

Abstract
WNK1 [with no lysine (K)] is a serine-threonine kinase associated with a form of familial hypertension. WNK1 is at the top of a kinase cascade leading to phosphorylation of several cotransporters, in particular those transporting sodium, potassium, and chloride (NKCC), sodium and chloride (NCC), and potassium and chloride (KCC). The responsiveness of NKCC, NCC, and KCC to changes in extracellular chloride parallels their phosphorylation state, provoking the proposal that these transporters are controlled by a chloride-sensitive protein kinase. Here, we found that chloride stabilizes the inactive conformation of WNK1, preventing kinase autophosphorylation and activation. Crystallographic studies of inactive WNK1 in the presence of chloride revealed that chloride binds directly to the catalytic site, providing a basis for the unique position of the catalytic lysine. Mutagenesis of the chloride binding site rendered the kinase less sensitive to inhibition of autophosphorylation by chloride, validating the binding site. Thus, these data suggest that WNK1 functions as a chloride sensor through direct binding of a regulatory chloride ion to the active site, which inhibits autophosphorylation.
Introduction
Chloride ion is an important electrolyte involved in blood pressure maintenance, neuronal excitability and nociception, transcellular electrolyte transport, cell volume control, and airway fluid balance (1, 2). Chloride is uniquely and precisely regulated in diverse cell types; it is maintained at modest (30–60 mM) concentrations in most cells, and dips very low (10 mM) in actively transporting epithelia and neurons (3, 4). Misregulation of chloride is associated with diseases, such as hypertension and epilepsy. Chloride concentration can change radically as a function of osmotic stress (5), and low chloride induces cell cycle arrest (6). The main transmembrane proteins that set chloride concentrations are members of the solute carrier electroneutral cation-Cl− cotransporters (SLC12A family, also known as CCCs). Other molecules that influence cellular chloride concentration include members of the SLC26 gene family of exchangers, and chloride channels, such as the cystic fibrosis transmembrane conductance regulator (CFTR) and the γ-aminobutyric acid-gated chloride channel (GABAA) (7–9). However, the molecules that sense and respond to changes in intracellular chloride concentrations are largely unknown.
How chloride binds to proteins in any context is also poorly understood. Known chloride-binding sites tend to fall into two structural categories. One category is typified by the ClC family of chloride channels, which bind chloride through backbone-amide and hydrophobic interactions (10). A similar binding site is observed in the atrial naturetic peptide (ANP) receptor, a guanyl cyclase involved in volume regulation (11). Angiotensin converting enzyme (ACE) is an example of a second structural class of chloride-binding sites (12). The chloride-binding site in ACE has both hydrophobic and positively charged amino acids. Other proteins that have this binding mode include the serotonin transporter (SERT) and α-amylase (13, 14).
The regulation of transport and other processes through chloride has been demonstrated in various cellular assays, but often the structural and biochemical mechanisms are poorly defined. Although the chloride-mediated regulation of a few soluble proteins has been defined at both a biochemical and structural level, for example α-amylase and hemoglobin (13, 15), how changes in chloride concentration regulate other proteins including those in cellular membranes is unknown. For example, chloride regulates the voltage-gated potassium channel KCNB1, increasing the K+ current as a function of chloride in patch-clamp studies (16). The chloride-dependent Na+/H+ exchanger (Cl− NHE) is activated by chloride, as determined by intracellular pH measurements on exchanger-transfected cells (17). Both of these processes occur through unknown mechanisms.
Chloride concentrations are set primarily by the action of the specific members of the CCC family, in particular the transmembrane cotransporters NKCC1 (sodium-potassium-chloride cotransporter 1) and KCC1 (potassium-chloride cotransporter 1) (1, 18). The cotransporters are passive, acting according to the electrochemical gradient: NKCC1 mediates influx of ions, KCC1 mediates efflux. The activities of these transporters are regulated. Low intracellular chloride concentrations activate NKCC1 in secreting epithelia (19). Each of the cation-chloride contransporters is also regulated by phosphorylation. NKCCs, as well as sodium-chloride cotransporters (NCCs), are activated by phosphorylation (20, 21). KCCs are inhibited by phosphorylation (22). As a natural consequence of these combined observations, it was hypothesized that there should be a chloride-sensitive kinase (19, 20, 23). This putative kinase might act both as a primary sensor of chloride and transducer of this information to the cotransporters. NKCCs and KCCs also regulate cell volume, which has led to the further hypothesis that there should also be a chloride- and volume-sensitive protein kinase (24, 25).
Kinases of the WNK [with no lysine (K)] family are serine/threonine protein kinases noted for the unique placement one of the catalytic lysine residues (26, 27). Shortly after these kinases were first cloned, mutations in WNK1 or WNK4 genes were found to underlie pseudohypoaldosteronism II, a familial disease characterized by hypertension, hyperkalemia, and hyperchloremia (28). This disorder is often treated with thiazide diuretics, which are inhibitors of NCCs. Pseudohypoaldosteronism II is symptomatically the inverse of Gitelman’s syndrome, which is due to loss of the SLC12A3 gene encoding an NCC (29). This connection suggested that WNKs are involved in the pathway of SLC12A cotransporter phosphorylation. The linkage was established in studies of NKCC1 (SLC12A2). First, the kinases OSR (oxidative stress responsive kinase) and SPAK (sterile-20 proline alanine-rich kinase) were shown to interact with and phosphorylate the cotransporters NKCC1 and KCCs (30, 31). Subsequent studies showed that WNK1 interacts with and phosphorylates OSR and its close homolog SPAK (32–36). Apparently, there is a two-tiered kinase cascade; WNK phosphorylates and activates OSR and SPAK, which phosphorylate and regulate CCCs.
WNK isoforms exhibit pleiotropic involvement in ion transport regulation. WNK1 and WNK4 inhibit the epithelial sodium channel (ENaC), which is a major mediator of aldosterone-induced salt reabsorption (37, 38), by promoting ubiquitin-mediated degradation of the channel (39). The regulation of ENaC appears to be mediated by SGK1 (aldosterone-regulated serum- and glucocorticoid-induced kinase), which is activated by WNK1 (40). WNK1 and WNK4 also inhibit the renal outer medullary potassium channel (ROMK), a channel involved in salt reabsorption (41, 42). In contrast to the ENaC regulation, WNK-mediated inhibition of ROMK is due to clathrin-mediated endocytosis. Both effects on ENaC and ROMK appear to require regions outside the kinase domain of WNK, and the involvement of kinase activity is unknown. WNK1 and WNK4 also affect paracellular chloride flux (43) and maintenance of vascular tone (44).
WNK isoforms are activated in response to reduction in intracellular chloride concentrations (36, 45). Activation of NKCC2 by reduced intracellular chloride is blocked by WNK mutants that affect WNK-OSR docking interactions (45). Further, overexpression of WNK3 in HEK293 cells increases intracellular chloride concentrations. WNKs have been proposed to be chloride-sensitive kinases on the basis of these observations (45–49). Here, we show that the kinase domain of WNK1 directly binds a chloride ion, and we locate the chloride-binding site in the previously determined crystal structure of the inactive WNK1 kinase domain (27). Chloride binding inhibited the autophosphorylation of WNK1, thereby inhibiting kinase activity. Furthermore, we confirmed the residues involved in chloride binding through identification of chloride-insensitive mutants. Our results suggest that WNKs function as direct chloride sensors.
Results
WNK1 is stabilized by chloride salts
Because WNK1 is a regulator of CCCs and because those transporters are salt sensitive, we explored the relationship between WNK1 stability and salt concentration in vitro. We examined the stability of two forms of the WNK1 kinase domain: the inactive form of the kinase domain (WNK1-KDm/SA, in which activation loop phosphorylation site Ser382 was mutated to alanine) (27, 50), and the active kinase domain (WNK1-KDm/S*, where * denotes phosphorylation on Ser382, on the activation loop). We estimated stability from the temperature of protein melting (Tm) as indicated by binding of the dye Sypro Orange (fig. S1)(51). Both the active and inactive proteins exhibited an ~10°C increase in Tm with increasing NaCl concentration (Fig. 1A). These results indicated that that the kinase domain of WNK1 may interact directly with either the sodium or chloride ion, or both, and that the interaction was independent of Ser382.
Fig. 1. Kinase stability, activity, and autophosphorylation as a function of NaCl concentration.

(A) Change in melt temperature (Tm) of WNK constructs as a function of [NaCl]. Tm is taken at the point of greatest dRFU/dt during differential scanning fluorimetry from 0 to 80 °C, where RFU is relative fluorescence units. WNK1 193–482 (WNK1-KDm/S*) and inactive WNK1 193–482 S382A (WNK1-KDm/SA) are plotted. (B) and (C) Change in Tm of WNK1-KDm/S* upon exposure to different cation and anion salts. Stability was analyzed as in A. (D) Change in Tm of three control kinases (p38α, MEK6, and ASK1-KDm) as a function of [NaCl]. Stability was analyzed as in A. (E) Activity of WNK1 as measured by incorporation of 32P onto a model substrate (MBP) as a function of [NaCl]. The highest measured activity was normalized to 1; a control measured in the absence of MBP was taken as 0. (F) Activity of the indicated kinases against MBP as a function of [NaCl]. Activity was analyzed as in E. (G) Autophosphorylation by WNK1-KDm and control kinases as a function of [NaCl] measured by 32P autoincorporation. Solid lines and points indicate data normalized with 1 set to the highest recorded signal and 0 set based on analysis in the absence of kinase. Dotted lines and circles indicate data normalized to the number of phosphate atoms incorporated per protein molecule. In all panels, data are shown as the mean and standard error from three independent experiments.
To determine which ion WNK1-KDm bound, we measured the change in Tm as a function of different concentrations of cation-chloride salts (LiCl, NaCl, KCl) (Fig. 1B) and sodium-anion salts (NaCl, NaBr, NaI) (Fig. 1C). NaBr exposure stabilized WNK1-KDm, whereas NaI was destabilizing. LiCl and KCl had similar effects to that of NaCl on the stability of WNK1.
We also tested three kinases or catalytic domains from kinases that have not been implicated in chloride signaling: full-length mitogen- and extracellular kinase-regulated kinase kinase 6 (MEK6) and p38α and the catalytic domain of apoptosis signal-regulated kinase 1 (ASK1-KDm). None of these exhibited an increase in stability in response to increased NaCl; although the ASK1-KDm was destabilized as the NaCl concentration increased (Fig. 1D). Each of the salts destabilized MEK6, p38α, and ASK1-KDm to varying degrees, with iodide causing the greatest destabilization (fig. S2).
For the WNK1-KDm, halide ions (Cl− and Br−) stabilized the catalytic domain; whereas iodide had a destabilizing effect (Fig. 1C). Because any of the cation-chloride salts stabilized WNK1-Dm, cation binding appeared less important than chloride binding.
WNK1 activity is inhibited by salts
If WNK1 activity is required to initiate the WNK1 cascade, then we would expect chloride binding to have some effect on WNK1-KDm/S* activity. We measured the activity of WNK1 toward the model substrate, myelin basic protein (MBP), as a function of NaCl concentration and found that WNK1-KDm/S* activity decreased as a function of NaCl, with an apparent IC50 of 40 mM (Fig. 1E). However, two of the chloride-insensitive control kinases tested (ASK1-KDm and MEK6) also exhibited a decrease in activity in response to increasing NaCl concentrations with similar IC50 [30 mM for both (Fig. 1F)]. WNK1-KDm/S* activity toward the kinase domain of the physiological WNK1 substrate OSR (OSR 1–295) was also inhibited by increasing concentrations of NaCl, but the IC50 (530 mM) was greater than for activity toward MBP (fig. S3).
Because this inhibition of the active kinase was not unique, this mechanism seems unlikely to be responsible for the chloride-sensitive responsiveness of the WNK1 to CCC pathway.
Chloride inhibits WNK1 autophosphorylation
WNK1 requires phosphorylation at the activation loop site for activity (52). In the previous experiment, we used WNK1-KDm/S*, which is autophosphorylated as produced in E. coli. To determine if NaCl inhibited WNK1 autophosphorylation, we generated the unphosphorylated species, WNK1-KDm/S, by dephosphorylating WNK1-KDm/S* with protein phosphatase 1 (PP1cγ) and verified the dephosphorylated protein by mass spectrometry (fig. S4A). NaCl inhibited the autophosphorylation of WNK1-KDm/S, as assessed by autoradiography, with an apparent IC50 of 20 mM (Fig. 1G). This IC50 is consistent with the observed [Cl]Internal, which varies from 5 to 60 mM depending on the cell type (3, 4). We verified the inhibition of autophosphorylation by chloride by mass spectrometry (fig. S4A–C). Neither of the control kinases MEK6 nor p38α autophosphorylated appreciably under these conditions (Fig. 1G). Although ASK1-KDm can autophosphorylate (53), the effect of NaCl on ASK1 autophosphorylation was weak (apparent IC50 of 1 M) and opposite from the effect on WNK1. Thus, chloride has a strong, inhibitory effect on the autophosphorylation of WNK1-KDm, suggesting that changes in cellular chloride concentrations could modulate the rate of WNK1 autophosphorylation and thus its activity.
Halide ion binds the kinase domain of WNK1
Because bromide ion bound the WNK1-KDm (Fig. 1C) and because bromide gives a unique anomalous X-ray diffraction signal that can be used to identify its binding site (54), we crystallized the kinase-dead WNK-KDm/SA in the presence of 300 mM NaBr. Data were collected at the bromine K absorption edge (0.92 Å) (Table 1).
Table 1.
Data and refinement statistics for WNK1-KDm/SA
| WNK1-KDm/SA (3FPQ Re-refinement) | WNK1-KDm/SA Chloride Refinement |
WNK1-KDm/SA Bromide |
|
|---|---|---|---|
| Space Group | P 1 | P 1 | P 1 21 1 |
| Unit Cell |
a=38.3 Å,
b=57.8 Å,
c=65.7
Å α=91.3°, β=90.0°, γ=90.9° |
a=38.3 Å,
b=57.8 Å,
c=65.7
Å α=91.3°, β=90.0°, γ=90.9° |
a=38.0 Å,
b=58.3
Å c=65.0 Å, β=91.35° |
| Resolution (Å) | 50-1.8 (1.9-1.8)† | 50-1.8 (1.9-1.8)† | 43-3.5 (3.56-3.5)† |
| Observed Reflections | 217,158 | 217,158 | 47,703 |
| Unique Reflections | 50,614 | 50,614 | 6,887 |
| Completeness | 97.6 (96.9) | 97.6 (96.9) | 98.8 (96.1) |
| I/σI | 27.9 (10.7) | 27.9 (10.7) | 44.3(4.3) |
| Redundancy | 4.3 (4.3) | 4.3 (4.3) | 6.9 (6.6) |
| Rwork(%)‡ | 16.7 | 16.7 | 23.0 |
| Rfree(%) | 20.8 | 20.8 | 28.8 |
| Rmerge (%)† | 0.077 (0.144) | 0.077 (0.144) | 0.075 (0.517) |
| Number of groups | |||
| Protein Atoms | 4,706 | 4,706 | 2,180 |
| Water | 345 | 344 | 10 |
| Anion (Occupancy) | None | Chloride (1.0) | Bromide (0.48) |
| RMSD | |||
| Bond Length, Å | 0.02 | 0.02 | 0.09 |
| Bond Angle, ° | 1.97 | 1.98 | 5.22 |
| Average B factor, Å2 | 27.2 | 27.3 | 24.3 |
| Ramachandran Map (%) | |||
| Favorable | 90.7 | 96.1 | 93.7 |
| Allowed | 8.6 | 3.0 | 4.5 |
| Disallowed | 0.6 | 0.9 | 1.8 |
Values in Parentheses indicate the highest resolution shell
Rwork = Σ ||Fobs − |Fcalc||/Σ |Fobs|, where Fobs and Fcalc are the observed and calculated structure factors, respectively.
The previously determined structure of WNK1-KDm/SA (PDB ID: 3FPQ) was solved in the presence of 300 mM NaCl (27) (Fig. 2A). We compared our bromide-containing structure to the published chloride-bound structure. The WNK1-KDm/SA bromide crystals adopted a similar space group to the chloride-bound WNK1-KDm/SA (Table 1). The dataset contained 47,703 reflections with a maximum resolution of 3.5 Å, We refined the bromide-bound structure using 3FPQ as a starting model.
Fig. 2. WNK1 halide binding is observed crystallographically.

(A) Cartoon representation of WNK1-KDm/SA, PDB ID: 3FPQ. α-helices are pink, β-sheets are cyan, and loops are grey. The chloride position (green) was determined from a 5.5 σ peak in the |Fo − Fc| map. The 3/10 helix and hydrogen-bonding distances are shown in the inset. (B) Cartoon representation of WNK1-KDm/SA (blue) crystallized in the presence of NaBr (with the ion shown in yellow). Data were collected at the bromine K-edge (0.92 Å). The anomalous difference Fourier map contoured at 3.5 σ is shown in yellow. (C) Top-down view of the 3/10 helix from (B) with a bromide anion (yellow) modeled into the anomalous peak to show hydrogen-bonding distances. (D) Alignment of PDB ID: 3FPQ (Pink) and WNK1-sKDm/S* (Blue). The chloride of PDB ID: 3FPQ is shown in green. A depiction of the disordered activation loop of WNK1-sKDm/S* is shown using blue dots, with one dot for each Cα. Pymol was used to generate the figures (77).
The refined bromide structure superimposed on the previously defined WNK1 structure (PDB ID: 3FPQ) with an all-atom alignment RMSD of 0.5 Å. The anomalous difference Fourier map, indicative of a bromide ion, revealed a single strong peak, 5.2 σ, near the backbone amides of Leu369 of the DLG sequence and the adjacent residue Gly370 (Fig. 2B). The distances to the backbone amide nitrogen atoms were 4.0 and 3.5 Å, indicative of NH-Br− hydrogen bonds (55) (Fig. 2C). The bromide ion refined to an occupancy of 0.48 (Table 1).
To identify chloride ions in the previously determined structure of WNK1-KDm/SA (PDB ID: 3FPQ) (27), we calculated the map with coefficients |Fo − Fcalc| (λ= 1.0 Å) and identified a 5.5 σ peak in a position equivalent to the bromide ion found in the bromide-containing crystals. No other peak of similar intensity was observed in the map. We modeled this peak, which was originally assigned as water, as chloride (Fig. 2A, inset). Refined hydrogen-bonding distances to the backbone amides of Gly370 and Leu371 are 2.9 Å and 3.3 Å, typical of chloride hydrogen bonds (55). Assuming a chloride ion rather than water in that structure raised the B-factor from 14 Å2 to 28 Å2, more in line with the B-factors of the rest of structure (Table 1).
This comparison indicated that the chloride binds WNK1-KDm/SA through the same backbone amides as the bromide binds. These residues, Gly370 and Leu371, form the N-terminus of a short 3/10 helix, encompassing residues Gly370-Lys375. The 3/10 helix is at the N-terminus of the activation loop and continues from the DLG sequence (subdomain VII) (56). The helix contributes to the inactive configuration observed in the unphosphorylated molecule (27). Similar helices have been found in the inactive conformations of several kinases, and this helix has been noted as a common mechanism for building an inactive configuration (57). None of the other 3/10 helices, however, are capped by an anion, or even a water molecule. The chloride ion additionally forms hydrophobic interactions with the following residues: Phe283 in β3, Leu299 in β4, Leu369 in the DLG sequence, and Leu371 in the 3/10 helix (fig. S5).
The structure of the phosphorylated form of WNK1 reveals conformational changes
To better understand the structural basis of the chloride inhibition, we determined the structure of the doubly phosphorylated kinase domain. A shorter fragment was used, eliminating residues missing in the electron density of the WNK1-KDm/SA, to form WNK1 (209–483)/S382* (WNK1-sKDm/S*). The WNK1-sKDm/S* crystals produced a 1.8 Ǻ structure (table S1). The WNK1-sKDm/S* structure is similar to WNK1-KDm/SA, with an overall RMSD of 0.55 Å. However, several differences were observed. Although a small amount of chloride (50 mM) was in the crystallization buffer, the chloride-binding site found in WNK1-KDm/SA is empty in WNK1-sKDm/S*. The 3/10 helix is present, but exhibits increased disorder and conformational changes, which are largest at the C-terminus (Lys375) (Fig. 2D and table S2). Disorder is also observed along helix αC, and in the activation loop. The N-terminal domain is rotated approximately 8° relative to the inactive structure (PDB ID: 3FPQ).
Several structural cues indicated that WNK1-sKDm/S* adopted an inactive configuration in the crystal, in particular the partial maintenance of the 3/10 helix and mispositioning of two catalytic residues Thr386 and Glu268. Thr386 at the C-terminus of the activation loop (58) is not present in the active site. Glu268 in helix αC which forms a hydrogen bond with the catalytic lysine in subdomain III in the active conformation (59), is displaced out of the active site. The 3/10 helices found in the activation loop of other inactive structures also disrupt this interaction (57). Disorder in the activation loop could also indicate an inactive conformer, but may be influenced by the lattice. A set of hydrophobic residues has been identified as a hallmark of active kinases, and referred to as a ‘spine’ (60). The corresponding residues in WNK1 are Phe283, Leu272, Leu369, and His347. All contacts between these residues are maintained in both WNK1-KDm/SA and WNK1-sKDm/S*, although Leu272 is rotated out with the displacement of helix C, such that the spine is not straight. Thus, WNK1-sKDm/S* adopts an inactive configuration despite the activating phosphorylation.
WNK1 with mutations in the chloride-binding site autophosphorylate
If chloride binding inhibited WNK1-KDm autophosphorylation as proposed, mutations in the chloride-binding site may reduce chloride binding and increase autophosphorylation. We introduced mutations into the hydrophobic residues lining the WNK1-KDm chloride-binding site as observed in WNK1-KDm/S* (L299F, L371F, or L369F) (Fig. 3A). These mutants were active and phosphorylated when expressed in and purified from E. coli [70–100% phosphorylation as determined by mass spectrometry (table S3). Increasing NaCl concentration had the least stabilizing effect on the mutant L369F (2.6 °C at 500 mM NaCl compared with 6.8 °C for WNK1-KDm/SA) (Fig. 3B). Using PP1cγ-treated proteins, we assessed the effect of increasing NaCl concentrations on autophosphorylation and the L369F mutant (in the sequence DLG) was less inhibited by NaCl, with an apparent IC50 in the autophosphorylation experiment of ~50 mM (Fig. 3C). Even at concentrations in excess of 1M, we only observed 50% inhibition. The L371F mutant (in the 3/10 helix) also exhibited an increase in IC50; whereas the L299F mutant (in β4) exhibited only slight changes when compared with WNK1-KDm/SA. None of the mutations substantially affected the ability of increasing concentrations of NaCl to inhibit activity toward MBP (Fig. 3D). Thus, the effects of these mutations on the inhibition of autophosphorylation and kinase activity toward substrates by chloride-containing salt substantiate the crystallographic data for the chloride-binding site and provide a context for understanding effect of chloride binding on autophosphorylation.
Fig. 3. WNK1-KDm/S* mutants.

(A) F283, L299, L369, and L371 form a hydrophobic pocket surrounding the bound chloride. (B) Change in melt temperature (Tm) of WNK1 mutants as a function of [NaCl]. Tm is taken at the point of greatest dRFU/dt during differential scanning fluorimetry from 0 to 80 °C, where RFU is relative fluorescence units. WNK1-KDm (Red), WNK1-KDm/L371F (Green), WNK1-KDm/L299F (Purple), and WNK1-KDm/L369F (Blue) are plotted. (C) Autophosphorylation of WNK1-KDm mutants in response to varying NaCl concentration as measured by 32P incorporation. Autoincorporation normalized with 1 being the highest signal and a kinase-free background of 0. WNK1-KDm (Red), WNK1-KDm/L371F (Green), WNK1-KDm/L299F (Purple), and WNK1-KDm/L369F (Blue) are plotted. (D) Activity of WNK1 and WNK1 mutants as measured by incorporation of 32P onto the generic kinase substrate myelin basic protein (MBP) as a function of [NaCl]. The highest measured activity was normalized to 1, and an MBP-deprived control was taken as 0. WNK1-KDm (Red), WNK1-KDm/L371F (Green), WNK1-KDm/L299F (Purple), and WNK1-KDm/L369F (Blue) are plotted. Data in B–D are shown as mean and standard error from three independent experiments.
Discussion
Here, we showed that the kinase domain of WNK1 binds chloride and that chloride has an inhibitory effect on the autophosphorylation and activation of WNK1. Early analysis of NKCCs postulated the existence of a chloride-sensitive kinase to explain the dual activation of these transporters by phosphorylation and low chloride. (19, 20, 23). More recently, it has been suggested that WNKs perform this role (47, 49). Our data validated both the concept of a chloride-sensitive kinase and that WNK1 is such a kinase. We also showed that chloride inhibited autophosphorylation with an observed IC50 of approximately 20 mM. This is slightly lower than observed concentrations for chloride in the kidney (~50 mM), and slightly higher than those present in neurons (~10–15 mM) (3, 4). Thus, chloride appears to regulate WNK1 within a concentration milieu appropriate to the functions of distinct tissues. The results suggest that the chloride concentration in cells may be set in part by the chloride sensitivity of WNK isoforms or splice variants. Further data are required to test the chloride-based inhibition of other WNK isoforms. The observed chloride-binding site, comprised of a combination of main chain interactions and hydrophobic contacts, is typical of the first of the two classes of chloride-binding sites described above, which utilizes backbone-amide and hydrophobic interactions. The chloride-binding site is similar to that of the ClC-family chloride cotransporter (10). In ClC, the chloride ion caps the N-terminus of a helix, making both two 3.5 Ǻ hydrogen bonds and hydrophobic contacts with a phenylalanine, an isoleucine, and a tyrosine. The ANP receptor also binds chloride through backbone amides (at a conserved glycine-proline turn) and has a hydrophobic flank formed of a disulfide bond (11).
The structural basis for the chloride regulation of WNK1 makes WNKs unique among metabolite or ion-sensing kinases and can explain the special placement of the catalytic lysine in WNKs. Most metabolite-sensing kinases have separate domains or subunits that bind the ligand. Protein kinase A (PKA), for example, possesses cAMP-binding subunits, whereas calcium calmodulin-dependent kinase binds calcium through the calmodulin subunit (61, 62). In contrast, the WNK1 chloride-binding site is directly in the kinase domain (Fig. 4A). This design may contribute to the fast cellular responses to changes in chloride concentration (47). The location of the chloride-binding site overlaps with the canonical placement of catalytic lysines found in other kinases, such as Lys72 in PKA (Fig. 4B); the catalytic lysine (Lys233) in WNK1-KDm/SA (Fig. 4B) does not overlap, thus offering a possible reason for the unique position of the catalytic lysine in WNKs.
Fig. 4. WNK1 serves as an intracellular salt sensor.
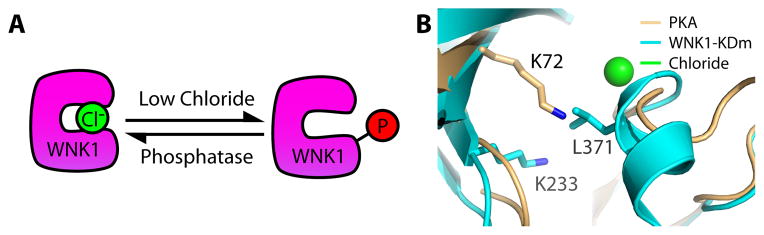
(A) WNK1 autoactivation is inhibited by chloride. In low chloride conditions, WNK1 autophosphorylates and becomes active. Phosphatase activity inactivates WNK1, but it can reactivate itself if the chloride concentration remains low. (B) WNK1-KDm/SA (PDB: 3FPQ) and PKA (PDB: 1ATP) were aligned in Pymol (77). The placement of L371 in WNK1-KDm/SA (cyan) overlaps spatially with the canonical kinase Lysine (Lys 72 of PKA) (gold, PDB: 1ATP).
By mutagenesis, we validated the chloride-binding site, which is situated at the DLG motif and the N-terminus of the activation loop. The mutant L369F, which is in the DLG motif, was the least sensitive to the inhibition of autophosphorylation by chloride. Most protein kinases have the sequence DFG (subdomain VII) at this position. Our data suggest that this leucine residue may destabilize the active structure. The question then arises whether kinases that contain a leucine in that position are regulated by ions or adopt inactive configurations similar to that adopted by WNK1. The β-adrenergic receptor kinase (βARK), among others, possesses a DLG motif (63). βARK negatively regulates the β-adrenergic receptor, a stimulator of chloride flux, through phosphorylation (64). Thus, βARK might be similarly regulated.
The present data offer a structural rationale for the low chloride activation of NKCCs by WNK1. However, the mechanism of hypertonic stress activation of NKCCs remains unclear (24). The dual activation of NKCCs by low chloride and extracellular hypertonicity led to the hypothesis that there exists a kinase sensitive not only to chloride but also to volume (23). The activation of this kinase in response to decreased cellular volume is thought to be mediated by macromolecular crowding (65). WNK1 is activated by hypertonic stress induced by either high external salt or sorbitol, as well as by low concentrations of chloride (26, 32, 36, 50, 66). The effects of hypertonic conditions on WNKs could occur on the kinase directly or on another factor that regulates the kinase. Here, we observed a phosphorylated form of WNK1-KDm that did not adopt a completely active configuration. This suggests a possible mechanism by which crowding effects could compete with chloride to promote an autophosphorylation-competent conformation. The observation that autophosporylation only potentiates activation has been made in other protein kinases (27). How a fully active conformation is induced and how hypertonicity activates WNK1 require further investigation.
WNK1-KDm/S* was dephosphorylated by PP1cγ in vitro. The activities of NKCCs and other CCC family members are likely regulated by the balance of kinases and phosphatases targeting these proteins. Because WNK1 can autophosphorylate and can be dephosphorylated suggests that the cellular balance of phosphatase and kinase activities may contribute to WNK1 regulation. Thus, we propose that as the chloride concentration decreases, autophosphorylation outstrips dephosphorylation and WNK1 activity increases, thereby stimulating downstream signaling (Fig. 4A). As the chloride concentration increases, dephosphorylation outstrips autophosphorylation, and WNK1 activity is inhibited, decreasing downstream signaling. The cellular phosphatase acting on WNKs is unknown, but both protein phosphatase-1 and 4 have been implicated (67, 68). The discovery that WNK1 is controlled by autophosphorylation, together with a method to induce constitutive activity of WNK1, may prove useful in elucidating the chloride sensitivity and signaling pathways involving WNK1 and other WNK isoforms.
Materials and Methods
Expression and purification of WNK1 and WNK1 mutants
Cloning, expression, and purification of the WNK1 kinase domain constructs of 194–483 and 209–483 was as described in Min et al. (27). The expression and purification of p38α was described in (69), MEK6 in (70), and ASK1-KDm in (53). Mutants were created using a Stratagene QuikChange® Site-Directed Mutagenesis kit and purified as WNK1 194–483.
Kinase function assays using MBP
WNK1-KDm/S*, p38α, MEK6, or ASK-KDm, or the WNK1 mutants was added at a final concentration of 2 μM to 10 molar excess of MBP in 30 μl of kinase reaction buffer (30 mM HEPES pH 8.0, 5 mM MgCl2, 1 mM DTT, 100 μM ATP) containing 10 μCi of [γ32P]-ATP. Increasing amounts of NaCl were added to the reaction mixture to make a series of final chloride concentrations in the range of 10–1000 mM. Reactions were incubated for 1 hour at 37 °C. Reactions were stopped by blotting 20 μL of each reaction on filter paper squares (1 cm × 1 cm) and plunging into a solution of 10% trichloroacetic acid (TCA). The squares were washed three times in TCA, blotted, and transferred to scintillation vials. The vials were filled with 5 ml Complete Counting Cocktail 3a70B (Research Products International Corp.) and counted on a Beckman LS 3801. Reactions were normalized to 100% highest signal and 0% for the sample lacking substrate (MBP). To confirm that all counts were the result of MBP phosphorylation, autophosphorylation was monitored by separation of the reaction mixture on an SDS-PAGE gel followed by scintillation counting of the band containing WNK1-KDm/S*. No autophosphorylation of the WNK1 kinase domain was detected in the MBP substrate assay. Mutants were assayed similarly.
Phosphorylation of OSR by WNK1-KDm/S*
WNK1-KDm/S* was added at a final concentration of 2 μM to 5 molar excess of OSR1 1–295 in 30 μl of kinase reaction buffer (30 mM HEPES pH 8.0, 5 mM MgCl2, 1 mM DTT, 100 μM ATP) containing 10 μCi of [γ32P]-ATP. Increasing amounts of NaCl were added to the reaction mixture to make a series of final chloride concentrations in the range of 30–1000 mM. Reactions were incubated for 1 hour at 37 °C. Reactions were stopped by blotting 20 μL of each reaction on filter paper squares (1 cm × 1 cm) and plunging into a solution of 10% trichloroacetic acid (TCA). The squares were washed three times in TCA, blotted, and transferred to scintillation vials. The vials were filled with 5 ml Complete Counting Cocktail 3a70B (Research Products International Corp.) and counted on a Beckman LS 3801. Reactions were normalized to 100% highest signal and 0% for the sample lacking substrate (OSR1 1–295).
Kinase Autophosphorylation
Autophosphorylation was measured similarly, using WNK1-KDm/S, p38α, MEK6, or ASK-KDm, or the WNK1 mutants generated from exposure of the protein to the serine-phosphatase PP1Cγ conjugated with a hexahistidine tag (5 μM) for 30 minutes at 37 °C in a solution of 100 mM Tris pH 8.0 and 10 mM MnCl2 in a total reaction volume of 4.5 ml. The reaction mixture was purified by passing over a Ni-agarose column (GE) to remove the phosphatase from kinase. The kinase was buffer exchanged into 50 mM Tris pH 8.0, 50 mM NaCl, and 1 mM DTT and assayed for phosphorylation by either 32P incorporation followed by scintillation counting as described above or by mass spectrometry. No other substrates except the dephosphorylated kinase were added to this reaction mixture, and the final WNK1-KDm/S or WNK1 mutant concentration was 2.5 μM for 32P incorporation or 55 μM for mass spectrometry. The final concentration of p38α, MEK6, or ASK-KDm was 2.5 μM for 32P incorporation. This solution was allowed to react for 30 minutes at 21 °C.
Mass spectrometry
WNK1 autophosphorylation reactions were performed and stopped by the addition of guanidine to 1 M final concentration at the time points indicated. WNK1 was digested in an 8:1 molar ratio with sequence-grade chymotrypsin (Roche) in the presence of 100 mM Tris pH 8.0, and 25 mM CaCl2 (100 μL total reaction volume) at 30 °C overnight. Following digestion, the peptide mixture was separated by HPLC (Agilent 1100) on a RP-C18 column (Phenomenex Aeris Widepore 150 × 2.1 mm) using an acetonitrile-water gradient from 4% to 28% with 0.2% formic acid. Mass spectrometric analysis was performed on an LCQ DECA XP ion-trap mass spectrometer (ThermoFinnigan) with the HPLC coupled inline to an orthogonal electrospray ionization source. MS detector responses were obtained by integration under ion traces corresponding to m/z ranges for activation loop peptides, then scaled to the integration of the total ion current for all eluted peptides. MS/MS spectra were acquired in a data dependent mode and analyzed using MASCOT software (Matrix Science Ltd.) (71, 72). Synthetic peptide standards (21st Century Biochemicals) were used to confirm HPLC elution times and for quantitation.
Differential scanning fluorimetry assays
5 μM of the kinase was incubated with 10 mM Tris pH 8.0, 5X SYPRO Orange, water, and the specified salt concentration to make a 20 μl reaction volume for a well on a 96-well clear-bottom plate. The 96-well plate was centrifuged at 805 × g to remove bubbles created by pipetting. The temperature in each well was cycled from an initial 4 °C to 80 °C using 0.5 °C increments in a BioRad CFX96 real-time PCR machine. The fluorescence intensity of SYPRO orange was probed using the fluorescein amidite channel of the PCR machine.
Crystallization and X-ray data collection
Crystallization of WNK1-KDm/SA (194–483 S382A) was performed with a variation on the condition used by Min et al (70). A mixture of 24% w/v PEG 2000 monomethyl ether (MME), 100 mM HEPES pH 8.0, and 300 mM NaBr was used to set up Nextal hanging drop vapor diffusion crystal trays (Qiagen). A 1 to 1 (2 μl total) ratio of protein to precipitant solution was used, at 7 mg/ml protein concentration. Crystals grew to 200 μm in one week at 16 °C. Five crystals from these plates were cryoprotected using a solution of 20% glycerol, 24% PEG 2000, MME, 100 mM HEPES (pH 8.0), 300 mM NaBr. Data were collected at the APS beamline 19-ID at the anomalous edge of bromine (0.92 Å). Integration and scaling were performed using the HKL2000 software suite (73).
Crystals of WNK1-sKDm/S* (209–483 S382*) were obtained by hanging drop vapor diffusion using a protein concentration of 8 mg/mL. Protein buffer was 50 mM NaCl, 50 mM HEPES 8.0, 1 mM DTT. A 1:1 ratio of protein and precipitant solution were mixed (2 μL each). Precipitant solution was 0.35 M KH2PO4 and 19% PEG 3350. Crystals were grown to a size of 200 μm at 16 °C. Crystals were cryoprotected in 0.35 M KH2PO4, 19% PEG 3350, and 30% PEG 400. 1.8 Ǻ resolution X-ray data were collected at the APS beamline 19-ID. Integration and scaling were performed using the HKL2000 software suite (73).
Structure determination and refinement
A 2.8 Ǻ resolution X-ray structure of WNK1-KDm/SA in the presence of NaBr was solved by molecular replacement utilizing the A subunit of the previously solved WNK1-KDm/SA [NaCl, PDB ID: 3FPQ, (27)] structure as a starting model with the MOLREP module of the CCP4 program suite (74). Anomalous difference Fourier maps generated using the CCP4 suite revealed the position of the bromide ion. Restrained refinement using REFMAC5 in the CCP4 suite for the WNK1-KDm/SA and including the bromide ion coordinates obtained above gave an initial R-factor of 0.22 and Rfree of 0.30. Model building using COOT (75) and subsequent rounds of restrained refinement in combination with TLS (Translation-Libration-Screw) and refinement of the occupancy of the bromide ion in PHENIX crystallographic software (76) yielded an R-factor and R-free of 0.20 and 0.29, respectively. The bromide ion occupancy was 0.48 with a 5.5 σ peak. All figures were made using Pymol (77).
The |Fo − Fc| map revealed a 5.5 σ peak in the active site. The original refinement based on the idea that water occupied this site resulted in a lower B-factor (14 Ǻ2) than the surrounding water molecules (30 Ǻ2 and 22 Ǻ2). Using the assumption that the peak corresponded to a chloride ion, the structure was rerefined, and the B-factor of the chloride ion rose to 28 Ǻ2, in line with the surrounding structure.
To determine the structure of WNK1-sKDm/S*, molecular replacement was carried out employing PDB ID: 3FPQ as a search model in the MOLREP module of the CCP4 program suite. One cycle of restrained refinement using REFMAC5/CCP4 program produced an R-factor of 0.24 and an Rfree of 0.30. Subsequent cycles of model building and restrained refinement in combination with TLS produced an R and Rfree of 0.23 and 0.27, respectively. The final model contained 233 water molecules, a phosphate ion, and a chloride ion.
Supplementary Material
Figure S1: Raw trace of WNK1-KDm/S* DSF using Sypro Orange
Figure S2: Control kinase stability as a function of salt concentration
Figure S3: Phosphorylation of OSR1 by WNK1-KDm/S*
Figure S4: WNK1 autophosphorylation measured by mass spectrometry
Figure S5: Interaction of bound chloride with hydrophobic residues
Table S1: Data and refinement statistics for WNK1-sKDm/S*
Table S2: B-Factors of the 3/10 helix residues in WNK1-KDm/SA and WNK1-sKDm/S*
Table S3: Phosphorylation state of WNK1-KDm/S* mutants after purification from E. coli
Acknowledgments
We thank the UTSW Structural Biology Lab members Diana Tomchick and Chad Brautigam for assistance with data collection. We also thank John Humphreys for mass spectrometry and Dominika Borek for help with structure analysis. Results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by U.Chicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357.
Funding: This research was supported by grants RO1DK092692 to EJG, I1128 from the Welch foundation to EJG and I1143 from the Welch Foundation and GM53032 to MHC.
Footnotes
Author contributions: T.M. made initial observation of chloride stability. A.P. designed and performed autophosphoylation assays. A.P and T.M. designed and performed kinase activity and stability assays. R.A. and T.M performed crystallographic studies. M.H.C. initiated WNK1 studies. E.J.G directed the study. E.J.G and A.P. wrote the manuscript.
Competing interests: the authors declare no competing interests.
Data and materials availability: WNK1-sKDm/S* is available in the Protein Data Bank as PDB ID: 4PWN, WNK1-KDm/SA with bromide is available as PDB ID: 4Q2A.
References
- 1.Russell JM. Sodium-potassium-chloride cotransport. Physiol Rev. 2000;80:211–276. doi: 10.1152/physrev.2000.80.1.211. [DOI] [PubMed] [Google Scholar]
- 2.Kahle KT, Rinehart J, Lifton RP. Phosphoregulation of the Na-K-2Cl and K-Cl cotransporters by the WNK kinases. Biochim Biophys Acta. 2010;1802:1150–1158. doi: 10.1016/j.bbadis.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Salomonsson M, Gonzalez E, Westerlund P, Persson AE. Chloride concentration in macula densa and cortical thick ascending limb cells. Kidney Int Suppl. 1991;32:S51–54. [PubMed] [Google Scholar]
- 4.Pellegrino C, Gubkina O, Schaefer M, Becq H, Ludwig A, Mukhtarov M, Chudotvorova I, Corby S, Salyha Y, Salozhin S, Bregestovski P, Medina I. Knocking down of the KCC2 in rat hippocampal neurons increases intracellular chloride concentration and compromises neuronal survival. J Physiol. 2011;589:2475–2496. doi: 10.1113/jphysiol.2010.203703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miyazaki H, Shiozaki A, Niisato N, Marunaka Y. Physiological significance of hypotonicity-induced regulatory volume decrease: reduction in intracellular Cl− concentration acting as an intracellular signaling. Am J Physiol Renal Physiol. 2007;292:F1411–1417. doi: 10.1152/ajprenal.00244.2006. [DOI] [PubMed] [Google Scholar]
- 6.Ohsawa R, Miyazaki H, Niisato N, Shiozaki A, Iwasaki Y, Otsuji E, Marunaka Y. Intracellular chloride regulates cell proliferation through the activation of stress-activated protein kinases in MKN28 human gastric cancer cells. J Cell Physiol. 2010;223:764–770. doi: 10.1002/jcp.22088. [DOI] [PubMed] [Google Scholar]
- 7.Guggino WB, Stanton BA. New insights into cystic fibrosis: molecular switches that regulate CFTR. Nat Rev Mol Cell Biol. 2006;7:426–436. doi: 10.1038/nrm1949. [DOI] [PubMed] [Google Scholar]
- 8.Chebib M, Johnston GA. The ‘ABC’ of GABA receptors: a brief review. Clin Exp Pharmacol Physiol. 1999;26:937–940. doi: 10.1046/j.1440-1681.1999.03151.x. [DOI] [PubMed] [Google Scholar]
- 9.Dorwart MR, Shcheynikov N, Yang D, Muallem S. The solute carrier 26 family of proteins in epithelial ion transport. Physiology. 2008;23:104–114. doi: 10.1152/physiol.00037.2007. [DOI] [PubMed] [Google Scholar]
- 10.Dutzler R, Campbell EB, Cadene M, Chait BT, MacKinnon R. X-ray structure of a ClC chloride channel at 3.0 A reveals the molecular basis of anion selectivity. Nature. 2002;415:287–294. doi: 10.1038/415287a. [DOI] [PubMed] [Google Scholar]
- 11.Ogawa H, Qiu Y, Philo JS, Arakawa T, Ogata CM, Misono KS. Reversibly bound chloride in the atrial natriuretic peptide receptor hormone-binding domain: possible allosteric regulation and a conserved structural motif for the chloride-binding site. Protein Sci. 2010;19:544–557. doi: 10.1002/pro.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tzakos AG, Galanis AS, Spyroulias GA, Cordopatis P, Manessi-Zoupa E, Gerothanassis IP. Structure-function discrimination of the N− and C− catalytic domains of human angiotensin-converting enzyme: implications for Cl− activation and peptide hydrolysis mechanisms. Protein Eng. 2003;16:993–1003. doi: 10.1093/protein/gzg122. [DOI] [PubMed] [Google Scholar]
- 13.Pokhrel R, McConnell IL, Brudvig GW. Chloride regulation of enzyme turnover: application to the role of chloride in photosystem II. Biochemistry. 2011;50:2725–2734. doi: 10.1021/bi2000388. [DOI] [PubMed] [Google Scholar]
- 14.Tavoulari S, Rizwan AN, Forrest LR, Rudnick G. Reconstructing a chloride-binding site in a bacterial neurotransmitter transporter homologue. J Biol Chem. 2011;286:2834–2842. doi: 10.1074/jbc.M110.186064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perutz MF, Shih DT, Williamson D. The chloride effect in human haemoglobin. A new kind of allosteric mechanism. J Mol Biol. 1994;239:555–560. doi: 10.1006/jmbi.1994.1394. [DOI] [PubMed] [Google Scholar]
- 16.Li X, Surguchev A, Bian S, Navaratnam D, Santos-Sacchi J. Extracellular chloride regulation of Kv2.1, contributor to the major outward Kv current in mammalian outer hair cells. Am J Physiol Cell Physiol. 2012;302:C296–306. doi: 10.1152/ajpcell.00177.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sangan P, Rajendran VM, Geibel JP, Binder HJ. Cloning and expression of a chloride-dependent Na+-H+ exchanger. J Biol Chem. 2002;277:9668–9675. doi: 10.1074/jbc.M110852200. [DOI] [PubMed] [Google Scholar]
- 18.Gamba G. Molecular physiology and pathophysiology of electroneutral cation-chloride cotransporters. Physiol Rev. 2005;85:423–493. doi: 10.1152/physrev.00011.2004. [DOI] [PubMed] [Google Scholar]
- 19.Lytle C, Forbush B., 3rd Regulatory phosphorylation of the secretory Na-K-Cl cotransporter: modulation by cytoplasmic Cl. Am J Physiol. 1996;270:C437–448. doi: 10.1152/ajpcell.1996.270.2.C437. [DOI] [PubMed] [Google Scholar]
- 20.Lytle C, Forbush B., 3rd Na-K-Cl cotransport in the shark rectal gland. II. Regulation in isolated tubules. Am J Physiol. 1992;262:C1009–1017. doi: 10.1152/ajpcell.1992.262.4.C1009. [DOI] [PubMed] [Google Scholar]
- 21.Pacheco-Alvarez D, Cristobal PS, Meade P, Moreno E, Vazquez N, Munoz E, Diaz A, Juarez ME, Gimenez I, Gamba G. The Na+:Cl− cotransporter is activated and phosphorylated at the amino-terminal domain upon intracellular chloride depletion. J Biol Chem. 2006;281:28755–28763. doi: 10.1074/jbc.M603773200. [DOI] [PubMed] [Google Scholar]
- 22.Jennings ML, Schulz RK. Okadaic acid inhibition of KCl cotransport. Evidence that protein dephosphorylation is necessary for activation of transport by either cell swelling or N-ethylmaleimide. J Gen Physiol. 1991;97:799–817. doi: 10.1085/jgp.97.4.799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haas M, Forbush B., 3rd The Na-K-Cl cotransporter of secretory epithelia. Annu Rev Physiol. 2000;62:515–534. doi: 10.1146/annurev.physiol.62.1.515. [DOI] [PubMed] [Google Scholar]
- 24.Lytle C, McManus T. Coordinate modulation of Na-K-2Cl cotransport and K-Cl cotransport by cell volume and chloride. Am J Physiol Cell Physiol. 2002;283:C1422–1431. doi: 10.1152/ajpcell.00130.2002. [DOI] [PubMed] [Google Scholar]
- 25.Haas M, McBrayer D, Lytle C. [Cl−]i-dependent phosphorylation of the Na-K-Cl cotransport protein of dog tracheal epithelial cells. J Biol Chem. 1995;270:28955–28961. doi: 10.1074/jbc.270.48.28955. [DOI] [PubMed] [Google Scholar]
- 26.Xu BE, English JM, Wilsbacher JL, Stippec S, Goldsmith EJ, Cobb MH. WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J Biol Chem. 2000;275:16795–16801. doi: 10.1074/jbc.275.22.16795. [DOI] [PubMed] [Google Scholar]
- 27.Min X, Lee BH, Cobb MH, Goldsmith EJ. Crystal structure of the kinase domain of WNK1, a kinase that causes a hereditary form of hypertension. Structure. 2004;12:1303–1311. doi: 10.1016/j.str.2004.04.014. [DOI] [PubMed] [Google Scholar]
- 28.Wilson FH, Disse-Nicodeme S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, Feely MP, Dussol B, Berland Y, Unwin RJ, Mayan H, Simon DB, Farfel Z, Jeunemaitre X, Lifton RP. Human hypertension caused by mutations in WNK kinases. Science. 2001;293:1107–1112. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- 29.Lemmink HH, Knoers NV, Karolyi L, van Dijk H, Niaudet P, Antignac C, Guay-Woodford LM, Goodyer PR, Carel JC, Hermes A, Seyberth HW, Monnens LA, van den Heuvel LP. Novel mutations in the thiazide-sensitive NaCl cotransporter gene in patients with Gitelman syndrome with predominant localization to the C-terminal domain. Kidney Int. 1998;54:720–730. doi: 10.1046/j.1523-1755.1998.00070.x. [DOI] [PubMed] [Google Scholar]
- 30.Piechotta K, Lu J, Delpire E. Cation chloride cotransporters interact with the stress-related kinases Ste20-related proline-alanine-rich kinase (SPAK) and oxidative stress response 1 (OSR1) J Biol Chem. 2002;277:50812–50819. doi: 10.1074/jbc.M208108200. [DOI] [PubMed] [Google Scholar]
- 31.Dowd BF, Forbush B. PASK (proline-alanine-rich STE20-related kinase), a regulatory kinase of the Na-K-Cl cotransporter (NKCC1) J Biol Chem. 2003;278:27347–27353. doi: 10.1074/jbc.M301899200. [DOI] [PubMed] [Google Scholar]
- 32.Zagorska A, Pozo-Guisado E, Boudeau J, Vitari AC, Rafiqi FH, Thastrup J, Deak M, Campbell DG, Morrice NA, Prescott AR, Alessi DR. Regulation of activity and localization of the WNK1 protein kinase by hyperosmotic stress. J Cell Biol. 2007;176:89–100. doi: 10.1083/jcb.200605093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anselmo AN, Earnest S, Chen W, Juang YC, Kim SC, Zhao YM, Cobb MH. WNK1 and OSR1 regulate the Na+, K+, 2Cl(−) cotransporter in HeLa cells. P Natl Acad Sci USA. 2006;103:10883–10888. doi: 10.1073/pnas.0604607103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richardson C, Rafiqi FH, Karlsson HKR, Moleleki N, Vandewalle A, Campbell DG, Morrice NA, Alessi DR. Activation of the thiazide-sensitive Na+-Cl− cotransporter by the WNK-regulated kinases SPAK and OSR1. J Cell Sci. 2008;121:675–684. doi: 10.1242/jcs.025312. [DOI] [PubMed] [Google Scholar]
- 35.Vitari AC, Deak M, Morrice NA, Alessi DR. The WNK1 and WNK4 protein kinases that are mutated in Gordon’s hypertension syndrome phosphorylate and activate SPAK and OSR1 protein kinases. Biochem J. 2005;391:17–24. doi: 10.1042/BJ20051180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moriguchi T, Urushiyama S, Hisamoto N, Iemura S, Uchida S, Natsume T, Matsumoto K, Shibuya H. WNK1 regulates phosphorylation of cation-chloride-coupled cotransporters via the STE20-related kinases, SPAK and OSR1. J Biol Chem. 2005;280:42685–42693. doi: 10.1074/jbc.M510042200. [DOI] [PubMed] [Google Scholar]
- 37.Ring AM, Cheng SX, Leng Q, Kahle KT, Rinehart J, Lalioti MD, Volkman HM, Wilson FH, Hebert SC, Lifton RP. WNK4 regulates activity of the epithelial Na+ channel in vitro and in vivo. Proc Natl Acad Sci U S A. 2007;104:4020–4024. doi: 10.1073/pnas.0611727104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu BE, Stippec S, Lazrak A, Huang CL, Cobb MH. WNK1 activates SGK1 by a phosphatidylinositol 3-kinase-dependent and non-catalytic mechanism. J Biol Chem. 2005;280:34218–34223. doi: 10.1074/jbc.M505735200. [DOI] [PubMed] [Google Scholar]
- 39.Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, Munster C, Chraibi A, Pratt JH, Horisberger JD, Pearce D, Loffing J, Staub O. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial Na(+) channel cell surface expression. EMBO J. 2001;20:7052–7059. doi: 10.1093/emboj/20.24.7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu BE, Stippec S, Chu PY, Lazrak A, Li XJ, Lee BH, English JM, Ortega B, Huang CL, Cobb MH. WNK1 activates SGK1 to regulate the epithelial sodium channel. Proc Natl Acad Sci U S A. 2005;102:10315–10320. doi: 10.1073/pnas.0504422102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang HR, Liu Z, Huang CL. Domains of WNK1 kinase in the regulation of ROMK1. Am J Physiol Renal Physiol. 2008;295:F438–445. doi: 10.1152/ajprenal.90287.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kahle KT, Wilson FH, Leng Q, Lalioti MD, O’Connell AD, Dong K, Rapson AK, MacGregor GG, Giebisch G, Hebert SC, Lifton RP. WNK4 regulates the balance between renal NaCl reabsorption and K+ secretion. Nat Genet. 2003;35:372–376. doi: 10.1038/ng1271. [DOI] [PubMed] [Google Scholar]
- 43.Ohta A, Yang SS, Rai T, Chiga M, Sasaki S, Uchida S. Overexpression of human WNK1 increases paracellular chloride permeability and phosphorylation of claudin-4 in MDCKII cells. Biochem Biophys Res Commun. 2006;349:804–808. doi: 10.1016/j.bbrc.2006.08.101. [DOI] [PubMed] [Google Scholar]
- 44.Park HW, Kim JY, Choi SK, Lee YH, Zeng W, Kim KH, Muallem S, Lee MG. Serine-threonine kinase with-no-lysine 4 (WNK4) controls blood pressure via transient receptor potential canonical 3 (TRPC3) in the vasculature. Proc Natl Acad Sci U S A. 2011;108:10750–10755. doi: 10.1073/pnas.1104271108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ponce-Coria J, San-Cristobal P, Kahle KT, Vazquez N, Pacheco-Alvarez D, de Los Heros P, Juarez P, Munoz E, Michel G, Bobadilla NA, Gimenez I, Lifton RP, Hebert SC, Gamba G. Regulation of NKCC2 by a chloride-sensing mechanism involving the WNK3 and SPAK kinases. Proc Natl Acad Sci U S A. 2008;105:8458–8463. doi: 10.1073/pnas.0802966105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kahle KT, Rinehart J, de Los Heros P, Louvi A, Meade P, Vazquez N, Hebert SC, Gamba G, Gimenez I, Lifton RP. WNK3 modulates transport of Cl− in and out of cells: implications for control of cell volume and neuronal excitability. Proc Natl Acad Sci U S A. 2005;102:16783–16788. doi: 10.1073/pnas.0508307102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cruz-Rangel S, Gamba G, Ramos-Mandujano G, Pasantes-Morales H. Influence of WNK3 on intracellular chloride concentration and volume regulation in HEK293 cells. Pflugers Arch. 2012;464:317–330. doi: 10.1007/s00424-012-1137-4. [DOI] [PubMed] [Google Scholar]
- 48.Rinehart J, Vazquez N, Kahle KT, Hodson CA, Ring AM, Gulcicek EE, Louvi A, Bobadilla NA, Gamba G, Lifton RP. WNK2 kinase is a novel regulator of essential neuronal cation-chloride cotransporters. J Biol Chem. 2011;286:30171–30180. doi: 10.1074/jbc.M111.222893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pacheco-Alvarez D, Gamba G. WNK3 is a putative chloride-sensing kinase. Cell Physiol Biochem. 2011;28:1123–1134. doi: 10.1159/000335848. [DOI] [PubMed] [Google Scholar]
- 50.Lenertz LY, Lee BH, Min X, Xu BE, Wedin K, Earnest S, Goldsmith EJ, Cobb MH. Properties of WNK1 and implications for other family members. J Biol Chem. 2005;280:26653–26658. doi: 10.1074/jbc.M502598200. [DOI] [PubMed] [Google Scholar]
- 51.Pantoliano MW, Petrella EC, Kwasnoski JD, Lobanov VS, Myslik J, Graf E, Carver T, Asel E, Springer BA, Lane P, Salemme FR. High-density miniaturized thermal shift assays as a general strategy for drug discovery. J Biomol Screen. 2001;6:429–440. doi: 10.1177/108705710100600609. [DOI] [PubMed] [Google Scholar]
- 52.Xu BE, Min X, Stippec S, Lee BH, Goldsmith EJ, Cobb MH. Regulation of WNK1 by an autoinhibitory domain and autophosphorylation. J Biol Chem. 2002;277:48456–48462. doi: 10.1074/jbc.M207917200. [DOI] [PubMed] [Google Scholar]
- 53.Bunkoczi G, Salah E, Filippakopoulos P, Fedorov O, Muller S, Sobott F, Parker SA, Zhang H, Min W, Turk BE, Knapp S. Structural and functional characterization of the human protein kinase ASK1. Structure. 2007;15:1215–1226. doi: 10.1016/j.str.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dauter Z, Dauter M, Dodson E. Jolly SAD. Acta Crystallogr, Sect D: Biol Crystallogr. 2002;58:494–506. doi: 10.1107/s090744490200118x. [DOI] [PubMed] [Google Scholar]
- 55.Steiner T. Hydrogen-bond distances to halide ions in organic and organometallic crystal structures: Up-to-date database study. Acta Crystallogr, Sect B: Struct Sci. 1998;54:456–463. [Google Scholar]
- 56.Hanks SK, Hunter T. Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB J. 1995;9:576–596. [PubMed] [Google Scholar]
- 57.Jura N, Zhang X, Endres NF, Seeliger MA, Schindler T, Kuriyan J. Catalytic control in the EGF receptor and its connection to general kinase regulatory mechanisms. Mol Cell. 2011;42:9–22. doi: 10.1016/j.molcel.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goldsmith EJ, Akella R, Min X, Zhou T, Humphreys JM. Substrate and Docking Interactions in Serine/Threonine Protein Kinases. Chem Rev. 2007;107:5065–5081. doi: 10.1021/cr068221w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.De Bondt HL, Rosenblatt J, Jancarik J, Jones HD, Morgan DO, Kim SH. Crystal structure of cyclin-dependent kinase 2. Nature. 1993;363:595–602. doi: 10.1038/363595a0. [DOI] [PubMed] [Google Scholar]
- 60.Taylor SS, Kornev AP. Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem Sci. 2011;36:65–77. doi: 10.1016/j.tibs.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim C, Cheng CY, Saldanha SA, Taylor SS. PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell. 2007;130:1032–1043. doi: 10.1016/j.cell.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 62.Kobe B, Heierhorst J, Feil SC, Parker MW, Benian GM, Weiss KR, Kemp BE. Giant protein kinases: domain interactions and structural basis of autoregulation. EMBO J. 1996;15:6810–6821. [PMC free article] [PubMed] [Google Scholar]
- 63.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 64.Pippig S, Andexinger S, Daniel K, Puzicha M, Caron MG, Lefkowitz RJ, Lohse MJ. Overexpression of beta-arrestin and beta-adrenergic receptor kinase augment desensitization of beta 2-adrenergic receptors. J Biol Chem. 1993;268:3201–3208. [PubMed] [Google Scholar]
- 65.Parker JC. In defense of cell volume? Am J Physiol. 1993;265:C1191–1200. doi: 10.1152/ajpcell.1993.265.5.C1191. [DOI] [PubMed] [Google Scholar]
- 66.Richardson C, Alessi DR. The regulation of salt transport and blood pressure by the WNK-SPAK/OSR1 signalling pathway. J Cell Sci. 2008;121:3293–3304. doi: 10.1242/jcs.029223. [DOI] [PubMed] [Google Scholar]
- 67.Darman RB, Flemmer A, Forbush B. Modulation of ion transport by direct targeting of protein phosphatase type 1 to the Na-K-Cl cotransporter. J Biol Chem. 2001;276:34359–34362. doi: 10.1074/jbc.C100368200. [DOI] [PubMed] [Google Scholar]
- 68.Glover M, Mercier Zuber A, Figg N, O’Shaughnessy KM. The activity of the thiazide-sensitive Na(+)-Cl(−) cotransporter is regulated by protein phosphatase PP4. Can J Physiol Pharmacol. 2010;88:986–995. doi: 10.1139/y10-080. [DOI] [PubMed] [Google Scholar]
- 69.Bukhtiyarova M, Northrop K, Chai X, Casper D, Karpusas M, Springman E. Improved expression, purification, and crystallization of p38alpha MAP kinase. Protein Expr Purif. 2004;37:154–161. doi: 10.1016/j.pep.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 70.Min XS, Akella R, He HX, Humphreys JM, Tsutakawa SE, Lee SJ, Tainer JA, Cobb MH, Goldsmith EJ. The Structure of the MAP2K MEK6 Reveals an Autoinhibitory Dimer. Structure. 2009;17:96–104. doi: 10.1016/j.str.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 72.Koenig T, Menze BH, Kirchner M, Monigatti F, Parker KC, Patterson T, Steen JJ, Hamprecht FA, Steen H. Robust prediction of the MASCOT score for an improved quality assessment in mass spectrometric proteomics. J Proteome Res. 2008;7:3708–3717. doi: 10.1021/pr700859x. [DOI] [PubMed] [Google Scholar]
- 73.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 74.N. Collaborative Computational Project. The CCP4 suite: programs for protein crystallography. Acta Crystallogr, Sect D: Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 75.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.DeLano WL. The PyMOL Molecular Graphics System. Delano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Raw trace of WNK1-KDm/S* DSF using Sypro Orange
Figure S2: Control kinase stability as a function of salt concentration
Figure S3: Phosphorylation of OSR1 by WNK1-KDm/S*
Figure S4: WNK1 autophosphorylation measured by mass spectrometry
Figure S5: Interaction of bound chloride with hydrophobic residues
Table S1: Data and refinement statistics for WNK1-sKDm/S*
Table S2: B-Factors of the 3/10 helix residues in WNK1-KDm/SA and WNK1-sKDm/S*
Table S3: Phosphorylation state of WNK1-KDm/S* mutants after purification from E. coli