

Abstract
The glomerulus, the filtering unit of the kidney, is a unique bundle of capillaries lined by delicate fenestrated endothelia, a complex mesh of proteins that serve as the glomerular basement membrane and specialized visceral epithelial cells that form the slit diaphragms between interdigitating foot processes. Taken together, this arrangement allows continuous filtration of the plasma volume. The dynamic physical forces that determine the single nephron glomerular filtration are considered. In addition, new insights into the cellular and molecular components of the glomerular tuft and their contribution to glomerular disorders are explored.
Keywords: renal physiology, glomerulus, glomerular filtration
The Glomerulus
The glomerulus, the filtering unit of the kidney, is a specialized bundle of capillaries that are uniquely situated between two resistance vessels (Figure 1). These capillaries are each contained within the Bowman’s capsule and they are the only capillary beds in the body that are not surrounded by interstitial tissue. Therefore, a unique support structure is needed to maintain flow in these essential capillary units. In fact, all of the major components of the filter itself are unique compared with related structures in other capillary beds. The proximal component layer of the glomerular filter itself is a fenestrated endothelium, characterized by the presence of individual fenestrae on the order of 70–100 nm in diameter. These cells drape the luminal aspect of the capillary and permit filtration. The second layer of the filter, the glomerular basement membrane (GBM), is a complex mesh of extracellular proteins, including type IV collagen, laminins, fibronectins, and proteoglycans. The distal layer of the glomerular filter is composed of the visceral epithelial cells, or podocytes. These remarkable cells help to create the filtration slit diaphragm and serve as support to help sustain the integrity of the free-standing capillary loops. A third cell type, the mesangial cells, also contributes to the integrity of the glomerular tuft and the dynamic nature of filtration. Together, this elegant structure permits the formation of the primary glomerular filtrate that enters a space delimited by the visceral and parietal epithelial cells before modification during transit through the tubule.
Figure 1.

Structure of the renal corpuscle, looking into the Bowman’s capsule at glomerular capillary tuft. The capsule is lined with parietal epithelium, which gives way to the cells of the proximal tubule at the urinary pole on the right. At the left, the vascular pole of the glomerulus includes both the afferent and efferent arterioles. In addition, the relationship between these arterioles and the specialized portion of the distal nephron called the macula densa is illustrated. (Inset) The layers that comprise the filtration barrier are displayed. The outermost layer is composed of the visceral epithelial cells, the podocytes, next the glomerular basement membrane (GBM) and finally the fenestrated endothelial cells.
Hemodynamic Control of Glomerular Filtration at the Single Nephron Level
Although there is considerable variability, the average human kidney is composed of approximately 1 million individual functioning nephrons, each containing a single glomerulus or filtering unit (1). The GFR for the entire organism is then the sum of the individual filtration rates of approximately 2 million glomeruli in two kidneys. There can be significant differences in the size and filtration rates of individual glomeruli in different regions of the kidney. For example, juxtamedullary nephrons tend to have a larger intraglomerular volume compared with superficial nephrons (2). Intrarenal blood flow distribution varies in physiologic and pathologic situations and this can also affect total GFR for the organism (3,4). Indeed, medullary blood flow appears to be greatest during a water diuresis and reduced in the setting of antidiuresis (4,5), and also appears to be better preserved in renal hypoperfusion. This discussion focuses on the regulation of filtration at the level of a single, typical glomerulus, by which the precise hemodynamic control of glomerular filtration is best understood. Although the majority of the experimental findings were gleaned from work done in Wistar rats (which have superficial glomeruli, in contrast with most mammals), the principles reviewed here are thought to apply to human glomeruli as well.
The Determinants of Glomerular Ultrafiltration
The transcapillary movement of water across the glomerular capillaries is controlled by the same Starling forces that control fluid movement across all capillary beds. Filtration occurs because there is an imbalance between the mean transcapillary hydraulic pressure gradient 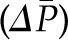 , which favors filtration, and the mean transcapillary oncotic pressure
, which favors filtration, and the mean transcapillary oncotic pressure 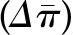 , which opposes filtration. (Of note, we use the term hydraulic, rather than hydrostatic, because the latter refers to pressure in a static liquid, whereas hydraulic pressure refers to pressure of fluid moving through tubes.) The resulting net pressure gradient integrated over the entire filtering portion of a single glomerular capillary network is the mean net ultrafiltration pressure
, which opposes filtration. (Of note, we use the term hydraulic, rather than hydrostatic, because the latter refers to pressure in a static liquid, whereas hydraulic pressure refers to pressure of fluid moving through tubes.) The resulting net pressure gradient integrated over the entire filtering portion of a single glomerular capillary network is the mean net ultrafiltration pressure 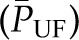 . This physiologic condition can be expressed mathematically by Equation 1:
. This physiologic condition can be expressed mathematically by Equation 1:
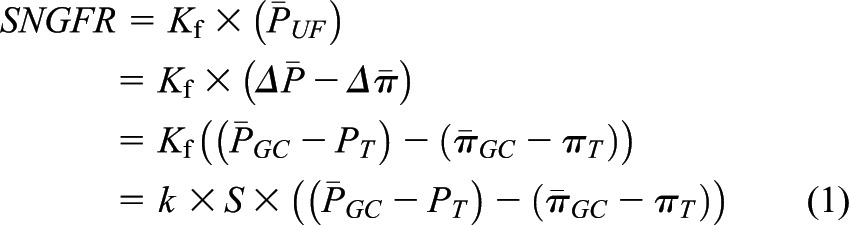 |
where SNGFR is the single nephron GFR, 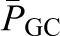 is the hydraulic pressure within the lumen of the glomerular capillary that is generated by the pumping action of the heart, and
is the hydraulic pressure within the lumen of the glomerular capillary that is generated by the pumping action of the heart, and 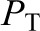 is the hydraulic pressure in Bowman’s space that is typically measured as the pressure within an early proximal tubule segment.
is the hydraulic pressure in Bowman’s space that is typically measured as the pressure within an early proximal tubule segment. 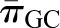 and
and 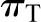 are the oncotic pressures within the glomerular capillary, generated by the plasma proteins, and in Bowman’s space. Because the sieving characteristics of the glomerular capillary wall that prevent transcapillary filtration of all but small, low molecular weight plasma proteins,
are the oncotic pressures within the glomerular capillary, generated by the plasma proteins, and in Bowman’s space. Because the sieving characteristics of the glomerular capillary wall that prevent transcapillary filtration of all but small, low molecular weight plasma proteins, 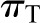 is effectively zero. Kf is determined by two factors: the hydraulic conductivity of the glomerular capillary wall, which depends on both its intrinsic characteristics and the concentration polarization of proteins (6) in the capillary (k), and the total capillary surface area available for filtration (S). For any given mean net filtration pressure,
is effectively zero. Kf is determined by two factors: the hydraulic conductivity of the glomerular capillary wall, which depends on both its intrinsic characteristics and the concentration polarization of proteins (6) in the capillary (k), and the total capillary surface area available for filtration (S). For any given mean net filtration pressure, 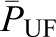 , the absolute amount of filtrate formed will also depend on Kf.
, the absolute amount of filtrate formed will also depend on Kf.
Glomerular Transcapillary Hydraulic and Oncotic Pressures, 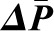 and
and 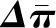 .
.
These relationships can also be represented graphically as shown in Figure 2. In this construct, the glomerular capillary network is imagined to be a continuous single tube beginning at the termination of the afferent arteriole, and ending at the origin of the efferent arteriole. The x axis represents fractional distance along the glomerular capillary. Both hydraulic and plasma oncotic pressures are shown on the y axis. The values provided in Figure 2 are typical of those observed in rats, which were first directly measured by Brenner et al. (7) and subsequently confirmed in many studies in rats (8) and squirrel monkeys (9). 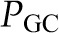 averages just <50 mmHg in normal rats and is nearly constant along the glomerular capillary network, a value significantly higher than in systemic capillaries. The oncotic pressure
averages just <50 mmHg in normal rats and is nearly constant along the glomerular capillary network, a value significantly higher than in systemic capillaries. The oncotic pressure 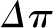 rises from approximately 18 to 34 mmHg by the efferent end of the network, a consequence of the filtration process during which water leaves the glomerular capillary lumen causing an increase in protein concentration.
rises from approximately 18 to 34 mmHg by the efferent end of the network, a consequence of the filtration process during which water leaves the glomerular capillary lumen causing an increase in protein concentration. 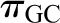 equals
equals 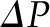 by the end of the capillary, resulting in a reduction of local filtration pressure from 17 to 0 mmHg (Figure 2).
by the end of the capillary, resulting in a reduction of local filtration pressure from 17 to 0 mmHg (Figure 2).
Figure 2.

Hydraulic and oncotic pressures along an idealized glomerular capillary and within Bowman’s space. The graph shows idealized hydraulic and oncotic pressure curves under conditions of filtration pressure equilibrium (curve A) when the mean net ultrafiltration pressure 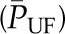 is equal to the shaded region between the
is equal to the shaded region between the 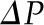 and the
and the 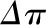 curves. Curve B is a hypothetical linear profile of the oncotic pressure that would occur with a minimum value of the Kf. By contrast, in curve C, filtration equilibrium is never reached. Modified from Taal MW, Chertow GM, Marsden PA, Skorecki K, Yu ASL, Brenner BM: Brenner and Rector's The Kidney, 9th Ed., Philadelphia, Elsevier Saunders, 2012, with permission.
curves. Curve B is a hypothetical linear profile of the oncotic pressure that would occur with a minimum value of the Kf. By contrast, in curve C, filtration equilibrium is never reached. Modified from Taal MW, Chertow GM, Marsden PA, Skorecki K, Yu ASL, Brenner BM: Brenner and Rector's The Kidney, 9th Ed., Philadelphia, Elsevier Saunders, 2012, with permission.
The oncotic pressure profile is highly nonlinear and is a consequence of (1) the greater local ultrafiltration pressure near the afferent end of the capillary leading to more rapid translocation of fluid and (2) the exponential relationship between the plasma protein concentration and the plasma oncotic pressure (8). In fact, under equilibrium conditions, the exact 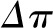 curve cannot be determined. Curve A in Figure 2 is just one of an infinite number of possibilities.
curve cannot be determined. Curve A in Figure 2 is just one of an infinite number of possibilities.
Ultrafiltration Coefficient, Kf.
As shown in Equation 1, SNGFR equals the Kf multiplied by the net ultrafiltration pressure integrated over the entire length of the glomerular capillary. The mean net ultrafiltration pressure is represented graphically in Figure 2 as the area between the 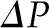 and the
and the 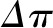 curves (shaded blue). As discussed above, because an exact
curves (shaded blue). As discussed above, because an exact 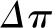 curve cannot be determined under the normal condition of filtration pressure equilibrium, the exact values for
curve cannot be determined under the normal condition of filtration pressure equilibrium, the exact values for 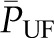 and Kf can also not be determined. However, a minimum value for Kf can be estimated using the dashed line in Figure 2 curve B, which gives a maximal possible value for
and Kf can also not be determined. However, a minimum value for Kf can be estimated using the dashed line in Figure 2 curve B, which gives a maximal possible value for 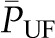 . Applying this approximation to values for SNGFR and
. Applying this approximation to values for SNGFR and 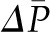 obtained in rats, Kf has been estimated to range from at least 3.5 to 8 nl/min per mmHg. Using the value of S obtained for rat glomeruli, the calculated value of k is approximately 2500 nl/min per mmHg per cm2. This value is one to two orders of magnitude greater than reported for other capillary beds (8), and this allows for the high rate of glomerular filtration despite a net driving pressure of <10 mmHg, on average, along the capillary.
obtained in rats, Kf has been estimated to range from at least 3.5 to 8 nl/min per mmHg. Using the value of S obtained for rat glomeruli, the calculated value of k is approximately 2500 nl/min per mmHg per cm2. This value is one to two orders of magnitude greater than reported for other capillary beds (8), and this allows for the high rate of glomerular filtration despite a net driving pressure of <10 mmHg, on average, along the capillary.
The Effects of Selective Alterations in the Primary Determinants of SNGFR
The four principal determinants of glomerular filtration at the single nephron level are the Kf, the transcapillary hydraulic pressure difference 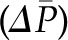 , the initial capillary oncotic pressure
, the initial capillary oncotic pressure 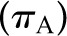 , and the initial glomerular plasma flow rate (QA) (Figure 3). Selective alteration in any of these four primary determinants has predictable effects on SNGFR, has been examined by mathematical modeling (10) and experimentally (8), and is described here. However, physiologic and pathophysiologic states often engender complex combinations of alterations in multiple determinants that may be additive or offsetting. Indeed, these determinants are not truly independent variables; rather, they tend to have complex and often reciprocal relationships.
, and the initial glomerular plasma flow rate (QA) (Figure 3). Selective alteration in any of these four primary determinants has predictable effects on SNGFR, has been examined by mathematical modeling (10) and experimentally (8), and is described here. However, physiologic and pathophysiologic states often engender complex combinations of alterations in multiple determinants that may be additive or offsetting. Indeed, these determinants are not truly independent variables; rather, they tend to have complex and often reciprocal relationships.
Figure 3.

The single nephron GFR (SNGFR) can be affected by alterations in each of the four major determinants of ultrafiltration. These affects can be expressed in a mathematical model, which helps illustrate the predicted effects of each change on the SNGFR. Normal values are indicated with a dotted line in each graph. Unless otherwise indicated in the graph, the input values are Kf = 0.095 nl/s·mmHg,  = 35 mmHg,
= 35 mmHg, 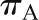 = 18 mmHg, and QA = 135 nl/min. (A) Alterations in Kf; (B) alterations in
= 18 mmHg, and QA = 135 nl/min. (A) Alterations in Kf; (B) alterations in 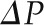 ; (C) alterations in ; (D) alterations in QA. Modified from Brenner BM, Dworkin LD, Ichikawa I: Glomerular filtration. In: Brenner and Rector’s The Kidney, 3rd Ed., edited by Brenner BM, Rector FC Jr, Philadelphia, Elsevier, 1986, with permission.
; (C) alterations in ; (D) alterations in QA. Modified from Brenner BM, Dworkin LD, Ichikawa I: Glomerular filtration. In: Brenner and Rector’s The Kidney, 3rd Ed., edited by Brenner BM, Rector FC Jr, Philadelphia, Elsevier, 1986, with permission.
Glomerular Capillary Ultrafiltration Coefficient, Kf.
Under conditions of filtration pressure equilibrium, which are typically present when QA is normal to low, reductions in Kf will not affect SNGFR (Figure 3A). Rather, because less fluid exits the capillary at each point along the network, the oncotic pressure curve is shifted down and to the right, moving the equilibrium point closer to the end of the capillary, as if going from curve A to curve B in Figure 2. This change in the oncotic pressure profile results in an increase in the area between the 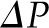 and
and 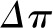 , which is equivalent to an increase in the mean net ultrafiltration pressure for the entire capillary network
, which is equivalent to an increase in the mean net ultrafiltration pressure for the entire capillary network 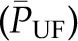 . This increase in
. This increase in 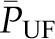 exactly opposes and offsets the decrease in Kf, resulting in constancy in SNGFR. Once Kf is reduced enough to produce disequilibrium, typically by ≥50%, further reductions in Kf will be associated with declines in SNGFR.
exactly opposes and offsets the decrease in Kf, resulting in constancy in SNGFR. Once Kf is reduced enough to produce disequilibrium, typically by ≥50%, further reductions in Kf will be associated with declines in SNGFR.
Because Kf is the product of the surface area available for filtration and hydraulic conductivity, reductions in either can affect Kf. Reductions in glomerular surface area may occur in glomerular diseases, such as membranous nephropathy (11) or physiologically, as a result of vasoconstrictors, such as NE (12). Hydraulic conductivity may also decline in glomerular diseases. This has been well characterized in a model for membranous nephropathy (13) and in anti-GBM–induced nephritis (14). In addition, studies in patients with diabetic nephropathy suggest that the hydraulic conductivity is progressively decreased (15). Furthermore, changes in 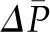 (16) or a change in the plasma protein concentration (16) (see below) have also been shown to alter hydraulic conductivity.
(16) or a change in the plasma protein concentration (16) (see below) have also been shown to alter hydraulic conductivity.
Transcapillary Hydraulic Pressure Difference, 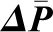 .
.
Until ΔP exceeds 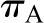 , no filtration will occur (Figure 3B). Once the hydraulic pressure exceeds the oncotic pressure, filtration rises with increases in
, no filtration will occur (Figure 3B). Once the hydraulic pressure exceeds the oncotic pressure, filtration rises with increases in 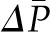 . This relationship is not linear because elevations in
. This relationship is not linear because elevations in 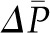 are associated with more fluid filtration earlier in the capillary as well as a more rapid rise in
are associated with more fluid filtration earlier in the capillary as well as a more rapid rise in 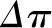 , although not enough to entirely offset the increase in
, although not enough to entirely offset the increase in 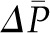 . However, because of the offsetting increase in oncotic pressure and because alterations in
. However, because of the offsetting increase in oncotic pressure and because alterations in 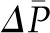 of approximately >10 mmHg are unusual under physiologic conditions (10), changes in
of approximately >10 mmHg are unusual under physiologic conditions (10), changes in 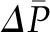 generally result in relatively minor changes in SNGFR.
generally result in relatively minor changes in SNGFR.
Oncotic Pressure, 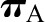 .
.
SNGFR is predicted to vary inversely with selective changes in 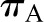 (10) (Figure 3C). This is because increases in oncotic pressure should reduce
(10) (Figure 3C). This is because increases in oncotic pressure should reduce 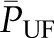 and, thereby, SNGFR. However, changes in
and, thereby, SNGFR. However, changes in 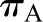 do not appear to be an important factor in physiologic regulation of glomerular filtration. First, the plasma protein concentration is relatively stable except in disease states that alter either the production (e.g., in severe liver disease) or degradation or loss of plasma proteins (e.g., in nephrotic syndrome). In addition, changes in plasma protein concentration in mammals are associated with reciprocal and offsetting alterations in Kf, such that GFR is often unchanged. This is thought to be a result of variation in the hydraulic conductivity of the GBM in response to changes in serum albumin concentration (16), but the exact mechanism is unknown. By contrast, GFR may be reduced in pathologic states in which the oncotic pressure is very high and exceeds hydraulic driving pressure. Case reports of AKI from dextran infusions (17) and massive albumin infusions (18) support this possibility.
do not appear to be an important factor in physiologic regulation of glomerular filtration. First, the plasma protein concentration is relatively stable except in disease states that alter either the production (e.g., in severe liver disease) or degradation or loss of plasma proteins (e.g., in nephrotic syndrome). In addition, changes in plasma protein concentration in mammals are associated with reciprocal and offsetting alterations in Kf, such that GFR is often unchanged. This is thought to be a result of variation in the hydraulic conductivity of the GBM in response to changes in serum albumin concentration (16), but the exact mechanism is unknown. By contrast, GFR may be reduced in pathologic states in which the oncotic pressure is very high and exceeds hydraulic driving pressure. Case reports of AKI from dextran infusions (17) and massive albumin infusions (18) support this possibility.
Glomerular Plasma Flow Rate, QA.
Because glomerular filtrate is essentially free of larger plasma proteins, conservation of mass requires that the total amount of protein entering the capillary is equal to the amount leaving it (Figure 3D), which is expressed mathematically in Equation 2:
 |
where QA and QE are the afferent and efferent arteriolar plasma flow rates and CA and CE are the respective afferent and efferent arteriolar blood plasma protein concentrations, respectively. Because QE=(QA–SNGFR), Equation 2 can be rewritten as follows (Equation 3):
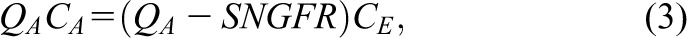 |
which can be rearranged as follows (Equation 4):
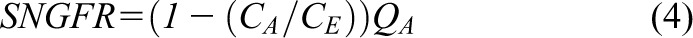 |
Although the relationship between the plasma protein concentration C and the oncotic pressure 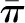 is not linear, Equation 4 is approximately equal to the following (Equation 5):
is not linear, Equation 4 is approximately equal to the following (Equation 5):
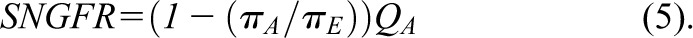 |
Because under conditions of filtration pressure equilibrium, 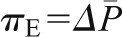 as follows (Equation 6):
as follows (Equation 6):
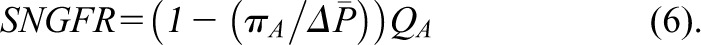 |
Therefore, if 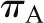 and
and  are held constant, SNGFR will vary directly with QA.
are held constant, SNGFR will vary directly with QA.
This relationship can also be understood graphically by examining Figure 2. As the plasma flow rate increases, there will be less contact time between the plasma flowing through the capillary and any given point along the capillary network. In that case, for any given pressure gradient at any given point, less fluid will leave the capillary as filtrate. As a result, the oncotic pressure profile will be shifted down and to the right, as if moving from curve A to curve B. The equilibrium point is shifted closer to the efferent end of the capillary, resulting in an increase in 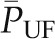 and SNGFR. On the other hand, once QA is sufficiently large, disequilibrium will occur, leading to less and less dependence of SNGFR on changes in QA. In fact, for animals or under conditions that are far from equilibrium, changes in QA are not predicted to have major effects on SNGFR.
and SNGFR. On the other hand, once QA is sufficiently large, disequilibrium will occur, leading to less and less dependence of SNGFR on changes in QA. In fact, for animals or under conditions that are far from equilibrium, changes in QA are not predicted to have major effects on SNGFR.
Afferent and Efferent Arteriolar Resistances, RA and RE
In most physiologic settings, alterations in  , Kf, and
, Kf, and 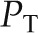 contribute little to the minute-to-minute regulation of SNGFR. Instead, the filtration rate is regulated by changes in
contribute little to the minute-to-minute regulation of SNGFR. Instead, the filtration rate is regulated by changes in 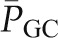 and QA, and they in turn are controlled by alterations in afferent and efferent arteriolar resistances (RA and RE, respectively). The glomerular capillaries are unique in that they are arranged in series between two resistance vessels, the afferent and efferent arterioles. Selective modulation of the relative resistances in these two vessels allows for precise and largely independent regulation of
and QA, and they in turn are controlled by alterations in afferent and efferent arteriolar resistances (RA and RE, respectively). The glomerular capillaries are unique in that they are arranged in series between two resistance vessels, the afferent and efferent arterioles. Selective modulation of the relative resistances in these two vessels allows for precise and largely independent regulation of 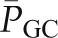 and
and 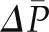 , QA, and SNGFR. In addition, a large number of hormones, vasoactive substances, growth factors, cytokines, and drugs alter resistance in these vessels, selectively or in concert (12).
, QA, and SNGFR. In addition, a large number of hormones, vasoactive substances, growth factors, cytokines, and drugs alter resistance in these vessels, selectively or in concert (12).
Renal Autoregulation
One important characteristic of the glomerular circulation is autoregulation, by which not only QA but also 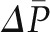 and SNGFR are held constant over a wide range of renal artery pressures (Figure 4). Robertson et al. examined the precise glomerular hemodynamic mechanisms that account for renal autoregulation (10) by applying micropuncture techniques to rats. Graded reductions in renal artery pressure were first associated with large declines in RA, followed by significant increases in RE. By contrast, an increase in renal plasma flow from vasodilatory substances was associated with a decline in RE and an increase in QA but was offset by a decrease in Kf. These combinations are predicted to be associated with near constancy of
and SNGFR are held constant over a wide range of renal artery pressures (Figure 4). Robertson et al. examined the precise glomerular hemodynamic mechanisms that account for renal autoregulation (10) by applying micropuncture techniques to rats. Graded reductions in renal artery pressure were first associated with large declines in RA, followed by significant increases in RE. By contrast, an increase in renal plasma flow from vasodilatory substances was associated with a decline in RE and an increase in QA but was offset by a decrease in Kf. These combinations are predicted to be associated with near constancy of 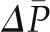 , QA, and SNGFR, which were indeed observed by these authors for renal artery pressures from approximately 80 to 120 mmHg.
, QA, and SNGFR, which were indeed observed by these authors for renal artery pressures from approximately 80 to 120 mmHg.
Figure 4.

The effects of graded reduction in renal artery perfusion pressure on glomerular hemodynamics in the rat. Glomerular blood flow (GBF) and glomerular capillary pressure 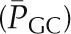 remained relatively constant over the range of pressure from 80 to 120 mmHg, in response to a marked decrease in afferent arteriolar resistance (RA). With further reduction in perfusion pressure to 60 mmHg, GBF declined proportionally more than
remained relatively constant over the range of pressure from 80 to 120 mmHg, in response to a marked decrease in afferent arteriolar resistance (RA). With further reduction in perfusion pressure to 60 mmHg, GBF declined proportionally more than 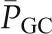 primarily because of an increase in RE. Reprinted from Taal MW, Chertow GM, Marsden PA, Skorecki K, Yu ASL, Brenner BM: Brenner and Rector’s The Kidney, 9th Ed., Philadelphia, Elsevier Saunders, 2012, with permission.
primarily because of an increase in RE. Reprinted from Taal MW, Chertow GM, Marsden PA, Skorecki K, Yu ASL, Brenner BM: Brenner and Rector’s The Kidney, 9th Ed., Philadelphia, Elsevier Saunders, 2012, with permission.
The mediators responsible for renal autoregulation include a myogenic response that is intrinsic to the blood vessels as well as tubuloglomerular feedback by which chloride uptake by the macula densa segment of the distal tubule is sensed and then elicits effector pathways that modulate RA and RE and maintain the SNGFR (3). In addition, changes in the filtration fraction will affect the oncotic pressure in peritubular capillaries and contribute to glomerular tubular balance (19).
Glomerular Filtration of Macromolecules
Classic studies on the separation of the urinary filtrate from the blood highlighted the restriction of serum proteins from the Bowman’s space. Detailed in vivo studies in both animals and humans have been able to characterize the sieving properties of the glomerular filter, which confers both size and charge selectivity. Studies using dextran molecules of varying sizes and charge demonstrated that neutral particles with a molecular radius >4.2 nm are restricted from the urinary space, whereas anion particles >3.4 nm have a fractional clearance that approaches zero. Thus, the charge of albumin, which is approximately 3.6 nm in size, explains the limited clearance observed (20). By contrast, in both experimental animals and humans with nephrotic syndrome, charge selectivity is lost with the alterations in normal glomerular architecture and this appears to contribute to the protein losses seen in these conditions (21).
Cellular and Molecular Components of the Glomerular Filter
Recent years have seen a rapid increase in knowledge about how the glomerulus and glomerular filtration are regulated at the cellular and molecular levels (Figure 5). Studies of diseases of the three basic components of the glomerular filter have provided a better understanding of the molecular physiology and pathophysiology of this structure. Much of this progress has come through studies of inherited disorders of glomerular function. The glomerular filter is both a charge and size barrier. Movement of anionic molecules across the filter is relatively restricted. Movement of large molecules across the filter is also restricted. The importance of all three components to normal charge and size selectivity is clear from the fact that disruption of any of these three components leads to proteinuric disease states (22).
Figure 5.

Major molecular components of the podocyte and slit diaphragm. Interdigitating podocytes from neighboring cells form the elaborate slit diaphragm that is composed of nephrin. Podocin helps regulate trafficking of nephrin to the slit diaphragm. Proteins α-actinin-4 and INF2 play important roles in the maintenance of the actin cytoskeleton, whereas integrins help anchor the podocytes to the glomerular basement membrane. Mutations in α-actinin-4, INF2, and TRPC6 channel all cause autosomal dominant forms of FSGS. Mutations in nephrin and podocin cause recessive forms of steroid-resistant nephrosis.
Disorders of Slit Diaphragm and Podocyte Structure and Function
The glomerular epithelial cell, or podocyte, has been the focus of a great deal of recent investigation. These cells, which are derived from metanephric mesenchyme, form the distal component of the glomerular filter and are both morphologically and functionally unique. Long intricate extensions, or primary processes, lead to secondary or foot processes, which form a complex interdigitating structure with the foot processes from adjacent podocytes. The serpentine cell–cell junction known as the slit diaphragm that bridges these podocytes can be considered a modified adherens junction with some unique additional molecular components (23).
Alterations in the structure and/or function of the visceral glomerular epithelial cell, or podocyte, lead to a spectrum of human disease. The best established examples are inherited disorders caused by changes in single genes. These inherited “podocytopathies” display a range of phenotypes. As a general rule, recessive forms of podocyte dysfunction are caused by mutations resulting in loss of the function of a normal protein.
The proteins nephrin and podocin are essentially podocyte limited in their expression. Humans, as well as mice, that lack normal copies of either protein develop severe glomerular disease. Both of these molecules were unknown until they were identified on the basis of human genetic studies. Nephrin, a transmembrane domain protein with a large extracellular domain, is required for both normal slit diaphragm structure and function. In Finland, two specific founder mutations in the NPHS1 nephrin gene make this form of congenital nephrotic syndrome much more common than in other parts of the world (24,25). In general, nephrin-mediated congenital nephrotic syndrome manifests as massive proteinuria (≥20 g) in the neonatal period. Defects in the NPHS2 podocin gene cause a wider spectrum of disease, ranging from congenital nephrosis to adult-onset FSGS (25,26). Podocin, like nephrin, is an integral membrane protein. Podocin interacts directly with nephrin and appears to help regulate its trafficking to the slit diaphragm (27).
The function of the podocyte in vivo, and consequently the function of the entire glomerulus, is very sensitive to mild perturbations in its actin cytoskeleton (28). Mutations in the widely expressed actin-regulating proteins α-actinin-4 and INF2 lead to human forms of podocyte dysfunction, FSGS, and progressive kidney disease, despite having little or no effect on other organ function (29,30). Mutations in the TRPC6 cation channel cause a phenotypically similar autosomal dominant form of FSGS (31).
A good deal of recent work has focused on signaling pathways critical to podocyte function. For example, fine regulation of small GTPase signaling appears essential for maintenance and repair of normal podocyte structure and function (32,33). Autophagy, a catabolic process of cell “self-eating,” whereby cells degrade unnecessary or dysfunctional intracellular components, is necessary for normal podocyte function. Transgenic mice deficient in autophagy show marked glomerular pathology (34). The communication between slit diaphragm proteins and the cytoskeleton is similarly of critical importance. Proper functioning of the cytoskeletal apparatus is required for organization of the slit diaphragm complex, and normal slit diaphragm function (as both a structural apparatus and signaling center) regulates actin dynamics (34,35).
The functions of some glomerular disease genes and gene products remain unclear. Variants in the APOL1 gene, encoding apoL1, explain the high rate of FSGS and other forms of nondiabetic kidney disease in African Americans. Its function in normal human physiology, if any, is unclear. APOL1 does not exist in rats, mice, and other nonprimates, and is present in only a few primate species.
The GBM
The importance of the GBM to normal glomerular physiology is made evident by the existence of several human diseases characterized by abnormalities in the GBM structure. The GBM is formed by the fusion of basement membranes from both the endothelial and epithelial cells that form the glomerular corpuscle. Morphologically, the GBM contains a dense inner layer called the lamina densa, flanked by the thinner laminae rara interna and rare externa. This morphology, as well as GBM function, is altered in the presence of mutations in type IV collagens, the major components of the GBM. Recessive forms of Alport syndrome can be caused by inheritance of mutations in the autosomal type 4 collagen genes COL3A4 and COL4A4, whereas the more common X-linked form is caused by mutations in COL5A4 (36). Infants born with two mutant copies of the gene encoding the GBM protein laminin β2 can exhibit congenital nephrotic syndrome (37).
Studies in several mouse models of altered GBM components (type IV collagen, laminin) show that the altered glomerular filters in these models can show increased permeability to large molecules (38). These alterations are seen to occur before ultrastructural abnormalities in the podocyte (39). Thus, altered GBM composition itself can lead to proteinuria. Recent studies have also readdressed the issue of whether glycosaminoglycans in the endothelium help regulate glomerular permeability, confirming the role of these large molecules in charge and size selectivity (40,41).
Despite extensive study, there are still open questions regarding the complex nature of this filter as well as how it allows such high rates of water flow while restricting the flow of large molecules and yet remains functional and “unclogged.” One recent hypothesis suggests that the glomerular filter, and the GBM in particular, acts more like a gel than a simple filter in that the size-selective properties of the glomerular filter are determined by permeation and diffusion properties of the GBM (42).
Endothelium
The glomerular endothelium represents the first layer in the glomerular filtration barrier and is in direct contact with the blood. Although the origin of these cells has been debated for some time, recent lineage tracing studies demonstrated that they may derive from the Foxd1-positive stromal cell lineage (43). Endothelial cells migrate from the metanephric mesenchyme into the developing glomerular tuft at the S-shape stage of glomerulogenesis. Glomerular endothelial cells are attracted by vascular growth factors, such as vascular endothelial growth factor (VEGF)-A, that are produced by adjacent podocytes (44–46). As glomerular development proceeds, the endothelial cells become progressively flattened and develop fenestrations (two features critical for maintenance of glomerular function), permitting high flux of water and small solutes. The endothelium is covered by a glycocalyx that likely restricts the passage of large macromolecules. Alterations of the glycocalyx were reported in various models of glomerular disease, including diabetic nephropathy (41). Studies in humans have been limited by incomplete knowledge of glycocalyx components and functions and by the need for specialized fixation and staining methods for proper visualization.
Diseases of the Glomerular Endothelium
Primary diseases of the glomerular endothelium may result in rapid loss of kidney function. Thrombotic microangiopathies (TMAs) represent a heterogeneous group of disorders characterized by varying degrees of endothelial swelling known as endotheliosis, fibrin and platelet deposition, splitting of the GBM, and red blood cell fragmentation (47,48). The molecular causes of a number of TMAs were recently elucidated. Although thrombotic thrombocytopenic purpura and hemolytic uremic syndrome (HUS) were once considered to represent a spectrum of the same disease (49), deficiency of ADAMTS13 enzymatic function as a result of congenital mutations or antibody-mediated inhibition is now known to be responsible for thrombotic thrombocytopenic purpura (50). By contrast, HUSs are associated with dysregulation of the alternate pathway of complement. Multiple mutations in genes that encode proteins required for the regulation of complement activation cause atypical HUS (48). Activation of complement leads to perturbation of otherwise thromboresistant renal endothelial cells with resultant local damage and an influx of inflammatory cells. Although the most common form of HUS—diarrheal HUS, caused by Shiga toxin—has not been clearly linked to complement dysregulation, several small series have demonstrated benefit from complement inhibition in patients during a recent large outbreak of diarrheal HUS in Germany (51,52). These data raise the intriguing possibility that complement dysregulation is responsible for renal injury in common forms of HUS as well. Thrombotic injury to the glomerular microvasculature is also a universal feature of the renal disease of preeclampsia and in subsets of patients treated with anti-VEGF agents. Importantly, podocytes produce large amounts of VEGF during development and in filtering adult glomeruli in order to nurture the endothelial cells despite the tide of glomerular filtrate. This paracrine factor appears to be important for maintenance of the normal endothelial cell structure on the basis of observations that inhibition of VEGF signaling within the glomerular endothelium may result in thrombotic injury. Human, mouse, and cell-based studies support this model (53,54). Finally, mutations in the D7E lipid enzyme were identified in families presenting with atypical HUS and/or FSGS (55,56). Endothelial and podocyte injury were observed in biopsies from these patients, suggesting that phosphorylation of lipid substrates is crucial for glomerular health.
In addition to TMAs, activation of the glomerular endothelium can result in pathologic angiogenesis, enhanced permeability and proteinuria, and upregulation of cell adhesion molecules involved in recruitment of inflammatory cells. These processes are documented in a large number of common glomerular diseases, including diabetic nephropathy and vasculitides. Future studies are needed to determine the molecular basis of endothelial dysfunction, which should provide interesting new therapeutic targets for glomerular disease.
Mesangium
The mesangium refers to the mesangial cells and the matrix they produce. During glomerular development, mesangial cells migrate into the developing tuft under the influence of PDGFβ produced by the glomerular endothelial cells acting on the PDGFβ receptor expressed by mesangial cells (57). Traditionally, mesangial cells were referred to as pericytes because they provide support to the adjacent capillary loops. However, podocytes also provide essential growth factors and support to the endothelium and they also function as specialized perivascular cells, making the glomerular microvasculature somewhat unique because it has intimate associations with two different supporting cell types. The mesangial processes are filled with bundles of actin and myosin-based microfilaments that extend to contact the GBM, in which they bind laminin α5 via integrin α3β1 and the basal cell adhesion molecule (58). These processes provide protection against glomerular pressure and may regulate glomerular capillary flow via contractile properties (59).
The mesangial matrix produced by mesangial cells is composed of a diverse group of proteins, including type III–VI collagens, heparin sulfate proteoglycans, and elastic fiber proteins including fibronectin, laminin, entactin, and fibrillin-1. Accumulation of the mesangial matrix and thickening of the GBM are features commonly observed in a number of glomerular diseases, including diabetic nephropathy. Under these settings, the matrix may include normal as well as new components such as collagen I. Proliferative glomerulonephritides may include mesangial cell proliferation and mesangial deposits.
Newer Methodologies
Much of the “classical” physiology of the mammalian glomerulus has been determined through studies in rats, particularly the extensive studies that have defined the control of filtration at the single nephron level. New experimental tools are expanding the ability to examine glomerular processes. These tools include new animal models, more sophisticated cellular models, and better methods to visualize glomerular function in existing rodent models. For example, multiphoton fluorescence microscopy enables examination of glomerular function in vivo, and is now being applied in analyses of models of disease (60). Such studies have confirmed that only a small amount of large proteins is filtered by the glomerulus.
Genetic manipulation of other model organisms, including mice and zebrafish, permits in vivo assessment of gene function. Using morpholino technology, it is possible to rapidly generate “gene knockdowns” in fish and analyze effects on the pronephros, which contains a single glomerulus. More precise modulation of the genome is possible with the multitude of cell and temporally controlled gene knockout strategies available in mice, and development of newer technologies, such as phenotype-driven N-ethyl-N-nitrosourea (ENU) screens, Clustered Regularly Interspaced Short Palindromic Repeats technology, and short hairpin RNA (shRNA) knockdowns. ENU screening is a technique in which the ENU alkylating is used to induce mutations and then the phenotype of the progeny can be studied and candidate genes from phenotypes of interest can be identified. Clustered Regularly Interspaced Short Palindromic Repeats technology can be used for gene “editing” to assist in the study of specific genetic targets. Finally, shRNA knockdown is a technique that utilizes shRNA sequences to silence a target gene of interest. Combining cell-based studies, model systems, and advances in “omics” will translate into tremendous growth in our knowledge about the physiology and regulation of the glomerulus.
Disclosures
None.
Footnotes
Published online ahead of print. Publication date available at www.cjasn.org.
References
- 1.Hinchliffe SA, Sargent PH, Howard CV, Chan YF, van Velzen D: Human intrauterine renal growth expressed in absolute number of glomeruli assessed by the disector method and Cavalieri principle. Lab Invest 64: 777–784, 1991 [PubMed] [Google Scholar]
- 2.Horster M, Thurau K: Micropuncture studies on the filtration rate of single superficial and juxtamedullary glomeruli in the rat kidney. Pflugers Arch Gesamte Physiol Menschen Tiere 301: 162–181, 1968 [DOI] [PubMed] [Google Scholar]
- 3.Gong R, Dworkin LD, Brenner BM, Maddox DA: The renal circulations and glomerular ultrafiltration. In: Brenner & Rector’s The Kidney, edited by Brenner BM, Philadelphia, Elsevier Saunders, 2008, pp 91–129 [Google Scholar]
- 4.Stein JH, Boonjarern S, Wilson CB, Ferris TF: Alterations in intrarenal blood flow distribution. Methods of measurement and relationship to sodium balance. Circ Res 32[Suppl 1]: 61–72, 1973 [PubMed] [Google Scholar]
- 5.Blantz RC, Wallin JD, Rector FC, Jr, Seldin DW: Effect of variation in dietary NaCl intake on the intrarenal distribution of plasma flow in the rat. J Clin Invest 51: 2790–2795, 1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Deen WM, Robertson CR, Brenner BM: Concentration polarization in an ultrafiltering capillary. Biophys J 14: 412–431, 1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brenner BM, Troy JL, Daugharty TM: The dynamics of glomerular ultrafiltration in the rat. J Clin Invest 50: 1776–1780, 1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maddox DA, Deen WM, Brenner BM: Glomerular Filtration. In: Handbook of Physiology Renal Physiology, edited by Windhager EE, Giebisch G, Baltimore, Williams and Wilkins, 1992 [Google Scholar]
- 9.Maddox DA, Deen WM, Brenner BM: Dynamics of glomerular ultrafiltration. VI. Studies in the primate. Kidney Int 5: 271–278, 1974 [DOI] [PubMed] [Google Scholar]
- 10.Deen WM, Robertson CR, Brenner BM: A model of glomerular ultrafiltration in the rat. Am J Physiol 223: 1178–1183, 1972 [DOI] [PubMed] [Google Scholar]
- 11.Squarer A, Lemley KV, Ambalavanan S, Kristal B, Deen WM, Sibley R, Anderson L, Myers BD: Mechanisms of progressive glomerular injury in membranous nephropathy. J Am Soc Nephrol 9: 1389–1398, 1998 [DOI] [PubMed] [Google Scholar]
- 12.Dworkin LD, Ichikawa I, Brenner BM: Hormonal modulation of glomerular function. Am J Physiol 244: F95–F104, 1983 [DOI] [PubMed] [Google Scholar]
- 13.Hladunewich MA, Lemley KV, Blouch KL, Myers BD: Determinants of GFR depression in early membranous nephropathy. Am J Physiol Renal Physiol 284: F1014–F1022, 2003 [DOI] [PubMed] [Google Scholar]
- 14.Blantz RC, Wilson CB: Acute effects of antiglomerular basement membrane antibody on the process of glomerular filtration in the rat. J Clin Invest 58: 899–911, 1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tomlanovich S, Deen WM, Jones HW, 3rd, Schwartz HC, Myers BD: Functional nature of glomerular injury in progressive diabetic glomerulopathy. Diabetes 36: 556–565, 1987 [DOI] [PubMed] [Google Scholar]
- 16.Daniels BS, Hauser EB, Deen WM, Hostetter TH: Glomerular basement membrane: In vitro studies of water and protein permeability. Am J Physiol 262: F919–F926, 1992 [DOI] [PubMed] [Google Scholar]
- 17.Moran M, Kapsner C: Acute renal failure associated with elevated plasma oncotic pressure. N Engl J Med 317: 150–153, 1987 [DOI] [PubMed] [Google Scholar]
- 18.Rozich JD, Paul RV: Acute renal failure precipitated by elevated colloid osmotic pressure. Am J Med 87: 359–360, 1989 [DOI] [PubMed] [Google Scholar]
- 19.Thomson SC, Blantz RC: Glomerulotubular balance, tubuloglomerular feedback, and salt homeostasis. J Am Soc Nephrol 19: 2272–2275, 2008 [DOI] [PubMed] [Google Scholar]
- 20.Brenner BM, Hostetter TH, Humes HD: Molecular basis of proteinuria of glomerular origin. N Engl J Med 298: 826–833, 1978 [DOI] [PubMed] [Google Scholar]
- 21.Guasch A, Deen WM, Myers BD: Charge selectivity of the glomerular filtration barrier in healthy and nephrotic humans. J Clin Invest 92: 2274–2282, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haraldsson B, Nyström J, Deen WM: Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev 88: 451–487, 2008 [DOI] [PubMed] [Google Scholar]
- 23.Reiser J, Kriz W, Kretzler M, Mundel P: The glomerular slit diaphragm is a modified adherens junction. J Am Soc Nephrol 11: 1–8, 2000 [DOI] [PubMed] [Google Scholar]
- 24.Kestilä M, Lenkkeri U, Männikkö M, Lamerdin J, McCready P, Putaala H, Ruotsalainen V, Morita T, Nissinen M, Herva R, Kashtan CE, Peltonen L, Holmberg C, Olsen A, Tryggvason K: Positionally cloned gene for a novel glomerular protein—nephrin—is mutated in congenital nephrotic syndrome. Mol Cell 1: 575–582, 1998 [DOI] [PubMed] [Google Scholar]
- 25.Tryggvason K, Patrakka J, Wartiovaara J: Hereditary proteinuria syndromes and mechanisms of proteinuria. N Engl J Med 354: 1387–1401, 2006 [DOI] [PubMed] [Google Scholar]
- 26.Boute N, Gribouval O, Roselli S, Benessy F, Lee H, Fuchshuber A, Dahan K, Gubler MC, Niaudet P, Antignac C: NPHS2, encoding the glomerular protein podocin, is mutated in autosomal recessive steroid-resistant nephrotic syndrome. Nat Genet 24: 349–354, 2000 [DOI] [PubMed] [Google Scholar]
- 27.Nishibori Y, Liu L, Hosoyamada M, Endou H, Kudo A, Takenaka H, Higashihara E, Bessho F, Takahashi S, Kershaw D, Ruotsalainen V, Tryggvason K, Khoshnoodi J, Yan K: Disease-causing missense mutations in NPHS2 gene alter normal nephrin trafficking to the plasma membrane. Kidney Int 66: 1755–1765, 2004 [DOI] [PubMed] [Google Scholar]
- 28.Faul C, Asanuma K, Yanagida-Asanuma E, Kim K, Mundel P: Actin up: Regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol 17: 428–437, 2007 [DOI] [PubMed] [Google Scholar]
- 29.Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, Mathis BJ, Rodríguez-Pérez JC, Allen PG, Beggs AH, Pollak MR: Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet 24: 251–256, 2000 [DOI] [PubMed] [Google Scholar]
- 30.Brown EJ, Schlondorff JS, Becker DJ, Tsukaguchi H, Tonna SJ, Uscinski AL, Higgs HN, Henderson JM, Pollak MR: Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat Genet 42: 72–76, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA, Howell DN, Vance JM, Rosenberg PB: A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 308: 1801–1804, 2005 [DOI] [PubMed] [Google Scholar]
- 32.Greka A, Mundel P: Cell biology and pathology of podocytes. Annu Rev Physiol 74: 299–323, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mouawad F, Tsui H, Takano T: Role of Rho-GTPases and their regulatory proteins in glomerular podocyte function. Can J Physiol Pharmacol 91: 773–782, 2013 [DOI] [PubMed] [Google Scholar]
- 34.Huber TB, Benzing T: The slit diaphragm: A signaling platform to regulate podocyte function. Curr Opin Nephrol Hypertens 14: 211–216, 2005 [DOI] [PubMed] [Google Scholar]
- 35.George B, Holzman LB: Signaling from the podocyte intercellular junction to the actin cytoskeleton. Semin Nephrol 32: 307–318, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hudson BG, Tryggvason K, Sundaramoorthy M, Neilson EG: Alport’s syndrome, Goodpasture’s syndrome, and type IV collagen. N Engl J Med 348: 2543–2556, 2003 [DOI] [PubMed] [Google Scholar]
- 37.Zenker M, Aigner T, Wendler O, Tralau T, Müntefering H, Fenski R, Pitz S, Schumacher V, Royer-Pokora B, Wühl E, Cochat P, Bouvier R, Kraus C, Mark K, Madlon H, Dötsch J, Rascher W, Maruniak-Chudek I, Lennert T, Neumann LM, Reis A: Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum Mol Genet 13: 2625–2632, 2004 [DOI] [PubMed] [Google Scholar]
- 38.Suh JH, Miner JH: The glomerular basement membrane as a barrier to albumin. Nat Rev Nephrol 9: 470–477, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jarad G, Cunningham J, Shaw AS, Miner JH: Proteinuria precedes podocyte abnormalities inLamb2-/- mice, implicating the glomerular basement membrane as an albumin barrier. J Clin Invest 116: 2272–2279, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fridén V, Oveland E, Tenstad O, Ebefors K, Nyström J, Nilsson UA, Haraldsson B: The glomerular endothelial cell coat is essential for glomerular filtration. Kidney Int 79: 1322–1330, 2011 [DOI] [PubMed] [Google Scholar]
- 41.Jeansson M, Haraldsson B: Morphological and functional evidence for an important role of the endothelial cell glycocalyx in the glomerular barrier. Am J Physiol Renal Physiol 290: F111–F116, 2006 [DOI] [PubMed] [Google Scholar]
- 42.Smithies O: Why the kidney glomerulus does not clog: A gel permeation/diffusion hypothesis of renal function. Proc Natl Acad Sci U S A 100: 4108–4113, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sims-Lucas S, Schaefer C, Bushnell D, Ho J, Logar A, Prochownik E, Gittes G, Bates CM: Endothelial progenitors exist within the kidney and lung mesenchyme. PLoS ONE 8: e65993, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eremina V, Sood M, Haigh J, Nagy A, Lajoie G, Ferrara N, Gerber HP, Kikkawa Y, Miner JH, Quaggin SE: Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest 111: 707–716, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eremina V, Cui S, Gerber H, Ferrara N, Haigh J, Nagy A, Ema M, Rossant J, Jothy S, Miner JH, Quaggin SE: Vascular endothelial growth factor a signaling in the podocyte-endothelial compartment is required for mesangial cell migration and survival. J Am Soc Nephrol 17: 724–735, 2006 [DOI] [PubMed] [Google Scholar]
- 46.Eremina V, Baelde HJ, Quaggin SE: Role of the VEGF—a signaling pathway in the glomerulus: Evidence for crosstalk between components of the glomerular filtration barrier. Nephron, Physiol 106: 32–37, 2007 [DOI] [PubMed] [Google Scholar]
- 47.Coppo P, Veyradier A: Thrombotic microangiopathies: Towards a pathophysiology-based classification. Cardiovasc Hematol Disord Drug Targets 9: 36–50, 2009 [DOI] [PubMed] [Google Scholar]
- 48.Noris M, Remuzzi G: Atypical hemolytic-uremic syndrome. N Engl J Med 361: 1676–1687, 2009 [DOI] [PubMed] [Google Scholar]
- 49.Remuzzi G: HUS and TTP: Variable expression of a single entity. Kidney Int 32: 292–308, 1987 [DOI] [PubMed] [Google Scholar]
- 50.Levy GG, Nichols WC, Lian EC, Foroud T, McClintick JN, McGee BM, Yang AY, Siemieniak DR, Stark KR, Gruppo R, Sarode R, Shurin SB, Chandrasekaran V, Stabler SP, Sabio H, Bouhassira EE, Upshaw JD, Jr, Ginsburg D, Tsai HM: Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature 413: 488–494, 2001 [DOI] [PubMed] [Google Scholar]
- 51.Lapeyraque AL, Malina M, Fremeaux-Bacchi V, Boppel T, Kirschfink M, Oualha M, Proulx F, Clermont MJ, Le Deist F, Niaudet P, Schaefer F: Eculizumab in severe Shiga-toxin-associated HUS. N Engl J Med 364: 2561–2563, 2011 [DOI] [PubMed] [Google Scholar]
- 52.Delmas Y, Vendrely B, Clouzeau B, Bachir H, Bui HN, Lacraz A, Helou S, Bordes C, Reffet A, Llanas B, Skopinski S, Rolland P, Gruson D, Combe C: Outbreak of Escherichia coli O104:H4 haemolytic uraemic syndrome in France: Outcome with eculizumab [published online ahead of print November 28, 2013]. Nephrol Dial Transplant 10.1093/ndt/gft470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, Libermann TA, Morgan JP, Sellke FW, Stillman IE, Epstein FH, Sukhatme VP, Karumanchi SA: Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 111: 649–658, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eremina V, Jefferson JA, Kowalewska J, Hochster H, Haas M, Weisstuch J, Richardson C, Kopp JB, Kabir MG, Backx PH, Gerber HP, Ferrara N, Barisoni L, Alpers CE, Quaggin SE: VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med 358: 1129–1136, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lemaire M, Frémeaux-Bacchi V, Schaefer F, Choi M, Tang WH, Le Quintrec M, Fakhouri F, Taque S, Nobili F, Martinez F, Ji W, Overton JD, Mane SM, Nürnberg G, Altmüller J, Thiele H, Morin D, Deschenes G, Baudouin V, Llanas B, Collard L, Majid MA, Simkova E, Nürnberg P, Rioux-Leclerc N, Moeckel GW, Gubler MC, Hwa J, Loirat C, Lifton RP: Recessive mutations in DGKE cause atypical hemolytic-uremic syndrome. Nat Genet 45: 531–536, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ozaltin F, Li B, Rauhauser A, An SW, Soylemezoglu O, Gonul II, Taskiran EZ, Ibsirlioglu T, Korkmaz E, Bilginer Y, Duzova A, Ozen S, Topaloglu R, Besbas N, Ashraf S, Du Y, Liang C, Chen P, Lu D, Vadnagara K, Arbuckle S, Lewis D, Wakeland B, Quigg RJ, Ransom RF, Wakeland EK, Topham MK, Bazan NG, Mohan C, Hildebrandt F, Bakkaloglu A, Huang CL, Attanasio M: DGKE variants cause a glomerular microangiopathy that mimics membranoproliferative GN. J Am Soc Nephrol 24: 377–384, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bjarnegård M, Enge M, Norlin J, Gustafsdottir S, Fredriksson S, Abramsson A, Takemoto M, Gustafsson E, Fässler R, Betsholtz C: Endothelium-specific ablation of PDGFB leads to pericyte loss and glomerular, cardiac and placental abnormalities. Development 131: 1847–1857, 2004 [DOI] [PubMed] [Google Scholar]
- 58.Kerjaschki D, Ojha PP, Susani M, Horvat R, Binder S, Hovorka A, Hillemanns P, Pytela R: A beta 1-integrin receptor for fibronectin in human kidney glomeruli. Am J Pathol 134: 481–489, 1989 [PMC free article] [PubMed] [Google Scholar]
- 59.Ghayur MN, Krepinsky JC, Janssen LJ: Contractility of the renal glomerulus and mesangial cells: Lingering doubts and strategies for the future. Med Hypotheses Res 4: 1–9, 2008 [PMC free article] [PubMed] [Google Scholar]
- 60.Peti-Peterdi J, Sipos A: A high-powered view of the filtration barrier. J Am Soc Nephrol 21: 1835–1841, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]