstein H, Kane JP, Wright SD. Lipopolysaccharide (LPS)-binding protein is carried on lipoproteins and acts as a cofactor in the neutralization of LPS. J Exp Med. 1994;180:1025–1035. doi: 10.1084/jem.180.3.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lepper PM, Triantafilou M, Schumann C, Schneider EM, Triantafilou K. Lipopolysaccharides from Helicobacter pylori can act as antagonists for Toll-like receptor 4. Cell Microbiol. 2005;7:519–528. doi: 10.1111/j.1462-5822.2005.00482.x. [DOI] [PubMed] [Google Scholar]
- 80.Bäckhed F, Rokbi B, Torstensson E, Zhao Y, Nilsson C, Seguin D, Normark S, Buchan AM, Richter-Dahlfors A. Gastric mucosal recognition of Helicobacter pylori is independent of Toll-like receptor 4. J Infect Dis. 2003;187:829–836. doi: 10.1086/367896. [DOI] [PubMed] [Google Scholar]
- 81.Smith MF, Mitchell A, Li G, Ding S, Fitzmaurice AM, Ryan K, Crowe S, Goldberg JB. Toll-like receptor (TLR) 2 and TLR5, but not TLR4, are required for Helicobacter pylori-induced NF-kappa B activation and chemokine expression by epithelial cells. J Biol Chem. 2003;278:32552–32560. doi: 10.1074/jbc.M305536200. [DOI] [PubMed] [Google Scholar]
- 82.Kawahara T, Teshima S, Oka A, Sugiyama T, Kishi K, Rokutan K. Type I Helicobacter pylori lipopolysaccharide stimulates toll-like receptor 4 and activates mitogen oxidase 1 in gastric pit cells. Infect Immun. 2001;69:4382–4389. doi: 10.1128/IAI.69.7.4382-4389.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yokota S, Okabayashi T, Rehli M, Fujii N, Amano K. Helicobacter pylori lipopolysaccharides upregulate toll-like receptor 4 expression and proliferation of gastric epithelial cells via the MEK1/2-ERK1/2 mitogen-activated protein kinase pathway. Infect Immun. 2010;78:468–476. doi: 10.1128/IAI.00903-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yokota S, Amano K, Nishitani C, Ariki S, Kuroki Y, Fujii N. Implication of antigenic conversion of Helicobacter pylori lipopolysaccharides that involve interaction with surfactant protein D. Infect Immun. 2012;80:2956–2962. doi: 10.1128/IAI.00345-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rudnicka K, Grebowska A, Moran AP, Matusiak A, Walencka M, Miszczyk E, Bąk-Romaniszyn L, Czkwianianc E, Płaneta-Małecka I, Rudnicka W, et al. Different effectiveness of Helicobacter pylori lipopolysaccharides with or without LewisXY determinants in stimulating the secretion of proinflammatory cytokines IL-8 and TNF-α by peripheral blood mononuclear leukocytes. Przeg Gastroenterol. 2011;6:401–408. [Google Scholar]
- 86.Nigou J, Zelle-Rieser C, Gilleron M, Thurnher M, Puzo G. Mannosylated lipoarabinomannans inhibit IL-12 production by human dendritic cells: evidence for a negative signal delivered through the mannose receptor. J Immunol. 2001;166:7477–7485. doi: 10.4049/jimmunol.166.12.7477. [DOI] [PubMed] [Google Scholar]
- 87.Geijtenbeek TB, Engering A, Van Kooyk Y. DC-SIGN, a C-type lectin on dendritic cells that unveils many aspects of dendritic cell biology. J Leukoc Biol. 2002;71:921–931. [PubMed] [Google Scholar]
- 88.Appelmelk BJ, van Die I, van Vliet SJ, Vandenbroucke-Grauls CM, Geijtenbeek TB, van Kooyk Y. Cutting edge: carbohydrate profiling identifies new pathogens that interact with dendritic cell-specific ICAM-3-grabbing nonintegrin on dendritic cells. J Immunol. 2003;170:1635–1639. doi: 10.4049/jimmunol.170.4.1635. [DOI] [PubMed] [Google Scholar]
- 89.Miszczyk E, Rudnicka K, Moran AP, Fol M, Kowalewicz-Kulbat M, Druszczyńska M, Matusiak A, Walencka M, Rudnicka W, Chmiela M. Interaction of Helicobacter pylori with C-type lectin dendritic cell-specific ICAM grabbing nonintegrin. J Biomed Biotechnol. 2012;2012:206463. doi: 10.1155/2012/206463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bergman MP, Engering A, Smits HH, van Vliet SJ, van Bodegraven AA, Wirth HP, Kapsenberg ML, Vandenbroucke-Grauls CM, van Kooyk Y, Appelmelk BJ. Helicobacter pylori modulates the T helper cell 1/T helper cell 2 balance through phase-variable interaction between lipopolysaccharide and DC-SIGN. J Exp Med. 2004;200:979–990. doi: 10.1084/jem.20041061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ford JW, McVicar DW. TREM and TREM-like receptors in inflammation and disease. Curr Opin Immunol. 2009;21:38–46. doi: 10.1016/j.coi.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Watanabe T, Asano N, Kitani A, Fuss IJ, Chiba T, Strober W. NOD1-Mediated Mucosal Host Defense against Helicobacter pylori. Int J Inflam. 2010;2010:476482. doi: 10.4061/2010/476482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Svensson H, Hansson M, Kilhamn J, Backert S, Quiding-Järbrink M. Selective upregulation of endothelial E-selectin in response to Helicobacter pylori-induced gastritis. Infect Immun. 2009;77:3109–3116. doi: 10.1128/IAI.01460-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Allen LA. Phagocytosis and persistence of Helicobacter pylori. Cell Microbiol. 2007;9:817–828. doi: 10.1111/j.1462-5822.2007.00906.x. [DOI] [PubMed] [Google Scholar]
- 95.Ramarao N, Gray-Owen SD, Backert S, Meyer TF. Helicobacter pylori inhibits phagocytosis by professional phagocytes involving type IV secretion components. Mol Microbiol. 2000;37:1389–1404. doi: 10.1046/j.1365-2958.2000.02089.x. [DOI] [PubMed] [Google Scholar]
- 96.Chmiela M, Czkwianianc E, Wadstrom T, Rudnicka W. Role of Helicobacter pylori surface structures in bacterial interaction with macrophages. Gut. 1997;40:20–24. doi: 10.1136/gut.40.1.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Grebowska A, Moran AP, Matusiak A, Bak-Romaniszyn L, Czkwianianc E, Rechciński T, Walencka M, Płaneta-Małecka I, Rudnicka W, Chmiela M. Anti-phagocytic activity of Helicobacter pylori lipopolysaccharide (LPS)--possible modulation of the innate immune response to these bacteria. Pol J Microbiol. 2008;57:185–192. [PubMed] [Google Scholar]
- 98.Wright SD, Tobias PS, Ulevitch RJ, Ramos RA. Lipopolysaccharide (LPS) binding protein opsonizes LPS-bearing particles for recognition by a novel receptor on macrophages. J Exp Med. 1989;170:1231–1241. doi: 10.1084/jem.170.4.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Horwitz AH, Williams RE, Nowakowski G. Human lipopolysaccharide-binding protein potentiates bactericidal activity of human bactericidal/permeability-increasing protein. Infect Immun. 1995;63:522–527. doi: 10.1128/iai.63.2.522-527.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Walz A, Odenbreit S, Mahdavi J, Borén T, Ruhl S. Identification and characterization of binding properties of Helicobacter pylori by glycoconjugate arrays. Glycobiology. 2005;15:700–708. doi: 10.1093/glycob/cwi049. [DOI] [PubMed] [Google Scholar]
- 101.Slomiany BL, Piotrowski J, Slomiany A. Gastric mucosal inflammatory responses toHelicobacter pylori lipopolysaccharide: suppression of caspase-3 and nitric oxide synthase-2 by omeprazole and sucralfate. Inflammopharmacology. 1999;7:163–177. doi: 10.1007/BF02918388. [DOI] [PubMed] [Google Scholar]
- 102.Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22:633–640. doi: 10.1016/s1471-4906(01)02060-9. [DOI] [PubMed] [Google Scholar]
- 103.O’Keeffe J, Gately CM, O’Donoghue Y, Zulquernain SA, Stevens FM, Moran AP. Natural killer cell receptor T-lymphocytes in normal and Helicobacter pylori-infected human gastric mucosa. Helicobacter. 2008;13:500–505. doi: 10.1111/j.1523-5378.2008.00641.x. [DOI] [PubMed] [Google Scholar]
- 104.Lindgren A, Yun CH, Lundgren A, Sjöling A, Ohman L, Svennerholm AM, Holmgren J, Lundin SB. CD8- natural killer cells are greatly enriched in the human gastrointestinal tract and have the capacity to respond to bacteria. J Innate Immun. 2010;2:294–302. doi: 10.1159/000286238. [DOI] [PubMed] [Google Scholar]
- 105.Tarkkanen J, Kosunen TU, Saksela E. Contact of lymphocytes with Helicobacter pylori augments natural killer cell activity and induces production of gamma interferon. Infect Immun. 1993;61:3012–3016. doi: 10.1128/iai.61.7.3012-3016.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yun CH, Lundgren A, Azem J, Sjöling A, Holmgren J, Svennerholm AM, Lundin BS. Natural killer cells and Helicobacter pylori infection: bacterial antigens and interleukin-12 act synergistically to induce gamma interferon production. Infect Immun. 2005;73:1482–1490. doi: 10.1128/IAI.73.3.1482-1490.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rudnicka K, Włodarczyk M, Moran AP, Rechciński T, Miszczyk E, Matusiak A, Szczęsna E, Walencka M, Rudnicka W, Chmiela M. Helicobacter pylori antigens as potential modulators of lymphocytes’ cytotoxic activity. Microbiol Immunol. 2012;56:62–75. doi: 10.1111/j.1348-0421.2011.00399.x. [DOI] [PubMed] [Google Scholar]
- 108.Rudnicka K, Włodarczyk M, Miszczyk E, Matusiak A, Walencka M, Chmiela M. NK and NKT cell responses to H. pylori liopopolysaccharide in relation to lymphocyte cytotoxic activity and H. pylori status. Proceedings of the 3rd Workshop on Microbiology in Health and Environmental Protection-MIKROBIOT 2013; 2013 Sep 17-20; Lodz, Poland. Adv Microbiol. 2013;52 Suppl 1:S90 III–P 17. [Google Scholar]
- 109.Rudnicka K, Matusiak A, Miszczyk E, Rudnicka W, Tenderenda M, Chmiela M. Immunophenotype of peripheral blood natural killer cells and IL-10 serum levels in relation to Helicobacter pylori status. APMIS. 2013;121:806–813. doi: 10.1111/apm.12120. [DOI] [PubMed] [Google Scholar]
- 110.Rudnicka K, Miszczyk E, Matusiak A, Walencka M, Moran AP, Rudnicka W, Chmiela M. Helicobacter pylori - driven modulation of NK cell expansion, intracellular cytokine expression and cytotoxic activity. Innate Immunity. 2014:In press. doi: 10.1177/1753425913518225. [DOI] [PubMed] [Google Scholar]
- 111.Goodier MR, Londei M. Lipopolysaccharide stimulates the proliferation of human CD56+CD3- NK cells: a regulatory role of monocytes and IL-10. J Immunol. 2000;165:139–147. doi: 10.4049/jimmunol.165.1.139. [DOI] [PubMed] [Google Scholar]
- 112.Miszczyk E, Rudnicka K, Kwiecień J, Walencka M, Matusiak A, Rudnicka W, Chmiela M. Helicobacter pylori - dependent modulation of lymphocyte cytotoxic activity in a guinea pig model. Proceedings of the 3rd Workshop on Microbiology in Health and Environmental Protection-MIKROBIOT 2013; 2013 Sep 17-20; Lodz, Poland. Adv Microbiol. 2013;52 Suppl 1:S89 III–P 13. [Google Scholar]
- 113.Ulevitch RJ, Tobias PS. Recognition of gram-negative bacteria and endotoxin by the innate immune system. Curr Opin Immunol. 1999;11:19–22. doi: 10.1016/s0952-7915(99)80004-1. [DOI] [PubMed] [Google Scholar]
- 114.Vogel SN, Hilfiker ML, Caulfield MJ. Endotoxin-induced T lymphocyte proliferation. J Immunol. 1983;130:1774–1779. [PubMed] [Google Scholar]
- 115.Mattern T, Thanhäuser A, Reiling N, Toellner KM, Duchrow M, Kusumoto S, Rietschel ET, Ernst M, Brade H, Flad HD. Endotoxin and lipid A stimulate proliferation of human T cells in the presence of autologous monocytes. J Immunol. 1994;153:2996–3004. [PubMed] [Google Scholar]
- 116.Tough DF, Sun S, Sprent J. T cell stimulation in vivo by lipopolysaccharide (LPS) J Exp Med. 1997;185:2089–2094. doi: 10.1084/jem.185.12.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rudnicka W, Jarosinska A, Bak-Romaniszyn L, Moran A, Planeta-Malecka I, Wadstrom T, Chmiela M. Helicobacter pylori lipopolysaccharide in the IL-2 milieu activates lymphocytes from dyspeptic children. FEMS Immunol Med Microbiol. 2003;36:141–145. doi: 10.1016/S0928-8244(03)00023-3. [DOI] [PubMed] [Google Scholar]
- 118.Bosco MC, Espinoza-Delgado I, Rowe TK, Malabarba MG, Longo DL, Varesio L. Functional role for the myeloid differentiation antigen CD14 in the activation of human monocytes by IL-2. J Immunol. 1997;159:2922–2931. [PubMed] [Google Scholar]
- 119.Grebowska A, Moran AP, Bielanski W, Matusiak A, Rechcinski T, Rudnicka K, Baranowska A, Rudnicka W, Chmiela M. Helicobacter pylori lipopolysaccharide activity in human peripheral blood mononuclear leukocyte cultures. J Physiol Pharmacol. 2010;61:437–442. [PubMed] [Google Scholar]
- 120.Caramalho I, Lopes-Carvalho T, Ostler D, Zelenay S, Haury M, Demengeot J. Regulatory T cells selectively express toll-like receptors and are activated by lipopolysaccharide. J Exp Med. 2003;197:403–411. doi: 10.1084/jem.20021633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Raghavan S, Holmgren J. CD4+CD25+ suppressor T cells regulate pathogen induced inflammation and disease. FEMS Immunol Med Microbiol. 2005;44:121–127. doi: 10.1016/j.femsim.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 122.Sundrud MS, Torres VJ, Unutmaz D, Cover TL. Inhibition of primary human T cell proliferation by Helicobacter pylori vacuolating toxin (VacA) is independent of VacA effects on IL-2 secretion. Proc Natl Acad Sci USA. 2004;101:7727–7732. doi: 10.1073/pnas.0401528101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Meyer F, Wilson KT, James SP. Modulation of innate cytokine responses by products of Helicobacter pylori. Infect Immun. 2000;68:6265–6272. doi: 10.1128/iai.68.11.6265-6272.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Taylor JM, Ziman ME, Huff JL, Moroski NM, Vajdy M, Solnick JV. Helicobacter pylori lipopolysaccharide promotes a Th1 type immune response in immunized mice. Vaccine. 2006;24:4987–4994. doi: 10.1016/j.vaccine.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 125.Gray BM, Fontaine CA, Poe SA, Eaton KA. Complex T cell interactions contribute to Helicobacter pylori gastritis in mice. Infect Immun. 2013;81:740–752. doi: 10.1128/IAI.01269-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Shi Y, Liu XF, Zhuang Y, Zhang JY, Liu T, Yin Z, Wu C, Mao XH, Jia KR, Wang FJ, et al. Helicobacter pylori-induced Th17 responses modulate Th1 cell responses, benefit bacterial growth, and contribute to pathology in mice. J Immunol. 2010;184:5121–5129. doi: 10.4049/jimmunol.0901115. [DOI] [PubMed] [Google Scholar]
- 127.Shiomi S, Toriie A, Imamura S, Konishi H, Mitsufuji S, Iwakura Y, Yamaoka Y, Ota H, Yamamoto T, Imanishi J, et al. IL-17 is involved in Helicobacter pylori-induced gastric inflammatory responses in a mouse model. Helicobacter. 2008;13:518–524. doi: 10.1111/j.1523-5378.2008.00629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Amedei A, Munari F, Bella CD, Niccolai E, Benagiano M, Bencini L, Cianchi F, Farsi M, Emmi G, Zanotti G, et al. Helicobacter pylori secreted peptidyl prolyl cis, trans-isomerase drives Th17 inflammation in gastric adenocarcinoma. Intern Emerg Med. 2014;9:303–309. doi: 10.1007/s11739-012-0867-9. [DOI] [PubMed] [Google Scholar]
- 129.Caruso R, Fina D, Paoluzi OA, Del Vecchio Blanco G, Stolfi C, Rizzo A, Caprioli F, Sarra M, Andrei F, Fantini MC, et al. IL-23-mediated regulation of IL-17 production in Helicobacter pylori-infected gastric mucosa. Eur J Immunol. 2008;38:470–478. doi: 10.1002/eji.200737635. [DOI] [PubMed] [Google Scholar]
- 130.Luzza F, Parrello T, Monteleone G, Sebkova L, Romano M, Zarrilli R, Imeneo M, Pallone F. Up-regulation of IL-17 is associated with bioactive IL-8 expression in Helicobacter pylori-infected human gastric mucosa. J Immunol. 2000;165:5332–5337. doi: 10.4049/jimmunol.165.9.5332. [DOI] [PubMed] [Google Scholar]
- 131.Okada T, Ayada K, Usui S, Yokota K, Cui J, Kawahara Y, Inaba T, Hirohata S, Mizuno M, Yamamoto D, et al. Antibodies against heat shock protein 60 derived from Helicobacter pylori: diagnostic implications in cardiovascular disease. J Autoimmun. 2007;29:106–115. doi: 10.1016/j.jaut.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 132.Appelmelk BJ, Simoons-Smit I, Negrini R, Moran AP, Aspinall GO, Forte JG, De Vries T, Quan H, Verboom T, Maaskant JJ, et al. Potential role of molecular mimicry between Helicobacter pylori lipopolysaccharide and host Lewis blood group antigens in autoimmunity. Infect Immun. 1996;64:2031–2040. doi: 10.1128/iai.64.6.2031-2040.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.D’Elios MM, Bergman MP, Amedei A, Appelmelk BJ, Del Prete G. Helicobacter pylori and gastric autoimmunity. Microbes Infect. 2004;6:1395–1401. doi: 10.1016/j.micinf.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 134.Taylor DE, Fedorak RN, Sherburne R. Antigenic mimicry between Helicobacter pylori and gastric mucosa: failure to implicate heat-shock protein Hsp60 using immunoelectron microscopy. Helicobacter. 1999;4:148–153. doi: 10.1046/j.1523-5378.1999.99295.x. [DOI] [PubMed] [Google Scholar]
- 135.Chmiela M, Jurkiewicz M, Wiśniewska M, Czkwianianc E, Płaneta-Małecka I, Rechciński T, Rudnicka W. Anti-Lewis X IgM and IgG in H. pylori infections in children and adults. Acta Microbiol Pol. 1999;48:277–281. [PubMed] [Google Scholar]
- 136.Chmiela M, Wadstrom T, Folkesson H, Płaneta Małecka I, Czkwianianc E, Rechciński T, Rudnicka W. Anti-Lewis X antibody and Lewis X-anti-Lewis X immune complexes in Helicobacter pylori infection. Immunol Lett. 1998;61:119–125. doi: 10.1016/s0165-2478(98)00004-2. [DOI] [PubMed] [Google Scholar]
- 137.Rudnicka W, Czkwianianc E, Płaneta-Małecka I, Jurkiewicz M, Wiśniewska M, Cieślikowski T, Rózalska B, Wadström T, Chmiela M. A potential double role of anti-Lewis X antibodies in Helicobacter pylori-associated gastroduodenal diseases. FEMS Immunol Med Microbiol. 2001;30:121–125. doi: 10.1111/j.1574-695X.2001.tb01559.x. [DOI] [PubMed] [Google Scholar]
- 138.Nagashima R, Maeda K, Yuda F, Kudo K, Saitoh M, Takahashi T. Helicobacter pylori antigen in the glomeruli of patients with membranous nephropathy. Virchows Arch. 1997;431:235–239. doi: 10.1007/s004280050094. [DOI] [PubMed] [Google Scholar]
- 139.Guruge JL, Falk PG, Lorenz RG, Dans M, Wirth HP, Blaser MJ, Berg DE, Gordon JI. Epithelial attachment alters the outcome of Helicobacter pylori infection. Proc Natl Acad Sci USA. 1998;95:3925–3930. doi: 10.1073/pnas.95.7.3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Heneghan MA, McCarthy CF, Janulaityte D, Moran AP. Relationship of anti-Lewis x and anti-Lewis y antibodies in serum samples from gastric cancer and chronic gastritis patients to Helicobacter pylori-mediated autoimmunity. Infect Immun. 2001;69:4774–4781. doi: 10.1128/IAI.69.8.4774-4781.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Alexander C, Rietschel ET. Bacterial lipopolysaccharides and innate immunity. J Endotoxin Res. 2001;7:167–202. [PubMed] [Google Scholar]
- 142.Laurila A, Bloigu A, Näyhä S, Hassi J, Leinonen M, Saikku P. Association of Helicobacter pylori infection with elevated serum lipids. Atherosclerosis. 1999;142:207–210. doi: 10.1016/s0021-9150(98)00194-4. [DOI] [PubMed] [Google Scholar]
- 143.Hildebrandt E, McGee DJ. Helicobacter pylori lipopolysaccharide modification, Lewis antigen expression, and gastric colonization are cholesterol-dependent. BMC Microbiol. 2009;9:258. doi: 10.1186/1471-2180-9-258. [DOI] [PMC free article] [PubMed] [Google Scholar]