

Abstract
Many viruses have developed mechanisms to evade the IFN response. Here, HIV-1 was shown to induce a distinct subset of IFN-stimulated genes (ISGs) in monocyte-derived dendritic cells (DCs), without detectable type I or II IFN. These ISGs all contained an IFN regulatory factor 1 (IRF-1) binding site in their promoters, and their expression was shown to be driven by IRF-1, indicating this subset was induced directly by viral infection by IRF-1. IRF-1 and -7 protein expression was enriched in HIV p24 antigen-positive DCs. A HIV deletion mutant with the IRF-1 binding site deleted from the long terminal repeat showed reduced growth kinetics. Early and persistent induction of IRF-1 was coupled with sequential transient up-regulation of its 2 inhibitors, IRF-8, followed by IRF-2, suggesting a mechanism for IFN inhibition. HIV-1 mutants with Vpr deleted induced IFN, showing that Vpr is inhibitory. However, HIV IFN inhibition was mediated by failure of IRF-3 activation rather than by its degradation, as in T cells. In contrast, herpes simplex virus type 2 markedly induced IFNβ and a broader range of ISGs to higher levels, supporting the hypothesis that HIV-1 specifically manipulates the induction of IFN and ISGs to enhance its noncytopathic replication in DCs.
Introduction
Langerhans cells and lamina propria dendritic cells (DCs) in the anogenital and cervical mucosa and the male foreskin are key target cells for sexual transmission of HIV-11–3 and probably facilitate access to CD4+ T cells in the submucosa and lymph nodes, resulting in a productive infection and subsequent dissemination.4–7 After HIV-1 binding to C-type lectin receptors on monocyte-derived DCs (MDDC) the majority (> 95%) of HIV-1 is endocytosed and subject to acid proteolytic digestion over 6-12 hours8 or taken up in tetraspanin-rich caves. A minority is transferred to CD4/CCR5, resulting in fusion of the virus envelope with the plasma membrane and de novo infection, apparent only at a later phase > 24 hours after infection. After contact between DCs and T cells, HIV-1 is transferred to the latter in 2 phases, first from “caves” and then later from the cytosol.5,9–12
However, how HIV-1 manipulates DC biology to use the cell for viral transfer to T cells without marked cytopathic effects is still unclear. Viruses often shape their intracellular environment through alterations to host cell gene transcription, protein translation, and posttranslational modification, often initiating these changes by signaling through cell surface receptors or at subsequent stages in their replication cycle.13,14
To determine the effects of HIV-1BaL on the DC transcriptome, we have previously performed rigorous microarray experiments with the use of highly purified, high-titer HIV-1BaL virus stocks and purified recombinant gp120 in a single replication cycle over 48 hours. We showed that HIV-1BaL induces changes in expression of several distinct gene clusters in 2 major groups in 2 transient and sequential phases, one group corresponding to HIV binding/entry and endocytosis over 6 hours after infection and the second group corresponding to the later stages of de novo replication (after reverse transcription) at 24-96 hours after infection.15 A minor group of genes showed persistent up-regulated expression across both phases. In the second phase HIV induced partial maturation of DCs which leads to enhanced migration and T-cell stimulation5 and also reduction in lysosomal enzyme expression and function.15 In this study with the use of a similar approach we show that a specific cluster of IFN-stimulated genes (ISGs) is up-regulated in response to HIV-1BaL, but there was no detectable type I or II IFN induction. Most of this subset showed the kinetics of the minor group of up-regulated genes. We demonstrate that this ISG subset can be driven by IFN regulatory factor 1 (IRF-1) and that this is the case in HIV infection of DCs. In addition, we show that deletion of the IRF-1/7 binding site from the HIV-1 long terminal repeat (LTR) results in a virus with decreased growth kinetics, indicating that HIV-1 induces IRF-1 and -7 expression early after infection of MDDCs to aid its own replication. Recent reports have indicated that the HIV-1 accessory proteins Vpr and Vif are required for the inhibition of an IFN response in T cells by targeted degradation of constitutively expressed IRF-3, the major IFN-inducing IRF.16,17 However, in MDDCs, HIV-1 infection had no effect on IRF-3 expression, but it inhibited its activation and translocation to the nucleus. Here, we show that Vpr but not Vif is required to inhibit the IFN response in MDDCs by an alternative mechanism to IRF-3 degradation.
Conversely, infection of DCs with HSV-2186, a virus known to productively infect MDDCs, resulted in IFN induction and increased expression of a broader range of ISGs to higher levels. Because ISGs potentially modulate effects on cell proliferation, activation, differentiation, and survival as well as restricting viral replication, the nature of the ISG subset regulated probably has an important role in determination of the outcome of HIV trafficking in myeloid DCs, their infection, and transfer to CD4+ lymphocytes.
Thus, HIV-induced defects in myeloid DCs probably allow the virus to obtain a toehold in the genital tract before the infiltration of other IFN-secreting cells, in particular plasmacytoid DCs. Furthermore, the induction of IRF-1 in myeloid DCs in the genital tract might provide part of the explanation for reduced susceptibility to HIV infection of Kenyan prostitutes with certain IRF-1 polymorphisms.18
Methods
Preparation of MDDCs
MDDCs were generated from CD14+ monocytes isolated from PBMCs with the use of CD14 magnetic beads (Miltenyi Biotech) as described previously.5
Viral stock preparation and construction of HIV-1 deletion mutants
Purified high-titer HIV-1BaL stocks in the order of 5 × 1010 50% tissue culture infective dose/mL were produced with the use of tangential filter concentration as described previously.5,10,19 Virus content was determined by p24 gag ELISA (Beckman-Coulter) and as values of 50% tissue culture infective dose generated in TZM-bl cells (NIH AIDS Research and Reference Reagent Program, contributed by John Kappes and Xiaoyun Wu) measured by LTR β-galactosidase reporter gene expression after a single round of infection.20 The endotoxin levels of these virus stocks were below the detectable limit of 0.005 U/mL or 0.0005 ng/mL (Limulus amebocyte lysate assay; Sigma), and testing for residual TNF-α, IFNα, IFNβ, and IFNγ by ELISA (R&D Systems) was negative. An IRF-1/7 binding site (or IFN-stimulated response element; ISRE) deletion mutant virus (pBaLISRE(Mut) was constructed by replacing the ISRE with the 18 bp Zeichner linker sequence,21 containing NdeI, XhoI, and SalI restriction sites. Vesicular stomatitis virus glycoprotein (VSVG)–pseudotyped HIV-1 deletion mutants were produced by transfection of either NL43ΔVpr or NL43ΔVif plasmid constructs. NL43ΔVpr and NL43ΔVif were generated by the addition of stop codons into the protein open reading fame by site-directed mutagenesis with the use of specific PCR primers. NL43ΔVpr was generated by the insertion of a stop codon at amino acid position 21 in the Vpr open reading frame. NL43ΔVif was generated by the insertion of a stop codon at amino acid position 18 in the Vif open reading frame. Overlapping open reading frames were not affected by these mutations. Stocks of HSV-2 (strain 186) were generated as described previously.22
Treatment of cultured cells with HIV-1, HSV-2, and Sendai virus
MDDCs were seeded at 1 × 106 cells/mL and treated with HIV-1BaL at a MOI of 10, 3, or 1 or with HSV-2186 at an MOI of 3 at day 6 or at day 2 with VSVG-pseudotyped HIV-1 virus stocks. TZM-bl cells were infected with Sendai virus at 150 hemagglutinin units/mL.
Microarray hybridization and data analysis
Total RNA derived from HIV-1BaL– or HSV-2186–treated MDDCs was prepared for hybridization to Human ResGen 8k (Australian Genome Research Facility) glass microarrays or to Sentrix Human-6 (Version 2) expression chips (Illumina) as described previously.15
Confirmation of differential gene expression by qPCR
Total unamplified RNA was treated with DNase I (Promega) and then reverse transcribed with oligo d(T) and superscript III (Invitrogen). The cDNA was then subject to quantitative PCR (qPCR) with the use of defined primers (Sigma) and SYBR Green (Invitrogen). The relative quantitation method (ΔΔCT)23 was used to evaluate the expression of selected genes with the GAPDH as an internal control and the normalizer for all data.24
In silico promoter analysis
Ensembl and RefSeq gene identifiers were obtained for all differentially regulated genes, and proximal promoter sequences were extracted with the use of the UCSC Genome Browser25 and the Ensembl Genome Browser.26 Sequence regions 3000 and 1500 bp immediately 5′ upstream from the transcription start site and the 5′ untranslated region were extracted. All differentially regulated genes were analyzed for IFN signatures with the use of the INTERFEROME database (http://www.interferome.org).27 The Java software, Toucan2, was used in the comparative promoter analysis. Twenty genes that were not differentially regulated during this experiment and 20 random IFN-regulated genes were chosen as controls for promoter analysis. The Transfac professional database (Version 11.4)28 was used to obtain the vertebrate transcription factor binding site (TFBS) matrices. The Toucan2 tool MotifScanner (Gibbs sampler) was used in identifying potential TFBSs in the sets of selected sequences. The prior (stringency level) for motif prediction was set to a value of either 0.05 or 0.1, and the human promoter set from the Eukaryotic Promoter Database29 was chosen as a third-order background model to determine overrepresented TFBSs. Toucan2 statistical tool was applied to the data obtained by MotifScanner to identify overrepresented TFBSs (those showing positive significance values) in the selected gene set.30
Transfection of 293T cells
293T cells were plated at 1.2 × 106 cells per well in a 6-well plate overnight (80% confluence). Cells were then transfected with polyethyleneimine as described previously31 with 1, 5, or 10 μg of the IRF-1–expressing plasmid CMVBL-IRF1-HNK (kindly provided by from Dr Angella Battistini, Istituto superiore di sanita) or empty vector. Cells were harvested at 24 and 48 hours after transfection and assayed for ISG expression by qPCR.
ChIP assay
ChIP experiments were performed in MDDCs with the use of a ChIP assay kit according to the manufacturer's instructions (Upstate Biotechnology) with an IRF-1 antibody (Santa Cruz Biotechnology Inc; sc-497x) as described previously.32
ELISA
Levels of secreted IFNα, IFNβ, and IFNγ from viral inoculum and MDDCs treated with HIV-1BaL, HSV-2186, or mock treated for 6-48 hours were determined by ELISA (R&D Systems) according to the manufacturer's instructions.
Flow cytometry
PE-conjugated p24 (clone KC57-RD1) and IgG1 mouse coulter clone monoclonal antibodies were obtained from Beckman Coulter. Purified mouse polyclonal antibodies directed toward IRF2, IRF7, and IRF3 were obtained from AbD Serotech, BD, Biosciences, and Dr Michael Gale (University Washington), respectively. Rabbit polyclonal antibodies directed toward IRF1 and IRF8 were obtained from Abcam. Alexa 488–conjugated goat anti–mouse and FITC-conjugated goat anti–rabbit secondary antibodies were obtained from Invitrogen and Sigma-Aldrich, respectively. HIV-1BaL or mock-treated MDDCs were fixed and permeablized in Cytofix/Cytoperm (BD). All antibody incubations were performed in permwash buffer (1% human AB serum, 0.1% saponin, 0.1% sodium azide, made up in PBS). IgG isotype control antibodies were incubated with cells to control for nonspecific binding. Cells were then analyzed with a FACSCanto flow cytometer (Becton Dickinson) and FlowJo software (TreeStar Inc).
Western blot
HIV-1BaL or mock-treated MDDCs were lysed with Berman lysis buffer (SDS (0.1%), sodium deoxyxcholate (0.5% w/v), NP-40 (1% v/v) containing a Protease inhibitor cocktail (Roche), separated in a 12% poly-acrylamide gel by electrophoresis and transferred to polyvinylidene difluoride membrane (Bio-Rad). Membranes were blocked overnight (5% w/v skim milk, 0.1% v/v Tween, and 1× PBS) and probed with a 1/500 dilution of monoclonal IRF-1 or IRF-3 primary antibody (BD PharMingen, Cell Signaling), followed by a secondary biotin conjugated α-mouse IgG antibody (Sigma-Aldrich). The antibody-reactive IRF proteins were visualized with the use of the 5-Bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium liquid substrate system (Sigma-Aldrich).
Confocal microscopy
HIV- or Sendai virus–treated MDDCs or TZM-bl cells were stained using a mouse IRF-3 monoclonal antibody (M.G.) and Alexa flour 488–conjugated goat anti–mouse secondary antibody (Invitrogen). Nuclear staining was carried out using ToPro3 (Invitrogen). Confocal microscopy was undertaken using a DMRE florescence microscope from Leica Microsystems with a 63×/1.4 numeric aperture oil objective lense. Images were analysed using Leica SP2 confocal system software.
Results
HIV-1 treatment triggers the expression of a subset of IFN-stimulated genes in MDDCs
cDNA microarrays were used to determine the genes differentially expressed in MDDCs in response to a purified high-titer HIV-1BaL stock compared with mock treatment at 6, 24, and 48 hours after treatment, initially with Human Resgen 8k arrays and then later checked with Illumina Sentrix Human-6 arrays. As previously reported, HIV uptake by DCs up-regulated expression of 2 major groups of genes in corresponding temporal phases: expression of 255 genes was transiently enhanced in the first phase (6 hours after exposure) and 385 in the second phase (48 hours after exposure).15 A third small group of ∼ 30 genes bridged these 2 phases. Eighteen genes associated with IFN induction (ISGs) were shown to be up-regulated in their expression by viable HIV-1 (Table 1), and one-half formed part of this minor group; that is, one-half of these genes were up-regulated by 6 hours after virus treatment, and all of them were up-regulated at 48 hours with the magnitude of up-regulation of all being higher at this later time. AT2-inactivated HIV-1 had little effect on ISGs; only Mx1 expression was consistently up-regulated at 6-48 hours after infection (data not shown).
Table 1.
Microarray- and qPCR-derived differential expression data for genes encoding ISGs in HIV-1–treated MDDCs
| Symbol | Accession no. | 6 Hours |
24 Hours |
48 Hours |
|||
|---|---|---|---|---|---|---|---|
| Array | qPCR | Array | qPCR | Array | qPCR | ||
| IFNA | All isoforms | No change | No change | No change | No change | No change | No change |
| IFNB | NM_002176 | No change | No change | No change | No change | No change | No change |
| IFNG | NM_000619 | No change | No change | No change | No change | No change | No change |
| MX1* | NM_002462 | 2.3 | 3.2 | 10.6 | 7.0 | 6.8 | 7.8 |
| OAS1* | NM_016816 | 1.8 | 2.0 | 3.7 | 5.5 | 1.9 | 3.0 |
| IRF-1* | NM_002198 | 2.0 | 2.6 | 3.4 | 4.7 | 2.0 | 7.4 |
| IRF-2* | NM_002199 | No change | No change | 1.3 | 1.6 | 1.9 | 2.7 |
| IRF-4* | NM_002460 | No change | No change | 1.5 | 4.0 | 1.3 | 4.4 |
| ISG15* | NM_005101 | 2.3 | 1.8 | 9.9 | 9.5 | 4.7 | 8.0 |
| SOCS3 | NM_003955 | 1.6 | 1.8 | 2.0 | 2.0 | 3.1 | 8.0 |
| IL10RB | NM_000628 | — | No change | 1.8 | 3.9 | 1.5 | 2.6 |
| STAT3* | NM_139276 | 1.7 | 1.3 | 1.7 | 3.5 | 1.7 | 2.9 |
| STAT1 | NM_139266 | No change | † | 2.1 | † | 2.3 | † |
| IFI35* | NM_005533 | 1.3 | 1.8 | 3.1 | 2.1 | 1.9 | 5.1 |
| IL18 | NM_001562 | No change | † | 1.5 | † | 1.4 | † |
| PSMB9 | NM_002800 | No change | † | 1.8 | † | 1.7 | † |
| WARS | NM_004184 | No change | † | 2.8 | † | 3.7 | † |
| NMI | NM_004688 | — | † | 1.7 | † | 1.7 | † |
| MNDA | NM_002432 | — | † | 1.5 | † | † | † |
| IFIT5* | NM_012420 | No change | No change | 2.1 | 2.0 | 2.1 | 3.8 |
| IFITM3* | NM_021034 | 1.4 | 2.3 | 4.4 | 4.8 | 2.1 | 4.4 |
| OAS3 | NM_006187 | — | 2.3 | — | 4.8 | — | 5.5 |
| DDX58* | NM_014314 | — | No change | — | 6.2 | — | 18.4 |
| IFIH1* | NM_022168 | — | No change | — | 8.3 | — | 12.9 |
| PKR | NM_002759 | — | 1.6 | — | 3.1 | — | 2.5 |
| IFIT1* | NM_001548 | — | 4.4 | — | 6.0 | — | 3.8 |
| IFIT2* | NM_001547 | — | 4.5 | — | 6.0 | — | 5.0 |
| IFIT3* | NM_001549 | — | 3.8 | 9.7 | — | 18 | |
| IFITM1* | NM_003641 | — | 2.2 | — | 5.7 | — | 15.2 |
| RSAD2* | NM_080658 | — | 1.8 | — | 2.6 | — | 4.7 |
Day 6 MDDCs were treated with HIV-1BaL (MOI, 10), and ISG expression was determined by qPCR or 8K cDNA microarrays. Differential expression data are presented for HIV-1– versus mock-treated cells at 6, 24, and 48 hours after treatment from 4 independent experiments.
The — indicates that no microarray data were available.
Expression of this gene was shown to be driven by IRF-1 in 293T cells (see Table 4).
qPCR was not performed to confirm the expression of this gene.
Ten genes from this list were chosen for confirmation of their differential expression by qPCR. In every case qPCR confirmed that the gene was up-regulated, although qPCR usually showed a larger magnitude of up-regulation than the corresponding microarray values. qPCR was also used to investigate the expression of 9 additional IFN-associated genes that were not present on the microarrays (Table 1 bottom 9 rows), which were also shown to be up-regulated in their expression. However, no genes that encode type I or II IFNs were increased in their expression as detected by microarray or qPCR (Table 1 top 3 rows).
HIV-1 treatment of MDDCs does not trigger IFN induction
To determine whether the HIV-1BaL–treated MDDCs produced IFNs, cells were infected with virus for 6-72 hours. qPCR was used to determine the expression levels of mRNAs encoding all IFNα isoforms, IFNβ, and IFNγ. In addition, secreted IFNα and IFNβ levels were determined in infected supernatant fluids with the use of ELISA. HIV-1–treated MDDCs showed no up-regulation of any IFN genes (Figure 1; data not shown) and no IFNα, IFNβ, or IFNγ proteins were detected in the supernatant fluids or the HIV inoculum (data not shown).
Figure 1.
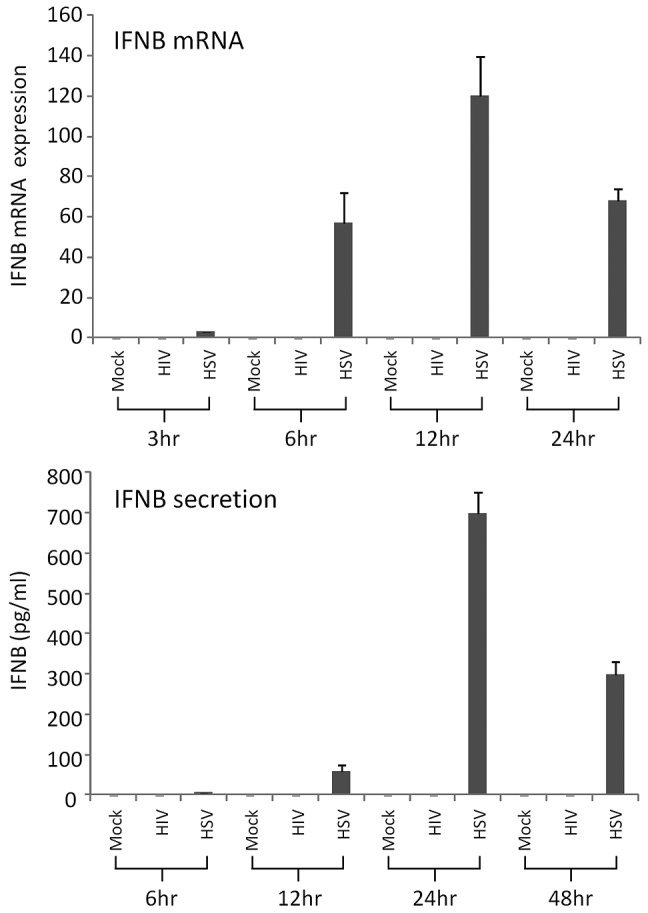
HIV-1 and HSV-2 induction of IFNβ in MDDCs. Day 6 MDDCs were exposed to purified HIV-1BaL or HSV-2186 at MOI of 3 for 3-96 hours. (A) IFNβ mRNA expression was determined by qPCR at 3, 6, 12, and 24 hours after infection. (B) The level of IFNβ secreted into the supernatant fluid was determined by ELISA at 6, 12, 24, and 48 hours after infection. The mean data from 3 experiments are shown with standard error bars.
IRF gene expression in response to HIV-1 treatment of MDDCs
Because 3 IRFs showed altered expression, we next used qPCR to determine the gene expression profiles of 4 other key IRF family members in MDDCs infected with HIV-1BaL for 6-48 hours (Table 2). By 6 hours the IRF-1, -7, and -8 genes were all increased in their expression compared with mock-treated cells. Expression of IRF-1 steadily increased to 48 hours, whereas IRF-8 steadily decreased. IRF-7 expression rose at 24 hours and then remained constant at 48 hours after infection. At 24 hours the IRF-2, -4, and -9 genes were also increased in their expression. IRF-2 gene expression was increased further at 48 hours, whereas that for IRF-4 remained constant. IRF-9 gene expression, however, was up-regulated to a lesser degree at 48 hours.
Table 2.
Differential expression data for genes encoding key IRFs in HIV-1–treated MDDCs
| Symbol | Accession no. | 6 Hours | 24 Hours | 48 Hours |
|---|---|---|---|---|
| IRF-1 | NM_002198 | 2.7 ± 0.9 | 4.6 ± 1.0 | 7.4 ± 1.2 |
| IRF-2 | NM_002199 | No change | 1.7 ± 0.2 | 2.7 ± 0.7 |
| IRF-3 | NM_001571 | No change | No change | No change |
| IRF-4 | NM_002460 | No change | 3.9 ± 1.0 | 4.4 ± 0.9 |
| IRF-7 | NM_004031 | 3.9 ± 0.25 | 5.0 ± 0.7 | 4.9 ± 1.6 |
| IRF-8 | NM_002163 | 8.9 ± 3.7 | 2.7 ± 0.7 | 1.2 ± 0.1 |
| IRF-9 | NM_006084 | No change | 3.8 ± 0.6 | 1.8 ± 0.7 |
Day 6 MDDCs were mock-treated or treated with HIV-1BaL (MOI, 10), and IRF gene expression was determined by qPCR. Data are mean fold change ± SE in the expression of IRF genes in HIV-1–treated cells at 6, 24, and 48 hours after treatment from 4 independent experiments.
HIV-1 triggers the expression of IRF-1, -2, -7, and -8 proteins in MDDCs
Both microarrays and qPCR showed significantly increased gene expression of IRF-1 and IRF-7 at all time points (Tables 1–3). Given the previously identified role of the IRF-1 protein in HIV-1 infection of T cells33–35 and the concurrent up-regulation of gene expression of its inhibitors, IRF-2 (at later time points) and -8 (at earlier time points), we focused on this protein and its inhibitors and next determined whether they were increased in their expression in response to HIV-1 treatment in DCs with the use of Western blot and flow cytometric analyses (Figure 2). In addition, we focused on the IRF-7 protein. The IRF-1 protein was up-regulated by 6 hours after treatment and was still increased in its expression at 48 hours (Figure 2A,D). In addition, the IRF-8 protein was up-regulated early after infection but not at later time points (no up-regulation by 120 hours), and the IRF-2 protein was up-regulated in the later stages of infection but not at early time points (Figure 2E), which closely matched the kinetics of the gene expression data. At 48 and 120 hours after infection when HIV-1 infectivity could be determined by p24 staining and flow cytometry (Figure 2B), the HIV-1–infected population showed a consistent 2- to 3-fold increase in IRF-1 expression compared with bystander cells (Figure 2C). Similar results were also observed for IRF-2, -7, and -8 (Figure 2E). Because the MOI of HIV-1 was sufficient for all DCs to be exposed to HIV, which is then endocytosed and destroyed by 48 hours, these results suggest that, although exposure to HIV-1 may be enough to trigger the expression of IRFs, productive infection has a much greater effect.
Table 3.
Differential expression data for genes encoding key IRFs in HSV-2–treated MDDCs
| Symbol | Accession no. | 3 Hours | 6 Hours | 12 Hours |
|---|---|---|---|---|
| IRF-1 | NM_002198 | 9.5 | 41.5 | 34.1 |
| IRF-2 | NM_002199 | 30.5 | 60.9 | 181.2 |
| IRF-3 | NM_001571 | 3.4 | 25.2 | 20.2 |
| IRF-4 | NM_002460 | 10.6 | 28.0 | 27.7 |
| IRF-7 | NM_004031 | 2.5 | 6.4 | 0.1 |
| IRF-8 | NM_002163 | 1.38 | 8.26 | 13.64 |
| IRF-9 | NM_006084 | ND | ND | ND |
Day 6 MDDCs were mock-treated or treated with HSV-2186 (MOI, 3), and IRF gene expression was determined by qPCR. Data are mean fold change in the expression of IRF genes in HSV-2– versus mock-treated cells for 3, 6, and 12 hours after treatment from 4 independent experiments.
ND indicates not determined.
Figure 2.

IRF protein expression after exposure to HIV. Day 6 MDDCs were treated with HIV-1BaL (MOI, 3) or mock-treated for 6-120 hours. (A) IRF-1 intracellular expression levels were determined by flow cytometry; the isotype control is shown as filled curve, mock-treated cells are shown with a solid line and HIV-1–treated cells are shown with a broken line. (B) The percentage of HIV-1–infected cells was determined by flow cytometry with the use of a PE-conjugated p24 antibody and (C) peak IRF-1 expression was determined in p24− cells and p24+ cells separately (means of 5 experiments shown with standard error bars). (D) IRF-1 expression was also determined by Western blot. (E) Panels A to C were repeated for IRF2, IRF-7, and IRF-8. Representative data are shown from 1 of 3 experiments.
Genes up-regulated in response to HIV-1 in MDDCs contain a strong IFN signature and are enriched for IRF-1 binding sites in their promoters
In silico promoter analysis was performed on the (microarray) up-regulated subset of 18 ISGs and compared with 18 genes selected randomly from those in the microarray dataset that were not differentially regulated and also with 18 other ISGs. The up-regulated subset was found to be highly enriched (16 of 18) for statistically significant IRF-1 binding sites within their proximal promoters (P = 4.17 × 10−4 when prior P = .1), suggesting they form an IRF-1–regulated subset of ISGs that are induced during HIV-1 infection (Figure 3).
Figure 3.

In silico promoter analysis of identified up-regulated IFN-associated genes in MDDCs. RefSeq IDs and promoters were extracted with the use of the UCSC genome browser (www.genome.ucsc.edu) and sequences 3000 bp 5′ upstream from the transcription start site, plus the 5′ untranslated region were extracted. Transfac database28 was then used to obtain vertebrate TFBS matrices, and the Toucan2 tool MotifScanner was used to detect potential TFBSs in the sets of selected sequences. The prior (stringency level) was set to a value of 0.05. A small box indicates the location of identified potential IRF-1 binding sites.
IRF-1 is able to drive the expression of HIV-stimulated ISGs
To confirm that IRF-1 is able to drive the expression of the HIV-1–induced ISGs, we transfected 293T cells with the IRF-1 expression vector CMVBL-IRF1-HNK or with empty vector. IRF-1 was highly expressed in the IRF-1–transfected cells only and was able to drive the expression of all 17 HIV-1–stimulated ISGs tested in a dose-dependent manner with the exception of IRF8 (Table 4). Some highly up-regulated examples include ISG15, IFIT2, IFIT3, and RSAD2 (aka viperin). In addition, the expression of 2 ISGs (IFITM3 and IFITM5) that were not up-regulated by HIV treatment in MDDCs was not affected by IRF1 transfection.
Table 4.
qPCR-derived ISG gene expression data for 293T cells transfected with an IRF-1 expression vector
| 1 μg IRF-1 | 5 μg IRF-1 | 10 μg IRF-1 | |
|---|---|---|---|
| IRF-1 | 6.4 × 105 | 6.4 × 107 | 1.0 × 108 |
| OAS1 | 2.7 | 32 | 49 |
| MXA | 11 | 12 | 23 |
| ISG15 | 3781 | 6152 | 1.2 × 104 |
| STAT3 | — | 3.8 | 7.6 |
| DDX48 | 4.5 | 29 | 57 |
| IFIH1 | 25 | 56 | 111 |
| RSAD2 | 2.7 | 27 | 149 |
| IRF-2 | 5.3 | 10 | 53 |
| IRF-4 | 5.1 | 276 | 377 |
| IRF-7 | 38 | 95 | 118 |
| IFIT1 | 6.2 | 21 | 115 |
| IFIT2 | 42 | 1506 | 2572 |
| IFIT3 | 33 | 620 | 709 |
| IFIT5 | 5.5 | 3.2 | 9.8 |
| IFITM1 | 12 | 462 | 468 |
| IFITM3 | 2.4 | 15 | 22 |
| IFI35 | 8.0 | 143 | 105 |
| IFN-α* | — | — | — |
| IFN-β* | — | — | — |
| IFN-γ* | — | — | — |
| IFITM2* | — | — | — |
| IFITM5* | — | — | — |
| IRF-8* | — | — | — |
293T cells were either untreated or transfected with 1, 5 and 10 μg of the IRF1 expression vector CMVBL-IRF1-HNK or empty vector or treated with PEI transfection reagent only. ISG expression was determined by qPCR 48 hours after treatment. The values shown represent fold change in gene expression compared with untransfected cells. Cells treated with PEI or transfected with 1, 5 and 10 μg of empty vector had a fold change < 2.0.
The — indicates a fold change < 2.0.
Genes not up-regulated in their expression in response to IRF1 transfection.
IRF-1 binds to the promoters of HIV-1–induced IFN-regulatory genes in MDDCs
To determine whether IRF-1 binds to the promoters of HIV-1–induced IFN-regulatory genes in vivo a ChIP assay was performed in MDDCs exposed to HIV-1BaL. IRF-1 was shown to bind to the promoters of 2 of the up-regulated ISGs that were chosen for validation (Figure 4), IRF-2 and IFIT5, with the use of an IRF-1–specific antibody and PCR amplification of promoter regions identified in silico to contain IRF-1–binding sites, in MDDCs treated with either HIV-1BaL (lane 2) or IFNγ, a known inducer of IRF-1 (lane 4).
Figure 4.
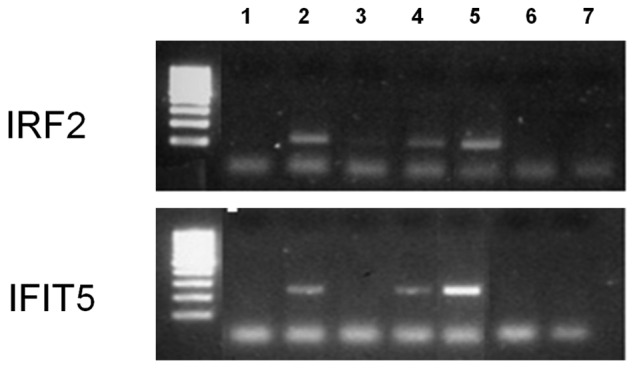
IRF-1 ChIP assay. Day 6 MDDCs were treated with HIV-1BaL or IFNγ for 48 hours. A ChIP assay was then performed with an IRF-1 antibody, and qPCR primers were directed toward IRF-2 or IFIT5 promoter sequences to determine IRF-1 binding. (Lane 1) HIV-1–treated MDDCs no IRF-1 antibody, (lane 2) HIV-1–treated MDDCs plus IRF-1 antibody, (lane 3) IFNγ-treated MDDCs no IRF-1 antibody, (lane 4) IFNγ-treated MDDCs plus IRF-1 antibody, (lane 5) cell input DNA, (lane 6) PCR negative no DNA control, and (lane 7) ChIP reagents only negative control.
Deletion of the ISRE from the HIV LTR region results in reduced infectivity
HIV-1 contains an ISRE in its LTR region with a known binding sequence for IRF-1/7. To determine whether HIV-1 requires IRF-1 binding to this for efficient replication in MDDCs, we next constructed a mutant virus with this ISRE deleted. This virus showed significantly reduced replication kinetics compared with the parental wild type (Figure 5A). To confirm that the virus with the ISRE deleted was less replication competent in DCs, we titrated the ratio of plasmids encoding the wild-type and mutant viruses and infected MDDCs with the progeny virions. The proportion of infected cells decreased as the ratio of HIV mutant plasmid increased compared with wild-type (Figure 5B).
Figure 5.

Deletion of the IRF-1/7 binding site (IRF-1 bs) from the HIV-1 LTR results in reduced infectivity. Day 2 MDDCs were treated with VSVG-pseudotyped HIV-1BaL-IRF-1 bs or HIV-1BaLwt at an MOI of 1 for 6-134 hours either alone in combination at various ratios. (A) The percentage of HIV-1–infected cells was determined by flow cytometry between 48 and 134 hours after infection. The mean data from 3 independent experiments are shown with standard error bars. There was a statistically significant difference between the rates of increase of the percentage of infected cells over time for HIV-1BaLwt versus HIV-1BaL-IRF-1 bs (P = .021 linear mixed-effects model). The mean difference in these infection rates was 0.028% per hour. (B) The greater the ratio of wild-type compared with mutant plasmid transfected, the greater the proportion of cells infected at 48 hours after infection. The mean data from 3 independent experiments are shown with standard error bars.
Exposure of MDDCs to HSV-2 leads to IFN induction
To determine whether the block in (or lack of) IFN induction and the distinct pattern of ISGs and IRFs in DCs was virus specific, we next exposed MDDCs to viable or UV-inactivated HSV-2186 for 30 minutes or 6 hours. Microarray analysis was conducted with the Illumina system to determine differential expression of genes, including ISGs (Table 5). In addition, IFNα and IFNβ levels from the infected cell supernatant fluids were determined by ELISA (Figure 2B). No ISGs were differentially expressed in MDDCs in response to treatment with UV-inactivated HSV-2186 or with viable HSV-2186 for 30 minutes. However, 34 ISGs were up-regulated in MDDCs treated with viable HSV-2186 for 6 hours (including IFNβ) and 20 with UV-inactivated HSV-2186. Nine genes from the list were chosen for validation by qPCR, which confirmed up-regulation in every case, although in most cases qPCR indicated a much greater degree of up-regulation (Table 5). Although many of the detected up-regulated genes were also increased in their expression in MDDCs treated with HIV-1BaL, the fold change increases were far higher in HSV-2186–treated cells. In contrast to cells treated with HIV-1BaL (in which no IFN-encoding genes were up-regulated) the IFNβ gene was markedly up-regulated by 600-fold by qPCR. No IFNα was detected. This was confirmed by ELISA on tissue culture supernatant fluids, which detected the presence of IFNβ but not IFNα in HSV-2 infected MDDC supernatant fluids (Figure 1), indicating induction of de novo IFNs by viral infection. Neither IFNα nor IFNβ was detected in the viral inoculum used for infection (data not shown). Furthermore, the pattern of IRFs induced by HSV-2 was broader than HIV-1, and the early IRF-1/8 and late IRF-1/2 pairings were not observed (Table 3).
Table 5.
ISG expression in HSV-2–treated MDDCs
| Symbol | Accession no. | Microarray (6 hours) | qPCR (6 hours) |
|---|---|---|---|
| IFNB1 | NM_002176 | 14.34/14.39 | 600 |
| OAS1* | NM_016816 | 8.06/1.04 | 18 |
| OAS2 | NM_016817 | 7.96/0.20 | — |
| OAS3 | NM_006187 | 9.25/2.94 | — |
| MX1* | NM_002462 | 6.40/−4.06 | 105 |
| MX2 | NM_002463 | 3.88/1.03 | — |
| PKR* | NM_002759 | 6.11/3.7 | 20 |
| IRF-1* | NM_002198 | 5.45/0.82 | 10.3 |
| IRF-9* | NM_006084 | 2.46/1.62 | 5.5 |
| GBP1 | NM_002053 | 11.90/3.46 | — |
| HERC5 | NM_016323 | 22.74/4.87 | — |
| IFI16 | NM_005531 | 2.27/6.22 | — |
| IFI35* | NM_005533 | 4.93/2.18 | — |
| IFI44 | NM_006417 | 11.99/3.99 | — |
| IFI44L | NM_006820 | 24.35/4.17 | — |
| IFIH1 | NM_022168 | 7.51/4.14 | — |
| IFIT1* | NM_001548 | 30.47/2.94 | 30 |
| IFIT2* | NM_001547 | 30.09/2.26 | 9 |
| IFIT3* | NM_001549 | 38.83/5.78 | — |
| IFIT5* | NM_012420 | 2.38/5.03 | — |
| IFITM1* | NM_003641 | 14.27/2.54 | — |
| IFITM2 | NM_006435 | 5.67/3.41 | — |
| IFITM3* | NM_021034 | 6.75/3.65 | — |
| IL27 | NM_145659 | 4.28/1.59 | — |
| ISG15* | NM_005101 | 13.72/2.40 | — |
| ISG20 | NM_002201 | 33.32/6.48 | — |
| JAK2 | NM_004972 | 2.40/0.82 | — |
| LOC400759 | XR_000992 | 6.05/3.19 | — |
| PSMB9* | NM_002800 | 2.25/4.46 | — |
| SP110* | NM_080424 | 2.40/2.45 | — |
| STAT1* | NM_139266 | 7.34/2.41 | — |
| UBA7 | NM_003335 | 2.52/2.09 | — |
| DDX58* | NM_014314 | 11.78/3.54 | — |
| DDX60 | NM_017631 | 5.29/3.45 | — |
| DHX58 | NM_017631 | 4.44/0.65 | — |
| WARS* | NM_004184 | 2.71/1.00 | — |
Day 6 MDDCs were treated with HSV-2186 (MOI, 3) for 30 minutes or 6 hours, and ISG expression was determined by 48K Illumina microarrays or qPCR (6 hours only). Expression data from 3 independent experiments are presented for HSV-2– versus mock-treated cells. In cells that were treated for 30 minutes, the ISG expression was determined by qPCR. Data are fold change ± B value.
The — indicates that qPCR was performed to confirm the expression of this gene.
Genes were also detected to be increased in their expression in MDDCs exposed to HIV-1BaL (Table 1).
Suppression of IFN induction is mediated by Vpr and by inhibiting activation but not degradation of IRF3
Recently, the HIV accessory proteins Vpr16,17 and Vif17 have been shown to be key mediators in the suppression of IFN induction in T cells by inducing the degradation of IRF-3. To determine whether these virally encoded proteins are also involved in suppression of IFN induction in MDDCs, we prepared HIV-1 viral stocks with the Vpr, Vif, or both Vpr and Vif genes deleted. As shown in Figure 6A, the Vpr single deletion mutant and the Vpr, Vif double deletion mutant induced IFNβ mRNA expression in MDDCs at both 48 and 96 hours. In contrast, no IFNβ was induced in response to wild-type HIV-1 or to the Vif deletion mutant.
Figure 6.

IFN induction in MDDCs by HIV viruses with Vpr but not Vif deleted from their genome. (A) Day 2 MDDCs were treated with VSVG-pseudotyped HIV-1NLAD8ΔVpr, HIV-1NLAD8ΔVif, HIV-1NLAD8ΔVpr,Vif, or HIV-1NLAD8 at an MOI of 1 for 48 and 96 hours. (A) IFNβ mRNA expression was determined by qPCR. The mean data from 3 independent donors are shown with standard error bars. (B-D) Day 6 MDDCs were treated with HIV-1BaL or mock-treated for 24 and 48 hours. (B) IRF-3 intracellular expression levels were determined by flow cytometry; the isotype control is shown as filled curve, mock treated cells are shown with a solid line, and HIV-1–treated cells are shown with a broken line. (C) The percentage of HIV-1–infected cells was determined by flow cytometry with the use of a PE-conjugated p24 antibody, and IRF-3 expression was determined in p24− cells and p24+ cells separately (mean of 5 experiments shown with standard error bars). (D-E) IRF-3 expression in response to HIV-1BaL was also determined by Western blot in both MDDCs (D) and SupT1 cells (E). (F) IRF3 cellular localization was determined by confocal microscopy in mock- and HIV-1–treated cells compared with Sendai virus–treated TZM-bl cells.
To examine the mechanism of inhibition of IFNβ production, we next determined whether IRF-3 was degraded in MDDCs infected with HIV-1 as in T cells or, alternatively whether its activation was impaired, as shown by failure to translocate to the nucleus. No down-regulation of IRF-3 was shown either by Western blot or flow cytometry (Figure 6B). However, IRF-3 did not translocate to the nucleus of MDDCs after HIV-1 infection in contrast to positive Sendai virus–infected TZM-bl cells36 (Figure 6C).
Discussion
HIV-1 capture and infection by DCs induces changes in clusters of genes that represent either responses of the cell to the virus or viral manipulations of the intracellular environment. The latter assist in viral replication and, in the case of DCs, transfer to T cells. We and others have found that the clusters of genes induced by HIV-1 in the major target cells, T cells, macrophages, and DCs, differ substantially between each cell type, although there is greater similarity between DCs and macrophages5,15,37–40 (Susan Maddocks, unpublished observations, August 2005). As recently reported, expression of 2 major groups of genes were transiently and sequentially up-regulated in DCs by HIV, the first peaking at 6-24 hours after infection and the second at 48 hours after infection, correlating with and probably because of the 2 phases of viral trafficking: the vesicular endosomal phase with declining intracellular HIV concentrations over 24 hours and de novo viral replication with HIV DNA appearing at 24 hours after infection and plateauing at 48-96 hours after infection.15 A minor group of 30 genes with up-regulated expression extends across the 2 phases. In this study we have shown that HIV-1 induces a specific subset of ISGs originally identified by DNA microarray studies and confirmed and broadened by downstream analysis initially by qPCR and then by protein studies, including Western blot and flow cytometry.
Induction of ISGs is a common feature of the innate response of many cells to viral infection, but here there were unusual features: First, up-regulation of more than one-half of the ISG subset was not transient but extended across the 2 phases of trafficking (ie, part of the minor group), suggesting a sustained effect. Second, no type I or type II IFNs could be detected, either in the supernatant fluid by ELISA or by extensive qPCR assays of all IFNα subtypes, as well as IFNβ and IFNγ from infected cell lysates at serial time points. Third, a specific subset of ISGs was induced (especially compared with HSV-2 infection), which contained an unusual pattern of up-regulation of key IRFs, particularly at early time points, consisting of IRF-1, -7, and -8 at 6 hours after infection and later IRF-1, -2, -7, -4, and -9. Induction of IRF-1 and -8 and later IRF-2 constitute an unusual pattern in that IRF-1 is much less well characterized than IRF-3 or -7 for IFN and ISG induction. IRF-2 and -8 can both inhibit IRF-1–mediated induction of transcription. IRF-2 competes with IRF-1 for its binding to the cellular promoter binding site,41,42 whereas IRF-8 does not bind DNA but rather forms a complex with IRF-1 that inhibits its transcriptional activation activity by blocking protein:protein interactions.34
Comparisons between the effect of HIV-1 and HSV-2 infection of DCs was conducted to determine whether these effects were virus specific. HSV-2 was chosen because it had been shown in the laboratory to productively infect MDDCs and induce IFN production as also shown here.43 Furthermore, there is a pathogenetic relationship between HIV-1 and HSV-2 in that both have been shown to coexist in the same recurrent herpetic lesion.44 Thus, coinfection or adjacent infection of epidermal and perhaps dermal DCs in these lesions with these viruses is probable. In these studies highly purified viral preparations and similar multiplicities of each virus were used over a range of 1-10 per cell. In contrast to HIV-1, HSV-2 infection of DCs induced functional IFNβ protein in the infected DC supernatant fluids, and IFNβ transcripts were induced as early as 3 hours after infection. Furthermore, a much broader range of ISGs was induced and at a much higher level. IFN-induced chemokines such as CXCL10 and CXCL1145,46 were also more prominent. These findings of marked induction of a type I IFN in HSV-2–infected MDDCs build on those of Pollara et al.43
The unusual subset of ISGs and particularly the unusual pattern of IRFs induced by viable HIV-1 in DCs raises several questions. What is the mechanism of the induction of such ISGs and of the failure of induction of type I or II IFNs? Does this altered pattern of ISGs benefit the host, the virus, or both? What components of the virus and cellular signaling pathways does HIV-1 use to induce such an altered pattern? A clue to the mechanism of induction of the subset of ISGs induced by HIV-1 in DCs was shown by heavy and significant weighting toward those containing an IRF-1/7 binding site in their promoters, initially identified by the INTERFEROME database and associated bioinformatics tools and later confirmed by ChIP assays.27 The ability of IRF1 to induce the expression of this specific ISG subset was shown after transfection of an IRF-1 expression vector into a 293T cell line in the absence of any IFN induction. The only exception was IRF-8, which is also induced early after infection similar to IRF-1, so this is not surprising. By analogy VSV has recently been reported to induce the antiviral ISG viperin via IRF1 and in the absence of type I IFNs (as we show here in DCs). In contrast Newcastle disease virus induces viperin by a non–IRF1-dependent pathway.47
The early kinetics of induction of IRF-1, -2, -7, and -8 provide a clue to the role of the specific IRFs induced by HIV-1. The combination of IRF-1, -2, and -8 may negate any IFN-inducing effects of IRF-1, because IRF-2 and -8 can both act to inhibit the transcriptional activity of IRF-1 and appear to be complimentary in their kinetics, one being induced early (IRF-8) and the other late (IRF-2). Furthermore, the LTR of most HIV-1 isolates has an IRF-1/7 binding site (the ISRE) adjacent to the U5 region between the NF-AT and SP-1 sites.48 In T cells Sgarbanti et al34 have shown that IRF-1 and -2 can bind to this site and that IRF-1 also forms complexes with the p65/50 NF-κB heterodimer49 to bind to upstream sites of their target genes. They showed that early IRF-1 induction by HIV-1 in de novo infection of T cells stimulates early HIV-1 transcription before Tat induction and also that IRF-2 is not inhibitory to this effect, in contrast to cellular gene promoters, rather it was IRF-8 that was responsible for the inhibitory effects on IRF-1.34,35,49 The failure of a cellular ISRE to substitute functionally for the HIV (LTR) ISRE suggests that they are differentially regulated by IRF-1 or IRF-2/8.49 Thus, in DCs the IRF-1/2/8 combination might selectively stimulate the HIV-1 LTR by either the IRF-1 or NF-κB binding site. To test the functional requirements of the HIV-1 IRF-1/7 binding site for viral replication we constructed an HIV-1 mutant with this site deleted. This resulted in a significant 20%-30% decrease in infectivity in DCs by 120 hours after infection. Combination of wild type and deletion mutant in various ratios further showed and confirmed the lower infectivity of the mutant virus. This system may be especially important in DCs in which de novo infection occurs at lower levels than in macrophages or T cells partly because most virus binding occurs by C-type lectin receptors on the surface of DCs and is rapidly endocytosed and degraded by the endocytic pathway. IRF-1 may also provide early stimulation in DCs before later production of Tat as in T cells. IRF-1 also appears to be driving the induction of the ISG subset observed in this study, explaining their induction without the detectible presence of IFN. A strong correlation between reduced susceptibility to HIV infection and IRF-1 polymorphisms and consequent aberrant patterns of IRF-1 induction in PBMCs by IFN-γ in Kenyan sex workers has been reported recently.50 In particular transient rather than sustained IRF-1 patterns were shown. If this is also occurring in the genital mucosal DCs after HIV exposure, it may contribute to the observed reduction in HIV acquisition.
The early and sustained induction of IRF-7 is also probably important. Its induction in the absence of IFNs suggests a parallel with dengue virus, which also induces IRF-7 in the absence of type 1 IFNs.51 IRF-3 and -7 are the 2 most important inducers of type I IFNs in most cell types and are activated by interactions of viral RNA with Toll-like receptors in the endosome or by cytosolic retinoic acid–inducible gene I–like receptor pathways, resulting in their phosphorylation, homodimerization, or heterodimerization with IRF-3 and translocation to the nucleus. In myeloid DCs IRF-7 concentrations are constitutively low, perhaps explaining the need for up-regulation by HIV-1. However, the reason for early induction of IRF-7 is not clear. Does IRF-7 also stimulate HIV replication? Furthermore, if IRF-7 is activated normally, why is there no IFN induction? In T cells the HIV-1 accessory proteins Vpr and Vif have been shown to ubiquitinate IRF-3, which leads to it being targeted to the proteasome and degraded, resulting in the inhibition of type I IFN induction.16,17 Thus, we pursued the hypothesis that HIV vpr or vif proteins may also be involved in the inhibition of IFN induction in DCs. We therefore constructed Vpr and Vif deletion mutants and showed that deletion of Vpr (but not Vif) did indeed result in a virus that had the ability to induce IFNβ in DCs. However, we did not observe down-regulation of IRF-3 protein, as has been shown in T cells.16,17 This strongly suggests that Vpr might be enhancing ubiquitination and degradation of other enzymes involved in phosphorylation and activation of IRF-3 or -7 or both. Because IRF3 did not translocate to the nucleus, this indicates inhibition of its activation most probably by a failure of phosphorylation of key upstream enzymes such as TANK-binding kinase 1 or IκB kinase ϵ.
Some of the HIV-induced ISGs have intrinsic unique antiviral actions, such as MxA, OAS1-3, and ISG15, and may substitute for absent type I IFNs by restricting HIV-1 replication to low levels, as previously characterized.52–54 The much higher levels and broader range of ISGs induced in DCs infected by HSV-2 are clearly a response to the very high levels of IFNβ induced by ISGF3/STAT pathways, not solely a response to IRF-1. Although the biology of HIV and HSV infection of DCs is quite different, they provide a reasonable comparison and contrast with the HIV-1–induced subset of ISGs. Interestingly, the subset of ISGs induced by HIV-1 in DCs is similar but not identical to that observed in macrophages in which IRF-1 is similarly induced but IRF-2 and -8 were not prominent (data not shown). The pattern of antiviral ISGs also showed some differences. However, the pattern of ISGs induced in T cells was more markedly different15,37–40 (Susan Maddocks, unpublished observations, August 2005).
The direct stimulation of a unique ISG subset by HIV-1 contrasts with different patterns of ISG stimulated by other viruses and by HIV in other cell types (Maddocks et al, unpublished data).47 This suggests direct viral modulation to enable successful HIV-1 transfer to T cells, their primary target cell, by viral synapses in the genital mucosa and other body sites. The IRF-1/2/8 combination may provide an early stimulus to the small inoculum of HIV-1 delivered into the cytoplasm of DCs while restricting viral replication through the antiviral ISGs to retain the integrity of the cell until it reaches the submucosal T lymphocytes or lymph nodes. Here, virus is transferred by the viral synapses. Now that this novel ISG and IFN pattern, distinct from that in T cells, has been identified, their mechanisms and role in maintaining the HIV-DC equilibrium can also be further investigated, focusing on IRF-1/2/8 and IRF-3/7 induction and activation and also inhibition of IFN induction. The patterns of ISG and IRF induction in DCs from patients with “resistant” and “sensitive” IRF1 genotypes also require urgent investigation.
Acknowledgment
We thank Chris Bye for his help with microarray analysis of cDNA microarrays.
This work was supported by the National Health and Medical Research Council (NHMRC; program grant 358399).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: A.N.H. prepared the manuscript and conducted the bulk of the experimental work with technical assistance from S.M.; J.L. conducted all experiments involving HSV-2; S.T. prepared HIV deletion mutant virus stocks; S.S. conducted the in silico promoter analysis; L.G. generated and provided the HIV-1 ISRE site deletion mutant; M.C. provided academic input into the design of the IRF-1 binding site HIV-1 deletion mutant; V.M. prepared all HSV-2 virus stocks and performed confocal microscopy; K.J. constructed and provided the HIV-1 Vpr and Vif deletion mutants; J.M. provided academic input into the design of the HIV-1 Vpr and Vif deletion mutant studies; N.N. grew many of the HIV stocks used in much of this study; H.C. conducted the IRF-1 ChIP assay; H.D. conducted interferon ELISA assays and provided intellectual input; M.G. and A.R. provided key reagents developed in their laboratory (anti IRF3 antibody) and M.G. provided key intellectual input; P.H. provided academic input into the design of the in silico promoter analysis and ChIP assays; and A.L.C. conceived and supervised the study and jointly prepared the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Anthony L. Cunningham, Centre for Virus Research, Westmead Millennium Institute, PO Box 412, Darcy Rd, Westmead, NSW 2145, Australia; e-mail: tony_cunningham@wmi.usyd.edu.au.
References
- 1.Hu J, Gardner MB, Miller CJ. Simian immunodeficiency virus rapidly penetrates the cervicovaginal mucosa after intravaginal inoculation and infects intraepithelial dendritic cells. J Virol. 2000;74(13):6087–6095. doi: 10.1128/jvi.74.13.6087-6095.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sewell AK, Price DA. Dendritic cells and transmission of HIV-1. Trends Immunol. 2001;22(4):173–175. doi: 10.1016/s1471-4906(01)01866-x. [DOI] [PubMed] [Google Scholar]
- 3.Knight SC. Dendritic cells and HIV infection: immunity with viral transmission versus compromised cellular immunity? Immunobiology. 2001;204(5):614–621. doi: 10.1078/0171-2985-00100. [DOI] [PubMed] [Google Scholar]
- 4.Turville SG, Cameron PU, Arthos J. Bitter-sweet symphony: defining the role of dendritic cell gp120 receptors in HIV infection. J Clin Virol. 2001;22(3):229–239. doi: 10.1016/s1386-6532(01)00194-9. [DOI] [PubMed] [Google Scholar]
- 5.Harman AN, Wilkinson J, Bye CR, et al. HIV induces maturation of monocyte-derived dendritic cells and Langerhans cells. J Immunol. 2006;177(10):7103–7113. doi: 10.4049/jimmunol.177.10.7103. [DOI] [PubMed] [Google Scholar]
- 6.de Witte L, Nabatov A, Pion M, et al. Langerin is a natural barrier to HIV-1 transmission by Langerhans cells. Nat Med. 2007;13(3):367–371. doi: 10.1038/nm1541. [DOI] [PubMed] [Google Scholar]
- 7.Kawamura T, Koyanagi Y, Nakamuara Y, et al. Significant virus replication in Langerhans cells following application of HIV to abraded skin: relevance to occupational transmission of HIV. J Immunol. 2008;180(5):3297–3304. doi: 10.4049/jimmunol.180.5.3297. [DOI] [PubMed] [Google Scholar]
- 8.Turville SG, Arthos J, Donald KM, et al. HIV gp120 receptors on human dendritic cells. Blood. 2001;98(8):2482–2488. doi: 10.1182/blood.v98.8.2482. [DOI] [PubMed] [Google Scholar]
- 9.Sugaya M, Lore K, Koup RA, Douek DC, Blauvelt A. HIV-infected Langerhans cells preferentially transmit virus to proliferating autologous CD4+ memory T cells located within Langerhans cell-T cell clusters. J Immunol. 2004;172(4):2219–2224. doi: 10.4049/jimmunol.172.4.2219. [DOI] [PubMed] [Google Scholar]
- 10.Turville SG, Santos JJ, Frank I, et al. Immunodeficiency virus uptake, turnover, and 2-phase transfer in human dendritic cells. Blood. 2004;103(6):2170–2179. doi: 10.1182/blood-2003-09-3129. [DOI] [PubMed] [Google Scholar]
- 11.Hladik F, Sakchalathorn P, Ballweber L, et al. Initial events in establishing vaginal entry and infection by human immunodeficiency virus type-1. Immunity. 2007;26(2):257–70. doi: 10.1016/j.immuni.2007.01.007. 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McDonald D, Wu L, Bohks SM, KewelRamani VN, Unutmaz D, Hope TJ. Recruitment of HIV and its receptors to dendritic cell-T cell junctions. Science. 2003;300(5623):1295–1297. doi: 10.1126/science.1084238. [DOI] [PubMed] [Google Scholar]
- 13.Weissman D, Rabin RL, Arthos J, et al. Macrophage-tropic HIV and SIV envelope proteins induce a signal through the CCR5 chemokine receptor. Nature. 1997;389(6654):981–985. doi: 10.1038/40173. [DOI] [PubMed] [Google Scholar]
- 14.Sodhi A, Montaner S, Gutkind JS. Viral hijacking of G-protein-coupled-receptor signalling networks. Nat Rev Mol Cell Biol. 2004;5(12):998–1012. doi: 10.1038/nrm1529. [DOI] [PubMed] [Google Scholar]
- 15.Harman AN, Kraus M, Bye CR, et al. HIV-1-infected dendritic cells show 2 phases of gene expression changes, with lysosomal enzyme activity decreased during the second phase. Blood. 2009;114(1):85–94. doi: 10.1182/blood-2008-12-194845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doehle BP, Hladik F, McNevin JP, et al. Human immunodeficiency virus type 1 mediates global disruption of innate antiviral signaling and immune defenses within infected cells. J Virol. 2009;83(20):10395–10405. doi: 10.1128/JVI.00849-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Okumura A, Alce T, Lubyova B, Ezelle H, Strebel K, Pitha PM. HIV-1 accessory proteins VPR and Vif modulate antiviral response by targeting IRF-3 for degradation. Virology. 2008;373(1):85–97. doi: 10.1016/j.virol.2007.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ball TB, Ji H, Kimani J, et al. Polymorphisms in IRF-1 associated with resistance to HIV-1 infection in highly exposed uninfected Kenyan sex workers. AIDS. 2007;21(9):1091–1101. doi: 10.1097/QAD.0b013e3280ef6ae1. [DOI] [PubMed] [Google Scholar]
- 19.Chertova E, Bess JW, Jr, Crise BJ, et al. Envelope glycoprotein incorporation, not shedding of surface envelope glycoprotein (gp120/SU), Is the primary determinant of SU content of purified human immunodeficiency virus type 1 and simian immunodeficiency virus. J Virol. 2002;76(11):5315–5325. doi: 10.1128/JVI.76.11.5315-5325.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koup RA, Ho DD, Poli G, Fauci AS. Isolation and quantitation of HIV in peripheral blood. Curr Protoc Immunol. 2001 doi: 10.1002/0471142735.im1202s05. Chapter 12:Unit 12 2. [DOI] [PubMed] [Google Scholar]
- 21.Zeichner SL, Kim JY, Alwine JC. Linker-scanning mutational analysis of the transcriptional activity of the human immunodeficiency virus type 1 long terminal repeat. J Virol. 1991;65(5):2436–2444. doi: 10.1128/jvi.65.5.2436-2444.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Donaghy H, Bosnjak L, Harman AN, et al. A role for plasmacytoid dendritic cells in the immune control of human recurrent herpes simplex. J Virol. 2009;83(4):1952–1961. doi: 10.1128/JVI.01578-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Watson S, Mercier S, Bye C, Wilkinson J, Cunningham AL, Hraman AN. Determination of suitable housekeeping genes for normalisation of quantitative real time PCR analysis of cells infected with human immunodeficiency virus and herpes viruses. Virol J. 2007;4:130. doi: 10.1186/1743-422X-4-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Genome Bioinformatics Group of UC Santa Cruz. UCSC Genome Browser. [Accessed August 2008]. http://www.genome.ucsc.edu.
- 26.European Bioinformatics Institute and Wellcome Trust Sanger Institute. Ensembl Genome Browser. [Accessed August 2008]. http://www.ensembl.org.
- 27.Samarajiwa SA, Forster S, Auchettl K, Hertzog PJ. INTERFEROME: the database of interferon regulated genes. Nucleic Acids Res. 2008;37:D852–D857. doi: 10.1093/nar/gkn732. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wingender E, Chen X, Fricke E, et al. The TRANSFAC system on gene expression regulation. Nucleic Acids Res. 2001;29(1):281–283. doi: 10.1093/nar/29.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Swiss Institute of Bioinformatics. [Accessed August 2008];Eukaryotic Promoter Database. http://www.epd.isb-sib.ch/ [Google Scholar]
- 30.Aerts S, Van Loo P, Thijs G, et al. TOUCAN 2: the all-inclusive open source workbench for regulatory sequence analysis. Nucleic Acids Res. 2005;33:W393–W396. doi: 10.1093/nar/gki354. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Turville SG, Aravantinou M, Stossel H, Romani N, Robbiani M. Resolution of de novo HIV production and trafficking in immature dendritic cells. Nat Methods. 2008;5(1):75–85. doi: 10.1038/nmeth1137. [DOI] [PubMed] [Google Scholar]
- 32.Gough DJ, Messina NL, Hii L, et al. Functional crosstalk between type I and II interferon through the regulated expression of STAT1. PLoS Biol. 2010;8(4):e1000361. doi: 10.1371/journal.pbio.1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marsili G, Remoli AL, Sqarbanti M, Battistina A. Role of acetylases and deacetylase inhibitors in IRF-1-mediated HIV-1 long terminal repeat transcription. Ann N Y Acad Sci. 2004;1030:636–643. doi: 10.1196/annals.1329.074. [DOI] [PubMed] [Google Scholar]
- 34.Sgarbanti M, Borsetti A, Moscufo N, et al. Modulation of human immunodeficiency virus 1 replication by interferon regulatory factors. J Exp Med. 2002;195(10):1359–1370. doi: 10.1084/jem.20010753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sgarbanti M, Marsili G, Remoli AL, et al. Analysis of the signal transduction pathway leading to human immunodeficiency virus-1-induced interferon regulatory factor-1 upregulation. Ann N Y Acad Sci. 2004;1030:187–195. doi: 10.1196/annals.1329.024. [DOI] [PubMed] [Google Scholar]
- 36.Loo YM, Fornek J, Crochet N, et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82(1):335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coberley CR, Kohler JJ, Brown JN, et al. Impact on genetic networks in human macrophages by a CCR5 strain of human immunodeficiency virus type 1. J Virol. 2004;78(21):11477–11486. doi: 10.1128/JVI.78.21.11477-11486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geiss GK, Bumgarner RE, An MC, et al. Large-scale monitoring of host cell gene expression during HIV-1 infection using cDNA microarrays. Virology. 2000;266(1):8–16. doi: 10.1006/viro.1999.0044. [DOI] [PubMed] [Google Scholar]
- 39.Izmailova E, Bertley FM, Huang Q, et al. HIV-1 Tat reprograms immature dendritic cells to express chemoattractants for activated T cells and macrophages. Nat Med. 2003;9(2):191–197. doi: 10.1038/nm822. [DOI] [PubMed] [Google Scholar]
- 40.van 't Wout AB, Lehrman GK, Mikheeva SA, et al. Cellular gene expression upon human immunodeficiency virus type 1 infection of CD4(+)-T-cell lines. J Virol. 2003;77(2):1392–1402. doi: 10.1128/JVI.77.2.1392-1402.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harada H, Willison K, Sakakibara J, Miyamoto M, Fujita T, Taniquchi T. Absence of the type I IFN system in EC cells: transcriptional activator (IRF-1) and repressor (IRF-2) genes are developmentally regulated. Cell. 1990;63(2):303–312. doi: 10.1016/0092-8674(90)90163-9. [DOI] [PubMed] [Google Scholar]
- 42.Hida S, Ogasawara K, Sato K, et al. CD8(+) T cell-mediated skin disease in mice lacking IRF-2, the transcriptional attenuator of interferon-alpha/beta signaling. Immunity. 2000;13(5):643–655. doi: 10.1016/s1074-7613(00)00064-9. [DOI] [PubMed] [Google Scholar]
- 43.Pollara G, Jones M, Handley ME, et al. Herpes simplex virus type-1-induced activation of myeloid dendritic cells: the roles of virus cell interaction and paracrine type I IFN secretion. J Immunol. 2004;173(6):4108–4119. doi: 10.4049/jimmunol.173.6.4108. [DOI] [PubMed] [Google Scholar]
- 44.Schacker T, Zeh J, Hu H, Shaughnessy M, Corey L. Changes in plasma human immunodeficiency virus type 1 RNA associated with herpes simplex virus reactivation and suppression. J Infect Dis. 2002;186(12):1718–1725. doi: 10.1086/345771. [DOI] [PubMed] [Google Scholar]
- 45.Nakaya T, Sata M, Hata N, et al. Gene induction pathways mediated by distinct IRFs during viral infection. Biochem Biophys Res Commun. 2001;283(5):1150–1156. doi: 10.1006/bbrc.2001.4913. [DOI] [PubMed] [Google Scholar]
- 46.Ogawa S, Lozach J, Benner C, et al. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell. 2005;122(5):707–721. doi: 10.1016/j.cell.2005.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stirnweiss A, Ksienzyk A, Klages K, et al. IFN regulatory factor-1 bypasses IFN-mediated antiviral effects through viperin gene induction. J Immunol. 2010;184(9):5179–5185. doi: 10.4049/jimmunol.0902264. [DOI] [PubMed] [Google Scholar]
- 48.Van Lint C, Amella CA, Emiliani S, John M, Jie T, Verdin E. Transcription factor binding sites downstream of the human immunodeficiency virus type 1 transcription start site are important for virus infectivity. J Virol. 1997;71(8):6113–6127. doi: 10.1128/jvi.71.8.6113-6127.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sgarbanti M, Remoli AL, Marsili G, et al. IRF-1 is required for full NF-kappaB transcriptional activity at the human immunodeficiency virus type 1 long terminal repeat enhancer. J Virol. 2008;82(7):3632–3641. doi: 10.1128/JVI.00599-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Su RC, Sivro A, Kimani J, Jaoko W, Plummer FA, Ball TB. Epigenetic control of IRF1 responses in HIV-exposed seronegative versus HIV-susceptible individuals. Blood. doi: 10.1182/blood-2010-10-312462. Prepublished on January 3, 2011, as DOI 10.1182/blood-2010-10-312462. (Now available as Blood. 2011;117(9):2649-2657) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Warke RV, Becerra A, Zawadzka A, et al. Efficient dengue virus (DENV) infection of human muscle satellite cells upregulates type I interferon response genes and differentially modulates MHC I expression on bystander and DENV-infected cells. J Gen Virol. 2008;89(Pt 7):1605–1615. doi: 10.1099/vir.0.2008/000968-0. [DOI] [PubMed] [Google Scholar]
- 52.Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol. 2008;8(7):559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Trapp S, Derby NR, Singer R, et al. Double-stranded RNA analog poly(I:C) inhibits human immunodeficiency virus amplification in dendritic cells via type I interferon-mediated activation of APOBEC3G. J Virol. 2009;83(2):884–895. doi: 10.1128/JVI.00023-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pion M, Granelli-Piperno A, Mangeat B, et al. APOBEC3G/3F mediates intrinsic resistance of monocyte-derived dendritic cells to HIV-1 infection. J Exp Med. 2006;203(13):2887–2893. doi: 10.1084/jem.20061519. [DOI] [PMC free article] [PubMed] [Google Scholar]