

Abstract
Developing countries have a low incidence of inflammatory bowel disease (IBD), perhaps prevented by the high prevalence of helminth infections and other alterations in intestinal flora and fauna. Helminth infections prevent colitis in various murine models of IBD. IBD may be driven by an aberrant immune response to luminal antigen(s). We therefore developed a murine model of IBD in which gut injury was induced by a specific antigen to better simulate the IBD disease process and to determine if helminth infections could abolish gut injury induced by an orally administered antigen. The model features pan-enterocolitis triggered by feeding ovalbumin. The intestinal inflammation is antigen-specific and generates IL-17 and IFN-γ, but not IL-4. Full expression of the disease required T cells with defective capacity to make IL-10 and treatment with a non-injurious, low dose of a non-steroidal anti-inflammatory drug. Exposure to H. polygyrus abrogated this antigen-induced gut injury. H. polygyrus colonization induced Foxp3+ Tregs and mucosal production of IL-10 from non-T cells. Lamina propria mononuclear cells from H. polygyrus infected mice released less IL-17 and IFN-γ constitutively and when stimulated with OVA or anti-CD3/CD28 mAbs. In conclusion, we developed a murine IBD model featuring antigen-specific enterocolitis and demonstrated for the first time that gut inflammation induced by an antigen could be abrogated by H. polygyrus infection. Protection was associated with suppressed IL-17 and IFN-γ production, induction of Foxp3+ Tregs and elevated secretion of non-T cells derived IL-10, all of which could be part of the protective processes.
Introduction
Helminthic colonization has been proposed as a new type of immunotherapy for the treatment of immunological disorders like inflammatory bowel disease (IBD). Protective effects of helminths have been demonstrated in various animal models of IBD 1,2 and in placebo-controlled human clinical trials.3,4 IBD are chronic relapsing immune-mediated disorders that presumably result from an aberrant response to gut luminal antigens (Ags) in genetically susceptible patients.
It is difficult to study the specific immune responses to intraluminal Ags in the gut because a multitude of poorly defined Ags shape mucosal immunity. To overcome this limitation, we recently modified the Rag IL10-/- T cell transfer model of IBD by also injecting OT-II transgenic T cells at the same time mice are reconstituted with IL10-/- T cells. The OT-II T cells recognize ovalbumin (OVA) in a class II-dependent fashion. In this model (IL-10KO/OT-II IBD model),5 the injected OT-II cells populate the gut, and isolated laminar propria mononuclear cells (LPMCs) stimulated in vitro with OVA produce large amounts of IFN-γ and IL-17 in an antigen-specific and dose-dependent manner. This antigenic response to in vitro OVA stimulation is abolished by colonizing the animals with H. polygyrus. While this model was of interest, the pathology was not driven by either the OT-II cells or OVA.
For this study, this IBD model was modified further such that the gut inflammation was dependent on intestinal OT-II T cells and luminal exposure to OVA. This allowed us to test the hypothesis that H. polygyrus exposure can abrogate antigen-induced gut injury. This study now shows that exposure to H. polygyrus suppresses gut inflammation triggered by an antigen and that the abrogation of this gut inflammation is associated with suppressed intestinal IL-17 and IFN-γ production, induction of Foxp3+ Tregs and increased gut IL-10 secretion from non-T cell sources.
Materials and Methods
Animals
This study used wide-type (WT), IL10-/-, OT-II (Thy1.2+), OT-II (Thy 1.1+) and Rag mice all on C57BL/6 background. OT-II (Thy1.1+) mice with TCR recognizing OVA peptide 323–339 were provided by A. Luster (Massachusetts General Hospital, Charlestown, MA). All mice were housed under pathogen-free conditions. Animals were housed and handled appropriately following National Institutes of Health guidelines and as approved by our Animal Review Committee.
IL-10KO/OT-II IBD model
Five-week-old Rag mice were reconstituted with 4 × 106 IL-10−/− and 2 × 106 OT-II (Thy1.1+ or Thy1.2+) splenic T cells via i.p. injection. A week later, 20 mg of piroxicam (Sigma-Aldrich)/250 g feed (NIH-31M) were administered for 2 wk. Piroxicam was stopped, and mice then received 25g OVA (Sigma-Aldrich)/250g feed for 1 wk before sacrifice. For some experiments, mice did not receive OT-II T cells, OVA or piroxicam. In other experiments, they were reconstituted with WT instead of IL-10-/- T cells.
H. polygyrus infection
In some experiments, mice were colonized with H. polygyrus, beginning 6 days after T cell transfer (figure 6A). Colonization was initiated with 150 third stage larvae (L3) of H. polygyrus given by gastric gavage. Infective, ensheathed H. polygyrus L3 (U.S. National Helminthological Collection no. 81930) were obtained (from fecal cultures of eggs) by the modified Baermann method and stored at 4°C until used.
Figure 6.

Hp protect mice from severe pan-enterocolitis upon oral OVA challenge in our non-IgE food hypersensitivity mode. A, Experimental scheme: Rag recipient of IL10-/- and OT-II T cells were infected with Hp before feeding low dose of piroxicam for 2 wks followed by OVA for 1 wk. B, OVA-fed mice pre-colonized with Hp had markedly less gut inflammation tan non-infected OVA-fed animals, with inflammatory scores similar to uninfected animals that were not fed OVA. C, Representative photomicrographs od duodenum, ileum and colon in Hp-infected and non-infected mice were shown in the left panel. Hp-infected mice had pronounced villi shortening, elongation of the crypts, and frequent crypt abscesses in the duodenum and ileum. There was deep lymphocyctic infiltraction of the colonic wall with ulceration, muscle thickening, epithelial hypertrophy. Non-infected mice had much milder inflammation. Magnification, ×40; H&E staining. Data are mean ± SE from 3 separate experiments (n=4-5 per group). * p<0.05.
Cell isolation and T cell enrichment
Single-cell suspensions of splenocytes were prepared by gentle teasing in RPMI 1640 medium (Life Technologies, Grand Island, NY). The cells were washed three times in RPMI 1640. Splenic T cells were isolated by negative selection using the EasySep mouse T cell enrichment kit, as outlined by the manufacturer (19751; StemCell Technologies, Vancouver, Canada). Viability was determined using exclusion of trypan blue dye.
Gut lamina propria mononuclear cells (LPMC) were isolated as described below. Intestinal tissue (terminal ileum or colon) was washed extensively with RPMI, and all visible Peyer's patches were removed with a scissors. The intestine was opened longitudinally, cut into 5-mm pieces, and then incubated in 0.5 mM EDTA in calcium- and magnesium-free HBSS for 20 min at 37°C with shaking to release intraepithelial lymphocytes and epithelial cells. This was repeated after thorough washing. Tissue then was incubated 20 min at 37°C in 20 ml RPMI containing 10% FCS, 25 mM HEPES buffer, 2 mM L-glutamine, 100 U/ml penicillin, 5 mg/ml gentamicin, and 100 mg/ml streptomycin (all GIBCO), and 1 mg/ml collagenase (catalog no. CO130; Sigma). At the end of the incubation, the tissue was subjected to further mechanical disruption with a 1-ml syringe. To remove debris, the LPMC preparations were washed through dampened gauze layered in a funnel with RPMI. Then LPMC were sieved through a prewetted 2-cm nylon wool column gently packed into a 10-ml syringe. After being washed, cells (up to 2 × 107) were layered onto a column of Percoll with a 30:70% gradient. Cells were spun at 2,200 g at room temperature for 20 min. The LPMC collected from the 30:70 interface were washed and maintained on ice until being used. Cell viability was 90% as determined by eosin Y exclusion For some experiments, LPMC were fractionated into T cell-enriched and T cell-depleted fractions using paramagnetic beads (DYNAL, Invitrogen Corp, Carlsbad, CA) as suggested by the manufacturer.
Cell culture
LPMC were cultured 48 h in 96-well microtiter plates (Corning Glass) with 200 μl of medium (5 × 105 cells/well) at 37°C. The culture medium was RPMI 1640 containing 10% FCS, 25 mM HEPES buffer, 2 mM L-glutamine, 5 × 10−5 M 2-ME, 1 mM sodium pyruvate, 100 U/ml penicillin, 5 mg/ml gentamicin, and 100 mg/ml streptomycin (all Life Technologies). LPMC were cultured alone, with OVA at 50 or 100 μg/ml (Sigma), or with anti-CD3 (2C11; 1 μg/ml; American Type Culture Collection) and anti-CD28 mAb (PV1; 1 μg/ml; American Type Culture Collection).
Cytokines ELISAs
ELISA was used to measure the concentrations of various cytokines in the supernatants. To measure IFN-γ, plates were coated with a mAb to IFN-γ (HB170, ATCC) and incubated with supernatant. IFN-γ was detected with polyclonal rabbit anti-IFN-γ (gift from Dr. Mary Wilson, University of Iowa) followed by biotinylated goat anti-rabbit IgG (Accurate Chemical Co., Westbury, NY). Color development used streptavidin-horseradish peroxidase (Zymed San Francisco, CA) and TMB substrate (Endogen, Woburn, MA), and plates were read at 450 nm. IL-4 was captured with 11B11 (HB191, DNAX Research Institute, Palo Alto, CA) and detected with biotinylated BVD6 (provided by Kevin Moore and John Abrams, DNAX). IL-17 was captured and detected with MAB721 and biotinylated BAF421 respectively (both R&D Systems). ELISAs were sensitive to 30 pg/ml.
Inflammatory scores
To grade intestinal inflammation, duodenum, ileum and colons were removed at the indicated time point, rolled, fixed, and embedded in paraffin. The inflammation of small bowel and colon was scored from 0 to 4. Inflammation of the small bowel were scored using the following criteria: grade 0, no change from normal tissue; grade 1, patchy mononuclear cell infiltrates in the lamina propria and normal villus; grade 2, more uniform mononuclear cell inflammation involving both the epithelium and lamina propria, minimal reduction of cryst/villus ratio (2-3); grade 3, diffuse lymphocytic infiltrate, <3 crypt abscesses /100× HPF and moderately reduced C/V ratio (1.0-1.9) and grade 4, villus blunting with C/V < 0.9, ≥3 crypt abscess/100× H. POLYGYRUSF and severe transmural lymphocytic infiltrate. Colitis was graded using scoring system we have previously described.5
FACS analysis
Cells were washed twice and adjusted to 107 cells/ml in FACS buffer (HBSS containing 1% FCS and .02% sodium azide). The cell suspensions then were dispensed into microcentrifuge tubes each containing 106 cells in 100 μl FACS buffer and stained with saturating amounts of conjugated antibodies for 30 min at 4°C. Following staining, cells were washed twice and re-suspended for analysis on a Becton Dickinson FACS 440 flow cytometer (Becton Dickinson, Mountain View, CA). Before adding labeled mAb, each tube received 1 μg 2.4G2 antibody (anti-FcγR; ATCC) to block nonspecific binding of conjugated antibodies to Fc receptors. The other mAb used for staining were anti-CD4-FTIC (RM2511; CalTag, Burlingame, CA), anti-CD8a-APC (53-6.7; Sigma), anti-Thy 1.2-PECy5 (TS; Sigma), anti-thy1.1-PECy7 (TS; Sigma) and anti-CD25-PE (PC61; PharMingen).
Statistical analysis
Data are means ± SE of multiple determinations. Difference in cytokine levels between two groups was compared using Student t test. Mann-Whitney was used to test for differences among inflammation scores. The difference between the mean of two groups were considered significant when the value of p values <0.05.
Results
OVA feeding induced severe enterocolitis
Rag mice were reconstituted with IL-10-/- T cells mixed with OT-II T cells bearing MHC class II-dependent TCR that recognizes OVA. The animals then were given a low dose of piroxicam orally for 2 wks. After discontinuing the piroxicam, groups of animals were fed standard mouse chow with or without OVA. After one wk of OVA administration, animals were sacrificed, and the duodenum, ileum and colon were examined for inflammation (Fig. 1). Animals fed OVA displayed intense inflammation involving all examined regions of small bowel and colon (Fig. 2). Control mice fed the usual mouse chow without OVA developed little or no inflammation in the small bowel and only mild colitis.
Figure 1.

Protocol used for mouse model of antigen-specific non-IgE food hypersensitivity. 4×106 IL10-/- and 2×106 OT-II splenic T cells were adaptively transferred into 5-6 wk old Rag mice via i.p. injection. A wk later, mice received 20 mg piroxicam/ 250g chow for 2wk. Piroxicam was stopped and mice were then fed 25mg OVA/250g chow 1 wk.
Figure 2.

OVA feeding induced non-IgE mediated severe enterocolitis. Rag recipients of IL10-/- & OT-II cells T cells T cells were treated with a low dose NSAID. Some then received OVA while some received regular chow for 1 wk before the gut were removed for histological examination, The inflammation was scored on a 0–4 point scale. Data are the mean ± SE from 10 or 12 animals studied in three separate experiments. A-C, Mice not fed OVA had minimal to no inflammation in the duodenum and ileum. There was mild colitis. Mice fed OVA had pronounced villi shortening, elongation of the crypts, and frequent crypt abscesses in the duodenum and ileum. There was deep lymphocytic infiltration of the colonic wall with ulceration, muscle thickening, and epithelial hypertrophy. Magnification ×40; H&E staining. *p<0.05
The intestinal inflammation generates IL-17 and IFN-γ, but not IL-4
We then determine the effect of OVA feeding on mucosal IL-17, IFN-γ and IL-4 production. Rag mice reconstituted with IL10-/- and OT-II T cell were treated with piroxicam for 2 wk as described above (Fig. 1). Some mice then were fed OVA for one wk while some only received standard mouse chow. LPMC isolated from the terminal ileum were cultured in vitro with or without OVA or anti-CD3/CD28 mAbs.
Compared to LPMC isolated from animals not fed OVA, LPMC from OVA fed animals produced more IL-17 and IFN-γ constitutively, as well as when they were stimulated in vitro with OVA or anti-CD3/CD28 mAbs. Only LPMC isolated from OVA fed animals responded to in vitro OVA stimulation by producing more IL-17 and IFN-γ. LPMC from animals not fed OVA did not respond to in vitro OVA stimulation (Fig. 3). IL-4 was not detected in cell cultures from either group.
Figure 3.
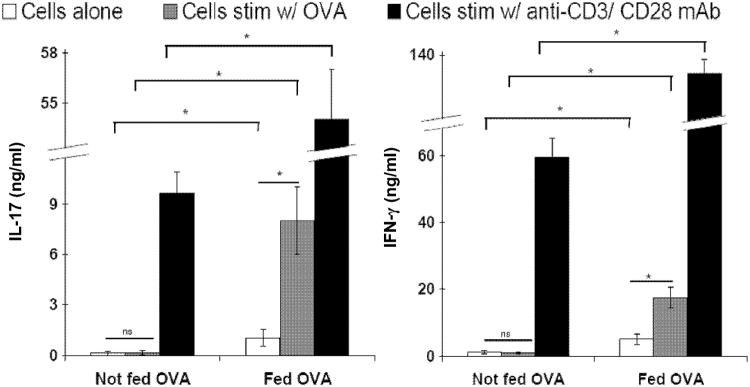
The intestinal inflammation generates IL-17A and IFN-γ. Rag-1 recipients of IL10KO and OT-II splenic T calls were treated with a low dose NSAID for 2 wk. some were then fed OVA while some received regular chow for a wk. LPMC were then isolated from the terminal ileum of bot groups. They were cultured for 48 hours with or witout OVA (100ug/ml) or anti-CD3/CD28 mAb. Supematant was analyzed with ELISA for cytokine production. LPMC from animals fed OVA produced more IL-17A and IFN-γ constitutively, and when stimulated with OVA or anti-CD3/CD28 mAb. LPMC from animals fed OVA responded to in vitro OVA stimulation while LPMC from animals not fed OVA did not respond. Data are mean ± SEM from 3 independent experiments wit more then five animals per group. * p<0.05. ns, not significant.
OT-II, IL10-/- T cell and piroxicam are necessary for the full expression of antigen-specific gut inflammation
The above experiment showed that OVA feeding can induce severe gut injury. To explore the importance of OT-II T cells, IL10-/- T cells and piroxicam in the disease process, animals were treated similarly to above except some did not receive OT-II T cells, or they were given WT T cells instead of IL10-/- T cells, or they did not receive piroxicam. Mice lacking the OVA-specific OT-II T cells had only mild colitis and minimal to no inflammation in the duodenum or ileum (Fig. 4). The appearance of the gut was similar to that of animals that did not receive OVA. Other mice received WT T cells in place of IL10-/- T cells, while receiving the usual OT-II T cells and piroxicam treatment. Mice reconstituted with WT T cells displayed significantly less gut inflammation after OVA feeding compared to mice given IL10-/- T cells. We also explored if piroxicam treatment was necessary for the induction of gut inflammation. Mice were handled in the usual fashion except some did not receive treatment with piroxicam. Mice that did not receive piroxicam had only mild colitis and nearly no inflammation in the duodenum or ileum (Fig. 4).
Figure 4.
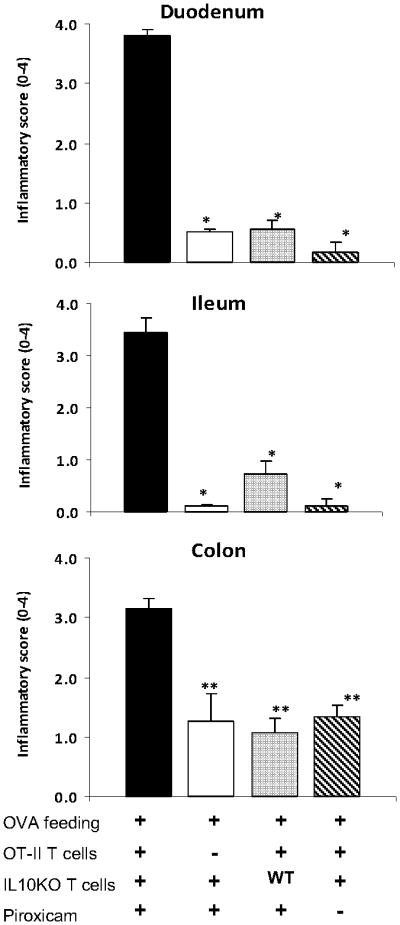
OT-II, IL10-/- T cells and piroxicam are necessary for the full expression of antigen-specific gut inflammation. To determine the importance of OT-II T cells, IL10KO T calls and piroxicam, mice were treated similarly except some did not receive OT-II T calls, or they were given WT T cells instead of IL10KO T cells, or they did not receive piroxicam. Data are mean ± SE from 2 separate experiments (n=4-5 per group). * p < 0.01, ** p < 0.05
Adoptively transferred OT-II T cells migrate to the gut
The above experiments showed that OVA feeding induced injury in the gut of mice reconstituted with OT-II T cells. We subsequently determined if the lamina propria contained substantial numbers of OT-II T cells after this reconstitution, and whether there was a change in the relative number of such T cells in the gut after OVA feeding. Rag mice were reconstituted with OT-II T cells expressing the cell surface protein Thy1.1, and IL10-/- T cells displaying Thy1.2. Mice then were treated with piroxicam for 2 wk, and some were challenged with oral OVA or fed just normal mouse chow in the usual fashion. Spleen (SPL), mesenteric lymph nodes (MLN) and LPMC from ileum and colon then were isolated and examined for Thy1.1 and Thy1.2 expression using flow cytometry.
As expected, piroxicam-treated, Rag recipients of OT-II (Thy1.1+) and IL10-/- (Thy1.2+) T cells developed severe gut inflammation only after OVA feeding. The severity of gut inflammation was similar whether the mice were reconstituted with OT-II (Thy1.2+) or OT-II (Thy1.1+) T cells (data not shown). FACS analysis showed that Thy1.1+ T cells populated the SPL, MLN, TI and colon of the Rag-1 recipients after adoptive transfer (Fig. 5). There were proportionally more Thy1.1+ cells in the gut than in the other organs (p<0.05). OVA feeding did not substantially alter the proportion of T cells in the spleen, MLN, TI or colon that expressed Thy1.1.
Figure 5.

Relative frequency of OT-II T cells in the spleen, MLN, TI and colon of mice fed cow with or without OVA. Rag-2 mice were reconstituted with 4 × 106 IL10-/- (Thy 1.2+) and 2 × 106 (Thy 1.1+) splenic T cells. One wk later, mice received 20 mg piroxicam/250g chow for 1 wk. Piroxicam was stopped and mice were then fed chow with and witout OVA for a wk. spleen, MLN, TI and colon LPMC were isolated, stained (PE-Cy7 for Thy 1.1 & PE-Cy5 for Thy 1.2) and analyzed by flow cytometry; data representative of 2 individual experiments each with 3-4 mice.
H. polygyrus abrogates gut inflammation
Next, we used this model to test the hypothesis that the enteric helminth H. polygyrus can protect against enterocolitis that is driven by a single luminal antigen, OVA. Mice were infected with H. polygyrus 1 day before commencement of NSAID treatment, which continued for 2 wks. After stopping the piroxicam, the mice were fed OVA for 1 week before analysis (Fig. 6A). Fig. 6B shows H. polygyrus colonization protected mice from severe OVA-induced pan-enterocolitis.
H. polygyrus induced Foxp3 expression in T cells and more IL-10 production
To determine if H. polygyrus induces Foxp3+ Treg, mice were reconstituted with IL-10-/- T cells from eGFP-Foxp3 IL-10-/- reporter mice and OT-II T cells. They were given piroxicam and challenged with oral OVA in the usual fashion. SPL, MLN and LPMC were isolated and analyzed by flow cytometry. Fig. 7A shows there was a higher frequency of Thy+CD4+Foxp3+ T cells in the spleen, MLN and TI of H. polygyrus-infected animals than in un-infected controls.
Figure 7.

A, There is a higher frequency of Thy1.2+ CD4+ Foxp3+ cells in Hp-infected animals than in non-infected controls. Splenic IL10-/- T cells from GFP-Foxp3+ IL10-/- reporter mice and OT-II splenic T cells were used for adaptive transfer in the NIEF model. Both groups were treated with piroxicam and fed OVA in the usual fashion. LPMC were isolated from the TI of both groups and analyzed with flow cytometry. B, compared to non-infected animals, LPMC isolated from te TI of Hp-infected animals produced less IL-17 and IFN-γ constitutively production of IL-10 from LPMC was higher in the Hp-infected animals than in the non-infected controls. In contrast to IL-17 and IFN-γ, production of IL-10 was n ot enhanced by in vitro OVA or anti-CD3/CD28 mAb stimulation in either groups. Data are mean ± SE from 2 separate experiments (n=3-4 per group).
We compared the cytokine profile of LPMC isolated from H. polygyrus-infected and un-infected controls. LPMC from H. polygyrus-infected mice released less IL-17 and IFN-γ constitutively, and when stimulated with anti-CD3/CD28 mAbs (p<0.05). Unlike the LPMC from the uninfected control animals, they did not produce more IL-17 or IFN-γ when stimulated with OVA in vitro (Fig. 7B top two panels). Also examined was IL-10 production. LPMC from H. polygyrus-infected animals released substantially more IL-10 than LPMC from uninfected control animals (Fig. 7B lower panel). IL-10 production was not enhanced by in vitro OVA or anti-CD3/CD28 mAb stimulation, in both H. polygyrus-infected and un-infected groups.
Non-T cells produce majority of the IL-10 in H. polygyrus-infected animals
The above experiment showed that the amount of IL-10 released from the LPMC was not affected by either OVA or anti-CD3/CD28 mAb stimulation, suggesting that the origin of the IL-10 was independent of T cells. To further address this point, we isolated LPMC from H. polygyrus-infected animals and fractionated LPMC into T cell-enriched and T cell-depleted fractions using paramagnetic beads. Isolated LP T cells and LPMC devoid of T cells were cultured in vitro 48 h in RPMI complete medium separately, and IL-10 was measured in the supernatants by ELISA at the end of the cultures. LPMC devoid of T cells made large amounts of IL-10, while the isolated LP T cells released essentially none (1.47±0.41 vs. 0.05 ± 0.03, ng/ml, p < 0.05, data are from 2 separate experiments with 3-4 mice in each experiment).
Discussion
In the present study, we established a murine model of IBD that featured severe pan-enterocolitis driven by a single antigen, OVA. Induction of this antigen-specific entercolitis requires both the presence of OT-II T cells that express TCR specific for OVA, and luminal exposure to OVA.
One of the most important features that differentiate our model from other existing antigen-specific IBD models is the significant inflammatory changes induced in both small and large intestines. Iqbal et al and Yoshida et al described an antigen-specific IBD model using Escherichia coli-producing OVA (ECOVA) to induce colitis in SCID mice reconstituted with T cells expressing TCR specific for OVA.6,7 The induced inflammation was confined to the colon, which may be due to site of colonization of ECOVA.
Further investigation demonstrated that piroxicam administration was needed to drive full expression of this antigen-specific gut injury. How piroxicam potentiated antigen-induced mucosal injury was not explored in this study. Piroxicam is a NSAID that non-selectively inhibits cyclooxygenases. It blocks the production of prostaglandins (PGs)8 and promotes development of intestinal inflammation by upregulating inflammatory cytokine secretion,9 chemokine production and chemokine receptor expression. 10–13 Using IL-10-/- IBD model, Berg, et. al. showed that colitis induced by piroxicam was dependent on inhibition of prostaglandins.14 It is tempting to speculate that interference with mucosal prostaglandin production, which is an important process that helps maintain mucosal immune homeostasis, potentiated the disease.
The severity of the disease also was dependent on the presence of T cells not able to making IL-10. Severe Th1/Th17 polarized enterocolitis was not observed in animals that were reconstituted with WT instead of IL-10-/- T cells. IL-10 is a potent anti-inflammatory cytokine and many cell types produce IL-10.15 Of the T cells that produce IL-10, T regulatory type 1 (Tr1) cells are the most studied. Tr1 cells play a central role in the induction of oral as well as systemic tolerance.16 They have been shown to dampen Th1 responses in various disease models.17 Recently, Rubtsov, et. al. showed that T cell-derived IL-10 was essential for keeping the immune response in check at environmental interfaces such as the colon and lungs,18 providing further evidence that IL-10 producing T cells play a critical role in maintaining mucosal immune homeostasis.
The fact that both piroxicam and IL-10-/- T cells were required to allow full expression of the OVA antigen-specific gut inflammation suggests that either T cell IL-10 or intact prostaglandin circuitry can suffice to afford a high level of protection and that their mechanisms of mucosal immune regulation is not interdependent.
We showed that LPMC isolated from animals fed OVA produced more of the proinflammatory cytokines IL-17 and IFN-γ than did LPMC from animals not fed OVA, suggesting that the enterocolitis is Th1- and Th17-polarized. IL-17 and IFN-γ are proinflammatory cytokines incriminated in driving colitis in both human and many murine models of IBD.19 However, we have not yet determined the origin of these molecules in the mucosal inflammation. It needs to be determined if the pathogenic OT-II cells, stimulated by OVA feeding, are a direct source of these proinflammatory cytokines and if they are important in driving the OVA-specific enterocolitis.
We also showed for the first time that H. polygyrus abrogates antigen-induced gut injury. Animals infected with H. polygyrus displayed an increased frequency of CD4+Foxp3+ Tregs. Rausch, et. al., and others showed that H. polygyrus infection expands Foxp3+ Tregs in the spleen, MLN and the intestine of healthy WT mice.20,21 Induction of Foxp3+ Tregs are not limited to H. polygyrus infection. For instance, their frequency is increased in helminth infections caused by Onchocerca volvulus, 22 Litomosoides sigmodontis 23 or Schistosoma mansoni. 24 These induced Tregs were thought to help quell the host's immune system and thus promote parasite survival. In vitro, Foxp3+ Tregs from H. polygyrus infected mice have more potent suppressive capacity of T cell proliferation and activation than such cells derived from uninfected mice.20,25 Tregs play a pivotal role in preventing autoimmune diseases such as type 1 diabetes26 and in limiting chronic inflammatory diseases like asthma and IBD.27,28 While we did not directly show Foxp3+ Tregs had a role in the protective process, their presence in increased frequency and their known biological effects in other animal model systems1,2,29–31 support the contention that H. polygyrus afforded protection at least in part through induction of these cells.
We also observed markedly increased IL-10 production from the LPMC isolated from H. polygyrus-infected animals. Others have previously showed that production of IL-10 is substantially heightened in H. polygyrus infection.32,33 The only two likely sources for this IL-10 in the dispersed LPMC were OT-II T cells and non-T cell elements like macrophages and dendritic cells. Our data suggested that the vast majority of the IL-10 came from non-T cell sources, since the cells failed to respond to either OVA or anti-CD3 mAb in vitro with enhance IL-10 secretion. Fractionating the LPMC into T cell-enriched and T cell-depleted components before cell culture also supported the assumption that the IL10 was mostly of non-T cell origin.
Although helminth induction of IL-10 producing Tregs is an important part of the immune-regulatory process that protects animalson in various disease models,34,35 the role of IL-10 producing non-T cells in the protection process is less well defined. Hess, et al showed that non-T cell-derived IL-10 is pivotal in reducing morbidity and immunopathology in a murine model of schistosomiasis, 36 suggesting that IL-10 producing non-T cells have an important immuno-modulatory role. The anti-inflammatory effect of IL-10 producing non-T cells was also demostrated by Chen, et. al. using a model of C. rodentium induced colitis, in which H. polygyrus co-infection was shown to suppress colitis via IL-10 of non-T origin.37 It is tempting to speculate that helminth-primed non-T cell derived IL-10 may have a protective role in our model of antigen-specific IBD.
We showed that LPMC isolated from H. polygyrus-infected animals displayed a decrease in IL-17 and IFN-γ production when cultured without stimulation, as well as a dampened response when stimulated with OVA or anti-CD3/CD28 mAbs in vitro. After H. polygyrus infection, we failed to detect IL-4 production suggesting that the decrease in IFN-γ and IL-17 release did not simply reflect a shift to the Th2 pathway of inflammation. H. polygyrus infection can inhibit colitis and suppress mucosal IL-17 and IFN-γ production in mice through direct effects on mucosal dendritic cells 5, which could be an important mechanism for cytokine regulation in our model. Whether down-regulation of IFN-γ and IL-17 production is part of the mechanism thorough which H. polygyrus infection protected animals from OVA-induced inflammation remains to be determined.
In conclusion, we develop a novel murine model of IBD in which gut injury is triggered by an oral antigen. The antigen-specific injury is severe and involves the entire gastrointestinal tract. It is characterized by increased production of IFN-γ and IL-17, but not IL-4. We showed for the first time that H. polygyrus infection can abrogate an antigen-induced gut inflammation probably through the induction of immune regulatory circuits. The ability of H polygyrus to limit antigenic responses in the gut could have relevance for the treatment of human IBD.
Acknowledgments
Fundings: This work was supported by Tufts Medical Center Research Fund 2010, Grants DK007542, DK38327 and DK058755 from the National Institutes of Health, the Broad Foundation, the Schneider Family, the Friedman Family, and the Gilman Family.
Abbreviations used in this paper
- DC
dendritic cell
- IBD
inflammatory bowel disease
- LPMC
lamina propria mononuclear cell
- NSAID
non-steroidal anti-inflammatory drug
- TI
terminal ileum
- MLN
mesenteric lymph nodes
- Hp
Heligmosomoides polygyrus
Footnotes
Disclosure: The authors have no financial conflicts of interest
References
- 1.Elliott DE, Setiawan T, Metwali A, et al. Heligmosomoides polygyrus inhibits established colitis in IL-10-deficient mice. Eur J Immunol. 2004;34(10):2690–2698. doi: 10.1002/eji.200324833. [DOI] [PubMed] [Google Scholar]
- 2.Elliott DE, Li J, Blum A, et al. Exposure to schistosome eggs protects mice from TNBS-induced colitis. Am J Physiol Gastrointest Liver Physiol. 2003;284(3):G385–391. doi: 10.1152/ajpgi.00049.2002. [DOI] [PubMed] [Google Scholar]
- 3.Summers RW, Elliott DE, Qadir K, et al. Trichuris suis seems to be safe and possibly effective in the treatment of inflammatory bowel disease. Am J Gastroenterol. 2003;98(9):2034–2041. doi: 10.1111/j.1572-0241.2003.07660.x. [DOI] [PubMed] [Google Scholar]
- 4.Summers RW, Elliott DE, Urban JF, Thompson R, Weinstock JV. Trichuris suis therapy in Crohn's disease. Gut. 2005;54(1):87–90. doi: 10.1136/gut.2004.041749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hang L, Setiawan T, Blum AM, et al. Heligmosomoides polygyrus infection can inhibit colitis through direct interaction with innate immunity. J Immunol. 2010;185(6):3184–3189. doi: 10.4049/jimmunol.1000941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iqbal N, Oliver JR, Wagner FH, et al. T Helper 1 and T Helper 2 Cells Are Pathogenic in an Antigen-specific Model of Colitis. The Journal of Experimental Medicine. 2002;195(1):71–84. doi: 10.1084/jem.2001889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshida M, Watanabe T, Usui T, et al. CD4 T cells monospecific to ovalbumin produced by Escherichia coli can induce colitis upon transfer to BALB/c and SCID mice. International Immunology. 2001;13(12):1561–1570. doi: 10.1093/intimm/13.12.1561. [DOI] [PubMed] [Google Scholar]
- 8.Funk CD. Prostaglandins and Leukotrienes: Advances in Eicosanoid Biology. Science. 2001;294(5548):1871–1875. doi: 10.1126/science.294.5548.1871. [DOI] [PubMed] [Google Scholar]
- 9.Kunkel SL, Spengler M, May MA, et al. Prostaglandin E2 regulates macrophage-derived tumor necrosis factor gene expression. J Biol Chem. 1988;263(11):5380–5384. [PubMed] [Google Scholar]
- 10.Thivierge M, Le Gouill C, Tremblay MJ, Stanková J, Rola-Pleszczynski M. Prostaglandin E2 induces resistance to human immunodeficiency virus-1 infection in monocyte-derived macrophages: downregulation of CCR5 expression by cyclic adenosine monophosphate. Blood. 1998;92(1):40–45. [PubMed] [Google Scholar]
- 11.Janabi N, Hau I, Tardieu M. Negative feedback between prostaglandin and alpha- and beta-chemokine synthesis in human microglial cells and astrocytes. J Immunol. 1999;162(3):1701–1706. [PubMed] [Google Scholar]
- 12.Zeidler R, Csanady M, Gires O, et al. Tumor cell-derived prostaglandin E2 inhibits monocyte function by interfering with CCR5 and Mac-1. FASEB J. 2000;14(5):661–668. doi: 10.1096/fasebj.14.5.661. [DOI] [PubMed] [Google Scholar]
- 13.Takahashi S, Fujita T, Yamamoto A. Role of cyclooxygenase-2 in Helicobacter pylori- induced gastritis in Mongolian gerbils. Am J Physiol Gastrointest Liver Physiol. 2000;279(4):G791–798. doi: 10.1152/ajpgi.2000.279.4.G791. [DOI] [PubMed] [Google Scholar]
- 14.Berg DJ, Zhang J, Weinstock JV, et al. Rapid development of colitis in NSAID-treated IL-10-deficient mice. Gastroenterology. 2002;123(5):1527–1542. doi: 10.1053/gast.2002.1231527. [DOI] [PubMed] [Google Scholar]
- 15.Saraiva M, O'Garra A. The regulation of IL-10 production by immune cells. Nat Rev Immunol. 2010;10(3):170–181. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
- 16.Battaglia M, Gianfrani C, Gregori S, Roncaroloa M. IL–10–Producing T Regulatory Type 1 Cells and Oral Tolerance. Annals of the New York Academy of Sciences. 2004;1029(1):142–153. doi: 10.1196/annals.1309.031. [DOI] [PubMed] [Google Scholar]
- 17.Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. J Immunol. 2008;180(9):5771–5777. doi: 10.4049/jimmunol.180.9.5771. [DOI] [PubMed] [Google Scholar]
- 18.Rubtsov YP, Rasmussen JP, Chi EY, et al. Regulatory T Cell-Derived Interleukin-10 Limits Inflammation at Environmental Interfaces. Immunity. 2008;28(4):546–558. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 19.Elliott DE, Metwali A, Leung J, et al. Colonization with Heligmosomoides polygyrus suppresses mucosal IL-17 production. J Immunol. 2008;181(4):2414–2419. doi: 10.4049/jimmunol.181.4.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rausch S, Huehn J, Kirchhoff D, et al. Functional analysis of effector and regulatory T cells in a parasitic nematode infection. Infect Immun. 2008;76(5):1908–1919. doi: 10.1128/IAI.01233-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Finney CAM, Taylor MD, Wilson MS, Maizels RM. Expansion and activation of CD4+CD25+ regulatory T cells inHeligmosomoides polygyrus infection. Eur J Immunol. 2007;37(7):1874–1886. doi: 10.1002/eji.200636751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Satoguina J, Mempel M, Larbi J, et al. Antigen-specific T regulatory-1 cells are associated with immunosuppression in a chronic helminth infection (onchocerciasis) Microbes Infect. 2002;4(13):1291–1300. doi: 10.1016/s1286-4579(02)00014-x. [DOI] [PubMed] [Google Scholar]
- 23.Taylor MD, LeGoff L, Harris A, et al. Removal of regulatory T cell activity reverses hyporesponsiveness and leads to filarial parasite clearance in vivo. J Immunol. 2005;174(8):4924–4933. doi: 10.4049/jimmunol.174.8.4924. [DOI] [PubMed] [Google Scholar]
- 24.McKee AS, Pearce EJ. CD25+CD4+ cells contribute to Th2 polarization during helminth infection by suppressing Th1 response development. J Immunol. 2004;173(2):1224–1231. doi: 10.4049/jimmunol.173.2.1224. [DOI] [PubMed] [Google Scholar]
- 25.Finney CAM, Taylor MD, Wilson MS, Maizels RM. Expansion and activation of CD4(+)CD25(+) regulatory T cells in Heligmosomoides polygyrus infection. Eur J Immunol. 2007;37(7):1874–1886. doi: 10.1002/eji.200636751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sakaguchi S, Sakaguchi N, Shimizu J, et al. Immunologic tolerance maintained by CD25+ CD4+ regulatory T cells: their common role in controlling autoimmunity, tumor immunity, and transplantation tolerance. Immunol Rev. 2001;182:18–32. doi: 10.1034/j.1600-065x.2001.1820102.x. [DOI] [PubMed] [Google Scholar]
- 27.Xystrakis E, Boswell SE, Hawrylowicz CM. T regulatory cells and the control of allergic disease. Expert Opin Biol Ther. 2006;6(2):121–133. doi: 10.1517/14712598.6.2.121. [DOI] [PubMed] [Google Scholar]
- 28.Coombes JL, Maloy KJ. Control of intestinal homeostasis by regulatory T cells and dendritic cells. Semin Immunol. 2007;19(2):116–126. doi: 10.1016/j.smim.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 29.La Flamme AC, Ruddenklau K, Bäckström BT. Schistosomiasis decreases central nervous system inflammation and alters the progression of experimental autoimmune encephalomyelitis. Infect Immun. 2003;71(9):4996–5004. doi: 10.1128/IAI.71.9.4996-5004.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saunders KA, Raine T, Cooke A, Lawrence CE. Inhibition of Autoimmune Type 1 Diabetes by Gastrointestinal Helminth Infection. Infect Immun. 2007;75(1):397–407. doi: 10.1128/IAI.00664-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Z, Liu Q, Bleich D, Salgame P, Gause W. Regulation of type 1 diabetes, tuberculosis, and asthma by parasites. [Accessed December 12, 2009];Journal of Molecular Medicine. doi: 10.1007/s00109-009-0546-0. Available at: http://dx.doi.org/10.1007/s00109-009-0546-0. [DOI] [PMC free article] [PubMed]
- 32.Su Z, Segura M, Morgan K, Loredo-Osti JC, Stevenson MM. Impairment of protective immunity to blood-stage malaria by concurrent nematode infection. Infect Immun. 2005;73(6):3531–3539. doi: 10.1128/IAI.73.6.3531-3539.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilson MS, Taylor MD, Balic A, et al. Suppression of allergic airway inflammation by helminth-induced regulatory T cells. J Exp Med. 2005;202(9):1199–1212. doi: 10.1084/jem.20042572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kitagaki K, Businga TR, Racila D, et al. Intestinal Helminths Protect in a Murine Model of Asthma. The Journal of Immunology. 2006;177(3):1628–1635. doi: 10.4049/jimmunol.177.3.1628. [DOI] [PubMed] [Google Scholar]
- 35.Shi M, Wang A, Prescott D, et al. Infection with an intestinal helminth parasite reduces Freund’s complete adjuvant-induced monoarthritis in mice. Arthritis Rheum. 2011;63(2):434–444. doi: 10.1002/art.30098. [DOI] [PubMed] [Google Scholar]
- 36.Hesse M, Piccirillo CA, Belkaid Y, et al. The pathogenesis of schistosomiasis is controlled by cooperating IL-10-producing innate effector and regulatory T cells. J Immunol. 2004;172(5):3157–3166. doi: 10.4049/jimmunol.172.5.3157. [DOI] [PubMed] [Google Scholar]
- 37.Chen CC, Louie S, McCormick BA, Walker WA, Shi HN. Helminth-Primed Dendritic Cells Alter the Host Response to Enteric Bacterial Infection. The Journal of Immunology. 2006;176(1):472–483. doi: 10.4049/jimmunol.176.1.472. [DOI] [PMC free article] [PubMed] [Google Scholar]