

Abstract
In order to demonstrate the cell-surface localization of a putative transmembrane receptor in cultured neurons, we labeled the protein on the surface of live neurons with a specific primary antibody raised against an extracellular portion of the protein. Given that receptors are trafficked to and from the surface, if cells are permeabilized after fixation then both cell-surface and internal protein will be detected by the same labeled secondary antibody. Here, we adapted a method used to study protein trafficking (“antibody feeding”) to differentially label protein that had been internalized by endocytosis during the antibody incubation step and protein that either remained on the cell surface or was trafficked to the surface during this period. The ability to distinguish these two pools of protein was made possible through the incorporation of an overnight blocking step with highly-concentrated unlabeled secondary antibody after an initial incubation of unpermeabilized neurons with a fluorescently-labeled secondary antibody. After the blocking step, permeabilization of the neurons allowed detection of the internalized pool with a fluorescent secondary antibody labeled with a different fluorophore. Using this technique we were able to obtain important information about the subcellular location of this putative receptor, revealing that it was, indeed, trafficked to the cell-surface in neurons. This technique is broadly applicable to a range of cell types and cell-surface proteins, providing a suitable antibody to an extracellular epitope is available.
Keywords: Neuroscience, Issue 84, two-color fluorescence immunocytochemistry, trafficking, endocytosis, recycling endosome, neurons
Introduction
In establishing the function of newly identified proteins, investigation of the subcellular localization and trafficking of the protein in question can provide important clues about the likely role/s of the protein1,2. Bioinformatic analysis of the transcriptome of the developing neocortex3 provided us with a list of genes exhibiting altered expression during mouse brain corticogenesis. We then adopted a gene knockout approach to ascertain that the protein encoded by one of these genes, sez6, has a key role in neuron development. We observed that the Seizure-related gene 6, or Sez6, protein is located in developing dendrites and is also present in dendritic spines, specialized structures on dendrites that receive and integrate excitatory signals. Furthermore, when this protein is lacking, dendrites and excitatory synapses fail to form correctly4. The probable dominant isoform of the protein has features of a transmembrane receptor although, when the subcellular distribution of immunolabeled protein was examined by confocal microscopy or by immunoelectron microscopy most, if not all, of the signal appeared associated with small vesicles in the somatodendritic compartment with little, or no, protein labeled on the plasma membrane at the cell surface.
In order to definitively show that this putative receptor with a predicted large extracellular domain is trafficked to the plasma membrane, we adopted a live-cell approach using the antiserum we had generated to an extracellular portion of the protein to label protein on the cell surface. By combining this “antibody feeding” approach with two applications of differentially-labeled secondary antibody separated by an extensive blocking step and a permeabilization step, we were able to identify two different pools of protein distinguished by binding to fluorescently-labeled secondary antibodies bearing different fluorescent tags. Thus, we were able to distinguish protein that had been internalized by endocytosis during the antibody incubation step from protein that either remained on the cell surface or was trafficked to the surface during this period. Using this method, we established that the protein of interest is trafficked to and from the cell surface in neurons. Therefore, this relatively fast and simple technique proved more informative than traditional immunocytochemistry methods or pre-embedding immunogold electron microscopy, despite the fact that we used the same rabbit polyclonal antiserum for all these techniques. This technique is generally applicable to any transmembrane protein provided a good antibody recognizing extracellular domain epitopes is available. The technique has been used previously to study receptor trafficking of the glutamate receptor GluR1 subunit5.
Protocol
1. Dissociated Hippocampal Neuron Culture
- Prepare coverslips (borosilicate glass):
- Wash in 100% ethanol.
- Air dry under UV irradiation.
- Coat with Poly-D-lysine (0.5 mg/ml in 0.15 M borate buffer, overnight at 4 °C).
- On the next day (the day of culture) wash 3x in PBS then coat with laminin (2.5 μg/ml, natural mouse laminin) + 5% v/v heat-inactivated fetal calf serum (FCS) diluted in PBS for 2 hr at 37 °C.
Dissect embryonic day 18 (E18) rat hippocampi and collect into PBS containing calcium and magnesium, chilled on ice. NOTE: all experimental protocols involving animals were approved by the Animal Ethics Committee of the University of Melbourne.
- Prepare the papain and DNase I solutions from the Papain Dissociation Kit according to the manufacturer’s instructions.
- Add 50 μl DNase I solution to 1 ml papain solution.
- NOTE: Once first prepared, the papain solution and the DNase I solutions may be separately dispensed into aliquots (0.5 ml and 25 μl aliquots for papain and DNase I, respectively) and stored at -20 °C until needed.
Remove as much PBS as possible and incubate hippocampi (from one litter of embryos) with 1 ml papain/DNase I solution at 37 °C for 15-20 min. Gently flick the tube to mix the contents twice during the incubation period.
Triturate the hippocampal tissue gently (avoiding the generation of bubbles) with a flame-polished siliconized Pasteur pipette 10-15x until cell dispersion is obtained and few, if any, chunks of undissociated tissue remain.
- Carefully layer the dissociated cell suspension over a 3 ml cushion of 4% w/v bovine serum albumin (BSA) in Hanks Balanced Salt Solution plus additives (HBSS+; see Section 1.6.1).
- Prepare this step gradient solution by dissolving the BSA at room temperature without stirring in HBSS containing 2 mM CaCl2, 1 mM MgSO4, 4 mM NaHCO3 and 40 mM glucose (HBSS+) then sterile filter and store at 4 °C.
Centrifuge, 100 x g, 7 min in a bench top centrifuge with a swing-out rotor.
Remove the supernatant being careful not to aspirate the cell pellet.
Resuspend the pelleted cells gently with a flame-polished (siliconized) Pasteur pipette (or a 1 ml blue pipette tip) in 1 ml complete Neurobasal medium (with 2% B27, 0.5 mM L-glutamine supplemented with 1% FCS).
Count two 10 μl aliquots using a hemocytometer (N.B. only count phase-bright cells as “live” cells).
Plate primary neurons on preprepared coated glass coverslips (0.75-1 x 105/18 mm coverslip in 12-well plate). Immediately prior to plating, aspirate excess laminin/serum solution from coverslips and replace with primary neuron culture medium (see below). N.B. The required number of coverslips needs to be determined in advance as coverslip preparation is begun the day prior to culture (see step 1.1).
Culture rat primary E18 hippocampal neurons for up to 21 days in vitro (DIV) in Neurobasal medium, 2% B27, 0.5 mM L-glutamine supplemented with 1% FCS (heat inactivated). Perform a half medium change at 7 DIV and weekly thereafter. The anti-mitotic fluorodeoxyuridine/uridine is added at 7 DIV; 1/1,000 dilution of 10 mM stock of each nucleoside) to prevent glial overgrowth.
2. Antibody Incubation with Live Neurons
- During the first week in culture, neurons are developing dendritic arbors and by week 2-3, neurons have matured sufficiently to be undergoing synaptogenesis6,7.
- At selected experimental time point/s, apply the primary antibody to triplicate wells containing primary embryonic neurons on coverslips. Add an aliquot of the antibody directly into the culture medium of the primary neurons to the desired final dilution Note: In our example, a rabbit polyclonal antiserum raised against a recombinant secreted form of the protein was diluted 1/500 by adding 2 μl to 1 ml of culture medium in well; a centrifugation for 10 min, 13,000 x g, RT can be incorporated prior to taking an aliquot of antibody as this will remove any particulate matter and can help reduce background.
- For the preimmune serum controls, add an equivalent amount of preimmune serum (to the same final dilution). If no preimmune serum is available, add the equivalent volume of Neurobasal medium or PBS (no primary antibody control). Alternatively, if a suitable primary antibody is available that recognizes intracellular region/s of the protein, this antibody can be added to a control well in order to test specificity of the surface staining.
- Return the cells to the culture incubator for 1-4 hr (this incubation period may be determined empirically). In our experiments, the antibody was present for the entire period although the experimental design could incorporate a pulse of antibody followed by a further incubation period after wash-out (a pulse-chase experiment) to assess the time course of internalization. OPTIONAL STEP: This incubation can be carried out at room temperature or even on ice to slow the rate of basal protein internalization, if necessary. If performed in a non-CO2 controlled environment, the medium should be changed to one that is not bicarbonate buffered before addition of the antibody/antiserum. CAUTION: Mature neuron cultures (>14 days, and particularly cultured mouse embryonic neurons) do not tolerate full medium changes well.
We have used incubation times for this primary antibody internalization step of 1 hr, 2 hr, and 4 hr with good results although the optimal time will depend on abundance of the protein of interest as well as the dynamics of protein trafficking in the cells under study. NOTE: The dual-color images shown in Representative Results were obtained with an incubation of 1 hr at 37 °C in a tissue culture incubator.
After incubation for the desired period, aspirate off the antibody-containing medium. Wash the wells gently but rapidly once with PBS at room temperature.
3. Secondary Antibody Application to Fixed, Unpermeabilized Cells
Fix neurons with 4% paraformaldehyde in phosphate buffer pH 7.2, 5 min at room temperature Note: for best results, use freshly prepared fixative; alternatively use fix that has been stored at -20 °C and completely thawed so that no precipitated paraformaldehyde is evident. CAUTION: Paraformaldehyde fixative should be prepared and handled in a fume hood.
- Remove fix from wells (transfer to liquid waste container in fume hood) and rinse 3x with PBS.
- OPTIONAL STEP: If desired, to save on antibody reagents, the coverslips can be carefully removed from the 12-well plate with forceps and placed cell-side-up, onto a sheet of Parafilm laid on the base of a disposable culture plate.
- Solutions can then be pipetted gently onto the coverslips so that they are completely covered without the solution flooding over the edge of the coverslip onto the Parafilm.
- This technique is suitable for short-term incubations (1-2 hr) however longer incubations (e.g. overnight) should be carried out in a humidified chamber. Alternatively, the coverslips can be placed back into the wells of the 12-well plate for the overnight incubation and sufficient volume should be added to ensure that the coverslips do not dry out.
Block for 30 min at room temperature with 5% BSA in PBS (Note: Do not add detergent to the blocking solution at this stage as it is important that the cells are not permeabilized).
In order to label surface protein before proceeding with detection of internalized protein, apply the first fluorescently labeled 2° antibody of choice. Note: In the example described here, the primary antiserum was raised in rabbit (in-house antibody) to a recombinant secreted isoform4. Thus, for the first secondary antibody to detect the extracellular region of the transmembrane isoform on the surface of unpermeabilized neurons, we used donkey anti-rabbit Dylight 649 (1/200 diluted in PBS containing 5% BSA). It is recommended to centrifuge diluted secondary antibody solutions (10 min, 13,000 x g, RT) prior to use.
Incubate the coverslips for 2 hr at room temperature.
Wash coverslips (in wells), 2x 5 min with PBS.
4. Blocking with Excess Unlabeled 2° Antibody
- Block the unpermeabilized neurons with a high concentration (>0.1 mg/ml) of unlabeled 2° antibody.
- The unlabeled 2° antibody should be raised against the species in which the primary antibody was raised (in this case rabbit) by incubation overnight at room temperature.
- For this protocol, AffiniPure Fab fragment Goat anti-Rabbit IgG (H+L) was used at a concentration of 0.13 mg/ml. NOTE: The overnight incubation was found to be crucial as a shorter incubation period (2 hr) was insufficient for complete blocking of primary antibody that was not fully bound by the labeled secondary antibody.
Wash coverslips (in wells), 2x 5 min with PBS.
After this blocking step, post-fix the cells with 4% paraformaldehyde in phosphate buffer pH 7.2, 5 min at room temperature. Rinse with PBS (2x) after removal of fixative (transfer fixative to liquid waste container in fume hood).
5. Permeabilization and Application of the Second Fluorescently Conjugated 2° Antibody
Permeabilize and block the cells with 5% BSA in PBS containing 0.1% Triton-X-100 at room temperature for 30 min.
- Remove the blocking solution (taking care that the coverslips do not dry out). Add the second fluorescently-conjugated 2° antibody tagged with a different fluorophore.
- NOTE: It must be possible to distinguish this fluorophore tag from the one previously used, depending on the available excitation/emission filters on the confocal microscope (see below).
- For the example presented in this protocol, an Alexa Fluor 488-conjugated donkey anti-rabbit 2° antibody was used (1/200 in PBS, 5% BSA and 0.1% Triton-X-100). Incubate 2 hr at room temperature then remove the 2° antibody solution.
Wash coverslips 3x 5 min with PBS and, finally, wash briefly with deionized water.
6. Mounting and Imaging
Mount coverslips on glass slides with an aqueous mounting medium containing antifade (e.g. VECTASHIELD) and allow to dry. Store in the dark at 4 °C for optimal preservation of fluorescent signal intensity.
- Image immunostained cells on a confocal microscope.
- Appropriate excitation and emission filters for detection of the two fluorophore signals must be available.
- If different experimental conditions are to be compared (for example, internalization rates under depolarized8,9 or control conditions), ensure that all replicate coverslips from the various conditions are imaged with the same image acquisition parameters. The integrated density of standard regions of interest or the puncta attributes (e.g. number, size) from the resulting images can then be measured using standard image analysis software (e.g. Fiji/ImageJ, Metamorph).
Representative Results
The dual-color fluorescent immunostaining technique presented here is useful for labeling extracellular domains of transmembrane proteins in living cells (shown schematically in Figure 1). During the incubation period, the immunoglobulins bind accessible epitopes and a proportion of the population of protein molecules, together with bound antibody, is endocytosed. In addition, newly synthesized protein may reach the cell surface via forward trafficking and recycled protein molecules may be returned to the plasma membrane10.
This method has been optimized for the detection of two distinct protein pools, cell-surface and internalized, using the same primary antibody specific for the protein under study. By applying one fluorescently tagged secondary antibody to fixed cells prior to permeabilization, protein localized on the cell-surface at the time of fixation can be detected. The incorporation of a thorough blocking step to prohibit any binding of remaining protein-primary antibody surface complexes that have not been bound by the initial secondary antibody allows the subsequent detection (with secondary antibody conjugated to a different fluorophore) of protein-antibody complexes that were endocytosed during the incubation period (Figure 1).
Representative images of the dual-color labeling obtained with this method are shown in Figure 2. The incorporation of a blocking step of long duration (overnight incubation) with the relevant unlabeled secondary antibody, aimed at saturation binding of any remaining surface protein/primary antibody complexes, proved to be crucial to the success of this approach. The efficiency of this blocking step with unlabeled secondary antibody prior to permeabilization is demonstrated by the absence of double-stained puncta (with the exception of the small cell marked with an arrowhead which exhibits characteristic morphology of a dying cell, appearing rounded-up and condensed; Figure 2). Optimization of the conditions and combinations for the two labeled secondary antibodies may be necessary and the no-primary antibody condition is an important control. While working up this protocol, we found that it was not possible to completely block nonspecific binding of certain labeled secondary antibodies, particularly with blocking periods shorter than the overnight incubation described here.
Examples of single-color labeling of internalized protein in cultured neurons (Figure 3) highlight the characteristic punctate staining pattern of protein in the endosomal compartment10,11. To confirm that protein internalized during the antibody-feeding incubation and displaying punctate staining was localized in endosomes, neurons expressing an early/recycling endosome marker (transferrin-mCherry; TfR-mCh)12 were immunostained after permeabilization. The extensive overlap of the punctate staining with TfR-mCh expression is shown in Figure 4.
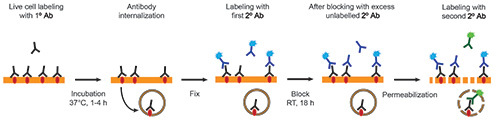 Figure 1. Schematic showing the dual-color labeling of cell-surface and internalized proteins after antibody feeding of live cultured neurons. Click here to view larger image.
Figure 1. Schematic showing the dual-color labeling of cell-surface and internalized proteins after antibody feeding of live cultured neurons. Click here to view larger image.
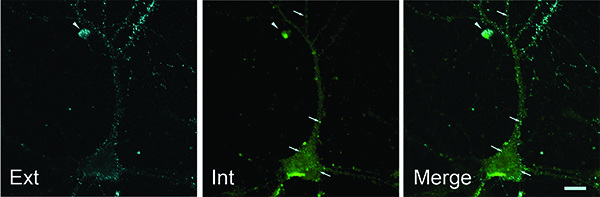 Figure 2. Labeling of cell-surface protein (cyan; EXT) and internalized protein (green; INT) on a cultured rat hippocampal neuron together with the merged image (MERGE). Double staining (arrowhead) was only observed in a cell that was apparently unhealthy. Scale bar = 10 μm. Click here to view larger image.
Figure 2. Labeling of cell-surface protein (cyan; EXT) and internalized protein (green; INT) on a cultured rat hippocampal neuron together with the merged image (MERGE). Double staining (arrowhead) was only observed in a cell that was apparently unhealthy. Scale bar = 10 μm. Click here to view larger image.
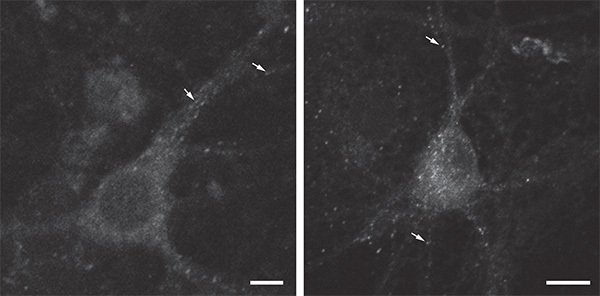 Figure 3. Higher power view of single-color (internalized protein) immunostaining showing the typical punctate pattern of endocytosed proteins. Scale bar = 10 μm. Click here to view larger image.
Figure 3. Higher power view of single-color (internalized protein) immunostaining showing the typical punctate pattern of endocytosed proteins. Scale bar = 10 μm. Click here to view larger image.
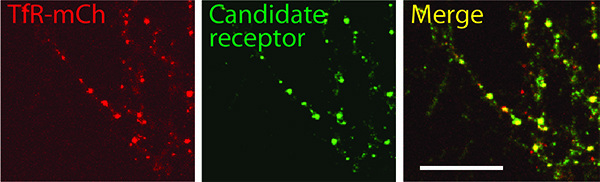 Figure 4. A region of the dendritic arbor of a neuron expressing a fluorescent marker for the recycling endosome (an expression construct for a transferrin receptor-mCherry fusion protein, TfR-mCh, was transfected using Lipofectamine 2000 [Invitrogen] according to the manufacturer’s instructions). The day after transfection, neurons were stained (after permeabilization) for endocytosed protein (candidate receptor), showing overlap with the recycling endosome compartment. Scale bar = 20 μm. Click here to view larger image.
Figure 4. A region of the dendritic arbor of a neuron expressing a fluorescent marker for the recycling endosome (an expression construct for a transferrin receptor-mCherry fusion protein, TfR-mCh, was transfected using Lipofectamine 2000 [Invitrogen] according to the manufacturer’s instructions). The day after transfection, neurons were stained (after permeabilization) for endocytosed protein (candidate receptor), showing overlap with the recycling endosome compartment. Scale bar = 20 μm. Click here to view larger image.
Discussion
The technique described here is complementary to that of cell-surface biotinylation (reviewed by Arancibia-Càrcamo et al.)12 and it is the method of choice for preserving information about the subcellular localization of the internalized protein, provided a suitable primary antibody to an extracellular epitope is available. In addition, quantitation of protein trafficking/internalization over time can be performed (by fixing coverslips at different times throughout the live cell incubation with primary antibody) without the need to prepare protein extracts.
Staining intensity or number and size of puncta in regions of interest (for example, the apical regions of pyramidal neuronal somata or the proximal apical dendrite at a set distance from the soma) in confocal images can be measured and compared under different conditions (for example, stimulated versus basal levels8,9). Internalized receptor signal intensity may then be normalized to that of surface receptors. Alternatively, the technique can be adapted for the quantitation of receptor recycling back to the cell surface through the inclusion of a stripping step to remove remaining antibody from the cell surface followed by an incubation to allow previously internalized, antibody-bound protein to return to the surface13.
Using this method, we were able to confirm that Sez6, the putative membrane receptor of interest, reaches and is localized on the cell-surface in neurons. Thus, this technique succeeded where more commonly-used immunofluorescence or immunoelectron microscopy (pre-embedding immunogold) protocols had previously failed. Despite predictions based on the primary amino acid sequence of this protein, definitive evidence of its presence at the cell-surface had been difficult to obtain. The protein we detected on the surface of nonpermeabilized cells was readily visible as distinct puncta possessing similar characteristics to those present intracellularly in the somatodendritic and axonal compartments. A possible explanation for this surface punctate staining is that the binding of antibody might stimulate internalization and, therefore, enable the detection of clustered cargo in nascent clathrin coated pits14. The overlap of the staining pattern of internalized protein puncta with that of the early/recycling endosome reporter TfR-mCherry lends indirect support to this concept. Additionally, we have evidence (unpublished) that a proportion of the protein is present in lipid rafts which also accounts for its clustered distribution on the surface.
While we have focused our attention on neurons in the current protocol, we observed evidence of uptake of immunoreactive protein (likely to represent the secreted and/or cleaved versions of the protein) in glial cells morphologically resembling astrocytes. This finding is of interest, firstly because it indicates that the method may be adapted for the study of the paracrine effects of secreted factors (for example, in cocultures) and, secondly, because it implies that the method will be applicable to other cell types.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
The authors thank Teele Palumaa for assistance with the figures. Funded by Project Grant 1008046 from the National Health and Medical Research Council, Australia.
References
- Lewis TL, Jr, Mao T, Arnold DB. A role for myosin VI in the localization of axonal proteins. PLoS Biol. 2010;9 doi: 10.1371/journal.pbio.1001021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Zastrow M, Williams JT. Modulating neuromodulation by receptor membrane traffic in the endocytic pathway. Neuron. 2012;76:22–32. doi: 10.1016/j.neuron.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunnersen JM, Augustine C, Spirkoska V, Kim M, Brown M, Tan S-S. Global analysis of gene expression patterns in developing mouse neocortex using serial analysis of gene expression. Mol. Cell. Neurosci. 2002;19:560–573. doi: 10.1006/mcne.2001.1098. [DOI] [PubMed] [Google Scholar]
- Gunnersen JM, Kim MH, et al. Sez-6 proteins affect dendritic arborization patterns and excitability of cortical pyramidal neurons. Neuron. 2007;56:621–639. doi: 10.1016/j.neuron.2007.09.018. [DOI] [PubMed] [Google Scholar]
- Kennedy MJ, Davison IG, Robinson CG, Ehlers MD. Syntaxin-4 Defines a Domain for Activity-Dependent Exocytosis in Dendritic Spines. Cell. 2010;141:524–535. doi: 10.1016/j.cell.2010.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen DK, Gilroy ME, Irons HR, Laplaca MC. Synapse-to-neuron ratio is inversely related to neuronal density in mature neuronal cultures. Brain Res. 2010;1359:44–55. doi: 10.1016/j.brainres.2010.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabrucker A, Vaida B, Bockmann J, Boeckers TM. Synaptogenesis of hippocampal neurons in primary cell culture. Cell Tissue Res. 2009. pp. 338–341. [DOI] [PubMed]
- Vaillant AR, Zanassi P, Walsh GS, Aumont A, Alonso A, Miller FD. Signaling mechanisms underlying reversible, activity-dependent dendrite formation. Neuron. 2002;34:985–998. doi: 10.1016/s0896-6273(02)00717-1. [DOI] [PubMed] [Google Scholar]
- Park M, Penick EC, Edwards JG, Kauer JA, Ehlers MD. Recycling endosomes supply AMPA receptors for LTP. Science. 2004;305:1972–1975. doi: 10.1126/science.1102026. [DOI] [PubMed] [Google Scholar]
- Kennedy MJ&, Ehlers MD. Organelles and trafficking machinery for postsynaptic plasticity. Annu. Rev. Neurosci. 2006;29:325–362. doi: 10.1146/annurev.neuro.29.051605.112808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasiecka ZM, Yap CC, Caplan S, Winckler B. Neuronal early endosomes require EHD1 for L1/NgCAM trafficking. J. Neurosci. 2010;30:16485–16497. doi: 10.1523/JNEUROSCI.3127-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arancibia-Cárcamo IL, Fairfax BP, Moss SJ, Kittler JT. Studying the localization, surface stability and endocytosis of neurotransmitter receptors by antibody labeling and biotinylation approaches. In: Kittler JT, Moss SJ, editors. The Dynamic Synapse: Molecular Methods in Ionotropic Receptor Biology. Boca Raton, FL: CRC Press; 2006. [PubMed] [Google Scholar]
- Petrini EM, Lu J, Cognet L, Lounis B, Ehlers MD, Choquet D. Endocytic trafficking and recycling maintain a pool of mobile surface AMPA receptors required for synaptic potentiation) Neuron. 2009;63:92–105. doi: 10.1016/j.neuron.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reider A, Wendland B. Endocytic adaptors – social networking at the plasma membrane. J. Cell Sci. 2011;124:1613–1622. doi: 10.1242/jcs.073395. [DOI] [PMC free article] [PubMed] [Google Scholar]