

Abstract
Aims: Apoptosis regulatory proteins, such as p53, play a pivotal role in neural differentiation, through mechanisms independent of cell death. In addition, p53 has been identified as an important regulator of mitochondrial survival response, maintaining mitochondrial DNA (mtDNA) integrity and oxidative protection. The aim of this study was to determine the role of mitochondrial p53 in organelle damage and neural differentiation. Results: Our results show that mitochondrial apoptotic events such as reactive oxygen species production, mitochondrial membrane permeabilization, and cytochrome c release are typical of early-stage mouse neural stem cell differentiation, which occurs 3–18 h after induction of differentiation, with no evidence of cell death. In addition, decreased mtDNA content, lipidated LC3 (LC3-II), colocalization of mitochondria and LC3-II puncta, and mitochondria-associated Parkin are consistent with activation of mitophagy. Importantly, at early stages of neural differentiation, p53 was actively translocated to mitochondria and attenuated mitochondrial oxidative stress, cytochrome c release, and mitophagy. Forced mitochondrial translocation of p53 increased neurogenic potential and neurite outgrowth. Innovation and Conclusion: In conclusion, our results reveal a novel role for mitochondrial p53, which modulates mitochondrial damage and apoptosis-related events in the context of neural differentiation, thus enhancing neuronal fate. Antioxid. Redox Signal. 21, 1009–1024.
Introduction
Poor survival and differentiation of neural stem cells (NSCs) have been a major problem for efficient use in neural replacement therapy. Therefore, a better understanding of the basic molecular mechanisms of long-term survival and differentiation of stem cells represents a step toward novel potential therapeutic approaches. Others and we have recently demonstrated that conserved elements of apoptosis, including p53, caspases, calpains, and apoptosis-associated microRNAs participate in neural differentiation, through mechanisms independent of cell death (3, 16, 18, 50, 53, 68). Thus, essential players of apoptosis executioner pathways may have a dual function in differentiation and cell death; they cannot be totally blocked to assure differentiation efficiency, but must be carefully regulated to avoid cell loss.
Recent evidence has suggested that mitochondria may also affect the proliferative and differentiation potential of stem cells (2, 43, 47). In fact, the density and activity of mitochondria vary in several types of terminally differentiated cells due to distinct levels of energy demand. The undifferentiated stage is usually associated with mitochondrial low oxygen consumption, needed to maintain the proliferative capacity (15), while aerobic metabolism, in association with increased reactive oxygen species (ROS) production, is crucial for successful differentiation (28, 62). Indeed, in response to changes of the cellular environment, such as those of differentiation, mitochondria may become a major producer of ROS and release prodeath proteins, rapidly changing into a death-promoting organelle. Curiously, the release of cytochrome c has also been associated with vital cell functions, including differentiation, suggesting that it does not always occur in an all-or-nothing fashion (20). A novel role for cytochrome c has also been described in remodeling chromatin at the nucleus compartment (38).
Innovation.
Conserved executioners of the apoptosis machinery, namely p53, have been shown to participate in neural differentiation through mechanisms independent of cell death. In addition, p53 has been identified as a key regulator of the mitochondrial survival response under stress conditions. Identifying the mechanisms by which mitochondrial p53 impacts on neural stem cell survival after differentiation-elicited mitochondrial damage is essential for a better understanding of how essential players of apoptosis may play a dual function in cell fate decision.
Surprisingly, in skeletal muscle and fibroblasts, p53 has been shown to mediate several nonapoptotic mitochondrial changes, including mitochondria aerobic metabolism (19, 33, 42), maintenance of mitochondrial DNA (mtDNA) stability, and oxidative protection (1, 29). Regulation of cellular function by p53 may occur through transcription-dependent and -independent mechanisms at the mitochondria, involving modulation of mitochondrial ROS production (23). Mitochondrial oxidative stress is thought to trigger mitophagy, a selective type of autophagy characterized by the engulfment of dysfunctional mitochondria in a multimembrane structure, the autophagosome, delivery, and subsequent degradation by the cells own lysosomal system (17, 56). In fact, autophagy plays multifunctional roles in cellular adaptation to stress, by maintaining mitochondrial integrity. Since ROS are required for this selective autophagic process, it is tempting to speculate that p53 may regulate mitochondrial quality control mechanisms, such as mitophagy, and differentiation-related oxidative damage.
In this study, we sought to investigate the role of mitochondrial p53 during NSC differentiation. We demonstrate that mitochondrial apoptosis-associated events, including mitochondrial membrane depolarization, ROS production, and cytochrome c release, as well as mitophagy are typical of early-stage mouse NSC differentiation. In contrast, active mitochondrial translocation of p53 prevented differentiation-induced mitochondrial alterations and enhanced neural fate, suggesting a novel role for p53-dependent mitochondrial control of NSC differentiation.
Results
Mitochondrial apoptosis-associated events in neural differentiation
Recent evidence suggests that mitochondrial apoptosis-associated events affect the proliferative and differentiation potential of NSCs (54). However, the precise mechanism by which differentiating cells can block preactivated apoptosis during differentiation is still unknown. To further understand mitochondrial alterations during neural differentiation, we used a NSC line. Induction of differentiation resulted in a tripotential cell population, favoring the conversion into the neuronal lineage compared with astroglial or oligodendroglial lineages (45, 52). In the present study, cells were allowed to differentiate up to 6 days and the expression of stemness (nestin), neuronal (βIII-tubulin), glial (GFAP), and oligodendrial (NG2) markers was analyzed throughout time by flow cytometry analysis. As expected, our results showed that under proliferative conditions, the majority of cells are positive for the proliferative marker nestin, and less than 3% are positive for βIII-tubulin, GFAP, or NG2. In addition, decreased proportions of nestin-positive cells and markedly increased βIII-tubulin-positive cells were observed in differentiating cells (p<0.01). Under these conditions, only few of the cells became GFAP- and NG2- positive at 1 and 6 days of differentiation (p<0.01), thus indicating that these in vitro differentiation conditions efficiently bias NSCs toward the neuronal lineage (Fig. 1A).
FIG. 1.

Mitochondrial apoptosis-associated events in neural differentiation. Mouse neural stem cells (NSCs) were expanded and induced to differentiate, then collected for flow cytometry and immunoblotting, as described in the Materials and Methods section. (A) Flow cytometry analysis of nestin, βIII-tubulin, GFAP, and NG2 expression at different times of differentiation. (B) Representative histogram of DiOC6(3)-positive cells at 18 h of differentiation (left) evaluated by flow cytometry and respective quantification data during neural differentiation (right). (C) Representative histogram (left) and quantification data (right) of mitochondrial reactive oxygen species (ROS) levels throughout differentiation by flow cytometry analysis using MitoSOX™ Red reagent. (D) Representative immunoblots of cytochrome c (top) and corresponding densitometry analysis (bottom) in both mitochondrial and cytosolic extracts. Results were normalized to endogenous voltage-dependent anion channel (VDAC) and GAPDH protein levels. Results are expressed as mean±SEM fold change for at least three different experiments. *p<0.01 from undifferentiated cells.
More importantly, mitochondrial membrane depolarized throughout neural differentiation, as assessed by a significant decrease in DiOC6 (3) mitochondrial incorporation (p<0.01) (Fig. 1B). Cells were also incubated with the MitoSOX™ Red reagent, which is oxidized by superoxide and exhibits red fluorescence, hence allowing the detection and quantification of mitochondrial ROS. Curiously, mitochondrial ROS levels were transiently increased at 1 h of differentiation, significantly decreasing at 6 and 18 h (p<0.01) and returning to basal levels thereafter (Fig. 1C). Moreover, NSC pretreatment with the cell-permeant 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) also revealed a similar trend in total intracellular ROS levels (data not shown). Since ROS-triggered mitochondrial membrane depolarization often results in mitochondrial cytochrome c release, the cellular distribution of cytochrome c was also evaluated throughout NSC differentiation. Notably, a transient but marked decrease of mitochondrial cytochrome c (p<0.01) was accompanied by a transient increase of cytosolic cytochrome c (p<0.01) (Fig. 1D). The relative purity of mitochondrial and cytosolic extracts was controlled using glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and voltage-dependent anion channel (VDAC) antibodies, respectively. In addition, to investigate whether differentiation-induced cytochrome c release results in increased cell death, cell viability was assessed by the Guava ViaCount assay and by measuring Annexin-V- and propidium iodide (PI)-negative cells by flow cytometry analysis (Supplementary Fig. S1A; Supplementary Data are available online at www.liebertpub.com/ars). No significant alterations, using both methods, were detected in cell death or cell viability during early-stage NSC differentiation. These findings indicate that neural differentiation induces mitochondrial apoptosis-associated events, which in turn do not involve cell death per se.
Mitophagy activation in neural differentiation
To further investigate the mechanisms by which differentiating cells block preactivated apoptosis during neural differentiation, we explored the presence of mitophagy, the major degradation pathway involved in mitochondrial quality control. In fact, recent studies have demonstrated that oxidative stress normally causes activation of mitochondrial quality control mechanisms leading to subsequent degradation of damaged mitochondria by mitophagy. The mtDNA content was quantified during neural differentiation by real-time polymerase chain reaction (PCR). Interestingly, the mtDNA copy number significantly decreased throughout NSC differentiation (p<0.01) (Fig. 2A). To better understand whether the decrease of the mitochondrial population was correlated to activation of mitophagy as a neural differentiation-induced mechanism of mitochondrial quality control, we assessed the conversion of endogenous LC3-I/-II as well as the mitochondrial translocation of Parkin. Recent studies have correlated the mitochondrial translocation of the cytosolic E3 ubiquitin ligase, Parkin, to the complete removal of depolarized and damaged mitochondria by mediating a quality control pathway of mitophagy (5, 37, 47, 56). The lipidated LC3 autophagic marker (Fig. 2B) and mitochondrial Parkin (Fig. 2C) increased after 6 h of neural differentiation (p<0.01), as assessed by immunoblotting analysis. The occurrence of mitophagy in this cellular context was confirmed by immunocytochemistry analysis, using confocal microscopy, where colocalization of LC3-II puncta and mitochondria was easily detected at 18 h of differentiation (Fig. 2D). Finally, the autophagic flux was controlled using 3-methyladenine (3-MA), an early phase inhibitor of autophagy that blocks autophagosome formation, and bafilomycin A1, a late phase inhibitor of autophagy that prevents the fusion between autophagosomes and lysosomes. As expected, at 18 h of differentiation, a significant decrease or accumulation in lipidated LC3 form was observed in 3-MA- and bafilomycin A1-treated NSCs, respectively (p<0.01). Immunocytochemistry analysis using confocal microscopy corroborated immunoblotting data (Fig. 2E). These results indicate that mitophagy may occur as a survival mechanism by which differentiating cells induce the clearance of damaged mitochondria and block preactivated mitochondrial apoptosis.
FIG. 2.

Mitophagy activation in neural differentiation. NSCs were induced to differentiate and collected for quantitative real-time PCR, immunoblotting, and immunocytochemistry analysis, as described in the Materials and Methods section. (A) Real-time PCR analysis of relative mtDNA copy number throughout NSC differentiation. (B) Representative immunoblots of LC3-I and LC3-II (top), and respective densitometric analysis (bottom) during neural differentiation. (C) Representative immunoblots of Parkin (top), and respective densitometric analysis (bottom) throughout differentiation. Results were normalized to endogenous β-actin or VDAC protein levels. (D) Representative confocal images of undifferentiated and differentiated NSCs, labeled with anti-LC3 antibody and MitoTracker® Red (top) and respective colocalization profile (bottom). (E) Immunoblot densitometric analysis of LC3-I and LC3-II turnover (left), in cells pretreated with 3-MA or bafilomycin A1, after 18 h of neural differentiation, and representative confocal images labeled with anti-LC3 antibody and MitoTracker Red (right). Nuclei were stained with Hoechst 33258. Scale bar, 5 μm. Results are expressed as mean±SEM fold change for at least three different experiments. *p<0.01 and §p<0.05 from undifferentiated cells; ‡p<0.01 from control. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
p53 mitochondrial translocation modulates oxidative stress, cytochrome c release, and mitophagy in neural differentiation
The tumor suppressor p53 protein has recently been identified as a critical regulator of mitochondrial survival response in several cell types, as it mediates mtDNA integrity and oxidative protection (1, 19, 23). Surprisingly, immunoblot analysis showed that p53 is actively but transiently translocated to mitochondria as early as 3 h after induction of neural differentiation (p<0.01), returning to basal levels after 2 days of differentiation induction (Fig. 3A). The relative purity of mitochondrial fractionation was controlled using the Lamin B1 antibody, which indicated the absence of nuclear contamination in the mitochondrial extracts. In addition, colocalization of p53 and mitochondria was easily monitored by confocal microscopy in NSCs, after 6 h of differentiation (Fig. 3B).
FIG. 3.
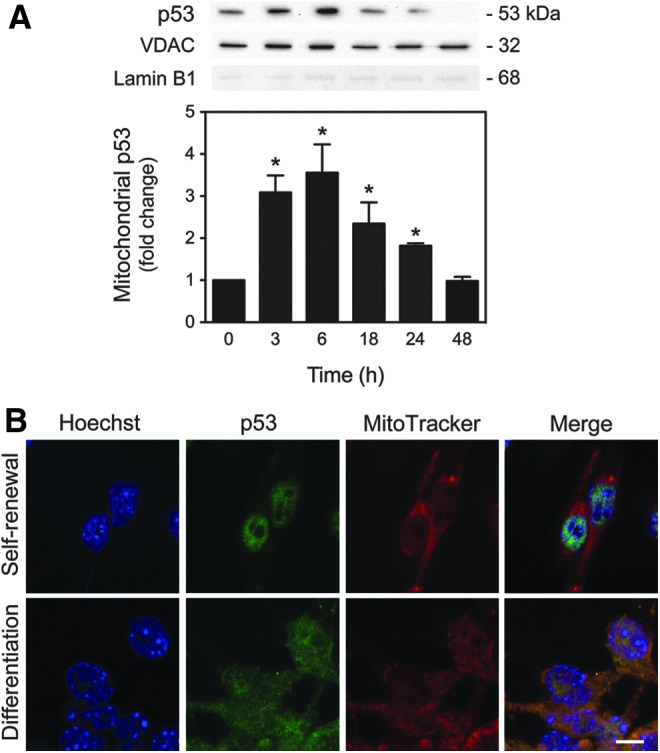
p53 mitochondrial translocation in neural differentiation. NSCs were induced to differentiate up to 48 h and collected for immunoblotting and immunocytochemistry, as described in the Materials and Methods section. (A) Representative immunoblots of p53 in mitochondrial extracts (top) and respective quantification data (bottom), throughout neural differentiation. Results were normalized to endogenous VDAC protein levels and nuclear contamination was assessed using the Lamin B1 antibody. (B) Representative confocal images of undifferentiated and differentiated NSCs for 6 h, labeled with Hoechst 33258, anti-p53 antibody, and MitoTracker Red. Scale bar, 5 μm. Results are expressed as mean±SEM fold change for at least three different experiments. *p<0.01 from undifferentiated cells. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
We then addressed the impact of p53 mitochondrial relocation on differentiation-induced oxidative stress, cytochrome c release, and mitophagy, by modulating p53 mitochondrial translocation in differentiating NSCs, rather than using a p53 null system that would also influence p53 nuclear function (53). For that, cells were transfected with the overexpressing plasmids, RECQL4-Wt and RECQL4-Δ84, which cannot translocate to the mitochondria, and RECQL4-GST, unable to physically interact with p53 (Fig. 4A) (13). The latter serves as a control to understand whether the effects are dependent on p53 or RECQL4 mitochondrial translocation. RECQL4, a nuclear protein member of the conserved RecQ family of DNA helicases (49), has been recently shown to physically interact with p53 to prevent its function as a transcription factor in the nucleus, and facilitate p53 mitochondrial localization (13). In accordance with p53 mitochondrial relocation, an increase of mitochondrial RECQL4 expression was observed at 3 and 6 h of NSC differentiation, markedly decreasing thereafter (at least p<0.05) (Supplementary Fig. S2). Higher levels of mitochondrial p53 were detected at 6 h of neural differentiation in RECQL4-Wt-transfected cells when compared with control (mock) (p<0.01), while p53 mitochondrial translocation significantly decreased in RECQL4-Δ84- and RECQL4-GST-transfected cells, when compared with wild type (p<0.01) (Fig. 4B). These results were also corroborated by immunocytochemistry analysis at 18 h of neural differentiation, using confocal microscopy (Fig. 4C). More importantly, modulation of p53 mitochondrial translocation during NSC differentiation revealed that forced mitochondrial relocation of p53 results in a marked decrease in oxidative stress at 6 h, also observed at later time points of neural differentiation (Fig. 5A). In addition, enhanced mitochondrial levels of p53 resulted in further retention of cytochrome c at the mitochondrial compartment at 6 h of differentiation induction (Fig. 5B). In fact, mitochondrial ROS (at least p<0.01) and cytosolic cytochrome c levels (p<0.05) were strongly diminished in RECQL4-Wt-transfected cells when compared with control or RECQL4-mutant-transfected cells. Curiously, when p53 was forced to translocate to mitochondria, the mtDNA copy number increased (p<0.05) (Fig. 6A) and both LC3-II (p<0.01) (Fig. 6B) and mitochondria-associated Parkin (Fig. 6C) decreased (at least p<0.05), when compared with mock or RECQL4-mutant-transfected cells. This suggests that regulation of mitochondrial damage by mitochondrial p53 is sufficient to increase the mitochondrial number and rule out the need for mitophagy activation. These results were further confirmed by decreased colocalization of LC3-II puncta and mitochondria, at 18 h of differentiation, in RECQL4-Wt-transfected cells when compared with control or RECQL4-mutant-transfected cells, using confocal microscopy. However, at 48 h of neural differentiation, the impact of mitochondrial p53 modulation, on these events, seems to be attenuated (Fig. 6D). Thus, our results reveal a novel role for mitochondrial p53, abrogating mitochondrial damage and preventing apoptosis-related events throughout neural differentiation.
FIG. 4.

Modulation of p53 mitochondrial translocation in neural differentiation. NSCs were induced to differentiate for 6 h, after transfection with RECQL4-Wt, RECQL4-Δ84, or RECQL4-GST plasmids, as described in the Materials and Methods section. Cells were then collected for immunoblotting and immunocytochemistry analysis, respectively. (A) Scheme representing RECQL4-Wt, RECQL4-Δ84, and RECQL4-GST protein function. (B) Representative immunoblot of p53 in mitochondrial extracts (top) and respective quantification data (bottom), after plasmid transfection and differentiation. Results were normalized to endogenous VDAC protein levels. (C) Representative confocal images of control, RECQL4-Wt-, RECQL4-Δ84-, and RECQL4-GST-transfected cells, labeled with Hoechst 33258, anti-p53 antibody, and MitoTracker Red, at 18 h of differentiation. Scale bar, 5 μm. Results are expressed as mean±SEM fold change for at least three different experiments. *p<0.01 from mock; ‡p<0.01 from RECQL4-Wt-transfected cells. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 5.

Mitochondrial p53 regulates differentiation-induced apoptotic events in neural differentiation. After transfection with RECQL4-Wt, RECQL4-Δ84, or RECQL4-GST plasmids, NSCs were induced to differentiate for 6 h and 48 h, and collected for flow cytometry and immunobloting analysis, as described in the Materials and Methods section. (A) Representative histogram of MitoSOX Red reagent fluorescence (left) and respective quantification data (right), after plasmid transfection and differentiation. (B) Representative immunoblots of cytochrome c (top) and corresponding densitometry analysis (bottom) in mitochondrial and cytosolic extracts of NSCs after 6 h of differentiation. Results are expressed as mean±SEM fold-change for at least three different experiments. *p<0.01 and §p<0.05 from mock; ‡p<0.01 and †p<0.05 from RECQL4-Wt-transfected cells.
FIG. 6.

Mitochondrial p53 regulates differentiation-induced mitophagy in neural differentiation. After transfection with RECQL4-Wt, RECQL4-Δ84, or RECQL4-GST plasmids, NSCs were induced to differentiate for 18 h and 48 h and collected for real-time PCR, immunoblotting, and immunocytochemistry analysis, as described in the Materials and Methods section. (A) Relative mtDNA content in NSCs after 18 h of differentiation. (B) Representative immunoblots of LC3-I and LC3-II expression (top), and the respective densitometric ratio (bottom) in NSCs after 18 h of differentiation. (C) Representative immunoblots of Parkin expression (top), and respective densitometry (bottom) in NSCs after 18 h of differentiation. Results were normalized to endogenous VDAC, GAPDH, or β-actin protein levels. (D) Representative confocal images of control, RECQL4-Wt-, RECQL4-Δ84-, and RECQL4-GST-transfected cells, labeled with anti-LC3 antibody and MitoTracker® Red (top) and respective colocalization profile (bottom), at 18 and 48 h of differentiation. Nuclei were stained with Hoechst 33258. Scale bar, 5 μm. Results are expressed as mean±SEM fold change for at least three different experiments. *p<0.01 and §p<0.05 from mock; ‡p<0.01 and †p<0.05 from RECQL4-Wt-transfected cells. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
p53 modulation of mitochondrial stress in neural differentiation is dependent on mitochondrial MnSOD activation
Superoxide dismutases (SOD) are the most commonly known scavengers of mitochondrial ROS in the cell (23). Although p53 has a protective effect on mitochondrial oxidative stress by regulating manganese superoxide dismutase (MnSOD) expression through the nucleus (23, 41), recent evidence suggests that mitochondrial p53 plays a role, along with MnSOD, in preventing mtDNA damage (1, 6, 7, 22, 32). Thus, we determined if the molecular mechanisms by which p53 prevents mitochondrial alterations during NSC differentiation are dependent on MnSOD. Interestingly, immunoblot analysis showed that MnSOD mitochondrial levels transiently increased at 3 and 6 h after induction of NSC differentiation (p<0.01), returning to basal levels after this time (Fig. 7A). More importantly, modulation of p53 mitochondrial translocation during NSC differentiation revealed that forced mitochondrial relocation of p53 markedly increased MnSOD mitochondrial levels at 6 h of differentiation, when compared with its levels in control or RECQL4-mutant-transfected cells (p<0.01) (Fig. 7B). Our results also showed that the effect of mitochondrial p53 in increasing mitochondrial levels of MnSOD may result, in part, from the ability of mitochondrial p53 to promote a more efficient import of this SOD, as demonstrated by a significant decrease of MnSOD cytosolic levels after forced mitochondrial translocation of p53 (p<0.01) (Fig. 7B). The effect of mitochondrial p53 on MnSOD protein stability was also investigated. For this purpose, the half-life of MnSOD was evaluated in NSCs after modulation of mitochondrial p53 levels by treating control and RECQL4-transfected NSCs with the protein synthesis inhibitor cycloheximide (CHX). Our results demonstrated that MnSOD was reduced in <3 h, in control and RECQL4-mutant-transfected cells (p<0.05). In contrast, MnSOD protein levels did not decrease until 6 h in RECQL4-Wt-transfected NSCs, demonstrating that p53 mitochondrial translocation also regulates the stability of MnSOD (Fig. 7C).
FIG. 7.

p53 requires MnSOD activity to prevent differentiation-induced mitochondrial stress. NSCs were induced to differentiate up to 48 h and collected for flow cytometry and immunoblotting analysis. After transfection with RECQL4-Wt, RECQL4-Δ84, or RECQL4-GST plasmids, NSCs were induced to differentiate for 6 h and collected for flow cytometry and immunoblotting analysis, as described in the Materials and Methods section. (A) Representative immunoblots of MnSOD in mitochondrial extracts (top) and respective densitometry data (bottom) throughout neural differentiation. (B) Representative immunoblots of MnSOD levels (top), and respective densitometry (bottom) in mitochondrial and cytosolic compartments after plasmid transfection and 6 h of neural differentiation. Results were normalized to endogenous VDAC protein levels. (C) Representative immunoblots of total MnSOD levels after transfection, where NSCs undergoing differentiation were treated with cycloheximide (CHX) for 3, 6, and 8 h before harvesting. Results were normalized to endogenous β-actin protein levels. (D) Mitochondrial ROS levels throughout differentiation using MitoSOX Red reagent by flow cytometry analysis, after treatment with cyclosporin A (CsA) for 1 h before harvest. (E) Evaluation of mitochondrial ROS levels, using MitoSOX Red reagent, after plasmid transfection and 6 h of neural differentiation, where cells were treated 1 h with CsA before harvest. Results are expressed as mean±SEM fold change for at least three different experiments. *p<0.01 from undifferentiated cells or mock; ‡p<0.01 and †p<0.05 from RECQL4-Wt-transfected cells or control.
Finally, to understand whether MnSOD was involved in mitochondrial p53 regulation of differentiation-induced mitochondrial ROS levels, we treated NSCs with cyclosporin A (CsA), a chemical inhibitor of mitochondrial MnSOD, and reevaluated mitochondrial oxidative stress. Surprisingly, CsA markedly induced mitochondrial ROS levels throughout neural differentiation (p<0.01) (Fig. 7D). More importantly, CsA-induced inhibition of MnSOD abolished the effect of mitochondrial p53 in modulating mitochondrial ROS levels (p<0.01) (Fig. 7E). Thus, our results reveal that p53-mediated mitochondrial oxidative protection during NSC differentiation is dependent on MnSOD activation.
p53 mitochondrial translocation enhances neuronal fate
To further investigate the precise role of mitochondrial p53 during neural fate, the impact of p53 mitochondrial translocation on NSC viability and differentiation was determined. Although modulation of p53 mitochondrial translocation did not elicit profound changes in cell death and viability throughout NSC differentiation (Supplementary Fig. S1B), the percentage of nestin- and βIII-tubulin-positive cells significantly decreased and increased, respectively, in RECQL4-Wt-transfected cells, when compared with mock or RECQL4 mutant plasmids (at least p<0.05), (Fig. 8A). We did not observe any changes in the percentage of GFAP- and NG2-positive cells in RECQL4-Wt-transfected cells, when compared with mock or RECQL4 mutant plasmids (data not shown). The proneurogenic effects of mitochondrial p53 were corroborated by immunocytochemistry analysis (Fig. 8B). In fact, in NSCs undergoing differentiation, forced mitochondrial p53 relocation promoted neuronal morphology, including neurite elongation and branching. The total neurite output, longest length, and number of neurites per cell were 45, 50, and 55% increased, respectively, after 1 day of neural differentiation when compared with mock or RECQL4 mutant plasmids (p<0.01). Interestingly, longer term studies revealed that the effect of mitochondrial p53 manipulation in enhancing neuronal fate remains detectable at 48 h of NSC differentiation (Fig. 8C). The changes elicited by forced p53 mitochondrial relocation on nestin and βIII-tubulin expression, as well as on the total neurite output and longest neurite length, were more evident after 6 days, when compared with 1 day of NSC differentiation. These results demonstrate that mitochondrial translocation of p53 strongly impacts on the neurogenic potential and on neurite elongation and branching of NSCs, with no changes on the survival rate.
FIG. 8.

Mitochondrial translocation of p53 modulates NSC fate. NSCs were induced to differentiate for 1 and 6 days, following transfection with RECQL4-Wt, RECQL4-Δ84, or RECQL4-GST plasmids, and collected for flow cytometry, immunocytochemistry, and bright field analysis as described in the Materials and Methods section. (A) Flow cytometry analysis of nestin (left) and βIII-tubulin (right). (B) Representative images of bright field and immunofluorescence detection of cells labeled with the anti-βIII-tubulin antibody. Nuclei were stained with Hoechst 33258. Scale bar, 10 μm. (C) Total neurite output, length of the longest neurite, and number of neurites per cell. Results are expressed as mean±SEM for at least three different experiments. *p<0.01 and §p<0.05 from mock; ‡p<0.01 and †p<0.05 from RECQL4-Wt-transfected cells. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Discussion
Apoptosis regulatory events may be involved in nonapoptotic functions, including differentiation, cell cycle progression, inflammation, metabolism, and autophagy (18, 53). However, the molecular checkpoints responsible for differentiation, as an alternative to cell death, remain poorly understood, despite the potential for therapeutic development. In this study, we suggest a novel role for mitochondrial p53 at early stages of neural differentiation. Our results indicate that mitochondrial translocation of p53 abrogates differentiation-induced mitochondrial alterations, including mitochondrial membrane depolarization, oxidative stress, and mitophagy, thus enhancing neuronal fate.
Mitochondria influence a wide range of cellular processes by mechanisms defined as retrograde signaling (9, 27). Indeed, the integrity and functional stage of mitochondria are crucial for cell proliferation, differentiation, apoptosis, and survival (34, 36, 43). During neural differentiation, profound mitochondrial morphologic and metabolic alterations occur to assure successful differentiation (26, 28, 40, 59, 62). On the other hand, the transition from anaerobic to aerobic metabolism implicates oxygen consumption, increased oxidative stress, and mitochondrial damage. In the present study, we have explored mitochondrial apoptosis-associated alterations during neural differentiation and showed that mitochondrial membrane depolarization and transient ROS production accompanied the neurogenic process. In fact, during the first 3 h of differentiation, neither p53 nor MnSOD has been sufficiently increased at the mitochondrial compartment to avoid mitochondrial stress. We believe that the subsequent decrease in mitochondrial ROS might be due to mitochondrial translocation of p53 and significant increases in MnSOD mitochondrial levels, which occur later during the differentiation process. Our data are in accordance with other studies highlighting the importance of mitochondrial dynamics and oxidative stress during routine neurogenesis (28, 62). In fact, a group of oxidation responsive genes was recently correlated to antioxidant administration, and was significantly elevated in genetic- and exercise-induced models of hyperactive hippocampal neurogenesis (61). Indeed, due to their ability to directly interact with specific receptors and to redox activate protein kinases and phosphatases, and transcription factors (51), controlled levels of ROS were shown to be pivotal as specific second messengers of signaling pathways involved in cell proliferation and differentiation. In addition, while mitochondrial fusion and fission are involved in regulating mitochondrial integrity during differentiation (34, 43, 66), they may also contribute to mitochondrial membrane depolarization, oxidative stress, and apoptosis (64). In agreement with neurogenesis-induced mitochondrial membrane permeabilization, we showed a transient release of cytochrome c from mitochondria during NSC differentiation. Nevertheless, increased ROS, mitochondrial membrane depolarization, and cytochrome c release did not enhance NSC death. Of note, in an apoptotic context, released cytochrome c was shown to gradually accumulate in the nucleus and induce chromatin condensation (38). Therefore, it is not surprising that neurogenesis-associated cytochrome c release may also be involved in chromatin remodeling required for the differentiation process. On the other hand, recent unpublished studies (Deshmukh M., personal communication) revealed that Hsp70 interacts with released cytochrome c to mediate its ubiquitination, thus preventing mitochondria-mediated cell death. Thus, it is also possible that Hsp70 might contribute for cell death resistance and neuronal survival in this cellular context. An increase in total levels of Hsp70 was also observed throughout NSC differentiation (data not shown). Nevertheless, additional mechanisms must be in place in differentiating cells to block preactivated apoptosis and avoid cell loss, which may involve p53-induced mitochondrial survival and mitophagy. Our results showed an increase of mitophagy throughout neural differentiation, as determined by increased lipidated LC3 and mitochondrial Parkin, colocalization of LC3-II puncta and mitochondria, as well as decreased mtDNA content. A decreased mitochondrial population may result from differentiation-induced engulfment of mitochondrial organelles through activation of mitophagy (25, 56), while Parkin translocation to the outer mitochondrial membrane of depolarized mitochondria is thought to ubiquitylate and recruit proteins that initiate this mechanism (10, 17, 25, 30, 65), ultimately ensuring mitochondrial quality control. Interestingly, autophagy has been shown to play a critical role in stem cell maintenance and in a variety of cell differentiation processes (4, 58, 67), also fulfilling NSC high-energy demands (57).
In the recent past, the proapoptotic protein p53 has been ascribed with an ever-increasing number of cellular functions, including the proneurogenic effect and mitochondrial survival response. The proneurogenic role of p53 is not entirely understood, but appears to be related with cell fate determination, actin cytoskeleton regulation, and control of specific signaling pathways (54). p53 has also been shown to regulate the cellular redox status by activating either anti- or pro-oxidant factors (19). In response to oxidative damage, p53 is translocated to mitochondria and physically interacts with mtDNA polymerase γ (Polγ), thereby promoting mtDNA replication and repair (1, 6, 22). Importantly, others have also demonstrated that p53 null mouse and p53 knockdown human primary fibroblasts exhibit mtDNA depletion and loss of mtDNA copy number under normal culture conditions (29). Based on the function of p53 and mitochondria during stem cell differentiation (31, 53, 68) as well as on the above evidence, we investigated the role of p53 in neurogenesis-induced mitochondrial alterations. Our results revealed that, following induction of NSC differentiation, p53 is actively translocated to mitochondria. More importantly, modulation of p53 mitochondrial relocation significantly altered differentiation-induced mitochondrial membrane depolarization, cytochrome c release, and oxidative stress, as well as the levels of mitophagy. Since mitochondrial changes only occur at early stages of neural differentiation, the manipulation of mitochondrial p53 did not promote major effects on mitochondria-associated events at later stages of differentiation. Our data support the idea that mitochondrial p53 is able to trigger a mitochondrial survival response and prevent apoptosis-associated events, under the early neural differentiation context. An exhaustive investigation on p53 submitochondrial distribution in different cell contexts would certainly help to further understand the variety of p53 roles in modulating mitochondrial apoptosis events. In fact, the localization of p53 at the surface of mitochondria is sufficient to induce apoptosis (39, 60, 63). In contrast, when p53 is localized to other mitochondrial compartments such as the mitochondrial matrix, it maintains mitochondrial genome integrity (1). In fact, while the antioxidant function of nonmitochondrial p53 has been well described (8, 11, 23, 24, 48), its antioxidative effect on the mitochondrial compartment has not been completely elucidated. Therefore, we further investigated the molecular mechanisms by which p53 prevents mitochondrial oxidative protection during NSC differentiation. Interestingly, our results support a functional coupling between the regulation of MnSOD, the major scavenger of mitochondrial ROS, and the decrease of mitochondrial damage by mitochondrial p53. In fact, although a negative regulation of MnSOD by mitochondrial p53 has already been demonstrated (23, 41), p53 may also play a role, along with MnSOD, in preventing mtDNA damage (7). The presence of mitochondrial p53 was shown to enhance the accuracy and repair efficiency of mtDNA by Polγ (1, 6, 22). On the other hand, mitochondrial MnSOD may also interact with Polγ to protect it from peroxynitrite-mediated inactivation (7, 32). Thus, it is possible that NSCs might use this novel step strategy to counteract differentiation-induced mitochondrial damage involving p53, MnSOD, and Polγ. In fact, our results also demonstrated that mitochondrial relocation of p53 promotes a more efficient mitochondrial import of MnSOD and its protein stabilization during the neural differentiation context, thus implicating p53, along with MnSOD, in preventing differentiation-induced oxidative stress. In fact, inhibition of MnSOD resulted in increased mitochondrial ROS levels, even after forced p53 mitochondrial translocation. This hypothesis is in agreement with our results demonstrating an increase in mtDNA content and reduction of mitophagy after forced mitochondrial p53 relocation. On the other hand, although mitophagy may be regulated by p53 (8, 35), it has never been directly associated with p53 mitochondrial relocation. The inhibition of neural differentiation-elicited mtDNA decrease and mitophagy that we observed in RECQL4-Wt-transfected cells might be partially explained by an indirect effect of mitochondrial p53 in regulating ROS and mtDNA damage. In fact, it appears that mitophagy acts downstream of p53 mitochondrial protection to avoid cell death. Forced mitochondrial localization of p53 during differentiation, by using RECQL4-Wt plasmids, enhanced mitochondrial integrity, as shown by increased mtDNA content and reduced mitochondrial ROS. Therefore, the activation of mitophagy, as a backup protection mechanism, decreases when compared with normal differentiating conditions. This is in accordance with no effect of mitochondrial p53 manipulation in cell viability.
Finally, we show that mitochondrial localization of p53, rather than promoting major effects on cell viability, has a long-term impact on neuronal fate and neurite outgrowth of NSCs. Previous studies showing the protective role of mitochondrial p53 in maintaining mtDNA integrity (1, 23, 42), and a strong linkage between mtDNA integrity and NSC fate (62), are also in agreement with the effect of mitochondrial p53 in increasing NSC neuronal fate. Indeed, 8-oxoguanine DNA glycosylase (OGG1) was shown to be pivotal for both mtDNA damage repair and NSC survival, being its expression associated with a higher neurogenic potential, while accumulation of mtDNA damage shifted NSC differentiation toward an astrocytic lineage. Additional experiments are eagerly required to elucidate p53-specific mitochondrial targets during NSC differentiation.
In conclusion, the present study provides a role for mitochondrial p53 in preventing NSC differentiation-elicited mitochondrial damage. In addition to influencing neuronal fate, p53 mitochondrial translocation is critical for the modulation of neuronal morphology, and neurite elongation and branching.
Materials and Methods
Ethics statement
The mouse NSC line used in this study was obtained from Dr. Smith's Laboratory, University of Cambridge, Cambridge, United Kingdom (52), and provided by Dr. Henrique, University of Lisbon, Lisbon, Portugal. The Animal Ethical Committee at the Faculty of Pharmacy, University of Lisbon, Portugal waived the need for approval.
Cell culture
NSCs were derived from 14.5-dpc mouse fetal forebrain. This cell line was established using a method that produces pure cultures of adherent NSCs, which continuously expand by symmetrical division and are capable of tripotential differentiation (12, 21, 44). NSCs were grown in monolayers as previously described (50, 55) and routinely maintained in an undifferentiation medium, comprising the Euromed-N medium (ECM0883L; EuroClone S.p.A., Pavia, Italy), supplemented with 1% N-2 supplement (17502-048; Invitrogen™, Life Technologies Corporation, Carlsbad, CA), 20 ng/ml epidermal growth factor (EGF, 315-09; PeproTech EC, London, United Kingdom), 20 ng/ml basic fibroblast growth factor (bFGF, 100-18B; PeproTech EC), and 1% penicillin–streptomycin (15140-122; Invitrogen, Life Technologies Corp.), in uncoated tissue culture plastic flasks at 37°C in a humidified atmosphere of 5% CO2. The medium was changed every 3 days and cells collected with accutase (A11105-01; Sigma-Aldrich Corporation, St. Louis, MO) when confluent. Neural differentiation was performed by first platting NSCs in the undifferentiation medium onto uncoated tissue culture plastic dishes at 6.5×105 cells/ml for 24 h, and changing the culture medium to a differentiation medium, comprising the Euromed-N medium supplemented with 10 ng/ml bFGF, 0.5% N-2 supplement, 1% B27 supplement (17504-044; Invitrogen, Life Technologies Corp.), and 1% penicillin–streptomycin. Cells were collected at 1, 3, 6, 18, 24, 48 h, and 6 days after induction of differentiation and processed for flow cytometry, immunoblotting, immunofluorescence, and quantitative real-time PCR assays. For autophagic flux control, NSCs were treated with 5 mM 3-MA (M9281; Sigma-Aldrich Corp.) or with 100 nM bafilomycin A1 (tlrl-baf1; InvivoGen Corporation, San Diego, CA) for 8 h before harvest. To inhibit the activity of MnSOD, NSCs were treated with 10 μM CsA (30024; Sigma-Aldrich Corp.) for 1 h before harvest. CsA induces nitration of MnSOD in tyrosine 34 residues, leading to MnSOD functional inactivation (32, 46). Finally, to evaluate the effects of mitochondrial p53 in modulating protein stability of MnSOD during differentiation, control and RECQL4-transfected NSCs were untreated (0 h) or treated with 100 μg/ml CHX (C4859; Sigma-Aldrich Corp.), an inhibitor of protein biosynthesis, for 3, 6, and 8 h before harvesting.
Transfection assays
Modulation of p53 mitochondrial translocation was performed by transfecting NSCs with the overexpression plasmids pcDNA3-Flag-RECQL4 (1-1208), pcDNA3-Flag-RECQL4 (Δ84), and RECQL4-GST, kindly provided by Dr. Sagar Sengupta (13). pcDNA3-Flag-RECQL4 (1-1208) (RECQL4-Wt) and pcDNA3-Flag-RECQL4 (Δ84) (RECQL4-Δ84) were obtained by cloning adenosine-5′-triphosphate-dependent DNA helicase Q4 (RECQL4) fragments between the NheI and XhoI sites of the FLAG-tagged pcDNA3 construct. The RECQL4-GST construct, in turn, was obtained by cloning the 270-400 fragment of RECQL4 into the EcoR1-XhoI sites of the pGEX4T-1 vector. Twenty-four hours after platting, NSCs were transfected with 5 μg construct per dish, using the Lipofectamine™ 2000 transfection reagent (11668-019; Invitrogen, Life Technologies Corp.), according to the manufacturer's instructions, and 24 h later the medium was changed to the differentiation medium. The efficiency of plasmid transfections was evaluated by monitoring RECQL4 expression in mitochondrial and cytosolic protein extracts by immunoblotting (Supplementary Fig. S3).
Analyses of NSC differentiation
For detection of nestin and βIII-tubulin expression levels, cells were fixed with paraformaldehyde (4%, w/v) in phosphate-buffered saline (PBS) for 20 min at 4°C, washed twice with a washing solution 0.1% saponin (47036; Sigma-Aldrich Corp.) in PBS, and incubated for 20 min in a blocking solution 0.25% saponin and 5% fetal bovine serum (FBS) in PBS. Subsequently, cells were washed and incubated for 30 min with mouse primary antibodies reactive to nestin (MAB353; Merck Millipore Corporation, Billerica, MA), βIII-tubulin (MMS-435P, TUJ1; Covance, Princeton, NJ), GFAP (NCL-GFAP-GA5; Novocastra, ©Leica Microsystems, Wetzlar, Germany), and NG2 (AB5320; Merck Millipore Corp.) at a dilution of 1:300, 1:500, 1:100, and 1:400, respectively. All antibodies were diluted in an antibody blocking solution, 0.1% saponin and 5% FBS in PBS. Cells were then washed twice and incubated with appropriate secondary antibodies, anti-mouse antibodies conjugated to Dylight 649 (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) at a dilution of 1:600, and anti-rabbit antibodies conjugated to Alexa Fluor® 594 (A-21207; Invitrogen, Life Technologies Corp.) at a dilution of 1:200, for 30 min. Cells were washed twice and resuspended in PBS with 2% FBS and analyzed using LSR Fortessa™ (Becton Dickinson, Mountain View, CA). Data were statistically evaluated using FlowJo software (Tree Star, Inc., Ashland, OR).
Mitochondrial ROS detection
NSCs were washed twice with Ca2+- and Mg2+-free PBS (Gibco®, Life Technologies Corp.), treated with accutase, and harvested with PBS. For mitochondrial ROS quantification, cells were pretreated with 5 μM MitoSOX Red mitochondrial superoxide indicator (M36008; Molecular Probes®, Life Technologies Corp.) in Hank's balanced salt solution (HBSS) (24020; Gibco, Life Technologies Corp.) at 37°C for 10 min. Several studies observed that MitoSOX Red reagent is quickly and mostly oxidized by superoxide rather than by other ROS- or reactive nitrogen species-generating systems, and oxidation of the probe is prevented by SOD. The oxidation of the probe becomes highly fluorescent upon binding to nucleic acids. Cells were then resuspended in PBS with 2% FBS (10082147; Invitrogen, Life Technologies Corp.) and emission of red fluorescence was analyzed in intact cells by cytometric analysis using Guava EasyCyte 5HT® (Merck Millipore Corp.). Data were statistically evaluated using GuavaSoft® software (Merck Millipore Corp.).
Measurement of mitochondrial transmembrane potential
Mitochondrial permeabilization was determined as the retention of the dye 3,3′-dihexyloxacarbocyanine iodide (DiOC6(3), D273; Molecular Probes, Life Technologies Corp.). NSCs were loaded with 50 nM DiOC6(3) for 30 min at 37°C and resuspended in PBS with 2% FBS. The emission of green fluorescence was analyzed by fluorescence-activated cell sorting analysis (FACS) using LSR Fortessa (Becton Dickinson). Data were statistically evaluated using FlowJo software (Tree Star, Inc.).
Evaluation of cell death and viability
Viability and cell death levels of NSCs were assessed by performing the ViaCount assay using the Guava easyCyte 5HT Flow cytometer (Guava Technologies, Inc., Merck Millipore Corp.). The ViaCount Assay distinguishes viable and nonviable cells based on differential permeability of two DNA-binding dyes present in the Guava ViaCount® Reagent. The nuclear dye only stains nucleated cells, while the viability dye brightly stains dying cells. Briefly, cells were resuspended in 500 μl PBS with 2% FBS. Subsequently, 20 μl of cell suspension was mixed with 180 μl of Guava ViaCount® reagent and incubated for 5 min. at room temperature. Sample acquisition and data analysis were performed using the ViaCount software module. Cell death and viability measurements were confirmed by staining NSCs with the Annexin-V-APC apoptosis detection kit (88-8007; eBioscience, Inc., San Diego, CA), according to the manufacturer's instructions. This kit allows the determination of phosphatidylserine exposure as well as the inclusion of the viability dye PI. Samples were analyzed by FACS using LSR Fortessa (Becton Dickinson). Data were statistically evaluated using FlowJo software (Tree Star, Inc.).
Total, mitochondrial, and cytosolic protein extraction
For isolation of total protein extracts, NSCs were collected and lysed using an ice-cold lysis buffer (10 mM Tris-HCl, pH 7.6, 5 mM MgCl2, 1.5 mM potassium acetate, 1% Nonidet P-40, 2 mM dithiothreitol) and protease inhibitor cocktail tablets Complete (Roche Applied Science, Mannheim, Germany) for 30 min. Samples were then sonicated, the lysate was centrifuged at 3,200 g at 4°C for 10 min, and the supernatant recovered. For isolation of mitochondrial protein extracts, cells were collected and lysed in an isolation buffer (20 mM HEPES, 10 mM KCl, 1.5 mM MgCl2.6H2O, 1 mM Na2EDTA, 1 mM EGTA, 250 mM Sucrose), supplemented with 1 mM DTT and Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, Inc., Rockford, IL), for 15 min. Cells were then disrupted by 40 strokes of a glass homogenizer, and homogenates were centrifuged at 2,500 g at 4°C for 10 min. The homogenate recovered was then centrifuged at 12,000 g at 4°C for 30 min to remove unbroken cells and nuclei. The mitochondrial fraction was centrifuged at 12,000 g for 30 min at 4°C, and the pellet resuspended in the isolation buffer and frozen at −80°C. The supernatant was removed and filtered through 0.2-μm and then 0.1-μm Ultrafree MC filters (Merck Millipore Corp.) to give cytosolic proteins. The protein content was measured by the Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA), according to the manufacturer's specifications, using bovine serum albumin as standard.
Immunoblotting
Levels of p53, cytochrome c, LC3, RECQL4, Parkin, and MnSOD were determined by western blot analysis. Briefly, 20–80 μg of total, mitochondrial, and cytosolic proteins were separated on 8%, 12%, or 15% sodium dodecyl sulfate–polyacrylamide electrophoresis gels and then subjected to immunoblotting using primary mouse monoclonal antibodies reactive to p53 (2524; Cell Signaling Technology®, Inc., Boston, MA) and cytochrome c (556433; BD Biosciences Pharmingen, San Diego, CA); primary rabbit polyclonal antibodies reactive to LC3 (PA1-16931; Thermo Fisher Scientific, Inc.), Parkin (sc-30130; Santa Cruz Biotechnology, Inc., Dallas, TX), and MnSOD (sc-30080; Santa Cruz Biotechnology, Inc.); or primary goat polyclonal antibodies reactive to mouse RECQL4 (sc-16927; Santa Cruz Biotechnology, Inc.) and human RECQL4 (sc-16924; Santa Cruz Biotechnology, Inc.). Blots were subsequently incubated with secondary antibodies conjugated with anti-mouse and anti-rabbit immunoglobulin G (IgG) conjugated with horseradish peroxidase (Bio-Rad Laboratories) and anti-goat IgG-HRP (sc-2020; Santa Cruz Biotechnology, Inc.) for 2 h at room temperature. Finally, membranes were processed for protein detection using Immobilon (Merck Millipore Corp.) or Super Signal™ substrate (Thermo Fisher Scientific, Inc.). VDAC (4866; Cell Signaling Technology, Inc.), GAPDH (sc-32233; Santa Cruz Biotechnology, Inc.), β-actin (A5441; Sigma-Aldrich Corp.), and Lamin B1 (ab16048; Abcam®, Cambridge, United Kingdom) antibodies were used as loading controls and/or to monitor the purity of mitochondrial and cytosolic fractionation.
Quantification of mtDNA copy number
Total cellular DNA was isolated using the QiaAmp DNA Mini Kit (51304; Qiagen, Hilden, Germany), following the manufacturer's protocols. Subsequently, quantitative real-time PCR was performed and a ratio of mtDNA to the nuclear 18S gene was generated for each sample, as previously described (14). mtDNA and 18S primer sequences used for amplification were as follows: for 18S rDNA nuclear gene 5′ TAG AGG GAC AAG TGG CGT TC 3′ (forward) and 5′ CGC TGA GCC AGT CAG TGT 3′ (reverse), and for mouse cytochrome oxidase-1 (CO-1) mitochondrial gene 5′ GCC CCA GAT ATA GCA TTC CC 3′ (forward) and 5′ GTT CAT CCT GTT CCT GCT CC 3′ (reverse). Two independent reactions for each primer set were assessed in a total volume of 25 μl containing 2×Power SYBR Green PCR master mix and 0.5 μM of each primer. The real-time PCR was run in the ABI 7300 (Applied Biosystems®, Life Technologies Corp.) sequence detection system. The relative mtDNA copy number was calculated based on the standard curve and the ratio of the amount of mtDNA versus 18S for each sample. The values were first expressed as percentage of total input and converted to fold change over control.
Immunocytochemistry and confocal microscopy
For visualization of p53 mitochondrial and LC3-II mitochondrial colocalizations in NSCs, cells were incubated with 0.5 μM MitoTracker® Red CMXRos (M-7512; Molecular Probes, Life Technologies Corp.), which preferentially accumulates in mitochondria, for 30 min at 37°C before cell harvesting. Cells were washed twice, fixed with paraformaldehyde (4%, w/v) in PBS, and then blocked for 1 h at room temperature in PBS, containing 0.1% Triton-X-100, 1% FBS, and 10% normal donkey serum (Jackson ImmunoResearch Laboratories, Inc.). Cells were incubated with a primary mouse monoclonal antibody reactive to p53 (2524; Cell Signaling Technology, Inc.) at a dilution of 1:1000 or with a primary rabbit polyclonal antibody reactive to LC3 (PA1-16931; Thermo Fisher Scientific, Inc.) at a dilution of 1:200, overnight at 4°C. After two washes, the secondary DyLight 488-conjugated anti-mouse antibody (Jackson ImmunoResearch Laboratories, Inc.) diluted 1:100 or the secondary Alexa Fluor 488 (A-21206; Invitrogen, Life Technologies Corp.) diluted 1:200, was added to cells for 2 h at room temperature. For evaluation of NSC differentiation, cells were incubated with the primary mouse monoclonal antibody reactive to βIII-tubulin (MMS-435P, TUJ1; Covance) at a dilution of 1:500 overnight at 4°C. After two washes, the secondary anti-mouse Alexa Fluor 568 (A-10037; Invitrogen, Life Technologies Corp.) was diluted 1:200 and added to cells for 2 h at room temperature. Mouse NSC nuclei were stained with Hoechst 33258 (861405; Sigma-Aldrich Corp.) at 50 μg/ml in PBS, for 3 min at room temperature. Samples were mounted using Fluoromount-G™ (731604; Beckman Coulter, Inc., Brea, CA). For p53 mitochondrial and LC3-II mitochondrial colocalization, the resulting fluorescent signals were imaged in a multitrack scanning mode using an inverted LSM 510 META confocal laser scanning microscope (Carl Zeiss, Jena, Germany) equipped with a 63x/1.4 oil differential interference contrast plan-apochromat objective. Single confocal planes were acquired with an optical slice thickness of 0.8 μm and the images were further processed using ImageJ. NSC differentiation was evaluated using bright field and fluorescence microscopy assessments performed with a Zeiss AX10 microscope (Carl Zeiss, Corp.), equipped with a 63x/1.4 oil plan-apochromat objective and a Leica DFC490 camera (Leica Microsystems). Images were processed using ImageJ.
Densitometry and statistical analysis
The relative intensities of protein bands were analyzed using the Quantity One Version 4.6.3 densitometric analysis program (Bio-Rad Laboratories). Results from different groups were compared using the Student's t test, two-way ANOVA, or one-way ANOVA followed by Bonferroni's or Dunnett's multiple comparison tests. Values of p<0.05 were considered statistically significant. All statistical analysis was performed with GraphPad Prism 5 software (GraphPad Software, Inc., San Diego, CA).
Supplementary Material
Abbreviations Used
- APC
allophycocyanin
- bFGF
basic fibroblast growth factor
- CO-1
cytochrome oxidase 1
- CHX
cycloheximide
- CsA
cyclosporin A
- DiOC6(3)
3,3 dihexyloxacarbocyanine iodide
- DNA
deoxyribonucleic acid
- dpc
days postcoitum
- DTT
dithiothreitol
- EGF
epidermal growth factor
- EGTA
ethylene glycol tetraacetic acid
- FACS
fluorescence activated cell sorting
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- HEPES
4-(2-hydroxyethyl) piperazine-1-ethanesulfonic acid
- HBSS
Hank's balanced salt solution
- HRP
horseradish peroxidase
- HSP
heat shock protein
- IgG
immunoglobulin G
- KCl
potassium chloride
- LC3
microtubule-associated protein light chain 3
- 3-MA
3-methyladenine
- Mg2+
magnesium
- MgCl2
magnesium chloride
- MgCl2.6H2O
magnesium chloride hexahydrate
- MnSOD
manganese superoxide dismutase
- mtDNA
mitochondrial DNA
- Na2EDTA
disodium ethylenediamine tetraacetic acid
- NSC
neural stem cell
- OGG1
8-oxoguanine DNA glycosylase
- PBS
phosphate-buffered saline
- pcDNA
plasmid construct DNA
- PCR
polymerase chain reaction
- PI
propidium iodide
- rDNA
ribosomal DNA
- RECQL4
ATP-dependent DNA helicase Q4
- RNA
ribonucleic acid
- ROS
reactive oxygen species
- SEM
standard error of the mean
- SOD
superoxide dismutase
- Tris-HCl
tris-hydrochloride
- VDAC
voltage-dependent anion channel
- Wt
wild type
Acknowledgments
The authors wish to thank Dr. Sagar Sengupta (National Institute for Immunology, New Delhi, India) for kindly providing the RECQL4 vector construct and Dr. Domingos Henrique for providing and giving assistance in establishing the NS cell line culture conditions (Instituto de Medicina Molecular, University of Lisbon, Lisbon, Portugal). We are also grateful to Dr. Maria Soares and Ana Isabel Vieira (Flow Cytometry Unit, Instituto de Medicina Molecular, University of Lisbon, Lisbon, Portugal) and Dr. José Rino, António Temudo, and Ana Margarida Nascimento (BioImaging Unit, Instituto de Medicina Molecular, University of Lisbon, Lisbon, Portugal) for the training and skillful technical assistance in designing accurate settings in LSR Fortessa cytometers and LSM 510 META confocal laser scanning microscopes, respectively. We would also like to thank all members of the laboratory for insightful discussions.
This work was supported by the grants PTDC/SAU-NMC/117877/2010, and PTDC/BIM-MED/0251/2012, and by the fellowships SFRH/BD/68368/2010 (JMX) and SFRH/BD/80060/2011 (ALM) from Fundação para a Ciência e Tecnologia, Portugal.
Author Disclosure Statement
The authors have declared that no competing interests exist.
References
- 1.Achanta G, Sasaki R, Feng L, Carew JS, Lu W, Pelicano H, Keating MJ, and Huang P. Novel role of p53 in maintaining mitochondrial genetic stability through interaction with DNA Pol gamma. EMBO J 24: 3482–3492, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antico Arciuch VG, Elguero ME, Poderoso JJ, and Carreras MC. Mitochondrial regulation of cell cycle and proliferation. Antioxid Redox Signal 16: 1150–1180, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aranha MM, Santos DM, Xavier JM, Low WC, Steer CJ, Sola S, and Rodrigues CM. Apoptosis-associated microRNAs are modulated in mouse, rat and human neural differentiation. BMC Genomics 11: 514, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Asanuma K, Tanida I, Shirato I, Ueno T, Takahara H, Nishitani T, Kominami E, and Tomino Y. MAP-LC3, a promising autophagosomal marker, is processed during the differentiation and recovery of podocytes from PAN nephrosis. FASEB J 17: 1165–1167, 2003 [DOI] [PubMed] [Google Scholar]
- 5.Ashrafi G. and Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 20: 31–42, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bakhanashvili M, Grinberg S, Bonda E, Simon AJ, Moshitch-Moshkovitz S, and Rahav G. p53 in mitochondria enhances the accuracy of DNA synthesis. Cell Death Differ 15: 1865–1874, 2008 [DOI] [PubMed] [Google Scholar]
- 7.Bakthavatchalu V, Dey S, Xu Y, Noel T, Jungsuwadee P, Holley AK, Dhar SK, Batinic-Haberle I, and St Clair DK. Manganese superoxide dismutase is a mitochondrial fidelity protein that protects Polgamma against UV-induced inactivation. Oncogene 31: 2129–2139, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bensaad K, Cheung EC, and Vousden KH. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J 28: 3015–3026, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Butow RA. and Avadhani NG. Mitochondrial signaling: the retrograde response. Mol Cell 14: 1–15, 2004 [DOI] [PubMed] [Google Scholar]
- 10.Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, and Chan DC. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 20: 1726–1737, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheung EC, Ludwig RL, and Vousden KH. Mitochondrial localization of TIGAR under hypoxia stimulates HK2 and lowers ROS and cell death. PNAS 109: 20491–20496, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conti L, Pollard SM, Gorba T, Reitano E, Toselli M, Biella G, Sun Y, Sanzone S, Ying QL, Cattaneo E, and Smith A. Niche-independent symmetrical self-renewal of a mammalian tissue stem cell. PLoS Biol 3: e283, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De S, Kumari J, Mudgal R, Modi P, Gupta S, Futami K, Goto H, Lindor NM, Furuichi Y, Mohanty D, and Sengupta S. RECQL4 is essential for the transport of p53 to mitochondria in normal human cells in the absence of exogenous stress. J Cell Sci 125: 2509–2522, 2012 [DOI] [PubMed] [Google Scholar]
- 14.Eaton JS, Lin ZP, Sartorelli AC, Bonawitz ND, and Shadel GS. Ataxia-telangiectasia mutated kinase regulates ribonucleotide reductase and mitochondrial homeostasis. J Clin Invest 117: 2723–2734, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Facucho-Oliveira JM, Alderson J, Spikings EC, Egginton S, and St John JC. Mitochondrial DNA replication during differentiation of murine embryonic stem cells. J Cell Sci 120: 4025–4034, 2007 [DOI] [PubMed] [Google Scholar]
- 16.Fernando P, Kelly JF, Balazsi K, Slack RS, and Megeney LA. Caspase 3 activity is required for skeletal muscle differentiation. Proc Natl Acad Sci U S A 99: 11025–11030, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frank M, Duvezin-Caubet S, Koob S, Occhipinti A, Jagasia R, Petcherski A, Ruonala MO, Priault M, Salin B, and Reichert AS. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim Biophys Acta 1823: 2297–2310, 2012 [DOI] [PubMed] [Google Scholar]
- 18.Galluzzi L, Kepp O, Trojel-Hansen C, and Kroemer G. Non-apoptotic functions of apoptosis-regulatory proteins. EMBO Rep 13: 322–330, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galluzzi L, Morselli E, Kepp O, Vitale I, Pinti M, and Kroemer G. Mitochondrial liaisons of p53. Antioxid Redox Signal 15: 1691–1714, 2011 [DOI] [PubMed] [Google Scholar]
- 20.Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, and Kroemer G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ 13: 1423–1433, 2006 [DOI] [PubMed] [Google Scholar]
- 21.Glaser T, Pollard SM, Smith A, and Brustle O. Tripotential differentiation of adherently expandable neural stem (NS) cells. PLoS One 2: e298, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heyne K, Mannebach S, Wuertz E, Knaup KX, Mahyar-Roemer M, and Roemer K. Identification of a putative p53 binding sequence within the human mitochondrial genome. Febs Lett 578: 198–202, 2004 [DOI] [PubMed] [Google Scholar]
- 23.Holley AK, Dhar SK, and St Clair DK. Manganese superoxide dismutase vs. p53: regulation of mitochondrial ROS. Mitochondrion 10: 649–661, 2010 [DOI] [PubMed] [Google Scholar]
- 24.Kang MY, Kim HB, Piao C, Lee KH, Hyun JW, Chang IY, and You HJ. The critical role of catalase in prooxidant and antioxidant function of p53. Cell Death Differ 20: 117–129, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim I, Rodriguez-Enriquez S, and Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch Biochem Biophys 462: 245–253, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kole AJ, Annis RP, and Deshmukh M. Mature neurons: equipped for survival. Cell Death Dis 4: 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koussevitzky S, Nott A, Mockler TC, Hong F, Sachetto-Martins G, Surpin M, Lim J, Mittler R, and Chory J. Signals from chloroplasts converge to regulate nuclear gene expression. Science 316: 715–719, 2007 [PubMed] [Google Scholar]
- 28.Le Belle JE, Orozco NM, Paucar AA, Saxe JP, Mottahedeh J, Pyle AD, Wu H, and Kornblum HI. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell 8: 59–71, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lebedeva MA, Eaton JS, and Shadel GS. Loss of p53 causes mitochondrial DNA depletion and altered mitochondrial reactive oxygen species homeostasis. Biochim Biophys Acta 1787: 328–334, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 8: 3–5, 2005 [DOI] [PubMed] [Google Scholar]
- 31.Lin T, Chao C, Saito S, Mazur SJ, Murphy ME, Appella E, and Xu Y. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol 7: 165–171, 2005 [DOI] [PubMed] [Google Scholar]
- 32.MacMillan-Crow LA, Crow JP, and Thompson JA. Peroxynitrite-mediated inactivation of manganese superoxide dismutase involves nitration and oxidation of critical tyrosine residues. Biochemistry 37: 1613–1622, 1998 [DOI] [PubMed] [Google Scholar]
- 33.Maddocks OD. and Vousden KH. Metabolic regulation by p53. J Mol Med (Berl) 89: 237–245, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McBride HM, Neuspiel M, and Wasiak S. Mitochondria: more than just a powerhouse. Curr Biol 16: R551–R560, 2006 [DOI] [PubMed] [Google Scholar]
- 35.Mikami K, Okita N, Tokunaga Y, Ichikawa T, Okazaki T, Takemoto K, Nagai W, Matsushima S, and Higami Y. Autophagosomes accumulate in differentiated and hypertrophic adipocytes in a p53-independent manner. Biochem Biophys Res Commun 427: 758–763, 2012 [DOI] [PubMed] [Google Scholar]
- 36.Nagley P, Higgins GC, Atkin JD, and Beart PM. Multifaceted deaths orchestrated by mitochondria in neurones. Biochim Biophys Acta 1802: 167–185, 2010 [DOI] [PubMed] [Google Scholar]
- 37.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, and Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8: e1000298, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nur EKA, Gross SR, Pan Z, Balklava Z, Ma J, and Liu LF. Nuclear translocation of cytochrome c during apoptosis. J Biol Chem 279: 24911–24914, 2004 [DOI] [PubMed] [Google Scholar]
- 39.Palacios G, Crawford HC, Vaseva A, and Moll UM. Mitochondrially targeted wild-type p53 induces apoptosis in a solid human tumor xenograft model. Cell Cycle 7: 2584–2590, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Panchision DM. The role of oxygen in regulating neural stem cells in development and disease. J Cell Physiol 220: 562–568, 2009 [DOI] [PubMed] [Google Scholar]
- 41.Pani G, Bedogni B, Anzevino R, Colavitti R, Palazzotti B, Borrello S, and Galeotti T. Deregulated manganese superoxide dismutase expression and resistance to oxidative injury in p53-deficient cells. Cancer Res 60: 4654–4660, 2000 [PubMed] [Google Scholar]
- 42.Park JY, Wang PY, Matsumoto T, Sung HJ, Ma W, Choi JW, Anderson SA, Leary SC, Balaban RS, Kang JG, and Hwang PM. p53 improves aerobic exercise capacity and augments skeletal muscle mitochondrial DNA content. Circ Res 105: 705–712, 711 p following 712, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parker GC, Acsadi G, and Brenner CA. Mitochondria: determinants of stem cell fate? Stem Cells Dev 18: 803–806, 2009 [DOI] [PubMed] [Google Scholar]
- 44.Pollard SM, Conti L, Sun Y, Goffredo D, and Smith A. Adherent neural stem (NS) cells from fetal and adult forebrain. Cereb Cortex 16Suppl 1: i112–i120, 2006 [DOI] [PubMed] [Google Scholar]
- 45.Pratt T, Sharp L, Nichols J, Price DJ, and Mason JO. Embryonic stem cells and transgenic mice ubiquitously expressing a tau-tagged green fluorescent protein. Dev Biol 228: 19–28, 2000 [DOI] [PubMed] [Google Scholar]
- 46.Redondo-Horcajo M, Romero N, Martinez-Acedo P, Martinez-Ruiz A, Quijano C, Lourenco CF, Movilla N, Enriquez JA, Rodriguez-Pascual F, Rial E, Radi R, Vazquez J, and Lamas S. Cyclosporine A-induced nitration of tyrosine 34 MnSOD in endothelial cells: role of mitochondrial superoxide. Cardiovasc Res 87: 356–365, 2010 [DOI] [PubMed] [Google Scholar]
- 47.Rugarli EI. and Langer T. Mitochondrial quality control: a matter of life and death for neurons. EMBO J 31: 1336–1349, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, and Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nat Med 11: 1306–1313, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sangrithi MN, Bernal JA, Madine M, Philpott A, Lee J, Dunphy WG, and Venkitaraman AR. Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund-Thomson syndrome. Cell 121: 887–898, 2005 [DOI] [PubMed] [Google Scholar]
- 50.Santos DM, Xavier JM, Morgado AL, Sola S, and Rodrigues CM. Distinct regulatory functions of calpain 1 and 2 during neural stem cell self-renewal and differentiation. PLoS One 7: e33468, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sauer H, Wartenberg M, and Hescheler J. Reactive oxygen species as intracellular messengers during cell growth and differentiation. Cell Physiol Biochem 11: 173–186, 2001 [DOI] [PubMed] [Google Scholar]
- 52.Silva J, Chambers I, Pollard S, and Smith A. Nanog promotes transfer of pluripotency after cell fusion. Nature 441: 997–1001, 2006 [DOI] [PubMed] [Google Scholar]
- 53.Sola S, Aranha MM, and Rodrigues CM. Driving apoptosis-relevant proteins toward neural differentiation. Mol Neurobiol 46: 316–331, 2012 [DOI] [PubMed] [Google Scholar]
- 54.Sola S, Morgado AL, and Rodrigues CM. Death receptors and mitochondria: Two prime triggers of neural apoptosis and differentiation. Biochim Biophys Acta 1830: 2160–2166, 2013 [DOI] [PubMed] [Google Scholar]
- 55.Spiliotopoulos D, Goffredo D, Conti L, Di Febo F, Biella G, Toselli M, and Cattaneo E. An optimized experimental strategy for efficient conversion of embryonic stem (ES)-derived mouse neural stem (NS) cells into a nearly homogeneous mature neuronal population. Neurobiol Dis 34: 320–331, 2009 [DOI] [PubMed] [Google Scholar]
- 56.Tolkovsky AM. Mitophagy. Biochim Biophys Acta 1793: 1508–1515, 2009 [DOI] [PubMed] [Google Scholar]
- 57.Vazquez P, Arroba AI, Cecconi F, de la Rosa EJ, Boya P, and de Pablo F. Atg5 and Ambra1 differentially modulate neurogenesis in neural stem cells. Autophagy 8: 187–199, 2012 [DOI] [PubMed] [Google Scholar]
- 58.Vessoni AT, Muotri AR, and Okamoto OK. Autophagy in stem cell maintenance and differentiation. Stem Cells Dev 21: 513–520, 2012 [DOI] [PubMed] [Google Scholar]
- 59.Voloboueva LA, Lee SW, Emery JF, Palmer TD, and Giffard RG. Mitochondrial protection attenuates inflammation-induced impairment of neurogenesis in vitro and in vivo. J Neurosci 30: 12242–12251, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walia V, Kakar S, and Elble R. Micromanagement of the mitochondrial apoptotic pathway by p53. Front Biosci 16: 749–758, 2011 [DOI] [PubMed] [Google Scholar]
- 61.Walton NM, Shin R, Tajinda K, Heusner CL, Kogan JH, Miyake S, Chen Q, Tamura K, and Matsumoto M. Adult neurogenesis transiently generates oxidative stress. PLoS One 7: e35264, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang W, Osenbroch P, Skinnes R, Esbensen Y, Bjoras M, and Eide L. Mitochondrial DNA integrity is essential for mitochondrial maturation during differentiation of neural stem cells. Stem Cells 28: 2195–2204, 2010 [DOI] [PubMed] [Google Scholar]
- 63.Wolff S, Erster S, Palacios G, and Moll UM. p53's mitochondrial translocation and MOMP action is independent of Puma and Bax and severely disrupts mitochondrial membrane integrity. Cell Res 18: 733–744, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu S, Zhou F, Zhang Z, and Xing D. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission-fusion proteins. FEBS J 278: 941–954, 2011 [DOI] [PubMed] [Google Scholar]
- 65.Yoshii SR, Kishi C, Ishihara N, and Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem 286: 19630–19640, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Youle RJ, and van der Bliek AM. Mitochondrial fission, fusion, and stress. Science 337: 1062–1065, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao Y, Huang Q, Yang J, Lou M, Wang A, Dong J, Qin Z, and Zhang T. Autophagy impairment inhibits differentiation of glioma stem/progenitor cells. Brain Res 1313: 250–258, 2010 [DOI] [PubMed] [Google Scholar]
- 68.Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, Perry SR, Tonon G, Chu GC, Ding Z, Stommel JM, Dunn KL, Wiedemeyer R, You MJ, Brennan C, Wang YA, Ligon KL, Wong WH, Chin L, and DePinho RA. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 455: 1129–1133, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.