

Summary
Elucidating the molecular mechanisms underlying quantitative neurocognitive phenotypes will further our understanding of the brain’s structural and functional architecture and advance the diagnosis and treatment of the psychiatric disorders that these traits underlie. Although many neurocognitive traits are highly heritable, little progress has been made in identifying genetic variants unequivocally associated with these phenotypes. A major obstacle to such progress is the difficulty in identifying heritable neurocognitive measures which are precisely defined, systematically assessed and represent unambiguous mental constructs, yet are amenable to the high-throughput phenotyping necessary to obtain adequate power for genetic association studies. In this perspective we compare the current status of genetic investigations of neurocognitive phenotypes to that of other categories of biomedically relevant traits and suggest strategies for genetically dissecting traits that may underlie disorders of brain and behavior.
Genetic investigations of neurocognitive phenotypes aim to elucidate mechanisms underlying cognitive function and behavior, and to further our understanding of the pathogenesis of neuropsychiatric disorders. A series of advances in genomics and bioinformatics have transformed human genetics over the past decade. For example, when genome wide association studies (GWAS) began, in about 2005, only a few genes associated with complex human traits had yet been identified. By 2010, GWAS had detected almost one thousand distinct genetic loci unequivocally associated with one or more such traits, i.e., having achieved a commonly accepted statistical threshold for genome wide significance (p < 5 × 10-8) and having been replicated in at least one independent sample (http://www.genome.gov/gwastudies/). During this period cognitive neuroscience has also experienced a technology-driven transformation; for example, the proliferation of various neuroimaging modalities has led to the delineation of a wide and still growing array of novel phenotypes. The apparent high heritability of many of these measures has in turn led to intense efforts to identify the responsible genetic loci. While several putative neurocognitive trait associations have been the focus of considerable attention, human geneticists have accepted almost none of them as having achieved validation, given the widely agreed upon statistical threshold noted above. At the same time, the genome wide approaches, which have been so clearly validated for a wide variety of complex traits, have not yet yielded clear associations for neurocognitive phenotypes. There is thus currently a disconnect between the fields of human genetics and cognitive neuroscience. In this perspective we discuss the basis for this gap and suggest strategies for bridging it. We illustrate key points by comparing and contrasting investigations of several prominent neurocognitive phenotypes with genetic studies of various “medical” traits. A comprehensive review of neurocognitive measures is, however, beyond our purview
Genetic architecture and genetic dissection of complex phenotypes
Understanding the challenges involved in genetic elucidation of neurocognitive phenotypes requires an appreciation of the current state of knowledge regarding the genetic architecture of human phenotypes generally. Among the greatest scientific advances of the past few decades has been the mapping of human genomes at increasingly high resolution, from the discovery of the first genetic markers, to the current prospect of complete sequencing on a population-wide scale. Accompanying this progress has been an increasingly nuanced appreciation of the relationship between human genotypic and phenotypic variation. In the late 1980’s and early 1990’s, genetic mapping studies identified mutations that precisely predicted risk for numerous rare disorders with Mendelian inheritance patterns, with Huntington’s disease being perhaps the most widely known example within neuropsychiatry (The Huntington’s Disease Collaborative Research Group, 1993). Mapping such mutations using whole genome linkage analysis and fine-mapping association analysis became straightforward once genomics efforts had identified sufficient numbers of polymorphic markers (Freimer and Sabatti, 2004). The spectacular successes of these studies created an expectation that similarly predictive mutations could explain risk for common diseases, and could be identified using similar methods. This expectation proved incorrect. Despite extensive efforts by numerous investigators, genetic mapping studies failed to implicate any such mutations in the causation of common disorders, with the notable exception of a few instances in which clearly unusual families demonstrated Mendelian transmission of a particularly distinctive phenotypic variant. Examples within neurogenetics include a rare familial form of migraine accompanied by additional phenotypes (hemiplegia and cerebellar atrophy) and accounted for by dominant mutations in the calcium channel gene CACNL1A4 (Ophoff et al., 1996) or Familial Alzheimer Disease. In the latter example, the discovery that dominant mutations in one of three genes (APP, PS1, and PS2) are implicated in early onset of the disorder in a small number of families has helped transform our understanding of biological processes underlying neurodegeneration (Selkoe and Podlisny, 2002).
The recognition in the 1990s that simple models of genetic causation could not explain the inherited risk for common diseases changed the direction of human genetics, emphasizing two ways of conceptualizing their investigation that remain central to the field today. On the one hand, it focused attention on characterizing the genetic architecture of common diseases, i.e. determining what number and combination of genetic variants of particular types are required to account for disease risk in the population (Sing et al., 1996; Singleton et al., 2010). On the other hand, it led to a realization that common diseases required genetic dissection, i.e. that they are etiologically heterogeneous conglomerations, which can be reduced to more homogeneous forms consequent to the discovery of risk variants (Ehrenreich et al., 2010; Lander and Schork, 1994). The process of genetic dissection thus characterizes the phenotypic architecture of common disorders.
In the last few years, genome-level investigations have generated for some common disorders, such as type 2 diabetes (T2D) and obesity, extraordinary advances in characterizing their genetic architecture and the initial steps in their genetic dissection. In a small number of pedigrees, disorders with strong clinical similarities to T2D and common forms of obesity have been associated unequivocally with Mendelian mutations in one of several genes; mature onset diabetes of the young (MODY) is a prominent example (O’Rahilly, 2009). For both phenotypes, GWAS of extensive population samples have uncovered significant replicated associations at a genome wide significance threshold, of common single nucleotide polymorphisms (SNPS) representing several dozen distinct genetic loci (O’Rahilly, 2009). Some of these loci (e.g. GCKR) show associations to both T2D and obesity (Diabetes Genetics Initiative, 2007), as well as to quantitative phenotypes such as serum lipid levels, which are highly correlated with them (Diabetes Genetics Initiative, 2007; Teslovich et al., 2010).
These numerous genome wide significant findings have served to validate the consensus standards which emerged within human genetics at the onset of the GWAS era: genetic association studies of complex traits should assay the entire genome, and require stringent genome wide significance thresholds as well as statistically significant replication in independent samples. However, the majority of published studies associating genetic variation with neurocognitive phenotypes has continued to rely upon investigations limited to one or a few hypothesis-based candidate genes, despite the well-known problems with this approach (Freimer and Sabatti, 2004; Glatt and Freimer, 2002; Potkin et al., 2010; Tabor et al., 2002). The insistence of human geneticists on genome wide significance thresholds for association studies reflects the extremely low prior probability that a single gene, or a single variant within that gene, out of the approximately 20,000 human genes (and their multiple variants) is significantly associated with the chosen phenotype (Freimer and Sabatti, 2004; Hariri, 2009). One justification of candidate gene approaches is that genes chosen on the basis of existing hypotheses could be considered to have a higher prior probability compared to random genes. Yet there is little to suggest that our current knowledge of the biological pathways involved in neurocognitive function provides a sufficient basis to make strongly motivated hypotheses regarding which genes are associated with these functions. Such hypothesizing is further complicated by an even greater lack of understanding of the potential effects of regulatory mechanisms, epigenetic influences, and environmental factors on variation in complex phenotypes. The notorious failures of replication in candidate gene association studies further emphasizes the fact that most such studies have been inadequately powered to detect associations that realistically would be expected to be of relatively small effect. We are not aware of any evidence that supports a contention that genome wide significance thresholds are less applicable to neuroimaging phenotypes than to other measures. Indeed the need for strict statistical control is if anything greater in genetic association studies using neuroimaging phenotypes, given the very high dimensionality of the phenotypic data, which potentially greatly increases the likelihood of false positives.
The fact that such a large proportion of trait associations in GWAS have occurred outside of previously proposed candidate genes, and in many cases outside of genes altogether, has further emphasizes the futility of investigations focused on individual hypothesis-based candidate genes. That the detection of these associations required in almost all instances samples of several thousand individuals also underscores how severely underpowered most previous association studies had been.
The many common variants now known to be associated with traits such as T2D and obesity reveal only a small proportion of the genetic architecture of these disorders. Intensive efforts are therefore now focused on identifying additional variants associated with these diseases, mainly by applying next-generation sequencing technologies to identify rare base-changes and copy number variants (CNV) which have been barely assayed in standard GWAS approaches. It is also likely that initial analyses of GWAS of these metabolic disorders –as with most current GWAS – have underestimated the contribution of common variants; recent analyses suggest that a substantial proportion of genetic risk for complex traits derives from large numbers of common variants, each of which explains such a minute proportion of variation that its effects do not achieve stringent significance thresholds (Yang et al., 2010). As metabolic disorders will be among the first sets of traits for which comprehensive genetic variation data are available (Mailman et al., 2007) in enormous samples (in some cases over 100,000 individuals), it is likely that they will be the testing ground for a variety of new analysis approaches, e.g. for assaying effects of gene-gene interactions.
The genetic architecture of the major psychiatric disorders, in stark contrast to that of metabolic disorders, remains largely unknown. To start with, for these disorders there are no unequivocal examples of Mendelian mutations implicated in unusual families with particularly distinctive phenotypic forms, although there are encouraging indications that rare CNVs contribute to risk, particularly for autism and schizophrenia (Manolio et al., 2009). The GWAS of psychiatric disorders have lagged behind those of common non-psychiatric disease phenotypes in identifying common risk variants. For example, of the nearly 800 published GWAS associations at genome wide significance thresholds for about 150 traits catalogued by the NIH (Hindorff et al., 2009), only a handful are for psychiatric phenotypes. Although, as discussed elsewhere in this issue, meta-analyses of multiple GWAS for bipolar disorder and schizophrenia have reported a few associations that could be considered genome wide significant and replicated (Psychiatric GWAS Consortium Coordinating Committee, 2009), these associations are sparser and show less striking significance levels and less convincing replications than the numerous associations observed for traits such as T2D and obesity.
Why has it been so difficult to unravel the genetic architecture of psychiatric disease traits? One possibility is that it has largely been a matter of inadequate sample size to identify variants that have only a small effect on population risk on a given phenotype (Psychiatric GWAS Consortium Coordinating Committee, 2009). There are now clear examples outside of psychiatry where replicated associations to common variants were not achieved until sample sizes reached tens of thousands (e.g. for blood pressure which required a meta-analysis of more than 30,000 individuals, to identify loci which individually explain less than 0.1% of variation in systolic or diastolic blood pressure (Newton-Cheh et al., 2009)).
Yet effect size in genetic association studies is not an intrinsic property of genetic variants but an expression of the biology of the variants in relation to the particular definition and measurement of phenotypes in that study. Extensive heterogeneity in phenotypes – as is presumed to characterize all of the major psychiatric disorders – diminishes the apparent effect of variants contributing to phenotypic variation. There is thus great value in any strategy that can reduce the heterogeneity of the phenotypes under investigation. One strategy for reducing heterogeneity has been to focus investigation on phenotypes hypothesized to underlie particular phenotypic features of disease classifications but which are more distinct, precisely and objectively measured, and more clearly related to specific biological mechanisms than the disorders themselves, so called endophenotypes (Gottesman and Gould, 2003).
Neurocognitive phenotypes
For complex neuropsychiatric disorders, attempts to identify endophenotypes have focused mainly on neurocognitive measures, which we define broadly as quantitative measures of inter-individual variation in brain structure and/or function that reflect specific mental constructs (including cognition, emotion, motivation, and personality). This focus does not imply that variations in these domains underlie all facets of psychopathology; for example, abnormalities in sleep and circadian activity likely contribute substantially to risk for bipolar disorder (Hasler et al., 2006). Nevertheless, overwhelming evidence links extreme values of neurocognitive variables with several common disorders (Bearden and Freimer, 2006; Bearden et al., 2009) and such measures contribute to heritable phenotypic variation in clearly Mendelian disorders as well, for example, several studies have used quantitative measures of neurocognitive performance in Huntington’s disease to obtain insight into the relationship between brain function and behavior (Lawrence et al., 1998).
Neurocognitive measures that have been proposed as endophenotypes for neuropsychiatric disorders include, among many others, self-report trait inventories; performance measures on tasks designed to assess executive function, attention, working memory, and language; neuroanatomic measures (of the volume of global grey and white matter, of specific structures, of cortical thickness, and of white matter structure); and functional imaging measures to assess task-induced and resting state activation patterns. As the number of potential neurocognitive phenotypes (what may be termed the phenomic space) is vast, it is necessary to employ systematic criteria to identify the neurocognitive measures that provide the best opportunity for detecting and replicating genetic associations that achieve acceptable thresholds of statistical significance.
Heritability
That traits must have a substantial heritable component to be genetically mapped is obvious, and at least some data supporting either familial aggregation or non-random twin concordance have been reported for most of the neurocognitive measures proposed as endophenotypes for neuropsychiatric disorders (Bearden and Freimer, 2006). Yet while the high heritability of the disorders themselves has been established through dozens of family and twin studies over several decades (McGue and Bouchard, 1998), the overall level of direct evidence supporting the heritability of self-reported trait inventories or performance measures (such as spatial processing or response inhibition) is limited and varies widely between traits (Bearden and Freimer, 2006; Glahn et al., 2010a). Often the evidence has derived from very small samples, and therefore the confidence intervals around reported heritability estimates are very large. Similarly, evidence that brain function is under genetic influence is variable and surprisingly sparse, although those few studies that have examined task-induced neural activation in twin pairs have provided support for the moderate heritability of functional brain activation in a limited set of regions of interest (ROIs) and during performance of specific tasks (Blokland et al., 2008; Koten et al., 2009; Matthews et al., 2007; Polk et al., 2007).
In contrast, there is substantial evidence for the high heritability of neuroanatomical measures (Glahn et al., 2010b; Peper et al., 2007; Schmitt et al., 2010; Thompson et al., 2001), and while some publications have reported high heritability for particular structures based on analyses of very small samples, the consistency of findings over numerous studies has given these estimates greater credence. Overall estimates suggest that perhaps 80% or more of variation in structural brain features is explained by genetic factors (Glahn et al., 2007), with similar heritability accounting for variation in white-matter tract microstructure (Chiang et al., 2009; Kochunov et al., 2010b). However, the evidence for genetic effects on structure vary across brain regions (Brun et al., 2009; Giedd et al., 2007), as well as types of measurements (Winkler et al., 2009).
It is important, however, to keep in mind that high heritability does not necessarily mean that a trait has a simple genetic architecture (Bearden et al., 2009). Furthermore, heritability is a population parameter, applies to the particular population and time studied (Visscher et al., 2008) and the variety of methods used to assess heritability may make it very difficult to compare heritability estimates across studies. For all of these reasons heritability should be considered a necessary but not sufficient criterion in determining the feasibility of genetically mapping a given trait. In our view characteristics of phenotype definition and measurement – such as precision, reliability, and objectivity, and the relationship to specific mental constructs – are more practically relevant criteria.
Phenotype definition
Comparisons of successful and unsuccessful GWAS show clearly the importance of precise definition of quantitative phenotypes, of the objectivity with which they are measured, and of the degree to which they can be related to well-characterized biological systems. For example, although obesity is clearly etiologically heterogeneous, it has a simple definition that reflects underlying biology (an abnormal increase in the total amount of triglyceride stored in adipose tissue) and relies on a measure that is simple and objective (the body mass index). Accordingly, GWAS investigating differences in adiposity have already unequivocally implicated about 30 distinct loci (O’Rahilly, 2009). An important factor in achieving these findings has been the use of essentially identical phenotype measures across studies, enabling well-powered meta-analyses.
In contrast, GWAS of neurobehavioral traits have often utilized ambiguous and subjective measures and have obtained disappointing results, even with substantial sample sizes. A good example is provided by investigations of self-reported inventories of temperament and personality. Self-report temperament scales are widely investigated endophenotypes based upon evidence for their heritability (Jang et al., 1996; Keller et al., 2005) and for their association with psychiatric disorders (Belsky and Pluess, 2009; De Pauw and Mervielde, 2010; Nigg, 2006). Yet uncertainty regarding the biological underpinnings of personality and continuing disagreement regarding the theoretical basis for personality and temperament constructs have created a situation in which several different measures have been applied in cohorts that have undergone GWAS. Currently reported GWAS samples for personality and temperament include more than 5,000 individuals assessed for Cloninger’s four temperament dimensions (Verweij et al., 2010) and more than 10,000 subjects assessed using personality scales based on the “Big 5” model (de Moor et al., 2009) and have uncovered no genome wide significant associations. Combination of such samples to increase statistical power is essentially precluded by the low correlations between these different scales.
One of the few examples reported so far of unequivocal genetic association for neurobehavioral measures – that between nicotine dependence and variants in a cluster of nicotinic cholinergic receptor loci – further underscores the decisive importance of precise and unambiguous phenotype definitions and assays which reflect underlying biology. Bierut and colleagues (Bierut, 2009) used simple measures obtained through subject recall that accurately quantify an individual’s smoking behavior and that reflect distinct phenotypes representing stages along the path to nicotine dependence (never using, initiating smoking, regular smoking, and nicotine dependence). The distinctiveness of these phenotypic stages with respect to the underlying biology of addiction provided an additional rationale for the use of these measures in genetic association analyses. In analyses distinguishing between individuals who smoked, but no more than 10 cigarettes a day (i.e., controls with a known exposure to nicotine), from individuals who smoked 20 or more cigarettes a day (Saccone et al., 2010; Saccone et al., 2007), they identified several associations, notably a genome wide significant and replicated association to variants influencing specifically the transition to dependence. Studies that have focused on any of a myriad of behavioral traits proposed as underlying a broader smoking phenotype (such as risk taking, impulsivity, and susceptibility to peer influences) have so far failed to identify such unambiguous associations (Saccone et al., 2007). One of the key features of the smoking phenotype employed by Bierut and colleagues is that its simplicity facilitated their being able to assess very large samples, and enhanced the opportunities for replicating their findings in other samples and incorporating their data in well-powered meta-analyses. For some neurocognitive phenotypes, however, it remains a source of debate as to whether strategies for phenotype simplification that enable the analysis of large samples may obscure the detection – using laborious approaches that may limit sample size – of distinct phenotypic features that maximize inter-individual variability. This tension is particularly salient for neuroimaging, where a plethora of methods are available for defining and measuring phenotypes.
In structural MRI analysis, some investigators continue to use long-established methods for manual delineation of anatomical regions, while others employ newer automated methods, which some evidence suggests may have comparable reliability (e.g., (Fischl et al., 2004)), and there are also a number of different schemes for defining specific anatomical phenotypes. Likewise, in functional MRI there is an almost unlimited set of analytic approaches and ways to define specific phenotypes. A recent review of reliability of fMRI measures underscores the limited information available across the range of tasks, preprocessing steps, and summary measures commonly used, despite the significant effect that each of these factors has on reliability estimates (Bennett and Miller, 2010) and reproducibility (Strother et al., 2002).
For example, although functional connectivity measures are believed to be more reflective of integrated cognitive function than is activity in any particular region (e.g., (McIntosh, 2000)), there has been very little examination of the reliability of connectivity results in task-based fMRI studies. One recent exception (Rowe et al., 2010) demonstrated that while the overall connectivity patterns estimated using dynamic causal modeling showed high test-retest reliability, quantitative estimates of connection strength were unreliable. Other recent work has begun to examine the accuracy of connectivity estimates more broadly using simulation methods (Smith et al., in press), and suggests that there are substantial differences in the ability of different analysis methods to accurately identify connectivity patterns. In contrast, resting state networks, which are believed to reflect consistent functional networks engaged across tasks and during rest, have been shown to have high test-retest reliability, to be highly replicable across samples and analysis methods, and to be significantly heritable (Glahn et al., 2010b; Shehzad et al., 2009).
The observation that inter-individual variability consistently exceeds intra-individual variability supports the continued use of fMRI measures as phenotypes in genetic investigations, as does growing evidence in support of the reliability and consistency of specific neural phenotypes (e.g., resting state networks). Yet variable reliability estimates derived across tasks, preprocessing steps, analysis methods, as well as scanners, create enormous complications for meta-analyses, limiting the possibility of obtaining genome wide significance levels much less replication.
Consistency and replication
As we have discussed, most neuroimaging studies to date have been severely underpowered to detect associations. Replicating association results requires even larger samples, making the need for phenotyping standardization and data sharing even greater. Early in the GWAS era it became apparent that the magnitude of initial genetic associations is systematically inflated, by what is known as the “winner’s curse” (Ioannidis, 2008). Underpowered studies are prone to large variation in risk estimates, and only those that are positively inflated (by noise) will be detected as significant; as a result, the initial apparent effect is inflated and decreases in subsequent replication studies (Chanock et al., 2007). This phenomenon is interesting in light of a meta-analysis of 81 association studies, which demonstrated that journals with high impact factors tended to publish studies with high bias scores (that is, studies that over-estimate the true effect size) and small sample sizes (Munafo et al., 2009). This bias is particularly problematic in imaging genetics studies, which may include insufficiently stringent statistical thresholds for both the genetic effect and the neural activation (Yarkoni, 2009; Yarkoni and Braver, 2010), thereby increasing the likelihood of bias.
Widespread data sharing – an essential component of the large-scale analyses that have proven so successful for traits such as T2D and obesity – is also a particularly acute problem for imaging data. The sharing of whole fMRI datasets is made challenging both by the immense size of the datasets and by the complexity of the metadata that are necessary to describe imaging acquisition and cognitive task paradigms. The most prominent previous attempt to database whole fMRI datasets, the fMRI Data Center (Van Horn et al., 2004), met with some resistance from the field but ultimately amassed over 100 full datasets. These datasets remain available, but the center has not accepted any new datasets since 2006, and there is no other current high-capacity repository for sharing of full fMRI datasets. The desperate need for data repositories has begun to be filled by bottom-up efforts, including the OASIS project for anatomical data (Marcus et al., 2007), the 1000 Functional Connectomes Project for resting state fMRI (Biswal et al., 2010), and the OpenfMRI project for task-based fMRI (www.openfmri.org). There is a substantial need for development of more effective data sharing resources within the neuroimaging community, which will also require the development of new frameworks for managing complex metadata. Efforts to develop large-scale data sharing have already begun to bear fruit in structural imaging studies, where standardization of phenotype assessment has enabled the pooling of data across large numbers of sites. For example, in genetic association studies of MRI measures in Alzheimer disease, such data pooling has permitted analyses of large samples which could not be realistically obtained at any single site (Jack et al., 2008; Petersen et al., 2010)).
The role of ontologies in neurocognitive phenotyping
The understanding of neurocognitive phenotypes is made particularly difficult by the indirect and often cloudy relation between the tasks used to measure mental function and the underlying mental processes that support performance on those tasks. For example, the “n-back” task is often considered to measure the construct of “working memory”, but detailed examination has shown that it does not exhibit convergent validity with other accepted measures of the working memory construct (Kane et al., 2007). The reliable use of cognitive task performance as phenotypes for genetic and neuroscientific investigation requires a more systematic characterization of the mental processes that underlie task performance. In other areas of biomedical science, formal knowledge bases known as ontologies have become crucial in the definition of phenotypes (Bard and Rhee, 2004). The best known and most successful such ontology is the Gene Ontology (www.geneontology.org (Ashburner et al., 2000)), which provides a formal characterization of cellular components, biological processes, and molecular functions. The availability of a standard vocabulary has enabled powerful new means of aggregation across studies.
The development of ontologies for neurocognitive function has lagged far behind those for genetics and systems biology, but recent work has begun to develop the semantic infrastructure needed to support systematic cognitive phenotype characterization (Bilder et al., 2009b). In particular, the Cognitive Atlas project (www.cognitiveatlas.org) has assembled an initial vocabulary of mental function and is currently developing a systematic database of mappings between mental functions and cognitive tasks. Other parallel efforts are currently underway to develop ontologies for the fine-grained description of cognitive tasks, such as the Cognitive Paradigm Ontology (www.cogpo.org). The availability of integrated knowledge bases will allow the principled and systematic aggregation of data across different tasks that measure the same mental constructs.
Phenomics, other systems level approaches, and the re-conceptualization of phenotypes
One of the main obstacles to progress in genetic dissection of brain and behavioral disorders is that our basic knowledge of the function of the human nervous system remains so incomplete in relation to that of other organ systems, making it difficult to design studies that map the comparative genetic architecture of particular disorders with specific neurocognitive phenotypes. In contrast, the known strong correlation between T2D and specific quantitative variables such as BMI and lipid levels has stimulated studies that have found differences between disease and normal samples in the sets of SNPs associated with various parameters of glucose metabolism (Dupuis, 2010). The lack of such established correlations for neurocognitive phenotypes has stimulated the development of several strategies for systematically conducting joint analyses of multiple different phenotypes in single study samples. These strategies can be considered attempts to expand – in a controlled manner – the phenotype space for phenotype-genotype studies. At the same time, functional genomics information may be used to increase the power of such studies by narrowing the genomic space that must be searched through. Together these phenomic and genomic strategies may be loosely categorized as systems level approaches.
Phenomics
What we have learned from both the successes and failures of recent genetic investigations has underscored the importance of coherent conceptual systems for organizing phenotypes. Insights into the genetic architecture of common diseases gained from several successful studies indicate that, in high dimensional phenotypic datasets, no single summary measure can account for the majority of phenotypic variation; certain combinations of traits will prove to be more informative than individual measures, or even the complete set of measures, alone (Bloss et al., 2010; Houle, 2010). By the conclusion of these studies highly specific correlations between genetic and phenotypic variation may be obvious (Oti et al., 2009; Xu et al., 2009), but for particularly complex traits such as neurocognitive phenotypes it is rarely evident at the outset which particular combinations of phenotypic measures should be considered together (Houle, 2010). Phenomics, the systematic standardization of measures hypothesized to represent the complete phenotypic space for a given biological system, and their assessment in all members of a study population, has been proposed as a framework for organizing genome-level phenotype-genotype association studies of complex traits (Bilder et al., 2009a).
[Figure 1] This approach further implies that the systematic assessment of multiple phenotypes in large population-based cohorts is conducted in a way that facilitates sharing of data across studies; establishing a common informatics infrastructure for combining phenotypic and genomic data is an important requirement for phenomics projects to succeed.
Figure 1. A phenomics approach provides a coherent conceptual system to align genomic and phenotypic space.
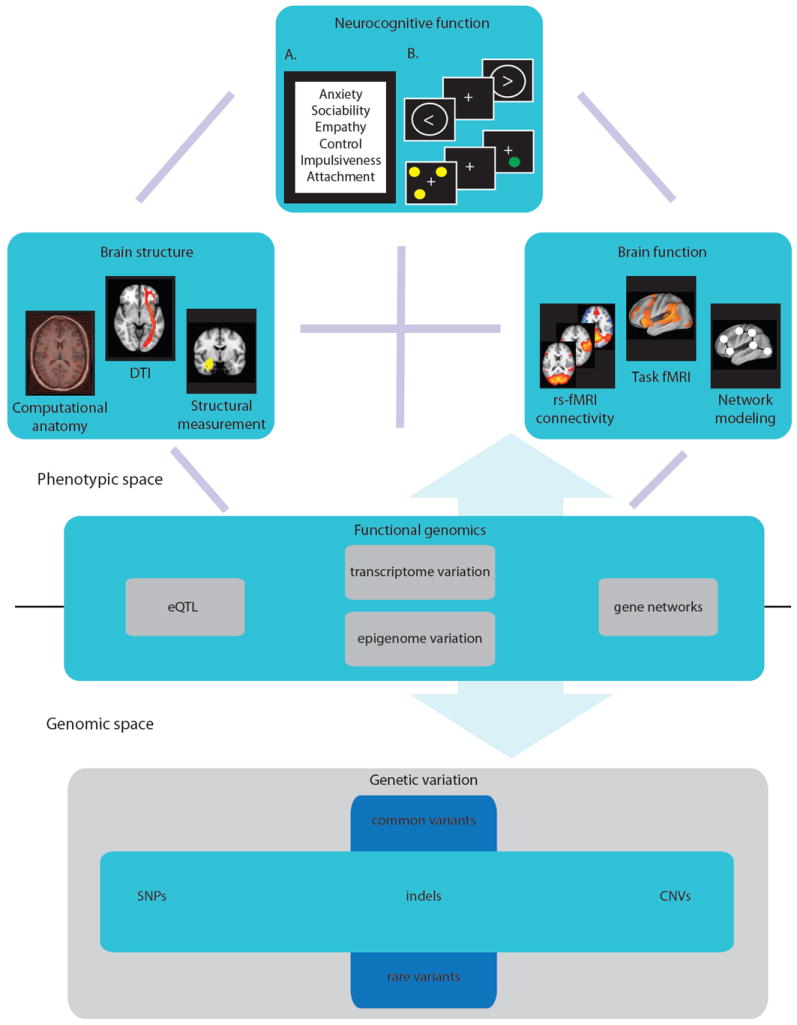
As this approach allows for the systematic collection of multiple phenotypes across large cohorts, it facilitates translational efforts, data sharing, and phenotype re-conceptualization. The phenotypic space is represented here by domains of brain structure (high-resolution anatomical and DTI scans provide measures of global grey and white matter volume, region of interest volume, and cortical thickness), and function (functional scans provide task-induced and resting state activation in regions of interest and across networks) and measures reflecting neurocognitive function (A. self-report inventories provide measures of personality and temperament dimensions, and B. neurocognitive tasks, such as the stop-signal task (upper) and spatial delayed response task (lower), measure neurocognitive performance, including executive function, attention, working memory, and language). Functional genomics assays provide measures of transcript level variation, which allow for transcript-SNP correlation and network analysis, as well as epigenome variation. The genomic space is represented by genome-wide genetic variation, which ranges in structural size (from SNPs to CNVs) and frequency (from common to rare variants). The phenotypic and genomic spaces are not exhaustively represented (e.g., neurophysiologic measures are not represented) and reflect a sample of the phenotypic space according to some of the main approaches. (Note: DTI, diffusion tensor imaging; rs-fMRI, resting state functional magnetic resonance imaging; eQTL, expression quantitative trait loci; SNP, single nucleotide polymorphism; indels, insertions or deletions; CNV, copy number variants.)
Perhaps the most formidable obstacle in using phenomics approaches for genetic dissection of human traits is the expense and logistical difficulty in obtaining comprehensive phenotypic data in samples that are large enough to support well-powered genotype-phenotype analyses. For example, efforts to conduct phenomic analyses using clinical databases have been largely frustrated by the paucity of phenotypic measures relevant to any set of disorders that they typically contain (Denny et al., 2010). More promising sources of phenomic data may be the databases maintained by longitudinal population cohort studies (Freimer and Sabatti, 2003; Pembrey, 2004). Cohort databases contain a rich array of data on diverse phenotypes and environmental exposures, although, because data collections have usually been overseen by different investigators over periods of up to several decades, such databases are rarely either systematic in the measures included or comprehensive in particular phenotypic domains. Recently initiated prospective studies, however aim to more systematically cover the phenotype space for genomic analysis of measures most relevant to psychiatric disorders (Bilder et al., 2009a).
Currently, projects in a variety of animal models better highlight than human studies the potential of phenomics approaches. For example, mouse researchers have accepted a standard set of protocols for extensive phenotype measurement and data sharing. As a result, the Mouse Phenome Database (MPD), which contains data from the systematic phenotyping of a number of strains, now serves as a rich and comprehensive resource for complex trait analysis (Grubb et al., 2009). While initial attempts have been made to compile “orthologous phenotypes” shared between mice and humans (Sardana et al., 2010), from the standpoint of the neurocognitive phenome, non-human primates (NHP) may provide more appropriate comparisons to humans, given the close phylogenetic, anatomic and functional relationship between these species, as we discuss later in this article.
Systems genomic approaches
Systems genomics – both experimental and bioinformatic – has become a central component of strategies for genetically dissecting neurocognitive traits, as it offers a means to reduce the genomic search space and provide biologically relevant, promising candidate genes for further genetic investigation. Measures of quantitative gene expression provide heritable, reliably assessed, and high-throughput phenotypes (Cheung et al., 2003; Cheung et al., 2005; Jasinska et al., 2009; Monks et al., 2004; Pickrell et al., 2010; van Nas et al., 2010), which may occupy a space intermediate between neurocognitive phenotypes and gene variants. Indeed, gene expression levels have proven among the most readily mapped quantitative phenotypes (Myers et al., 2007; Webster et al., 2009); the genetic variants that are linked or associated with variation in the level of expression of a given transcript are termed expression quantitative trait loci (eQTL). The availability of databases annotating genome wide SNPS with respect to their correspondence with eQTL in various types of tissue (Gamazon et al., 2010) is likely to facilitate elucidation of the genetic architecture of complex traits. For example, recent analyses of such databases together with GWAS data for a wide range of complex traits suggest that eQTL detected in studies of lymphoblast cell lines are much more likely to correspond to the SNPs that have demonstrated association with complex traits than with other SNPs in the database (Nicolae et al., 2010). Such use of eQTL-annotation databases not only may improve the precision of estimates of effect size for genetic associations, but provides information on the biology underlying particular associations.
One of the major questions in using gene expression profiles as tools in genetic dissection of neurocognitive traits concerns the applicability of gene expression measures in peripheral blood to expression patterns in less accessible tissue such as the brain. Recent evidence suggests that, for many genes, stable inter-individual variation in expression levels in peripheral blood correlates strongly with such variation in brain (Jasinska et al., 2009; Rollins et al., 2010). As data become available from investigations of transcriptome variation in increasingly large numbers of individuals and tissues, and as the sensitivity and specificity of such studies advance through the employment of direct RNA sequencing (Babak et al., 2010; Pickrell et al., 2010), we anticipate that transcriptome analysis will have a transformative impact on the genetic dissection of neurocognitive traits.
As genetic investigation of complex neurocognitive phenotypes increasingly turns to the collection of phenomic and genomic data, the integration of such data represents a substantial challenge for their joint analysis. Whereas current approaches mostly treat both neural and genetic data as large sets of independent observations, both brain function and gene expression are better characterized as complex interacting networks (Bullmore and Sporns, 2009; Geschwind and Konopka, 2009; Schadt, 2009) that are amenable to the broad set of methods developed for network analysis (Newman et al., 2006). These methods offer the ability to greatly reduce the dimensionality of the data for analysis, while still respecting the complex structure inherent in those data. For example, using network analysis methods it is possible to reliably identify neighborhoods of voxels across the brain that are functionally integrated and serve as nodes in a larger connected network (e.g., (Cohen et al., 2008; Mumford et al., 2010)); the aggregate signal from within these neighborhoods can then be used for subsequent analyses, greatly reducing the dimensionality of the data and thus the stringency of multiple comparison corrections. Recent work using such network analyses has shown that it is possible to detect individual differences in network connectivity that are not evident in overall activation (e.g.,(Fair et al., 2007)). Indeed, the use of network analysis approaches is likely to have particular value in genome-wide imaging genetics studies, as they provide a means of reducing the dimensionality of imaging data, thereby making genome-wide analyses feasible, while providing phenotypes which do not depend on a priori assumptions about brain structure yet can be related to what is known about brain function.
Network analysis approaches to gene expression data have also been successful in providing a systems-level understanding of gene function. Such analyses have revealed that the expression in the human cerebral cortex is organized into modules that reflect cell classes (Oldham et al., 2008) and distinct brain regions (Oldham et al., 2006), and that gene co-expression networks in human and chimpanzee brains can be used to quantify conservation across brain regions (Oldham et al., 2006). In this way, network analysis approaches to gene expression data are beginning to elucidate relationships between biology and function in ways not possible by considering genomic data alone.
The next step is to integrate data across levels of analysis. In this way, network analysis approaches can be used to embrace the high dimensionality of the data and provide a context in which to interpret genotype-phenotype associations (Schadt, 2009). That is, as opposed to univariate approaches, which require correction of non-independent traits, or data dimensionality reduction approaches that either result in indirect factors that are difficult to interpret or qualitative descriptors, recent multivariate approaches have been developed that reduce data dimensionality while still retaining information about complex phenotypes. These approaches include ways to account for information contained within quantitative trait networks for incorporation in genetic association analyses (Kim and Xing, 2009), as well as ways to allow for the simultaneous investigation of latent (factor) and specific (variable) tests (Medland and Neale, 2010). The recent work by Mumford et al. (2010), in which methods originally developed for the analysis of gene networks were profitably used to analyze fMRI activation networks, further suggests that it should be possible to integrate across these very different levels of analysis with the use of network modeling strategies.
Systems level investigations in nonhuman primates
Investigations of rodent models have played an important role in the genetic dissection of traits such as obesity (Yang et al., 2009), but have so far had less impact on our understanding of disorders of brain and behavior (Nestler and Hyman, 2010). Yet numerous limitations to investigations of the human brain necessitate the employment of animal genetic models, particularly for systems level investigations which may be infeasible in humans. Recent large-scale efforts aimed at characterizing genetic and functional variation in several non-human primate (NHP) genomes have made NHP models increasingly important as bridges between rodents and humans., Phenome-level investigations are now underway in several of the most widely employed NHP species, including rhesus macaques, vervet monkeys (also termed African green monkeys), and baboons. Advantages of these systems include the opportunities for longitudinal genetic and genomic studies incorporating a wide range of assays across the lifespan, as well as for studies assessing inter-individual variation in medically relevant interventions (e.g., drug response). In particular, the possibility of obtaining biological materials that are largely inaccessible in humans – for example, tissues that enable the investigation of functional genomic variation between brain regions, between the brain and peripheral organs, and across the lifespan – creates the opportunity for phenomic analyses that are truly comprehensive. An additional important factor in the genetic investigation of NHPs is that environmental contributions to trait variance (such as diet or social interactions) can be controlled or at least documented to a far greater degree than is possible in human studies; diminished variance in shared environments likely contributes to findings of higher heritability in NHPs compared to humans for traits such as the size of various brain structures (Fears et al., 2009). The precision of heritability estimates in NHPs has also been enhanced by the availability of large, well-powered pedigree samples for assessing complex phenotypes that could not readily be assayed in comparable human families. For example, Oler et al (2010) used high-resolution 18F-labelled deoxyglucose positron-emission tomography (FDG–PET) in more than 200 rhesus macaques from a single pedigree to demonstrate significant inter-individual differences in a behavioral trait (anxious temperament), which were correlated with brain region-specific variations in glucose metabolism (Oler et al., 2010). They were able to further determine that the heritability of the metabolic phenotype differed significantly between regions known to be important components of the circuitry underlying the behavioral trait.
The genetic dissection of complex neurocognitive phenotypes is already underway, through linkage and association studies now being conducted in well-powered NHP pedigree or population samples. Examples of such phenotypes include dimensions of impulsivity and working memory (James et al., 2007), features of brain structural variation (Fears et al., 2009; Kochunov et al., 2010a; Rogers et al., 2007; Rogers et al., 2010), and measures reflecting central dopamine turnover (Freimer et al., 2007). The restricted genetic and environmental heterogeneity that we postulate characterizes several NHP study samples likely accounts for the large effect size observed for QTL mapped in such samples, as illustrated by a locus for central dopamine turnover in the vervet monkey that accounts for about 60% of the heritable variance for this trait within the Vervet Research Colony (Freimer et al., 2007). Genetic investigations in NHP will soon be aided by the anticipated availability in such species of eQTL reflective of transcriptome variation in multiple tissue types (Gamazon et al., 2010; Jasinska et al., 2009). [Figure 2] Additionally, efforts to bank samples from large numbers of unrelated NHPs will soon generate opportunities to conduct well-powered studies that could detect associations to loci of smaller effect than those which can be identified in pedigree samples (http://www.genomequebec.mcgill.ca/compgen/vervet_research/; (Kanthaswamy et al., 2009)).
Figure 2. Nonhuman primate (NHP) model representing a comprehensive phenomic approach.
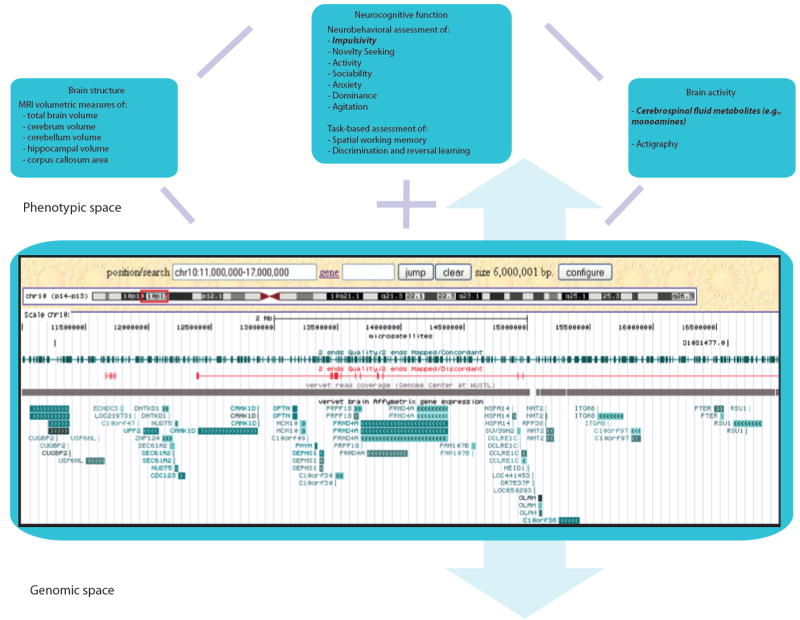
The assessment of multi-level sets of measures in extended NHP multigenerational pedigrees provides complete coverage of the phenotypic space and sufficient power for linkage and association analysis with comprehensive genome-level genetic variation data. Examples are shown here from the Integrated Vervet/African Green Monkey Research and Resources Consortium (http://www.genomequebec.mcgill.ca/compgen/vervet_research/). An illustration of the multi-level analysis of neurocognitive traits in this model system (see highlighted text) is illustrated by the genetic mapping of a QTL for the dopamine catabolite homovanillic acid (HVA) in an extended vervet pedigree (Freimer et al., 2007). Other neurocognitive phenotypes assessed in this pedigree that are hypothesized to be influenced by dopaminergic function include measures of impulsivity and reversal learning and are therefore candidates for inclusion in multivariate phenotype-genotype analyses along with HVA levels and relevant gene expression variation. The genomic resources available for such analyses (illustrated here through by a screenshot of a customized Genome Browser track) include genome wide sequence, genetic variation, and gene expression data. Details of the Genome Browser screenshot include nucleotide position; microsatellites; concordantly and discordantly mapped BAC clone ends; 454 short sequence read coverage from Genome Center at WUSTL; and vervet brain gene expression using Human Genome U133 Plus 2.0 Array from Affymetrix. (Note: MRI, magnetic resonance imaging; Actigraphy is the measurement of body movement patterns to infer sleep/wake and rest/activity cycles.)
Reconceptualization of phenotypes based on genetic dissection
The promise of genetic dissection is that it provides a more precise alignment of genotypic and phenotypic variation. As we have discussed already, obtaining sufficient power for the discovery phase in the genetic dissection of complex traits requires sample sizes that are unrealistic to obtain for certain phenotypic measures with inescapably low throughput, such as fMRI. Even relatively modest samples, however, can be sufficiently powerful for the hypothesis-based process of genetic dissection which must occur after the identification of significant and replicated associations to a related trait; because of such strong prior genetic evidence, a genome wide level of significance is not required in these studies. The follow-up of significant association signals using neuroimaging paradigms offers a means of elucidating mechanisms and, as has been recently argued, represents a new role of endophenotypes in the GWAS era, for functional characterization of common disorders (Hall and Smoller, 2010).
Examples of this use of neuroimaging phenotypes include several studies undertaken to better understand the effects of particular APOE genotypes in relation to cognitive performance. The e4 allele of the apolipoprotein E gene (APOE), which is the predominant risk variant for common forms of Alzheimer disease (AD), has been repeatedly associated with disease risk, as well as with differences in cognitive abilities in pre-morbid individuals (Bookheimer and Burggren, 2009). Given the reproducibility of the association between this variant and risk for AD in large samples, findings of genotype group differences in MRI measures of brain activation and structure in modestly sized samples represent a clear demonstration of the use of such low throughput measures to elucidate mechanisms (Burggren et al., 2008; Donix et al., 2010; Reiman et al., 2004). In particular, Wolk et al. demonstrated a dissociable effect of APOE genotype on cognitive and neuroanatomic phenotypes in a sample of patients with mild AD (Wolk, 2010). APOE e4 allele carriers were significantly impaired in episodic memory performance and had reduced cortical thickness in the medial temporal lobe as well as smaller hippocampal volumes as compared to the noncarriers, while the noncarriers were significantly impaired in tasks of attention and executive function, and had reduced cortical thickness in all other regions of interest previously implicated in AD.
The example of APOE and measures related to AD remains a singular one, given that the effect size on AD of the e4 allele is so large. The impact of GWAS findings in metabolic disorders, however, illustrates how even variants with a small apparent effect can contribute to the re-conceptualization of phenotypes. Some of the strongest associations observed for common metabolic disorders occur at loci whose presumed function centers on neurobehavioral contributions to obesity (O’Rahilly, 2009). Such findings have contributed to increased recognition that biological overlaps between metabolic and neurocognitive systems may be important in a wide range of human disorders. This possibility has also been raised by basic studies in model organisms (e.g.(Marcheva et al., 2010)) and by findings such as the discovery of high-penetrance deletions responsible for extreme obesity phenotypes, which were first uncovered in individuals with various cognitive disorders (Walters et al., 2010).
As discussed previously, there are not yet sufficient replicated associations for psychiatric disorders for such phenotypic reconceptualization to have occurred. However findings from initial large GWAS of such disorders have begun to suggest how systematic genetic dissection of neurocognitive phenotypes may be helpful in this regard. A recent meta-analysis of case-control GWAS data indicates the existence of a substantial, shared genetic component between schizophrenia and bipolar disorder that is not shared with several non-psychiatric diseases and that reflects common variants of small effect on these phenotypes (The International Schizophrenia The International Schizophrenia Consortium, 2009). It is unlikely, however, that the phenotypic data available in such case-control samples will permit detection of specific features that are responsible for such overlap, and that could suggest the underlying biological mechanisms. Genetic analyses focused on comprehensive sets of neurocognitive phenotypes, however, could be useful for this purpose.
Conclusion
Identification of variants associated with a wide range of neurocognitive phenotypes could provide a foundation for the systematic genetic dissection of normal brain function as well as brain dysfunction. Achieving this goal will require high-throughput assessment in large samples using measures that are precisely defined, reliably assayed, and unambiguously reflect underlying mental functions. Although neuroimaging variables remain among the most promising neurocognitive phenotypes for genetic investigation, the validation and standardization of imaging-based phenotypes remains a critical challenge. Implementation of systems-level approaches – phenomic as well as genomic – offers a potentially powerful approach for identification of genetic variants associated with neurocognitive traits, but may require application of strategies such as network analysis for reducing the dimensionality of immense data sets. Once we obtain unequivocal associations for neurocognitive phenotypes, characterizing the pathways by which these genetic variants affect mental function will produce additional challenges, It will be difficult in many cases to identify and characterize the function of the specific genetic variants implicated by these associations; comparative investigations including both traditional animal models as well as NHP systems may be important in this endeavor. Additionally, it will likely be a slow process to relate the genetic architecture identified for neurocognitive traits to phenotypic features in clinical samples that represent potential targets for either therapeutic interventions or prevention strategies. The development of validated and standardized phenotypes will provide the scaffold on which these difficult tasks can be performed.
Acknowledgments
This work was funded by research grants from the NIH (RL1MH083268, N. Freimer; R01MH075007, N. Freimer; R01RR016300, N. Freimer; R01MH082795, R. Poldrack; PL1MH083271, R. Bilder; UL1 DE019580-03, R. Bilder). We thank A. Jasinska for assistance with figure preparation and R. Bilder for helpful comments on the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babak T, Garrett-Engele P, Armour C, Raymond C, Keller M, Chen R, Rohl C, Johnson J, Attie A, Fraser H, Schadt E. Genetic validation of whole-transcriptome sequencing for mapping expression affected by cis-regulatory variation. BMC Genomics. 2010;11:473. doi: 10.1186/1471-2164-11-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard JBL, Rhee SY. Ontologies in biology: design, applications and future challenges. Nat Rev Genet. 2004;5:213–222. doi: 10.1038/nrg1295. [DOI] [PubMed] [Google Scholar]
- Bearden CE, Freimer NB. Endophenotypes for psychiatric disorders: Ready for primetime? Trends Genet. 2006;22:306–313. doi: 10.1016/j.tig.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Bearden CE, Jasinska AJ, Freimer NB. Methodological issues in molecular genetic studies of mental disorders. Annual Review of Clinical Psychology. 2009;5:49–69. doi: 10.1146/annurev.clinpsy.032408.153545. [DOI] [PubMed] [Google Scholar]
- Belsky J, Pluess M. Beyond diathesis stress: Differential susceptibility to environmental influences. Psychol Bull. 2009;135:885–908. doi: 10.1037/a0017376. [DOI] [PubMed] [Google Scholar]
- Bennett CM, Miller MB. How reliable are the results from functional magnetic resonance imaging? Ann N Y Acad Sci. 2010;1191:133–155. doi: 10.1111/j.1749-6632.2010.05446.x. [DOI] [PubMed] [Google Scholar]
- Bierut LJ. Nicotine dependence and genetic variation in the nicotinic receptors. Drug and Alcohol Dependence. 2009;104:S64–S69. doi: 10.1016/j.drugalcdep.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilder RM, Sabb FW, Cannon TD, London ED, Jentsch JD, Parker DS, Poldrack RA, Evans C, Freimer NB. Phenomics: The systematic study of phenotypes on a genome-wide scale. Neuroscience. 2009a;164:30–42. doi: 10.1016/j.neuroscience.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilder RM, Sabb FW, Parker DS, Kalar D, Chu WW, Fox J, Freimer NB, Poldrack RA. Cognitive ontologies for neuropsychiatric phenomics research. Cognitive Neuropsychiatry. 2009b;14:419–450. doi: 10.1080/13546800902787180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswal BB, Mennes M, Zuo X-N, Gohel S, Kelly C, Smith SM, Beckmann CF, Adelstein JS, Buckner RL, Colcombe S, et al. Toward discovery science of human brain function. Proceedings of the National Academy of Sciences. 2010;107:4734–4739. doi: 10.1073/pnas.0911855107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokland GA, McMahon KL, Hoffman J, Z G, Meredith M, Martin NG, Thompson PM, de Zubicaray GI, Wright MJ. Quantifying the heritability of task-related brain activation and performance during the N-back working memory task: A twin fMRI study. Biol Psychol. 2008;79:70–79. doi: 10.1016/j.biopsycho.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloss CS, Schiabor KM, Schork NJ. Human behavioral informatics in genetic studies of neuropsychiatric disease: Multivariate profile-based analysis. Brain Research Bulletin. 2010;83:177–188. doi: 10.1016/j.brainresbull.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bookheimer S, Burggren A. APOE-4 genotype and neurophysiological vulnerability to Alzheimer’s and cognitive aging. Annual Review of Clinical Psychology. 2009;5:343–362. doi: 10.1146/annurev.clinpsy.032408.153625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun CC, Leporé N, Pennec X, Lee AD, Barysheva M, Madsen SK, Avedissian C, Chou YY, de Zubicaray GI, McMahon KL, et al. Mapping the regional influence of genetics on brain structure variability--a tensor-based morphometry study. Neuroimage. 2009;48:37–49. doi: 10.1016/j.neuroimage.2009.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullmore E, Sporns O. Complex brain networks: Graph theoretical analysis of structural and functional systems. Nat Rev Neurosci. 2009;10:186–198. doi: 10.1038/nrn2575. [DOI] [PubMed] [Google Scholar]
- Burggren AC, Zeineh MM, Ekstrom AD, Braskie MN, Thompson PM, Small GW, Bookheimer SY. Reduced cortical thickness in hippocampal subregions among cognitively normal apolipoprotein E e4 carriers. Neuroimage. 2008;41:1177–1183. doi: 10.1016/j.neuroimage.2008.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanock SJ, Manolio T, Boehnke M, Boerwinkle E, Hunter DJ, Thomas G, Hirschhorn JN, Abecasis G, Altshuler D, Bailey-Wilson JE, et al. Replicating genotype-phenotype associations. Nature. 2007;447:655–660. doi: 10.1038/447655a. [DOI] [PubMed] [Google Scholar]
- Cheung VG, Conlin LK, Weber TM, Arcaro M, Jen KY, Morley M, Spielman RS. Natural variation in human gene expression assessed in lymphoblastoid cells. Nat Genet. 2003;33:422–425. doi: 10.1038/ng1094. [DOI] [PubMed] [Google Scholar]
- Cheung VG, Spielman RS, Ewens KG, Weber TM, Morley M, Burdick JT. Mapping determinants of human gene expression by regional and genome-wide association. Nature. 2005;437:1365–1369. doi: 10.1038/nature04244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang MC, Barysheva M, Shattuck DW, Lee AD, Madsen SK, Avedissian C, Klunder AD, Toga AW, McMahon KL, de Zubicaray GI, et al. Genetics of brain fiber architecture and intellectual performance. J Neurosci. 2009;29:2212–2224. doi: 10.1523/JNEUROSCI.4184-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen AL, Fair DA, Dosenbach NUF, Miezin FM, Dierker D, Van Essen DC, Schlaggar BL, Petersen SE. Defining functional areas in individual human brains using resting functional connectivity MRI. Neuroimage. 2008;41:45–57. doi: 10.1016/j.neuroimage.2008.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Moor MHM, Boomsma DI, de Geus EJC, Willemsen G, Hottenga J-J, Distel MA, Abecasis G, Terracciano A, McCrae R, Costa P, et al. Behavior Genetics Association. Minneapolis, MN: Springer Netherlands; 2009. Meta-analysis of genome-wide association results in > 10.000 individuals for the Big Five personality traits; p. 12. [Google Scholar]
- De Pauw S, Mervielde I. Temperament, personality and developmental psychopathology: A review based on the conceptual dimensions underlying childhood traits. Child Psychiatry & Human Development. 2010;41:313–329. doi: 10.1007/s10578-009-0171-8. [DOI] [PubMed] [Google Scholar]
- Denny JC, Ritchie MD, Basford MA, Pulley JM, Bastarache L, Brown-Gentry K, Wang D, Masys DR, Roden DM, Crawford DC. PheWAS: Demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics. 2010;26:1205–1210. doi: 10.1093/bioinformatics/btq126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diabetes Genetics Initiative, Broad Institute of Harvard, MIT, Lund University, Novartis Institutes of BioMedical Research. Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PIW, et al. Genome-wide association analysis identifies loci for Type 2 Diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- Donix M, Burggren AC, Suthana NA, Siddarth P, Ekstrom AD, Krupa AK, Jones M, Martin-Harris L, Ercoli LM, Miller KJ, et al. Family history of Alzheimer’s Disease and hippocampal structure in healthy people. Am J Psychiatry. 2010 doi: 10.1176/appi.ajp.2010.09111575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42:105–116. doi: 10.1038/ng.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenreich IM, Torabi N, Jia Y, Kent J, Martis S, Shapiro JA, Gresham D, Caudy AA, Kruglyak L. Dissection of genetically complex traits with extremely large pools of yeast segregants. Nature. 2010;464:1039–1042. doi: 10.1038/nature08923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fair DA, Dosenbach NUF, Church JA, Cohen AL, Brahmbhatt S, Miezin FM, Barch DM, Raichle ME, Petersen SE, Schlaggar BL. Development of distinct control networks through segregation and integration. PNAS. 2007;104:13507–13512. doi: 10.1073/pnas.0705843104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fears SC, Melega WP, Service SK, Lee C, Chen K, Tu Z, Jorgensen MJ, Fairbanks LA, Cantor RM, Freimer NB, Woods RP. Identifying Heritable Brain Phenotypes in an Extended Pedigree of Vervet Monkeys. Journal of Neuroscience. 2009;29:2867–2875. doi: 10.1523/JNEUROSCI.5153-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, van der Kouwe A, Destrieux C, Halgren E, Ségonne F, Salat DH, Busa E, Seidman LJ, Goldstein J, Kennedy D, et al. Automatically parcellating the human cerebral cortex. Cerebral Cortex. 2004;14:11–22. doi: 10.1093/cercor/bhg087. [DOI] [PubMed] [Google Scholar]
- Freimer NB, Sabatti C. The human phenome project. Nat Genet. 2003;34:15–21. doi: 10.1038/ng0503-15. [DOI] [PubMed] [Google Scholar]
- Freimer NB, Sabatti C. The use of pedigree, sib-pair and association studies of common diseases for genetic mapping and epidemiology. Nat Genet. 2004;36:1045–1051. doi: 10.1038/ng1433. [DOI] [PubMed] [Google Scholar]
- Freimer NB, Service SK, Ophoff RA, Jasinska AJ, McKee K, Villeneuve A, Belisle A, Bailey JN, Breidenthal SE, Jorgensen MJ, et al. A quantitative trait locus for variation in dopamine metabolism mapped in a primate model using reference sequences from related species. Proceedings of the National Academy of Sciences. 2007;104:15811–15816. doi: 10.1073/pnas.0707640104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamazon ER, Zhang W, Konkashbaev A, Duan S, Kistner EO, Nicolae DL, Dolan ME, Cox NJ. SCAN: SNP and copy number annotation. Bioinformatics. 2010;26:259–262. doi: 10.1093/bioinformatics/btp644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geschwind DH, Konopka G. Neuroscience in the era of functional genomics and systems biology. Nature. 2009;461:908–915. doi: 10.1038/nature08537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giedd JN, Schmitt JE, Neale MC. Structural brain magnetic resonance imaging of pediatric twins. Human Brain Mapping. 2007;28:474–481. doi: 10.1002/hbm.20403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glahn DC, Almasy L, Barguil M, Hare E, Peralta JM, Kent JWJ, Dassori A, Contreras J, Pacheco A, Lanzagorta N, et al. Neurocognitive endophenotypes for bipolar disorder identified in multiplex multigenerational families. Arch Gen Psychiatry. 2010a;67:168–177. doi: 10.1001/archgenpsychiatry.2009.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glahn DC, Thompson PM, Blangero J. Neuroimaging endophenotypes: Strategies for finding genes influencing brain structure and function. Human Brain Mapping. 2007;28:488–501. doi: 10.1002/hbm.20401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glahn DC, Winkler AM, Kochunov P, Almasy L, Duggirala R, Carless MA, Curran JC, Olvera RL, Laird AR, Smith SM, et al. Genetic control over the resting brain. PNAS. 2010b;107:1223–1228. doi: 10.1073/pnas.0909969107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glatt CE, Freimer NB. Association analysis of candidate genes for neuropsychiatric disease: The perpetual campaign. Trends in Genetics. 2002;18:307–312. doi: 10.1016/S0168-9525(02)02670-7. [DOI] [PubMed] [Google Scholar]
- Gottesman II, Gould TD. The endophenotype concept in psychiatry: Etymology and strategic intentions. Am J Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- Grubb SC, Maddatu TP, Bult CJ, Bogue MA. Mouse Phenome Database. Nucl Acids Res. 2009;37:D720–730. doi: 10.1093/nar/gkn778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall M-H, Smoller JW. A new role for endophenotypes in the GWAS era: Functional characterization of risk variants. Harvard Review of Psychiatry. 2010;18:67–74. doi: 10.3109/10673220903523532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hariri AR. The neurobiology of individual differences in complex behavioral traits. Annu Rev Neurosci. 2009;32:225–247. doi: 10.1146/annurev.neuro.051508.135335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasler G, Drevets WC, Gould TD, Gottesman II, Manji HK. Toward constructing an endophenotype strategy for bipolar disorders. Biological Psychiatry. 2006;60:93–105. doi: 10.1016/j.biopsych.2005.11.006. [DOI] [PubMed] [Google Scholar]
- Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proceedings of the National Academy of Sciences. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houle D. Numbering the hairs on our heads: The shared challenge and promise of phenomics. PNAS. 2010;107:1793–1799. doi: 10.1073/pnas.0906195106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis JP. Why most discovered true associations are inflated. Epidemiology. 2008;19:640–648. doi: 10.1097/EDE.0b013e31818131e7. [DOI] [PubMed] [Google Scholar]
- Jack CR, Bernstein MA, Fox NC, Thompson P, Alexander G, Harvey D, Borowski B, Britson PJ, L Whitwell J, Ward C, et al. The Alzheimer’s disease neuroimaging initiative (ADNI): MRI methods. Journal of Magnetic Resonance Imaging. 2008;27:685–691. doi: 10.1002/jmri.21049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James AS, Groman SM, Seu E, Jorgensen M, Fairbanks LA, Jentsch JD. Dimensions of impulsivity are associated with poor spatial working memory performance in monkeys. J Neurosci. 2007;27:14358–14364. doi: 10.1523/JNEUROSCI.4508-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang KL, Livesley WJ, Vernon PA. Heritability of the big five personality dimensions and their facets: A twin study. J Pers. 1996;64:577–591. doi: 10.1111/j.1467-6494.1996.tb00522.x. [DOI] [PubMed] [Google Scholar]
- Jasinska AJ, Service S, Choi OW, DeYoung J, Grujic O, Kong SY, Jorgensen MJ, Bailey J, Breidenthal S, Fairbanks LA, et al. Identification of brain transcriptional variation reproduced in peripheral blood: An approach for mapping brain expression traits. Hum Mol Genet. 2009;18:4415–4427. doi: 10.1093/hmg/ddp397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane MJ, Conway AR, Miura TK, Colflesh GJ. Working memory, attention control, and the N-back task: A question of construct validity. J Exp Psychol Learn Mem Cogn. 2007;33:615–622. doi: 10.1037/0278-7393.33.3.615. [DOI] [PubMed] [Google Scholar]
- Kanthaswamy S, Capitanio J, Dubay C, Ferguson B, Folks T, Ha J, Hotchkiss C, Johnson Z, Katze M, Kean L, et al. Resources for genetic management and genomics research on non-human primates at the National Primate Research Centers (NPRCs) Journal of Medical Primatology. 2009;38:17–23. doi: 10.1111/j.1600-0684.2009.00371.x. [DOI] [PubMed] [Google Scholar]
- Keller MC, Coventry WL, Heath AC, Martin NG. Widespread evidence for non-additive genetic variation in Cloninger’s and Eysenck’s personality dimensions using a twin plus sibling design. Behav Genet. 2005;35:707–721. doi: 10.1007/s10519-005-6041-7. [DOI] [PubMed] [Google Scholar]
- Kim S, Xing EP. Statistical estimation of correlated genome associations to a quantitative trait network. PLoS Genet. 2009;5:e1000587. doi: 10.1371/journal.pgen.1000587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochunov P, Glahn DC, Fox PT, Lancaster JL, Saleem K, Shelledy W, Zilles K, Thompson PM, Coulon O, Mangin JF, et al. Genetics of primary cerebral gyrification: Heritability of length, depth and area of primary sulci in an extended pedigree of Papio baboons. Neuroimage. 2010a;53:1126–1134. doi: 10.1016/j.neuroimage.2009.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochunov P, Glahn DC, Lancaster JL, Winkler AM, Smith S, Thompson PM, Almasy L, Duggirala R, Fox PT, Blangero J. Genetics of microstructure of cerebral white matter using diffusion tensor imaging. Neuroimage. 2010b;53:1109–1116. doi: 10.1016/j.neuroimage.2010.01.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koten JWJ, Wood G, Hagoort P, Goebel R, Propping P, Willmes K, Boomsma DI. Genetic contribution to variation in cognitive function: An FMRI study in twins. Science. 2009;323:1737–1740. doi: 10.1126/science.1167371. [DOI] [PubMed] [Google Scholar]
- Lander ES, Schork NJ. Genetic dissection of complex traits. Science. 1994;265:2037–2048. doi: 10.1126/science.8091226. [DOI] [PubMed] [Google Scholar]
- Lawrence AD, Sahakian BJ, Robbins TW. Cognitive functions and corticostriatal circuits: Insights from Huntington’s disease. Trends in Cognitive Sciences. 1998;2:379–388. doi: 10.1016/s1364-6613(98)01231-5. [DOI] [PubMed] [Google Scholar]
- Mailman MD, Feolo M, Jin Y, Kimura M, Tryka K, Bagoutdinov R, Hao L, Kiang A, Paschall J, Phan L, et al. The NCBI dbGaP database of genotypes and phenotypes. Nat Genet. 2007;39:1181–1186. doi: 10.1038/ng1007-1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–753. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcheva B, Ramsey KM, Buhr ED, Kobayashi Y, Su H, Ko CH, Ivanova G, Omura C, Mo S, Vitaterna MH, et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. 2010;466:627–631. doi: 10.1038/nature09253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus DS, Wang TH, Parker J, Csernansky JG, Morris JC, Buckner RL. Open Access Series of Imaging Studies (OASIS): Cross-sectional MRI data in young, middle aged, nondemented, and demented older adults. Journal of Cognitive Neuroscience. 2007;19:1498–1507. doi: 10.1162/jocn.2007.19.9.1498. [DOI] [PubMed] [Google Scholar]
- Matthews SC, Simmons AN, Strigo I, Jang K, Stein MB, Paulus MP. Heritability of anterior cingulate response to conflict: An fMRI study in female twins. Neuroimage. 2007;38:223–227. doi: 10.1016/j.neuroimage.2007.07.015. [DOI] [PubMed] [Google Scholar]
- McGue M, Bouchard TJ. Genetic and environmental influences on human behavioral differences. Annual Review of Neuroscience. 1998;21:1–24. doi: 10.1146/annurev.neuro.21.1.1. [DOI] [PubMed] [Google Scholar]
- McIntosh AR. Towards a network theory of cognition. Neural Networks. 2000;13:861–870. doi: 10.1016/s0893-6080(00)00059-9. [DOI] [PubMed] [Google Scholar]
- Medland SE, Neale MC. An integrated phenomic approach to multivariate allelic association. Eur J Hum Genet. 2010;18:233–239. doi: 10.1038/ejhg.2009.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monks SA, Leonardson A, Zhu H, Cundiff P, Pietrusiak P, Edwards S, Phillips JW, Sachs A, Schadt EE. Genetic inheritance of gene expression in human cell lines. Am J Hum Genet. 2004;75:1094–1105. doi: 10.1086/426461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumford JA, Horvath S, Oldham MC, Langfelder P, Geschwind DH, Poldrack RA. Detecting network modules in fMRI time series: A weighted network analysis approach. Neuroimage. 2010;52:1465–1476. doi: 10.1016/j.neuroimage.2010.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munafo MR, Stothart G, Flint J. Bias in genetic association studies and impact factor. Molecular Psychiatry. 2009;14:119–120. doi: 10.1038/mp.2008.77. [DOI] [PubMed] [Google Scholar]
- Myers AJ, Gibbs JR, Webster JA, Rohrer K, Zhao A, Marlowe L, Kaleem M, Leung D, Bryden L, Nath P, et al. A survey of genetic human cortical gene expression. Nat Genet. 2007;39:1494–1499. doi: 10.1038/ng.2007.16. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Hyman SE. Animal models of neuropsychiatric disorders. Nat Neurosci. 2010;13:1161–1169. doi: 10.1038/nn.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman M, Barabási A-L, Watts DJ. The Structure and Dynamics of Networks. 1. Princeton, New Jersey: Princeton University Press; 2006. [Google Scholar]
- Newton-Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, Coin L, Najjar SS, Zhao JH, Heath SC, Eyheramendy S, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet. 2009;41:666–676. doi: 10.1038/ng.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolae DL, Gamazon E, Zhang W, Duan S, Dolan ME, Cox NJ. Trait-associated SNPs are more likely to be eQTLs: Annotation to enhance discovery from GWAS. PLoS Genet. 2010;6:e1000888. doi: 10.1371/journal.pgen.1000888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg JT. Temperament and developmental psychopathology. J Child Psychol Psychiatry. 2006;47:395–422. doi: 10.1111/j.1469-7610.2006.01612.x. [DOI] [PubMed] [Google Scholar]
- O’Rahilly S. Human genetics illuminates the paths to metabolic disease. Nature. 2009;462:307–314. doi: 10.1038/nature08532. [DOI] [PubMed] [Google Scholar]
- Oldham MC, Horvath S, Geschwind DH. Conservation and evolution of gene coexpression networks in human and chimpanzee brains. PNAS. 2006;103:17973–17978. doi: 10.1073/pnas.0605938103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham MC, Konopka G, Iwamoto K, Langfelder P, Kato T, Horvath S, Geschwind DH. Functional organization of the transcriptome in human brain. Nat Neurosci. 2008;11:1271–1282. doi: 10.1038/nn.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oler JA, Fox AS, Shelton SE, Rogers J, Dyer TD, Davidson RJ, Shelledy W, Oakes TR, Blangero J, Kalin NH. Amygdalar and hippocampal substrates of anxious temperament differ in their heritability. Nature. 2010;466:864–868. doi: 10.1038/nature09282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SMG, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, et al. Familial Hemiplegic Migraine and Episodic Ataxia Type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543–552. doi: 10.1016/s0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- Oti M, Huynen MA, Brunner HG. The biological coherence of human phenome databases. The American Journal of Human Genetics. 2009;85:801–808. doi: 10.1016/j.ajhg.2009.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pembrey M. Genetic epidemiology: Some special contributions of birth cohorts. Paediatric and Perinatal Epidemiology. 2004;18:3–7. doi: 10.1111/j.1365-3016.2004.00530.x. [DOI] [PubMed] [Google Scholar]
- Peper JS, Brouwer RM, Boomsma DI, Kahn RS, Hulshoff Pol HE. Genetic influences on human brain structure: A review of brain imaging studies in twins. Human Brain Mapping. 2007;28:464–473. doi: 10.1002/hbm.20398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RC, Aisen PS, Beckett LA, Donohue MC, Gamst AC, Harvey DJ, Jack CRJ, Jagust WJ, Shaw LM, Toga AW, et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology. 2010;74:201–209. doi: 10.1212/WNL.0b013e3181cb3e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, Veyrieras J-B, Stephens M, Gilad Y, Pritchard JK. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature. 2010;464:768–772. doi: 10.1038/nature08872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polk TA, Park J, Smith MR, Park DC. Nature versus nurture in ventral visual cortex: A functional magnetic resonance imaging study of twins. J Neurosci. 2007;27:13921–13925. doi: 10.1523/JNEUROSCI.4001-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potkin SG, Macciardi F, Guffanti G, Wang Q, Turner JA, Lakatos A, Miles MF, Lander A, Vawter MP, Xie X. Identifying gene regulatory networks in schizophrenia. Neuroimage. 2010 doi: 10.1016/j.neuroimage.2010.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Psychiatric GWAS Consortium Coordinating Committee. Genomewide association studies: History, rationale, and prospects for psychiatric disorders. Am J Psychiatry. 2009;166:540–556. doi: 10.1176/appi.ajp.2008.08091354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, Saunders AM, Hardy J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. PNAS. 2004;101:284–289. doi: 10.1073/pnas.2635903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J, Kochunov P, Lancaster J, Shelledy W, Glahn D, Blangero J, Fox P. Heritability of brain volume, surface area and shape: An MRI study in an extended pedigree of baboons. Human Brain Mapping. 2007;28:576–583. doi: 10.1002/hbm.20407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J, Kochunov P, Zilles K, Shelledy W, Lancaster J, Thompson P, Duggirala R, Blangero J, Fox PT, Glahn DC. On the genetic architecture of cortical folding and brain volume in primates. Neuroimage. 2010;53:1103–1108. doi: 10.1016/j.neuroimage.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollins B, Martin MV, Morgan L, Vawter MP. Analysis of whole genome biomarker expression in blood and brain. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2010;153B:919–936. doi: 10.1002/ajmg.b.31062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe JB, Hughes LE, Barker RA, Owen AM. Dynamic causal modelling of effective connectivity from fMRI: Are results reproducible and sensitive to Parkinson’s disease and its treatment? Neuroimage. 2010;52:1015–1026. doi: 10.1016/j.neuroimage.2009.12.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccone NL, Culverhouse RC, Schwantes-An T-H, Cannon DS, Chen X, Cichon S, Giegling I, Han S, Han Y, Keskitalo-Vuokko K, et al. Multiple independent loci at chromosome 15q25.1 affect smoking quantity: A meta-analysis and comparison with lung cancer and COPD. PLoS Genet. 2010;6:e1001053. doi: 10.1371/journal.pgen.1001053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saccone SF, Hinrichs AL, Saccone NL, Chase GA, Konvicka K, Madden PAF, Breslau N, Johnson EO, Hatsukami D, Pomerleau O, et al. Cholinergic nicotinic receptor genes implicated in a nicotine dependence association study targeting 348 candidate genes with 3713 SNPs. Human Molecular Genetics. 2007;16:36–49. doi: 10.1093/hmg/ddl438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardana D, Vasa S, Vepachedu N, Chen J, Gudivada RC, Aronow BJ, Jegga AG. PhenoHM: Human-mouse comparative phenome-genome server. Nucl Acids Res. 2010;38:W165–174. doi: 10.1093/nar/gkq472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE. Molecular networks as sensors and drivers of common human diseases. Nature. 2009;461:218–223. doi: 10.1038/nature08454. [DOI] [PubMed] [Google Scholar]
- Schmitt JE, Wallace GL, Lenroot RK, Ordaz SE, Greenstein D, Clasen L, Kendler KS, Neale MC, Giedd JN. A twin study of intracerebral volumetric relationships. Behav Genet. 2010;40:114–124. doi: 10.1007/s10519-010-9332-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ, Podlisny MB. Deciphering the genetic basis of Alzheimer’s Disease. Annual Review of Genomics and Human Genetics. 2002;3:67–99. doi: 10.1146/annurev.genom.3.022502.103022. [DOI] [PubMed] [Google Scholar]
- Shehzad Z, Kelly AM, Reiss PT, Gee DG, Gotimer K, Uddin LQ, Lee SH, Margulies DS, Roy AK, Biswal BB, et al. The resting brain: Unconstrained yet reliable. Cereb Cortex. 2009;19:2209–2229. doi: 10.1093/cercor/bhn256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sing CF, Haviland MB, Reilly SL. Genetic architecture of common multifactorial diseases. Ciba Found Symp. 1996;197:211–229. doi: 10.1002/9780470514887.ch12. [DOI] [PubMed] [Google Scholar]
- Singleton AB, Hardy J, Traynor BJ, Houlden H. Towards a complete resolution of the genetic architecture of disease. Trends in Genetics. 2010;26:438–442. doi: 10.1016/j.tig.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SM, Miller KL, Salimi-Khorshidi G, Webster M, Beckmann CF, Nichols TE, Ramsey JD, Woolrich MW. Network modelling methods for fMRI. Neuroimage. doi: 10.1016/j.neuroimage.2010.08.063. in press. [DOI] [PubMed] [Google Scholar]
- Strother SC, Anderson J, Hansen LK, Kjems U, Kustra R, Sidtis J, Frutiger S, Muley S, LaConte S, Rottenberg D. The quantitative evaluation of functional neuroimaging experiments: The NPAIRS data analysis framework. Neuroimage. 2002;15:747–771. doi: 10.1006/nimg.2001.1034. [DOI] [PubMed] [Google Scholar]
- Tabor HK, Risch NJ, Myers RM. Candidate-gene approaches for studying complex genetic traits: Practical considerations. Nat Rev Genet. 2002;3:391–397. doi: 10.1038/nrg796. [DOI] [PubMed] [Google Scholar]
- Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S, Chasman DI, Willer CJ, et al. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010;466:707–713. doi: 10.1038/nature09270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- The International Schizophrenia Consortium. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460:748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PM, Cannon TD, Narr KL, van Erp T, Poutanen VP, Huttunen M, Lönnqvist J, Standertskjöld-Nordenstam CG, Kaprio J, Khaledy M, et al. Genetic influence on brain structure. Nat Neurosci. 2001;4:1253–1258. doi: 10.1038/nn758. [DOI] [PubMed] [Google Scholar]
- Van Horn JD, Grafton ST, Rockmore D, Gazzaniga MS. Sharing neuroimaging studies of human cognition. Nat Neurosci. 2004;7:473–481. doi: 10.1038/nn1231. [DOI] [PubMed] [Google Scholar]
- van Nas A, Ingram-Drake L, Sinsheimer JS, Wang SS, Schadt EE, Drake T, Lusis AJ. Expression quantitative trait loci: Replication, tissue- and sex-specificity in mice. Genetics. 2010;185:1059–1068. doi: 10.1534/genetics.110.116087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verweij KJH, Zietsch BP, Medland SE, Gordon SD, Benyamin B, Nyholt DR, McEvoy BP, Sullivan PF, Heath AC, Madden PAF, et al. A genome-wide association study of Cloninger’s temperament scales: Implications for the evolutionary genetics of personality. Biological Psychology. 2010 doi: 10.1016/j.biopsycho.2010.07.018. In Press, Corrected Proof. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visscher PM, Hill WG, Wray NR. Heritability in the genomics era--concepts and misconceptions. Nat Rev Genet. 2008;9:255–266. doi: 10.1038/nrg2322. [DOI] [PubMed] [Google Scholar]
- Walters RG, Jacquemont S, Valsesia A, de Smith AJ, Martinet D, Andersson J, Falchi M, Chen F, Andrieux J, Lobbens S, et al. A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature. 2010;463:671–675. doi: 10.1038/nature08727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster JA, Gibbs JR, Clarke J, Ray M, Zhang W, Holmans P, Rohrer K, Zhao A, Marlowe L, Kaleem M, et al. Genetic control of human brain transcript expression in Alzheimer disease. Am J Hum Genet. 2009;84:445–458. doi: 10.1016/j.ajhg.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler AM, Kochunov P, Blangero J, Almasy L, Zilles K, Fox PT, Duggirala R, Glahn DC. Cortical thickness or grey matter volume? The importance of selecting the phenotype for imaging genetics studies. Neuroimage. 2009 doi: 10.1016/j.neuroimage.2009.12.028. In Press, Corrected Proof. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolk DA, Dickerson BC Alzheimer’s Disease Neuroimaging Initiative. Apolipoprotein E (APOE) genotype has dissociable effects on memory and attentional-executive network function in Alzheimer’s disease. Proceedings of the National Academy of Sciences. 2010;107:10256–10261. doi: 10.1073/pnas.1001412107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Li W, James GM, Mehan MR, Zhou XJ. Automated multidimensional phenotypic profiling using large public microarray repositories. Proceedings of the National Academy of Sciences. 2009;106:12323–12328. doi: 10.1073/pnas.0900883106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, Madden PA, Heath AC, Martin NG, Montgomery GW, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–569. doi: 10.1038/ng.608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Deignan JL, Qi H, Zhu J, Qian S, Zhong J, Torosyan G, Majid S, Falkard B, Kleinhanz RR, et al. Validation of candidate causal genes for obesity that affect shared metabolic pathways and networks. Nat Genet. 2009;41:415–423. doi: 10.1038/ng.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarkoni T. Big correlations in little studies: Inflated fMRI correlations reflect low statistical power--Commentary on Vul et al. Perspectives on Psychological Science. 2009;4:294–298. doi: 10.1111/j.1745-6924.2009.01127.x. [DOI] [PubMed] [Google Scholar]
- Yarkoni T, Braver TS. Cognitive neuroscience approaches to individual differences in working memory and executive control: Conceptual and methodological issues. In: M G, Gruszka A, Szymura B, editors. Handbook of Individual Differences in Cognition. Springer; New York: 2010. pp. 87–107. [Google Scholar]