

Abstract
Congenital heart diseases are the most commonly observed human birth defects and are the leading cause of infant morbidity and mortality. Accumulating evidence indicates that transforming growth factor-β/bone morphogenetic protein signaling pathways play critical roles during cardiogenesis. Smad4 encodes the only common Smad protein in mammals, which is a critical nuclear mediator of transforming growth factor-β/bone morphogenetic protein signaling. The aim of this work was to investigate the roles of Smad4 during heart development. To overcome the early embryonic lethality of Smad4−/− mice, we specifically disrupted Smad4 in the myocardium using a Cre/loxP system. We show that myocardial-specific inactivation of Smad4 caused heart failure and embryonic lethality at midgestation. Histological analysis revealed that mutant mice displayed a hypocellular myocardial wall defect, which is likely the primary cause for heart failure. Both decreased cell proliferation and increased apoptosis contributed to the myocardial wall defect in mutant mice. Data presented in this article contradict a previous report showing that Smad4 is dispensable for heart development. Our further molecular characterization showed that expression of Nmyc and its downstream targets, including cyclin D1, cyclin D2, and Id2, were downregulated in mutant embryos. Reporter analysis indicated that the transcriptional activity of the 351-bp Nmyc promoter can be positively regulated by bone morphogenetic protein stimulation and negatively regulated by transforming growth factor-β stimulation. Chromatin immunoprecipitation analysis revealed that the Nmyc promoter can form a complex with Smad4, suggesting that Nmyc is a direct downstream target of Smad4. In conclusion, this study provides the first mouse model showing that Smad4 plays essential roles during cardiogenesis.
Keywords: Smad4, cardiogenesis, transforming growth factor β/bone morphogenetic protein, Nmyc, myocardium
Congenital heart diseases (CHDs) are the most commonly observed human birth defects and are the leading cause of infant morbidity and mortality,1,2 yet their molecular etiologies are poorly understood. Accumulating evidence from mouse genetic studies has suggested that members of the family of transforming growth factor (TGF)β/bone morphogenetic protein (BMP) cytokines play critical functions during heart development.3–5 Mouse embryos derived from epiblasts lacking Bmp1ra do not contain any observable heart rudiment, suggesting that BMP signaling is required for induction of heart formation from mesoderm.6,7 Myocardial depletion of Alk3 or inactivation of BMP10 causes heart failure and embryonic lethality at midgestation because of significantly elevated apoptosis and reduced proliferation in cardiomyocytes.8,9 Myocardial inactivation of BMP2 leads to abnormal specification of the atrioventricular canal (AVC) myocardium and failure of atrioventricular (AV) cushion formation.10,11 TGFβ2−/− embryos display a range of cardiac defects including double-outlet right ventricle, atrial or ventricular septal defect, and an overriding tricuspid valve.12,13 Myocardial expression of a constitutively active form of Tgfbr1 disrupts cardiac looping and leads to heart failure.14 Therefore TGFβ/BMP signaling plays critical roles during multiple steps and in many aspects of cardiogenesis.
TGFβ/BMP signals are transduced through heterodimeric complexes of type I and type II serine/threonine kinase receptors. After formation of the receptor/ligand complex, the type II receptor will phosphorylate the type I receptor, which in turn phosphorylates specific members of the receptor-activated Smads (R-Smads).15–18 Phosphorylated R-Smads associate with the co-Smad Smad4, and the complex translocates to the nucleus to regulate transcription of target genes. In addition to the “canonical” pathway of Smad-mediated transcription, TGFβ/BMP signaling may also transduce signals through “noncanonical” mitogen-activated protein kinase pathways.17,19
Smad4 encodes the only co-Smad in mammals that can interact with both BMP and TGFβ R-Smads. Although Smad4 was originally thought to be an essential component of all Smad transcriptional complexes, recent studies have shown that TGFβ/BMP-activated transcription of some genes can occur in the absence of Smad4.20,21 Furthermore, Smad2−/− embryos display more severe defects than Smad4−/− embryos.22,23 These data suggest that rather than acting as a general transcriptional coactivator of R-Smads, Smad4 mediates specific cellular responses induced by TGFβ/BMP signaling.
The aim of this study was to reveal specific functions of Smad4 during cardiogenesis. Inactivation of Smad4 using a conventional gene knockout approach leads to defective gastrulation, precluding investigation of its roles during heart development.22,24 Specific inactivation of Smad4 in epiblasts causes embryonic lethality at embryonic day (E)8.5; however, in mutant embryos, heart rudiments are formed, indicating that Smad4 is not required for induction of cardiomyocytes from lateral mesoderm.23 Recent work by Wang et al reported no observable embryonic heart defect following myocardial-specific inactivation of Smad4, suggesting that Smad4 is dispensable for heart development.25 In contrast to this published work, our study provides compelling mouse genetic evidence showing that Smad4 plays essential cardiogenic roles, because myocardial inactivation of Smad4 leads to heart failure and embryonic lethality at midgestation. We have further performed a detailed characterization of the novel mouse model at both the morphological and molecular levels.
Materials and Methods
Mouse and Embryo Manipulations
All procedures were approved by the Institutional Animal Care and Use Committee at the University of Alabama. The cTnt-Cre;Smad4loxp/+ mice were obtained by crossing the cTnt-Cre mice26 with the Smad4loxp/loxp mice,27 both of which were backcrossed with C57Bl6 mice for more than 5 generations. Embryo treatment was performed as described previously.28,29 The PCR conditions for amplifying Cre and unrecombined and recombined Smad4loxp alleles have been described previously.26,27
TUNEL, Nonradioactive Section In Situ Hybridization, and Western Blot Analysis
TUNEL assays were performed using the Dead End Colorimetric TUNEL system (Promega) following the instructions of the manufacturer. Nonradioactive section in situ hybridization and Western blot analysis were performed as described previously.29,30
Immunostaining Studies
Immunofluorescence studies were performed as described previously.28,29 Samples were examined with the Leica HC fluorescent microscope equipped with an RT SLIDER digital camera. Immunohistochemistry studies were performed using the Envision+ system (DakoCytomation). The primary antibodies used in this study included antibodies recognizing cyclin D1 (BD Biosciences), cyclin D2 (Santa Cruz Biotechnology), phospho-H3 (Upstate), Smad4, phospho-Smad1/5/8, phospho-Smad2/3 (Cell Signaling), NFATc1 (BD Biosciences), cardiac myosin heavy chain, cardiac troponin T (Iowa Hybridoma Bank), and p57Kip2 (Labvision).
Luciferase Reporter Analysis
The 351-bp promoter region of the mouse Nmyc gene was PCR amplified and cloned into the pGL2-basic vector to acquire the pNmyc-Luc construct. Site-directed mutagenesis was performed to disrupt the potential Smad4-binding site within the Nmyc promoter using the QuickChange II site-directed mutagenesis kit (Stratagene). The mutagenic primer sequence is 5′-CAAAGCGCAGCCAGTGACAGTCATCTCGGATCCCGCGCTGGGTGGATGCG, in which the 7 underlined bases are the mutations that disrupt the potential Smad4-binding site. Transient transfection was performed with Fugene6 (Roche) following the instructions of the manufacturer. Treatment of cells with hBMP4 and TGFβ1 (R&D) and measurement of luciferase activity were performed as previously described.30,31
Chromatin Immunoprecipitation Analysis
P19 cells were transfected with the pCMV-HA vector or pHA-Smad4, which expresses hemagglutinin (HA)-tagged Smad4.30 Chromatin immunoprecipitation (ChIP) analysis was performed as described previously31 using an anti-HA antibody (Covance). Rabbit IgG was used as a negative control. The 351-bp Nmyc promoter region was amplified with the forward primer 5′GAGAAAAG-CAAATGGCTTTTGGC and the reverse primer 5′-TCTCCGGGTGGGCTGAGGGAG An unrelated genomic DNA sequence (corresponding to the DNA sequence at the Tgfbr2 allele) was amplified with primers 5′TAAACAAGGTCCGGAGCCCA and 5′ACTTCTGCAAGAGGTCCCCT
Results
Inactivation of Smad4 in the Myocardium by cTnt-Cre
To reveal cardiogenic roles of Smad4 and to overcome the early lethality of Smad4−/− mouse embryos,22–24 we specifically inactivated Smad4 in the myocardium by crossing the male cTnTcre;Smad4loxp/+ mice with the female Smad4loxp/loxp mice.27 The cTnt-Cre induces recombination early in the cardiomyocyte lineage (E7.5) and efficiently inactivates target genes between E9.5 to E10.5.26,32,33 To confirm that Smad4 was efficiently inactivated in the myocardium, we performed PCR analysis using genomic DNA isolated from E10.5 embryos (Figure 1A and 1B). The signal for the unrecombined Smad4loxp allele in mutant hearts was reduced to less than 10% of the control level. As expected, the recombined allele was detected in only mutant hearts and not in control samples (Smad4loxp/loxp). To evaluate the reduction of Smad4 at the protein level, we performed immunohistochemistry analysis using a Smad4-specific antibody. Expression of Smad4 in the myocardium was dramatically reduced in mutant hearts, yet Smad4-positive cells were evident in the mutant AV cushions (Figure 1C through D′), confirming that cTnt-Cre specifically inactivated Smad4 in cardiomyocytes.
Figure 1.

Myocardial-specific inactivation of Smad4. A, Genomic DNA was extracted from yolk sacs of mouse embryos at E10.5 to determine their genotypes using PCR analysis with primers for Cre or for the unrecombined Smad4loxp allele. B, Genomic DNA was extracted from hearts of the control (Smad4loxp/loxp) and mutant (cTnt-Cre;Smad4loxp/loxp) embryos examined in A. Semiquantitative PCR analysis was performed with primers for the unrecombined Smad4loxp allele (top). Lanes 1 to 3 are control samples with 10%, 50%, and 100% of input DNA. Lane 4 is the mutant sample with 100% of input DNA. Tgfbr2 (an unrelated genomic DNA fragment) was used as a loading control (middle). Bottom, genomic DNA was subjected to PCR analysis using primers for the recombined Smad4loxp allele. The PCR product could be detected in only the mutant sample and not in the control. C through D′, Sections of control (C and C′) and mutant (D and D′) mouse hearts at E10.5 were immunostained with an anti-Smad4 antibody (brown). Samples were counterstained with hematoxylin (blue). C′ and D′ correspond to the boxed regions of C and D, respectively. Cko indicates cTnt-Cre;Smad4loxp/loxp; Ctrl, Smad4loxp/loxp; Cu, cushion.
To test whether depletion of Smad4 in the myocardium affects activation of BMP and TGFβ R-Smads, we performed immunohistochemistry analysis using antibodies against phospho-Smad1/5/8 or against phospho-Smad2/3 (Figure I in the online data supplement, available at http://circres.ahajournals.org). No obvious alteration in the level of activated R-Smads was observed in mutant hearts, consistent with the previously reported observation that activation of R-Smads is independent of the presence of Smad4.17,18
Requirement of Smad4 During Normal Cardiogenesis in Mouse Embryos
We did not recover any mutant (cTnt-Cre;Smad4loxp/loxp) neonates from the breeding of cTnTcre;Smad4loxp/+ mice and Smad4loxp/loxp mice (total of 7 litters), suggesting that myocardial inactivation of Smad4 causes embryonic lethality. To determine the stage at which embryonic death occurred, we isolated embryos from E9.5 to E16.5. The number of mutant embryos between E9.5 and E11.5 was not markedly different from the expected ratio (25%, supplemental Figure II, A). At the E12.5 stage, the number of living mutant embryos was reduced to ≈10% of total embryos, and very few mutant embryos survived beyond E13.5, indicating that the majority of embryonic lethality occurred between E11.5 and E13.5. At E12.5, all mutant embryos (including those that were alive) displayed internal hemorrhages (supplemental Figure II, B), suggesting that the embryonic lethality was caused by cardiovascular insufficiency.
To reveal heart defects caused by myocardial depletion of Smad4, we performed detailed histological examination of living embryos between E9.5 and E12.5. No obvious abnormality was observed in E9.5 and E10.5 mutant hearts (Figure 2). At E11.5, although the AV cushions were of normal size, the superior and inferior AV cushions were improperly aligned in mutant hearts. The myocardium of mutant embryos at E11.5 does not display any obvious morphological defects except that the number of cells in the trabeculation zone appeared to be higher in mutant hearts than in those of controls (Figure 2C and 2G). At E12.5, all mutant hearts displayed thin myocardial walls (Figure 2D, 2D′, H, and H′), suggesting that Smad4 is required for the proper morphogenesis of myocardial wall. The hypocellular myocardial wall can reduce contractility of embryonic hearts, resulting in cardioinsufficiency, and is likely the primary cause of the embryonic lethality observed in mutants. Despite the obvious misalignment defect between the superior and inferior AV cushions in E11.5 hearts, AV cushions in E12.5 mutants were properly fused to form the central mesenchymal mass, which correctly separated the AV canal into left and right channels. The intraventricular septum in mutant hearts was formed at the proper position separating left and right ventricles (E12.5), although it was thinner than that in controls (supplemental Figure II).
Figure 2.
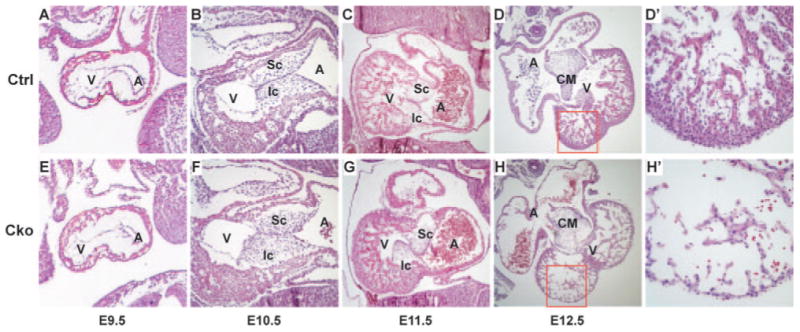
Myocardial depletion of Smad4 causes heart defects. Control (A through C) and mutant (E through G) embryos at E9.5 to E11.5 were sagittally sectioned and hematoxylin and eosin stained. Control (D and D′) and mutant (H and H′) embryonic hearts at E12.5 were cross sectioned and hematoxylin and eosin stained. D′ and H′ correspond to the boxed regions of D and H, respectively. A indicates atrium; CM, central mesenchymal mass; Cko, cTnt-Cre;Smad4loxp/loxp; Ctrl, Smad4loxp/loxp; Ic, inferior AV cushion; Sc, superior AV cushion; v, ventricle.
Molecular Examination of Mutant Embryonic Hearts
To determine the molecular defects in mutant (cTnt-Cre;Smad4loxp/loxp) embryonic hearts, we studied expression of multiple genes that play critical roles during cardiogenesis. We first examined general cardiogenic transcription factors including Nkx2.5, Mef2c, GATA4, and myocardin and showed that they were properly expressed in mutant embryos (Figure 3A and 3B; supplemental Figure III, A through F). Expression of Chisel, which was downregulated by BMP10 inactivation, was not altered in mutant hearts (supplemental Figure III, G and H). Furthermore, myocardial cells in mutants appear to be appropriately differentiated, as judged from normal expression of cardiomyocyte differentiation markers including cardiac troponin T (Figure 3C and 3D) and cardiac myosin heavy chain (supplemental Figure III, I and J). These data are consistent with previous results indicating that inactivation of Smad4 in epiblasts did not block specification and differentiation of cardiomyocytes.23 To test whether the AVC myocardium was properly specified, we examined expression of BMP2 and Tbx2 and found no abnormal expression of the 2 genes in mutant hearts (Figure 3E and 3F; supplemental Figure IIII, K and L), suggesting that the AV cushion misalignment defect was not caused by malspecification of the AVC region. We next examined expression of BMP10 and ANF and found that expression of both trabecular markers9,34 was increased in mutant hearts (Figure 3G and 3H; supplemental Figure III, M and N). These findings are consistent with our observation that mutant embryonic hearts contain more cells in their trabecular zones than do hearts from control embryos. We further showed that expression of NFATc135–38 was not altered in mutant hearts (supplemental Figure III, O and P), indicating that specification of endocardial cells was not compromised by myocardial depletion of Smad4.
Figure 3.
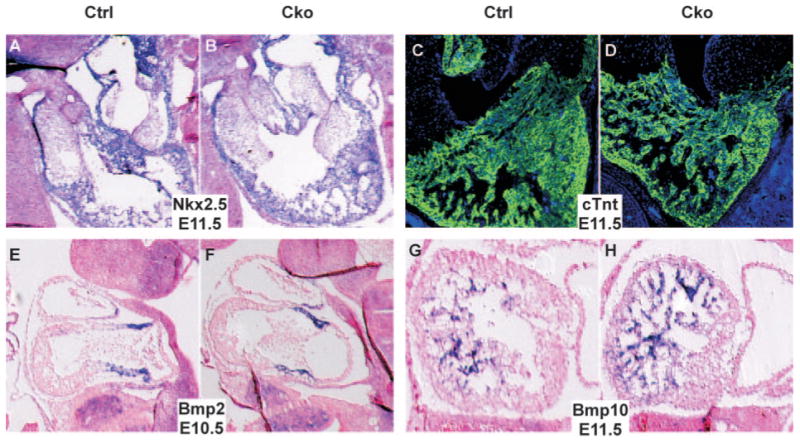
Molecular examination of mutant embryonic hearts. A and B, Control (A) and mutant (B) embryos at E11.5 were sagittally sectioned. Nonisotope section in situ hybridization analysis was performed with a probe against Nkx2.5 (purple). Samples were stained with nuclear fast red to visualize nuclei (red). C and D, Immunofluorescence analysis was performed on saggital sections of control (C) and mutant (D) embryos at E11.5 using an antibody against cardiac troponin T (green). Samples were stained with 4′,6-diamidino-2-phenylindole to visualize total nuclei (blue). E and F, Section in situ hybridization analysis was performed on saggital sections of control (E) and mutant (F) embryos at E10.5 using a BMP2 antisense probe (purple). G and H, Section in situ hybridization analysis was performed on saggital sections of control (G) and mutant (H) embryos at E11.5 using probes against BMP10. Cko indicates cTnt-Cre;Smad4loxp/loxp; Ctrl, Smad4loxp/loxp.
Decreased Cell Proliferation and Increased Apoptosis in Mutant Embryonic Hearts
To reveal the cellular mechanism accounting for the hypocellular myocardial wall defect observed in mutant hearts, we first examined cell proliferation by performing immunofluorescence studies using a primary antibody against phospho-H3. No reduction in cell proliferation was observed in mutant hearts until E10.5 (data not shown). In E11.5 mutant hearts, the cell proliferation rate was reduced in ventricles but not in other heart segments (Figure 4A through 4D), and quantitative analysis confirmed the statistical significance of the reduction (P<0.01, Figure 4E).
Figure 4.
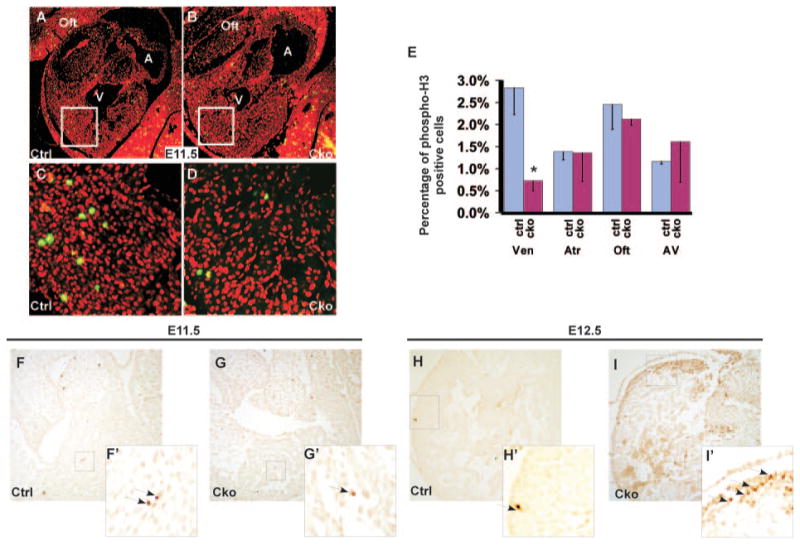
Myocardial depletion of Smad4 causes a reduction in cell proliferation and an increase in cell apoptosis. A through E, Saggital sections of control (A and C) and mutant (B and D) embryos at E11.5 were immunostained with a primary antibody against phospho-H3 (green), and total nuclei were visualized with propidium iodide staining (red). C and D correspond to the boxed regions of A and B, respectively. Quantitative analysis (E) indicates that the proliferation rate in mutant ventricles is significantly reduced compared with the control sample. At least 400 nuclei of each segment of each embryonic heart were counted. Data were averaged from 3 independent embryos with error bars indicating SD. *P<0.01 (Student t test). F through I′, TUNEL analysis was performed on sections of control (F and F′, H and H′), and mutant (G and G′, I and I′) embryonic hearts at E11.5 and at E12.5. Arrows indicate examples of TUNEL-positive cells. F′, G′, H′, and I′ correspond to the boxed regions of F, G, H, and I, respectively. A indicates atrium; Cko, cTnt-Cre;Smad4loxp/loxp; Ctrl, Smad4loxp/loxp; oft, outflow tract; V, ventricle.
We performed TUNEL assays to test the potential contribution of cell apoptosis to the thin-wall defect. We did not observe any obvious increase in cell death in mutant hearts until E11.5 (Figure 4F through G′). At E12.5, many cardiomyocytes in mutant hearts were undergoing apoptosis, whereas very few cardiac cells in control hearts were positive for TUNEL signals. Similar results were acquired from immunostaining studies using an anti-caspase 3 antibody (data not shown). We concluded that both decreased cell proliferation and increased apoptosis contribute to the hypocellular myocardial wall defect in mutant embryos.
Expression of Nmyc Is Downregulated in Mutant Hearts
We noticed that the thin-myocardial-wall defect of cTnt-Cre;Smad4loxp/loxp embryonic hearts is similar to the cardiac defects observed in embryos with compound heterozygous Nmyc alleles (Nmyc9a/BRP).39 Therefore, we decided to test whether expression of Nmyc is altered by myocardial inactivation of Smad4. At E10.5, expression of Nmyc was weak in both wild-type and mutant embryonic hearts, and no obvious reduction in Nmyc expression was observed in mutant embryos (Figure 5A and 5B). At E11.5, Nmyc expression in mutant hearts was dramatically reduced compared with that in control hearts (Figure 5C and 5D). To further investigate these findings, we performed Western blot analysis using an anti-Nmyc antibody and confirmed that cardiac expression of the Nmyc protein was reduced in mutants (Figure 5E).
Figure 5.
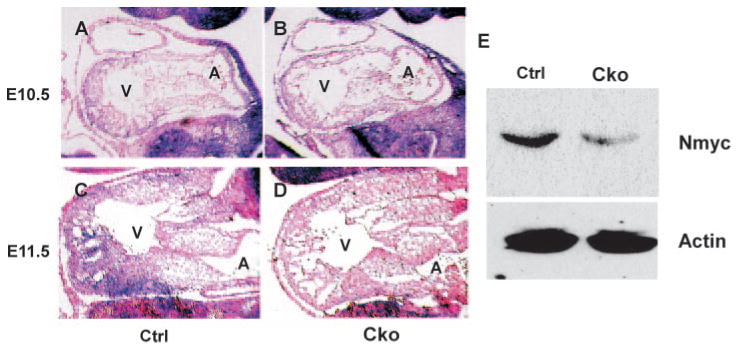
Expression of Nmyc is downregulated by myocardial depletion of Smad4. A through D, Section in situ hybridization analysis was performed on saggital sections of control (A and C) and mutant (B and D) embryos at E10.5 or at E11.5 using an Nmyc antisense probe (purple). Samples were counterstained with nuclear fast red (red). E, Proteins were extracted from control and mutant embryonic hearts at E11.5, and Western blot analysis was performed with an anti-Nmyc antibody. β-Actin was used as a loading control.
Smad4 Directly Activates Transcription of Nmyc
To test the possibility that Smad4 directly activates transcription of Nmyc, we analyzed the promoter regions of human and mouse Nmyc genes. Sequence alignment with the zPicture program (http://zpicture.dcode.org) revealed that the 351-bp promoter region (from bp-303 to bp +48, relative to the transcriptional start site) of Nmyc is highly conserved between human and mouse genes (supplemental Figure IV). Further sequence analysis using the rVista2.0 program (http://rvista.dcode.org) identified a potential Smad4-binding site within the 351-bp promoter region, which is 100% conserved between mouse and human genes. To test whether this 351-bp promoter region can respond to BMP/TGFβ stimulation, we generated an Nmyc-luciferase reporter construct and performed luciferase analysis. Our results showed that BMP4 stimulated, whereas TGFβ1 repressed, the transcriptional activity of the 351-bp Nmyc promoter in a dose-dependent manner (Figure 6A and 6B). Furthermore, the mutation disrupting the Smad4-binding site blocked the BMP4-induced upregulation of the reporter (Figure 6C), suggesting that the BMP ligand acts through the potential Smad4-binding site to simulate the Nmyc promoter. To test whether Smad4 directly binds to the 351-bp promoter region of Nmyc, we performed ChIP analysis using P19 cells transiently transfected with a plasmid expressing HA-Smad4. HA-Smad4 is specifically associated with the 351-bp Nmyc promoter but not with an unrelated genomic DNA sequence (Figure 6D).
Figure 6.
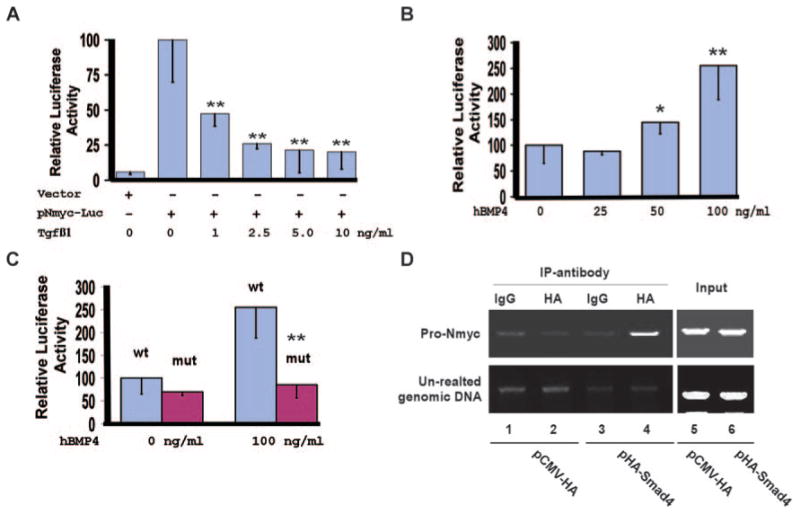
Nmyc is a direct downstream target of Smad4. A, NIH3T3 cells were transiently transfected with the reporter vector (pGL-basic) or pNmyc-luc, in which expression of luciferase is driven by the 351-bp Nmyc promoter. Cells were treated with 0 to 10 ng/mL of TGFβ1 for 48 hours, and luciferase activities were measured. The luciferase activity of pNmyc-luc without TGFβ1 stimulation was arbitrarily defined as 100 units. Data were averaged from 3 to 6 independent cultures, with error bars indicating SD. B, P19 cells were transiently transfected with pNmyc-Luc and were treated with 0 to 100 ng/mL hBMP4 for 48 hours. Luciferase analysis was then performed. The activity of the cell cultures without hBMP4 stimulation was defined as 100units. Data were averaged from 6 to 9 independent cultures. C, P19 cells were transfected with pNmyc-luc (wt) or pNmyc-luc-mut (mut), in which the potential Smad4-binding site within the Nmyc promoter was disrupted. Cells were treated with 0 or 100 ng/mL of hBMP4 for 48 hours, and luciferase activities were measured. BMP4 failed to upregulate expression of the reporter driven by the mutant Nmyc promoter. D, P19 cells were transiently transfected with pCMV-HA vector (negative control) or pHA-Smad4, which expresses HA-Smad4. ChIP analysis was performed with a rabbit IgG or an HA polyclonal antibody. HA-Smad4 formed a complex with the 351-bp Nmyc promoter but not with an unrelated genomic DNA sequence. The complex could be captured only by an anti-HA antibody and not by rabbit IgG. *P<0.1, **P<0.05.
Reduction of Nmyc Target Genes in Mutant Embryonic Hearts
The Nmyc gene promotes cell proliferation through activation of target genes involved in cell cycle control.40,41 To test whether myocardial inactivation of Smad4 reduced expression of genes located downstream of Nmyc, we examined expression of 3 known Nmyc target genes including Id2,42,43 cyclin D1,44 and cyclin D2.45,46 We first performed Western blot analysis using proteins extracted from wild-type and mutant embryonic hearts (E11.5) and found that expression of all 3 proteins was reduced in mutants (Figure 7A). The reduction in expression of these genes was further confirmed with immunofluorescence studies for cyclins D1 and D2 and with in situ hybridization analysis for Id2 (Figure 7). These data suggest that expression of at least a subset of Nmyc target genes was downregulated by myocardial depletion of Smad4. It was previously demonstrated that inactivation of BMP10 increased expression of p57Kip2, which negatively regulates the cell cycle9; however, we did not observe upregulation of p57Kip2 in cTnt-Cre;Smad4loxp/loxp embryonic hearts (E11.5; supplemental Figure III, Q and R).
Figure 7.
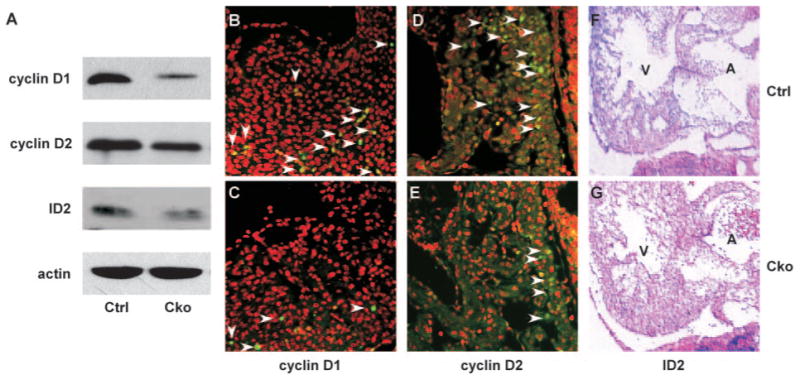
Downregulation of Nmyc target genes in mutant embryonic hearts. A, Proteins were extracted from control and mutant embryonic hearts at E11.5, and Western blot analysis was performed using antibodies against cyclin D1, cyclin D2, and Id2. B through E, Immunofluorescence analysis was performed on sections of control or mutant embryonic hearts at E11.5 using anti–cyclin D1 (B and C) or anti–cyclin D2 (D and E) antibodies (green). Total nuclei were visualized with propidium iodide staining (red). Arrow heads indicate examples of cells with positive signals. F and G, Saggital sections of control and mutant embryos at E11.5 were subjected to section in situ hybridization analysis using an Id2 antisense probe (purple). Samples were counterstained with nuclear fast red (red).
Discussion
This study provides the first mouse model demonstrating that Smad4 plays essential roles during mammalian heart development, myocardial depletion of Smad4 causes severe heart defects, and embryonic lethality at midgestation. Our results contradict a previously published study25 applying a similar conditional gene-activation approach to address functions of Smad4 during cardiogenesis. In that study, Wang et al inactivated Smad4 in the myocardium using an MHC-Cre line and showed that mutant mice can survive to adulthood without obvious congenital defects.25 The disparity between the 2 studies is unlikely caused by different mouse genetic backgrounds, as both our group and Wang and colleagues performed gene-conditional inactivation experiments on the C57BL/6 congenic background. We speculate that the discrepancy may be explained by the fact that 2 different Cre lines were used in these studies. The cTnt-Cre line used in this study can induce recombination of target genes starting from E7.5 and efficiently inactivate target genes at E10.5.26,32 We confirmed that Smad4 was efficiently inactivated in mutant embryonic hearts at both the DNA and protein levels at E10.5. The efficiency and specificity of Smad4 inactivation in mouse embryos using the MHC-Cre line generated by Wang et al were not clearly demonstrated.25 Therefore, inactivation of Smad4 with MHC-Cre used in the previous study may miss the critical time window in which the activity of Smad4 is essential for heart development.
The hypocellular myocardial wall defect at E12.5 in cTnt-Cre;Smad4loxp/loxp embryos is likely the major cause of embryonic lethality. Reduction in cell proliferation was initially observed at E11.5, when no abnormal apoptosis was detected, thus representing the primary cellular defect causing the hypocellular wall abnormality. Increased apoptosis at a later stage would further enhance this thin-myocardial-wall defect. Motivated by the similar cardiac phenotype observed with cTnt-Cre;Smad4loxp/loxp and Nmyc9a/BRP 39 embryos, we examined expression of Nmyc to gain further insight into the mechanism of Smad4 regulation of myocardial cell proliferation. Our data demonstrated that expression of Nmyc and its target genes, including cyclin D1, cyclin D2, and Id2, were all reduced in cTnt-Cre;Smad4loxp/loxp embryonic hearts, suggesting that Nmyc plays an important role in mediating Smad4-promoted myocardial cell proliferation. In further support of this idea, significant reduction in cardiomyocyte proliferation was observed primarily in the ventricles, the main cardiac segment where Nmyc is expressed (Figure 5 and Moens et al39). We observed a slightly increased number of cells in the trabeculation area of mutant hearts (Figure 2). This phenotype was repeatedly observed and was confirmed with molecular marker examination including BMP10 and ANF (Figure 3; supplemental Figure III). The mechanism underlying this defect warrants further investigation.
We provide strong evidence suggesting that Nmyc is a direct downstream target of Smad4 through sequence comparison, reporter analysis, and ChIP assays. This study is among the first to support that Myc genes can be directly upregulated by BMP stimulation. A recent study reported that Smad1 (a BMP R-Smad) can interact with β-catenin to upregulate expression of c-Myc.47 It is currently unclear whether upregulation of Nmyc by Smad4 also involves β-catenin. We conclude that in addition to the indirect regulation of Nmyc expression by BMP signaling through Tbx genes,48 BMP ligands may also directly stimulate Nmyc transcription through Smad4.
Because myocardial depletion of Smad4 downregulates Nmyc expression and reduces myocardial cell proliferation, we propose that Smad4 primarily mediates BMP signaling, rather than TGFβ signaling, during cardiogenesis. Among mouse models with mutations in BMP ligand genes, the myocardial wall defect of cTnt-Cre;Smad4loxp/loxp embryos most closely resembles BMP10−/− embryos, which also displayed reduced myocardial cell proliferation.9 However, depletion of BMP10 also dramatically decreased expression of some general cardiogenic transcription factors including Mef2c and Nkx2.5 (and its downstream target, Chisel) and increased expression of p57kip2.9 These defects were not observed in cTnt-Cre;Smad4loxp/loxp embryos. Therefore, the activities of BMP10 to promote expression of cardiogenic transcription factors and to repress expression of p57kip2 are mediated by Smad4-independent pathways.
In conclusion, this study provides convincing mouse genetic evidence showing that Smad4 is required for normal cardiogenesis. Functions of myocardial Smad4 are summarized in Figure 8. Our data support the notion that TGFβ/BMP signaling in the myocardium can be mediated through both Smad4-dependent and -independent pathways. Furthermore, we show that Nmyc and its downstream genes play major roles in mediating functions of Smad4 during cardiogenesis.
Figure 8.
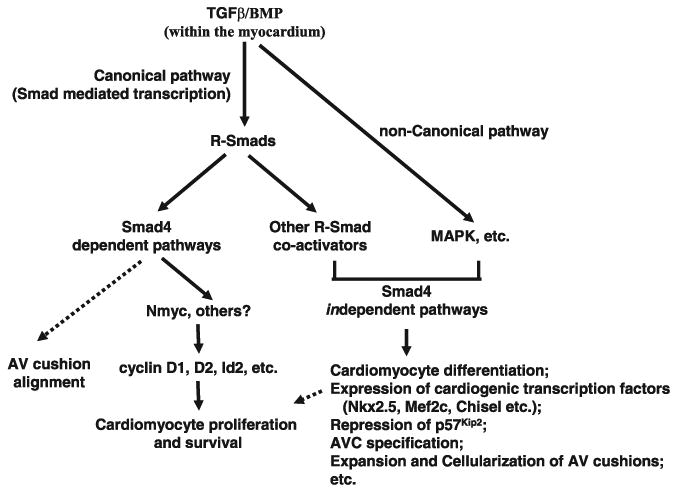
Mediation of TGFβ/BMP signaling through Smad4-dependent and Smad4-independent pathways. Our data showed that Smad4 is required for proper AV cushion alignment and myocardial cell proliferation and survival. Nmyc is a direct target of Smad4 in the myocardium during embryogenesis.
Supplementary Material
Fig. S1. Activation of R-Smads in mutant embryonic hearts. Sections of control (A, C) and mutant (B, D) embryonic hearts at E10.5 were immunostained with an anti-phospho-Smad 1/5/8 antibody (A, B) or an anti-phospho-Smad2/3 antibody (C, D) (brown). Samples were also stained with Hematoxylin (blue). No obvious difference was observed between control and mutant samples. Cko: cTnt-Cre;Smad4loxp/loxp; Ctrl: Smad4loxp/loxp.
Fig. S2. Myocardial depletion of Smad4 causes heart defects and embryonic lethality at midgestation. (A) The chart shows the percentage of living mutant (cTnt-Cre;Smad4loxp/loxp) embryos isolated at various stages from the cross between cTnt-Cre;Smad4Ioxp/+ mice and Smad4loxp/loxp mice. The expected ratio is 25%. The total numbers of embryos examined are 61, 97, 43 and 59 for E10.5, E11.5, E12.5 and El3.5 stages, respectively. (B) All mutant embryos (Cko) isolated at E12.5 displayed internal hemorrhage. (C, D) Control (C) and mutant (D) embryonic hearts at E12.5 were sectioned and HE stained. Cko: cTnt-Cre;Smad4loxp/loxp; Ctrl: Smad4loxp/loxp; la: left atrium; lv: left ventricle; vs: intraventricular septum.
Fig. S3. Molecular examination of mutant embryonic hearts. (A-H) Control (A, C, E, G) and mutant (B, D, F, H) embryos at E11.5 were sagittally sectioned. Non-isotope section in situ hybridization analysis was performed with anti-sense probes against GATA4 (A, B), Mef2c (C, D), Myocardin (E, F) and Chisel (G, H) (purple). Samples were stained with nuclear-fast-red to visualize nuclei (red). (I, J) Immunofluorescence analysis was performed on saggital sections of control (I) and mutant (J) embryos at E11.5 using a primary antibody against cardiac myosin heavy chain (green). Samples were stained with DAPI to visualize total nuclei (blue). (K-N) Non-isotope in situ hybridization analysis was performed on saggital sections of control (K, M) and mutant (L, N) embryos at E11.5 using a Tbx2 anti-sense probe (K, L) or a ANF anti-sense probe (M, N) (purple). Samples also were counterstained with nuclear-fast-red (red). (O-R) Immunofluorescence studies were performed on saggital sections of control (O, Q) and mutant (P, R) embryos at E11.5 using an anti-Nfatc1 primary antibody (O, P) or an anti-p57Kip2 primary antibody (green). Total nuclei were visualized with Propidiumiodide staining. Cko: cTnt-Cre;Smad4loxp/loxp; Ctrl: Smad4loxp/loxp.
Fig. S4. Sequence analysis of the Nmyc promoter. The 351bp mouse Nmyc promoter (bp-303 to bp+48 relative to the transcriptional start site) was aligned with the corresponding human Nmyc promoter using the zPicture program (http://zpicture.dcode.org/). A potential Smad4 binding site, which is highlighted in yellow, was identified using the rVista2.0 program (http://rvista.dcode.org/). This potential Smad4 binding site (bp-83 to bp-97) is 100% conserved between mouse and human genes.
Acknowledgments
We thank Dr Weinian Shou (Indiana University) for critically reading the manuscript. We thank Drs Brigid Hogan (Duke), Sylvia Evans (University of California, San Diego), Dazhi Wang (University of North Carolina at Chapel Hill), and Eric Olson (University of Texas Southwestern) for providing various in situ probes. We thank members of the laboratory of K.J. for suggestions on the project.
Sources of Funding: This project is supported by a Scientist Development Grant from the American Heart Association, NIH grant 1R21HL085510-01, and a Health Sciences Foundation–General Endowment Fund scholar award (to K.J.).
Footnotes
Disclosures: None
References
- 1.Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39:1890–1900. doi: 10.1016/s0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- 2.Clark KL, Yutzey KE, Benson DW. Transcription factors and congenital heart defects. Annu Rev Physiol. 2006;68:97–121. doi: 10.1146/annurev.physiol.68.040104.113828. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong EJ, Bischoff J. Heart valve development: endothelial cell signaling and differentiation. Circ Res. 2004;95:459–470. doi: 10.1161/01.RES.0000141146.95728.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Azhar M, Schultz Jel J, Grupp I, Dorn GW, 2nd, Meneton P, Molin DG, Gittenberger-de Groot AC, Doetschman T. Transforming growth factor beta in cardiovascular development and function. Cytokine Growth Factor Rev. 2003;14:391–407. doi: 10.1016/s1359-6101(03)00044-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schneider MD, Gaussin V, Lyons KM. Tempting fate: BMP signals for cardiac morphogenesis. Cytokine Growth Factor Rev. 2003;14:1–4. doi: 10.1016/s1359-6101(02)00053-9. [DOI] [PubMed] [Google Scholar]
- 6.Miura S, Tallquist MD, Soriano P, Mishina Y. BMP signaling is important for mesoderm induction and germ layer development in mouse embryogenesis. Dev Biol. 2002;247:505. [Google Scholar]
- 7.Miura S, Davis S, Klingensmith J, Mishina Y. BMP signaling in the epiblast is required for proper recruitment of the prospective paraxial mesoderm and development of the somites. Development. 2006;133:3767–3775. doi: 10.1242/dev.02552. [DOI] [PubMed] [Google Scholar]
- 8.Gaussin V, Van de Putte T, Mishina Y, Hanks MC, Zwijsen A, Huylebroeck D, Behringer RR, Schneider MD. Endocardial cushion and myocardial defects after cardiac myocyte- specific conditional deletion of the bone morphogenetic protein receptor ALK3. Proc Natl Acad Sci U S A. 2002;99:2878–2883. doi: 10.1073/pnas.042390499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen H, Shi S, Acosta L, Li W, Lu J, Bao S, Chen Z, Yang Z, Schneider MD, Chien KR, Conway SJ, Yoder MC, Haneline LS, Franco D, Shou W. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development. 2004;131:2219–2231. doi: 10.1242/dev.01094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma L, Lu MF, Schwartz RJ, Martin JF. Bmp2 is essential for cardiac cushion epithelial-mesenchymal transition and myocardial patterning. Development. 2005;132:5601–5611. doi: 10.1242/dev.02156. [DOI] [PubMed] [Google Scholar]
- 11.Rivera-Feliciano J, Tabin CJ. Bmp2 instructs cardiac progenitors to form the heart-valve-inducing field. Dev Biol. 2006;295:580–588. doi: 10.1016/j.ydbio.2006.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGFbeta2 knockout mice have multiple developmental defects that are nonoverlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bartram U, Molin DG, Wisse LJ, Mohamad A, Sanford LP, Doetschman T, Speer CP, Poelmann RE, Gittenberger-de Groot AC. Double-outlet right ventricle and overriding tricuspid valve reflect disturbances of looping, myocardialization, endocardial cushion differentiation, and apoptosis in TGF-beta(2)-knockout mice. Circulation. 2001;103:2745–2752. doi: 10.1161/01.cir.103.22.2745. [DOI] [PubMed] [Google Scholar]
- 14.Charng MJ, Frenkel PA, Lin Q, Yamada M, Schwartz RJ, Olson EN, Overbeek P, Schneider MD, Yumada M. A constitutive mutation of ALK5 disrupts cardiac looping and morphogenesis in mice. Dev Biol. 1998;199:72–79. doi: 10.1006/dbio.1998.8905. [DOI] [PubMed] [Google Scholar]
- 15.Massague J, Gomis RR. The logic of TGFbeta signaling. FEBS Lett. 2006;580:2811–2820. doi: 10.1016/j.febslet.2006.04.033. [DOI] [PubMed] [Google Scholar]
- 16.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 17.ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends Biochem Sci. 2004;29:265–273. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 18.Massague J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- 19.Moustakas A, Heldin CH. Non-Smad TGF-beta signals. J Cell Sci. 2005;118:3573–3584. doi: 10.1242/jcs.02554. [DOI] [PubMed] [Google Scholar]
- 20.Sirard C, Kim S, Mirtsos C, Tadich P, Hoodless PA, Itie A, Maxson R, Wrana JL, Mak TW. Targeted disruption in murine cells reveals variable requirement for Smad4 in transforming growth factor beta-related signaling. J Biol Chem. 2000;275:2063–2070. doi: 10.1074/jbc.275.3.2063. [DOI] [PubMed] [Google Scholar]
- 21.He W, Dorn DC, Erdjument-Bromage H, Tempst P, Moore MA, Massague J. Hematopoiesis controlled by distinct TIF1gamma and Smad4 branches of the TGFbeta pathway. Cell. 2006;125:929–941. doi: 10.1016/j.cell.2006.03.045. [DOI] [PubMed] [Google Scholar]
- 22.Sirard C, de la Pompa JL, Elia A, Itie A, Mirtsos C, Cheung A, Hahn S, Wakeham A, Schwartz L, Kern SE, Rossant J, Mak TW. The tumor suppressor gene Smad4/Dpc4 is required for gastrulation and later for anterior development of the mouse embryo. Genes Dev. 1998;12:107–119. doi: 10.1101/gad.12.1.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chu GC, Dunn NR, Anderson DC, Oxburgh L, Robertson EJ. Differential requirements for Smad4 in TGFbeta-dependent patterning of the early mouse embryo. Development. 2004;131:3501–3512. doi: 10.1242/dev.01248. [DOI] [PubMed] [Google Scholar]
- 24.Yang X, Li C, Xu X, Deng C. The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc Natl Acad Sci U S A. 1998;95:3667–3672. doi: 10.1073/pnas.95.7.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J, Xu N, Feng X, Hou N, Zhang J, Cheng X, Chen Y, Zhang Y, Yang X. Targeted disruption of Smad4 in cardiomyocytes results in cardiac hypertrophy and heart failure. Circ Res. 2005;97:821–828. doi: 10.1161/01.RES.0000185833.42544.06. [DOI] [PubMed] [Google Scholar]
- 26.Jiao K, Kulessa H, Tompkins K, Zhou Y, Batts L, Baldwin HS, Hogan BL. An essential role of Bmp4 in the atrioventricular septation of the mouse heart. Genes Dev. 2003;17:2362–2367. doi: 10.1101/gad.1124803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang X, Li C, Herrera PL, Deng CX. Generation of Smad4/Dpc4 conditional knockout mice. Genesis. 2002;32:80–81. doi: 10.1002/gene.10029. [DOI] [PubMed] [Google Scholar]
- 28.Jiao K, Langworthy M, Batts L, Brown CB, Moses HL, Baldwin HS. Tgf{beta} signaling is required for atrioventricular cushion mesenchyme remodeling during in vivo cardiac development. Development. 2006;133:4585–4593. doi: 10.1242/dev.02597. [DOI] [PubMed] [Google Scholar]
- 29.Song L, Fassler R, Mishina Y, Jiao K, Baldwin HS. Essential functions of Alk3 during AV cushion morphogenesis in mouse embryonic hearts. Dev Biol. 2007;301:276–286. doi: 10.1016/j.ydbio.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 30.Jiao K, Zhou Y, Hogan BL. Identification of mZnf8, a mouse Kruppel-like transcriptional repressor, as a novel nuclear interaction partner of Smad1. Mol Cell Biol. 2002;22:7633–7644. doi: 10.1128/MCB.22.21.7633-7644.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yan W, Chen X. GPX2, a direct target of p63, inhibits oxidative stress-induced apoptosis in a p53-dependent manner. J Biol Chem. 2006;281:7856–7862. doi: 10.1074/jbc.M512655200. [DOI] [PubMed] [Google Scholar]
- 32.Chen JW, Zhou B, Yu QC, Shin SJ, Jiao K, Schneider MD, Baldwin HS, Bergelson JM. Cardiomyocyte-specific deletion of the coxsackievirus and adenovirus receptor results in hyperplasia of the embryonic left ventricle and abnormalities of sinuatrial valves. Circ Res. 2006;98:923–930. doi: 10.1161/01.RES.0000218041.41932.e3. [DOI] [PubMed] [Google Scholar]
- 33.Ilagan R, Abu-Issa R, Brown D, Yang YP, Jiao K, Schwartz RJ, Klingensmith J, Meyers EN. Fgf8 is required for anterior heart field development. Development. 2006;133:2435–2445. doi: 10.1242/dev.02408. [DOI] [PubMed] [Google Scholar]
- 34.Christoffels VM, Habets PE, Franco D, Campione M, de Jong F, Lamers WH, Bao ZZ, Palmer S, Biben C, Harvey RP, Moorman AF. Chamber formation and morphogenesis in the developing mammalian heart. Dev Biol. 2000;223:266–278. doi: 10.1006/dbio.2000.9753. [DOI] [PubMed] [Google Scholar]
- 35.Ranger AM, Grusby MJ, Hodge MR, Gravallese EM, de la Brousse FC, Hoey T, Mickanin C, Baldwin HS, Glimcher LH. The transcription factor NF-ATc is essential for cardiac valve formation. Nature. 1998;392:186–190. doi: 10.1038/32426. [DOI] [PubMed] [Google Scholar]
- 36.Chang CP, Neilson JR, Bayle JH, Gestwicki JE, Kuo A, Stankunas K, Graef IA, Crabtree GR. A field of myocardial-endocardial NFAT signaling underlies heart valve morphogenesis. Cell. 2004;118:649–663. doi: 10.1016/j.cell.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 37.de la Pompa JL, Timmerman LA, Takimoto H, Yoshida H, Elia AJ, Samper E, Potter J, Wakeham A, Marengere L, Langille BL, Crabtree GR, Mak TW. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature. 1998;392:182–186. doi: 10.1038/32419. [DOI] [PubMed] [Google Scholar]
- 38.Zhou B, Wu B, Tompkins KL, Boyer KL, Grindley JC, Baldwin HS. Characterization of Nfatc1 regulation identifies an enhancer required for gene expression that is specific to pro-valve endocardial cells in the developing heart. Development. 2005;132:1137–1146. doi: 10.1242/dev.01640. [DOI] [PubMed] [Google Scholar]
- 39.Moens CB, Stanton BR, Parada LF, Rossant J. Defects in heart and lung development in compound heterozygotes for two different targeted mutations at the N-myc locus. Development. 1993;119:485–499. doi: 10.1242/dev.119.2.485. [DOI] [PubMed] [Google Scholar]
- 40.Adhikary S, Eilers M. Transcriptional regulation and transformation by Myc proteins. Nat Rev Mol Cell Biol. 2005;6:635–645. doi: 10.1038/nrm1703. [DOI] [PubMed] [Google Scholar]
- 41.Hurlin PJ. N-Myc functions in transcription and development. Birth Defects Res C Embryo Today. 2005;75:340–352. doi: 10.1002/bdrc.20059. [DOI] [PubMed] [Google Scholar]
- 42.Lasorella A, Noseda M, Beyna M, Yokota Y, Iavarone A. Id2 is a retinoblastoma protein target and mediates signalling by Myc oncoproteins. Nature. 2000;407:592–598. doi: 10.1038/35036504. [DOI] [PubMed] [Google Scholar]
- 43.Lasorella A, Boldrini R, Dominici C, Donfrancesco A, Yokota Y, Inserra A, Iavarone A. Id2 is critical for cellular proliferation and is the oncogenic effector of N-myc in human neuroblastoma. Cancer Res. 2002;62:301–306. [PubMed] [Google Scholar]
- 44.Kenney AM, Cole MD, Rowitch DH. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development. 2003;130:15–28. doi: 10.1242/dev.00182. [DOI] [PubMed] [Google Scholar]
- 45.Bouchard C, Dittrich O, Kiermaier A, Dohmann K, Menkel A, Eilers M, Luscher B. Regulation of cyclin D2 gene expression by the Myc/Max/Mad network: Myc-dependent TRRAP recruitment and histone acetylation at the cyclin D2 promoter. Genes Dev. 2001;15:2042–2047. doi: 10.1101/gad.907901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bouchard C, Thieke K, Maier A, Saffrich R, Hanley-Hyde J, Ansorge W, Reed S, Sicinski P, Bartek J, Eilers M. Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J. 1999;18:5321–5333. doi: 10.1093/emboj/18.19.5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hu MC, Rosenblum ND. Smad1, beta-catenin and Tcf4 associate in a molecular complex with the Myc promoter in dysplastic renal tissue and cooperate to control Myc transcription. Development. 2005;132:215–225. doi: 10.1242/dev.01573. [DOI] [PubMed] [Google Scholar]
- 48.Cai CL, Zhou W, Yang L, Bu L, Qyang Y, Zhang X, Li X, Rosenfeld MG, Chen J, Evans S. T-box genes coordinate regional rates of proliferation and regional specification during cardiogenesis. Development. 2005;132:2475–2487. doi: 10.1242/dev.01832. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Activation of R-Smads in mutant embryonic hearts. Sections of control (A, C) and mutant (B, D) embryonic hearts at E10.5 were immunostained with an anti-phospho-Smad 1/5/8 antibody (A, B) or an anti-phospho-Smad2/3 antibody (C, D) (brown). Samples were also stained with Hematoxylin (blue). No obvious difference was observed between control and mutant samples. Cko: cTnt-Cre;Smad4loxp/loxp; Ctrl: Smad4loxp/loxp.
Fig. S2. Myocardial depletion of Smad4 causes heart defects and embryonic lethality at midgestation. (A) The chart shows the percentage of living mutant (cTnt-Cre;Smad4loxp/loxp) embryos isolated at various stages from the cross between cTnt-Cre;Smad4Ioxp/+ mice and Smad4loxp/loxp mice. The expected ratio is 25%. The total numbers of embryos examined are 61, 97, 43 and 59 for E10.5, E11.5, E12.5 and El3.5 stages, respectively. (B) All mutant embryos (Cko) isolated at E12.5 displayed internal hemorrhage. (C, D) Control (C) and mutant (D) embryonic hearts at E12.5 were sectioned and HE stained. Cko: cTnt-Cre;Smad4loxp/loxp; Ctrl: Smad4loxp/loxp; la: left atrium; lv: left ventricle; vs: intraventricular septum.
Fig. S3. Molecular examination of mutant embryonic hearts. (A-H) Control (A, C, E, G) and mutant (B, D, F, H) embryos at E11.5 were sagittally sectioned. Non-isotope section in situ hybridization analysis was performed with anti-sense probes against GATA4 (A, B), Mef2c (C, D), Myocardin (E, F) and Chisel (G, H) (purple). Samples were stained with nuclear-fast-red to visualize nuclei (red). (I, J) Immunofluorescence analysis was performed on saggital sections of control (I) and mutant (J) embryos at E11.5 using a primary antibody against cardiac myosin heavy chain (green). Samples were stained with DAPI to visualize total nuclei (blue). (K-N) Non-isotope in situ hybridization analysis was performed on saggital sections of control (K, M) and mutant (L, N) embryos at E11.5 using a Tbx2 anti-sense probe (K, L) or a ANF anti-sense probe (M, N) (purple). Samples also were counterstained with nuclear-fast-red (red). (O-R) Immunofluorescence studies were performed on saggital sections of control (O, Q) and mutant (P, R) embryos at E11.5 using an anti-Nfatc1 primary antibody (O, P) or an anti-p57Kip2 primary antibody (green). Total nuclei were visualized with Propidiumiodide staining. Cko: cTnt-Cre;Smad4loxp/loxp; Ctrl: Smad4loxp/loxp.
Fig. S4. Sequence analysis of the Nmyc promoter. The 351bp mouse Nmyc promoter (bp-303 to bp+48 relative to the transcriptional start site) was aligned with the corresponding human Nmyc promoter using the zPicture program (http://zpicture.dcode.org/). A potential Smad4 binding site, which is highlighted in yellow, was identified using the rVista2.0 program (http://rvista.dcode.org/). This potential Smad4 binding site (bp-83 to bp-97) is 100% conserved between mouse and human genes.