

Abstract
Metals of interest for biomedical applications often need to be stably complexed and associated with a targeting agent before use. While the basics of the complexation of most transition metals have been thoroughly studied in that end, ZrIV has been somewhat neglected. Yet, this metal has received a growing attention in recent years, especially in nuclear medicine with the use of 89Zr, a β+-emitter with near ideal characteristics for cancer imaging. However, the best chelating agent known for this radionuclide is the tris-hydroxamate desferrioxamine B (DFB), whose ZrIV complex exhibits non-optimal stability resulting in the progressive release of 89Zr in vivo. Based on a recent report demonstrating the higher thermodynamic stability of the tetrahydroxamate complexes of ZrIV compared to the tris-hydroxamate complexes analogue to DFB, we designed a series of tetrahydroxamic acids of varying geometries for improved complexation of this metal. Three macrocycles differing by their cavity size (28 to 36-membered rings) were synthesized using a ring closing metathesis strategy, as well as their acyclic analogues. A solution study with 89Zr showed the complexation to be more effective with increasing size of cavity. Evaluation of the kinetic inertness of these new complexes in EDTA solution showed significantly improved stabilities of the larger chelates compared to 89Zr-DFB, whereas the smaller complexes exhibited insufficient stabilities. These results were rationalized by a quantum chemical study. The lower stability of the smaller chelates was attributed to the ring strain, whereas the better stability of the larger cyclic complexes was explained by the macrocyclic effect and structural rigidity. Overall, these new chelating agents open new perspectives for the safe and efficient use of 89Zr in nuclear imaging, with the best ones providing dramatically improved stabilities compared to the reference DFB.
Keywords: Chelates, Hydroxamic acids, Imaging agents, Quantum chemistry, Zirconium
Introduction
Positron emitted tomography (PET) is a powerful imaging technique for the detection of various pathologies, including cancers. 18F (T1/2 = 1.8 h) is currently the most commonly used β+-emitter in nuclear imaging, and continuous progress are being reported related to the chemistry and applications with this radionuclide.[1] In parallel, a growing number of investigations on the chemistry of longer-lived radionuclides such as 64Cu (T1/2 = 12.7 h), 86Y (T1/2 = 14.7 h) or 124I (T1/2 = 100.2 h) is ongoing, expanding the possible applications of PET imaging[2] Among these promising β+-emitters, 89Zr (T1/2 = 78.4 h) is particularly interesting when associated with higher molecular weight targeting agents such as cancer targeting antibodies[3]. Indeed, its half-life ideally matches their blood kinetics, providing high sensitive imaging of tumors for several days after injection.
Hydroxamic acids are bidentate ligands that are known to bind strongly to hard cations such as Zr4+ via their oxygen atoms,[4] and 89Zr is usually bound to antibodies via a conjugated tris-hydroxamate-based chelating agent, the natural siderophore desferrioxamine B (DFB) (Figure 1)5,6 Although DFB has been a useful tool to prove the high potential of 89Zr for PET-applications,[7] many animal studies have shown this chelator to be far from optimal to adequately sequester ZrIV for in vivo use, forming a complex that lacks kinetic inertness. This instability is visible via the progressive uptake of 89Zr in the bone as the metallic radionuclide is released from DFB over a few days&lsq[8]. Possible consequences of this instability are toxicity to the bone marrow with potentially high radiation doses of radioactivity that can be deposited due to the relatively long half-life of 89Zr associated with its 909 keV γ-emission, as well as the loss of image resolution due to lower signal/noise ratio, especially if the tumors are in the vicinity of bones. This instability issue stems from the lack of chelation chemistry studies directed specifically for ZrIV, leaving an important area of research ill-defined as compared to other transition metals. For instance, a review of recent literature of 89Zr labeling of proteins shows that studies were focused mostly towards the conjugation moiety and not on the ligand itself6,9,10. Only one article reports the use of alternative chelators with a series of phosphonates based ligands. However, results showed that these new chelators were less efficient than DFB for 89Zr complexation.[11]
Figure 1.
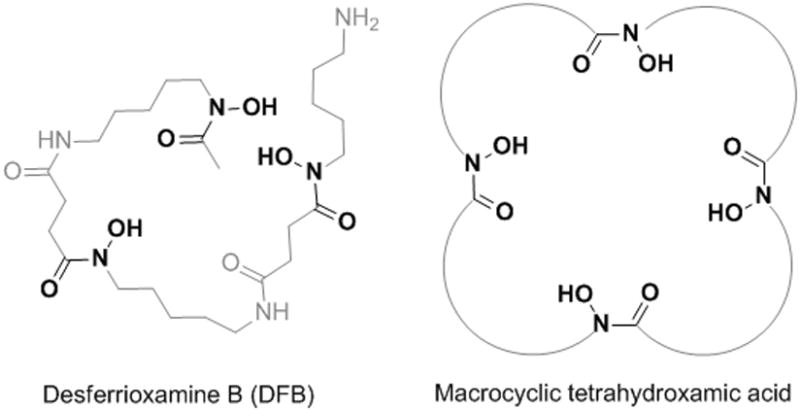
Structure of DFB and schematic representation of the tetrahydroxamic acid chelators C5, C6 and C7 considered in this study, with each hydroxamic acid linked by a chain of 5, 6 or 7 methylene groups, respectively.
To increase the stability of the complexation of ZrIV, our approach was to improve rationally the characteristics of DFB to fit the cation needs. The preference of ZrIV to form octa-coordinated complexes with 4 hydroxamate ligands was confirmed only recently,[12] suggesting that the chelating properties of DFB, which consists of only 3 hydroxamate subunits, could be improved by the inclusion of a fourth hydroxamate in a suitably pre-organized chelator. Whereas adequate pre-organization is essential to form thermodynamically stable chelates,[13] macrocyclic structures can also favor higher kinetic inertness of their metallic complex, a point clearly illustrated in Liu and Edwards' review.[14] This factor is crucial when considering in vivo applications since the complexes, which are highly diluted in their use, are exposed to endogenous competitive cations and natural chelators that may challenge the stability of the metal chelate.
For these reasons, we focused on the development of a macrocyclic tetrahydroxamic acid ligand of appropriate geometry for ZrIV. To probe the optimal ring size, 3 macrocycles made of 28, 32 or 36 atoms were designed, all of them symmetrical, with a 5, 6 or 7-carbon alkyl chain between each hydroxamic acid and denoted C5, C6 and C7, respectively (Figure 1). These ring sizes were deemed to be in the appropriate range to preserve the spatial orientation of the four hydroxamate ligands as observed in the X-ray structure of tetrakis(N-(hydroxyl)-N-methylacetamidato)-zirconium(IV).[12] Accordingly, comparison of these chelators was pursued to further refine the optimal ring size imparting both thermodynamic stability and kinetic inertness to the ZrIV complexes.
Results and Discussion
Preparation of the tetrahydroxamic acid chelators
The general synthetic approach was based on the formation of linear chains of four O-benzylated hydroxamates connected by alkyl chains and bearing an alkene group at both ends for macrocyclization by ring closing metathesis (RCM). At first, O-benzyl protected hydroxylamine building blocks (2a-c and 3a-c) were prepared by N-alkylation of N-Boc-O-benzylhydroxylamine with bromoalkyl esters of adequate chain lengths to form compounds 1a-c, followed by Boc removal or ester hydrolysis (Scheme 1). As depicted in Scheme 2, the O-benzylated tetrahydroxamates 8a-c were obtained by appropriate sequence of conjugation via mixed anhydride intermediates formed by using ethyl chloroformate as reported previously,[15] and ester or Boc deprotection. Macrocyclization was performed using the 2nd generation Grubbs catalyst in CH2Cl2. A minimum of 0.3 equivalent of catalyst was necessary to ensure complete conversion, and a maximum diene concentration of 1.2 mM was found to be optimal to avoid detectable formation of dimer (from the reaction mixture analyzed by mass spectrometry). After purification, the protected macrocyclic tetrahydroxamates 9a-c were obtained as Z/E mixtures with yields of 52-58% (Scheme 3). Finally, hydrogenation allowed for the simultaneous reduction of the resulting alkene and benzyl removal, affording the macrocyclic chelators C5, C6 and C7. In parallel, compounds 8a-c were directly hydrogenated to form the open chain tetrahydroxamic acid analogues L5, L6 and L7, their evaluation being essential to assess the impact of the macrocyclic effect on the stabilization of the ZrIV complexes. Detailed synthetic procedures are available in supporting information.
Scheme 1.
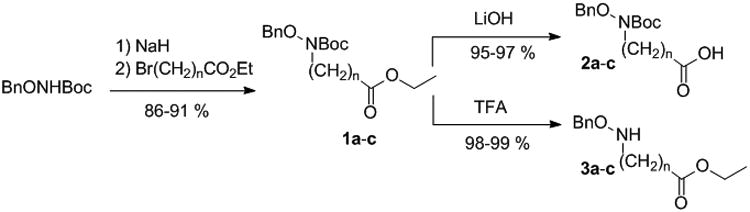
Preparation of the O-benzyl protected hydroxylamine building blocks 2a-c and 3a-c (with 1a: n = 5, 1b: n = 6 and 1c: n = 7).
Scheme 2.
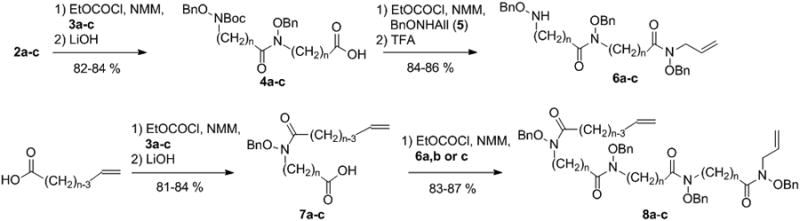
Preparation of the O-benzyl tetrahydroxamate precursors (NMM = N-methylmorpholine; TFA = trifluoroacetic acid).
Scheme 3.
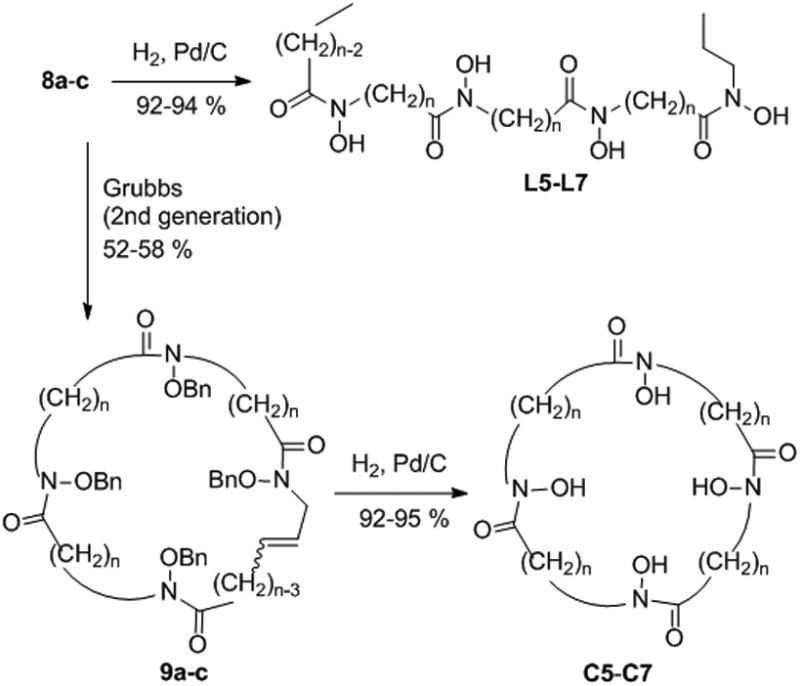
Formation of the cyclic and acyclic tetrahydroxamic acid chelators.
Little data is available on the aqueous stability and structural characterization of chelators complexed to ZrIV. For instance, even in the case of the Zr-DFB complex widely used in nuclear medicine, no thermodynamic or crystallographic data is available. The difficulty in obtaining such data is often related to solubility issues, and the complexes of the present study were no exception. When preparation of the complexes was attempted by ligand exchange from ZrIV acetylacetonate with the newly synthesized chelators, it was not possible to dissolve the product of reaction after isolation, precluding solution analysis or crystallization. Nonetheless, mass spectrometry analyses of the reaction solutions before isolation of the complexes were performed. It was thereby possible to confirm the formation of the expected ZrIV complexes with C7, L7 and L6. The absence of detection of the complexes with C6, C5 and L5 may be related to the lower stability of these smaller complexes in the conditions of the analysis. However, the FT-IR study showed the disappearance of NO-H stretching of the free ligands at ∼3170 cm-1 upon complexation as well as a red shift of the carbonyl stretching of the free ligands from ∼1600 cm-1 to ∼1585 cm-1 in all the complexes (Example with C7 in Figure 2). Additionally, ICP-EOS analyses of the isolated complexes confirmed the correct ratio of zirconium in the complexes.
Figure 2.

FT-IR spectra of free ligand C7 and its ZrIV complex.
Due to the high lipophilicity of the Zr complexes, precipitation issues prevented determination of the stability constants in aqueous solution or water/organic mixture using conventional methodologies such as potentiometry or spectrophotometry. Consequently, the comparison of their relative stability was performed at tracer concentrations with 89Zr as well as with quantum chemical calculations as discussed in the next sections.
Complexation of 89Zr and stability study
To assess the complexation ability of these new ligands at trace concentration of cation, a radiolabeling study was performed with 89Zr in aqueous solution. For this, the ligands were incubated with [89Zr] ZrIV oxalate at pH 7, at various temperatures and times, and analyzed by chromatography, with DFB included for comparison. DFB appeared to be the most rapid for complexing 89Zr, with a quantitative radiochemical yield (RCY) after 30 min at 20 °C. Although exhibiting slightly slower kinetics, the larger ligands C7, L7 and L6 provided excellent complexation abilities (> 99% complexation after 120 min at 20 °C). In contrast, the smaller ligands appeared less adequate with decreasing size of cavity, with higher temperatures required to obtain high complexation yields, and the smaller macrocycle C5 reaching only 29% even at 80 °C (Table 1). In the case of the smaller ligands, the actual RCY may be underestimated because of possible decomplexation during the conditions of analysis that consisted in chromatography using a 50 mM EDTA solution as eluent. In this system, EDTA is present in large excess and complexes rapidly the free 89Zr, forming a complex that is separated from the other 89Zr chelates. However transchelation can also occur with weak complexes during the elution, resulting in an underestimated RCY (see details in experimental section). Overall, L6, L7 and C7 appear to be the best ligands in the perspective of radiolabeling proteins, with nearly quantitative complexation yields obtained in 2 hours or less at 20 °C. The lower kinetics of complexation observed compared to DFB would probably be appropriate when considering labeling of proteins given the relatively long half-life of 89Zr that permits a comfortable timescale for radiochemical procedures.
Table 1.
Influence of time and temperature on the complexation yield of 89Zr by L5, L6, L7, C5, C6 and C7 and DFB.
| Ligand | 20 °C | 50 °C | 80 °C | |
|---|---|---|---|---|
|
| ||||
| 30 min | 120 min | 30 min | ||
| L5 | 60% | 71% | 73% | 87.3% |
| C5 | 17% | 22% | 23% | 29% |
| L6 | 82% | 95% | 96% | > 99% |
| C6 | 77% | 79% | 92% | 92% |
| L7 | 88% | >99% | > 99% | > 99% |
| C7 | 92% | > 99% | > 99% | > 99% |
| DFB | > 99% | > 99% | > 99% | > 99% |
Next, the kinetic inertness of these complexes was assessed in various media. The mechanisms by which 89Zr is released from DFB in vivo have never been described and remain hypothetical This is why we chose to assess the stability of these complexes in various conditions simulating environments likely to occur in vivo or during their preparation and storage. When incubated for 7 days in phosphate buffer (pH = 7.4) at 20 °C, or in human serum at 37 °C, virtually no release of 89Zr was detected for all complexes (stability > 99 %), similarly to 89Zr-DFB. However, when incubation was performed in phosphate buffer at pH 6.5 (a pH value that is representative of many tumoral environments) and 37 °C, significant decomplexation was observed for the smaller ligands L5, C5 and C6, while the larger ligands, including DFB showed very limited decomplexation with more than 97% of intact complex after 7 days (Table 2). Consequently, a more challenging test was necessary to observe significant release of 89Zr from the most stable complexes and be able to distinguish their respective stabilities. EDTA has been reported to form thermodynamically stable complexes with ZrIV with a Log K = 29,[16] but it exhibits insufficient kinetic inertness in vivo for medical use. For this reason, a 50 mM EDTA solution at pH = 7 was used to incubate the complexes at 37 °C, this concentration corresponding to a large excess of competing chelator compared to the tetrahydroxamate ligands (approximately a 1750 fold excess of EDTA). This test was chosen to simulate the transchelation that might occur to ZrIV in vivo while its complex is used in high dilution and exposed to endogen natural competing chelators. Over a week, varying levels of decomplexation were observed. While fast release of 89Zr occurred with C5 and L5 within the first minutes of incubation, it was significantly slower for the other ligands (Figure 3). Stability was higher with the larger tetrahydroxamates, and better results were obtained with the cyclic ligand C7 compared to its linear analogue L7. The inverse tendency was observed for the L5/C5 and L6/C6 pairs, illustrating a detrimental macrocyclic effect for the smaller rings, whereas it apparently becomes beneficial for the 36-member ring C7. Most importantly, greater stabilities than DFB were obtained with 2 of the new ligands (L7 and C7) with the highest kinetic inertness obtained for C7 compared to DFB (87 ± 3% vs. 47 ± 3% of intact complex, respectively, after 7 days), confirming the expected superiority of tetrahydroxamate-ligands of optimal geometric pre-organization.
Table 2.
Stability of the 89Zr-complexes in 0.1 M sodium phosphate buffer at pH 6.5 over 7 days.
| Fraction of intact complex | |||
|---|---|---|---|
|
|
|||
| Ligand | 1 Day | 4 Days | 7 Days |
| L5 | 93% | 89% | 77% |
| C5 | 69% | 46% | 42% |
| L6 | 97% | 94% | 94% |
| C6 | 89% | 79% | 61% |
| L7 | 99% | 98% | 98% |
| C7 | > 99% | 99% | 99% |
| DFB | 99% | 98% | 98% |
Figure 3.

Stability of the 89Zr-complexes in 50 mM EDTA at pH 7 and 37 °C over 7 days (n = 4).
Quantum chemical study
To provide a structural basis for the observed stabilities, quantum chemical calculations were carried out with density functional theory utilizing the M06L functional[17] with the pseudopotential LanL2DZ[18] for the Zr atom and the 6-31+G* basis set for the rest of the atoms as implemented in Gaussian 09 software.[19] To this end, we first calculated ΔG for the complexation reaction of Zr(OH)4 by Me-AHA (N-methyl-acetohydroxamic acid), whose resulting product Zr(Me-AHA)4 X-ray structure and stability constant in H2O (log β = 45.98 at 25°C corresponding to a ΔG of − 62.7 kcal/mol) was previously determined.[12] The calculated ΔG for the complexation (see Equation 1) in the reaction field of water was &minus 56.1 kcal/mol, underestimating the experimental ΔG by 6.6 kcal/mol.[20] The calculated ΔH and TΔS at 298.15 K were −68.9 kcal/mol and −12.8 kcal/mol, respectively, indicating that the reaction is all enthalpy driven.
| (1) |
Having captured ∼90 % of ΔG for the formation of Zr(Me-AHA)4, we used the same approach to further calculate the reaction energetic of Equation 2 with cyclic and acyclic ligands.
| (2) |
Cyclic (C5-C7) and acyclic (L5-L7) complexes of ZrIV were built by using the previously reported X-ray structure of PuIV complexed with the cyclic tris-hydroxamate ligand desferrioxamine E as starting point.[21] Both the energy minimized ZrIV complexes and ligands may not represent a global minimum for the system given the flexibility of each ligand. Nonetheless, the calculated ΔGs in the reaction field of water (Table 3) are all in good agreement with the relative stabilities of the 89Zr complexes observed in Figure 3. It is also noted that all cyclic and acyclic complex formations are both enthalpy and entropy driven.
Table 3.
Gibbs free energy, enthalpy, and entropy contribution (kcal/mol) for the complexation of Zr(OH)4 by the tetrahydroxamic acid ligands (Equation 2) at 298.15 K calculated at the M06L level with the pseudopotential LanL2DZ for the Zr atom and the 6-31+G* basis set for the rest of the atoms[a].
| Ligand | ΔG | ΔH | TΔS |
|---|---|---|---|
| C7 | −66.4 (−71.0) | −42.3 (−45.2) | 24.1 (25.8) |
| L7 | −67.4 (−70.6) | −43.7 (−47.3) | 23.7 (23.3) |
| C6 | −60.0 (−62.2) | −37.1 (−36.7) | 22.9 (25.5) |
| L6 | −66.4 (−68.0) | −43.3 (−44.9) | 23.1 (23.1) |
| C5 | −44.7 (−49.2) | −22.4 (−23.8) | 22.3 (25.4) |
| L5 | −47.5 (−53.2) | −24.1 (−28.9) | 23.4 (24.3) |
Numbers in parenthesis represent the values calculated in the reaction field of water with the polarizable continuum model (PCM) based on the gas phase optimized geometries.
For example, Zr-C7 is respectively 8.8 kcal/mol and 21.8 kcal/mol more stable than Zr-C6 and Zr-C5 in terms of ΔG calculated in the reaction field of water. This energy difference correlates well with the observation that 87 ± 3% of 89Zr remains complexed to C7 after a week while only 32 ± 4% is still complexed to C6, and nearly none by C5. In terms of kinetics, the release of ∼50 % of 89Zr from C5 and C6 occurred within the first minutes and a single day, respectively. This suggests that ZrIV complexes exhibiting better thermodynamic stabilities also have higher energy barriers for the ZrIV release. The stability differences among the three cyclic complexes are primarily due to a ring strain, as manifested by their ΔH values since the entropy contribution at 298.15 K for the three complexes are all comparable (∼ 23 to 26 kcal/mol). The calculated ring strain energy going from C7 to C6 is 8.8 kcal/mol, and further increases to 13.0 kcal/mol when going from C6 to C5, suggesting that reduction of the macrocycle cavity is detrimental to the stability of the resulting ZrIV complex.
The observed stability with the acyclic complexes also correlates well with their calculated ΔGs. A notable difference is that the ring strain energy going from L7 to L6 is only 2.6 kcal/mol as compared to the C7 to C6 transition being 8.8 kcal/mol. While a ring opening alleviates the ring strain of the L6 complex, it does not overcome the ring strain in L5 (14.8 kcal/mol for the L6 to L5 transition).
As to the relative stability of Zr-C7 over Zr-L7 observed after a week, the calculated ΔG difference between the two is only 0.4 kcal/mol, suggesting this stability difference rather to be related to kinetics. As shown in Figure 4, the two open alkyl chains in Zr-L7 can fluctuate more than the alkyl chains in the more rigid Zr-C7. This fluctuation is likely to lead to the weakening of the Zr-O bonds, resulting in a lower energy barrier for the ZrIV release from L7. On the other hand, since the L6 complex is more stable than the C6 analogue by 5.8 kcal/mol, the observed higher stability of Zr-L6 over Zr-C6 can be attributed to thermodynamics. Whereas better thermodynamic stability does not necessarily achieve higher kinetic inertness, the present thermochemical calculations indicate that such a correlation exists in the smaller cyclic and acyclic ligands complexed with ZrIV studied here. On the other hand, when similar thermodynamic stabilities are achieved with L7 and C7, a beneficial macrocyclic effect on the kinetic inertness of C7 was observed.
Figure 4.
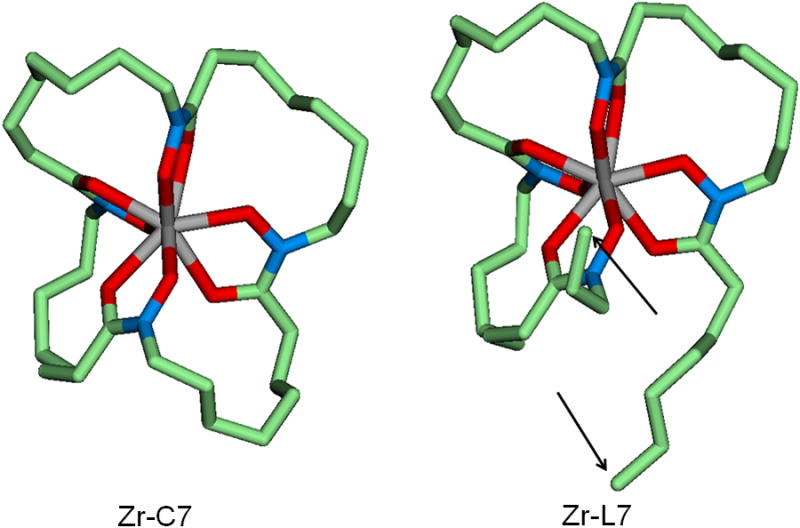
Geometry optimized cyclic Zr-C7 and acyclic Zr-L7 complexes. The arrows indicate the two open alkyl chains. Carbon in green, oxygen in red, nitrogen in blue and central zirconium in grey color (hydrogen atoms not shown). Average NO-Zr bond length is 2.169 Å in Zr-L7 and 2.167 Å in Zr-C7 and average CO-Zr bond length is 2.289 Å in Zr-L7 and 2.296 Å in Zr-C7. These calculated bond lengths are all comparable to those determined by X-ray diffraction of the crystalline Zr(Me-AHA)4 (NO-Zr = 2.183 Å and CO-Zr = 2.204 Å). The coordinates of the complexes and ligands discussed in this paper are all available in supporting information.
Overall, these calculations as well as the stability study, illustrate well the fact that the best pre-organization is reached when chains of 7 carbons separate the hydroxamate functions. At this optimal size, kinetic inertness was significantly enhanced with the macrocycle C7 compared to its acyclic analogue L7.
Conclusion
In this study, the rational design of a chelating agent fitted to the ZrIV cation was proposed by using an adequate set of ligands with varying pre-organization. A kinetic inertness evaluation in relatively harsh conditions using EDTA as a strong competing agent provided a classification of these ligands and proved that the use of 4 hydroxamic acids in a single chelator can significantly increase the kinetic inertness of the resulting complex which is essential for in vivo applications of radionuclides. The lack of stability data in aqueous solutions inherent to this kind of complex of low solubility was circumvented by resorting to quantum chemical calculations while using the crystallographic structures of ZrIV and PuIV complexed with hydroxamates as a template for the construction of the present ZrIV complexes. The relative stabilities of the investigated complexes were rationalized in terms of structures and energetics, and the good agreement seen in the present work indicate that quantum chemical calculations can provide an insight for the design of metal-ligand complexes of interest, particularly when aqueous solution studies are not applicable.
Finally, this is to our knowledge the first time complexes of ZrIV are reported which exhibit significantly superior stabilities compared to the reference DFB used in nuclear medicine for the complexation of 89Zr. The modification of the most promising ligands of this study for conjugation to antibodies and in vivo applications is in progress, especially L7 and C7 which exhibited the best stabilities in all conditions tested, as well as efficient complexation abilities of 89Zr at 20 °C.
Experimental Section
General procedure for the preparation of the cold ZrIV complexes
Example with Zr-L7: To zirconium(IV) acetylacetonate (81 mg, 162 μmol) dissolved in 10 mL dry methanol under nitrogen atmosphere was added ligand L7 (113 mg, 171 μmol) dissolved in 10 mL iso-propyl alcohol. The solution was heated for 14 h at 70 °C, resulting in a milky solution with the complex partially precipitated. Analysis of the solution by ESI(+)-MS confirmed the formation of 1:1 (metal:ligand) complex: [M+H]+ = 745.3 (+ Zr isotopic distribution peaks). The solution was evaporated, resulting in a pale yellow powder. Residual zirconium(IV) acetylacetonate and ligand were removed by boiling and filtering the solid successively in methanol and iso-propyl alcohol. After drying in vacuum, 91 mg of pale yellow solid were obtained (76%). A sample was analyzed by ICP-OES for Zr content. %Zrcalc = 12.23%, %Zrfound = 13.29 (within the experimental error limits of the analysis which is 10%).
All complexes were prepared using the same procedure. Only L6, L7 and C7 formed a complex that could be detected by ESI(+)-MS. All complexes had a Zr content within the error range of the ICP-OES analysis method, except for Zr:L6.
Zr-C5, yield = 92%, not detected in ESI(+)-MS. %Zrcalc = 15.11, found = 14.7.
Zr-L5, yield = 94%, not detected in ESI(+)-MS. %Zrcalc = 14.39, found = 13.38.
Zr-C6, yield = 82%, not detected in ESI(+)-MS. %Zrcalc = 13.82, found = 14.14.
Zr-L6, yield = 96%, ESI(+)-MS [M+H]+ = 689.2. %Zrcalc = 13.22, found = 15.11.
Zr-C7, yield = 78%, ESI(+)-MS [M+H]+ = 715.3. %Zrcalc = 12.74, found = 12.59.
89Zr production
89Zr was produced and purified at the National Institutes of Health, Bethesda, MD, USA, by the following procedure: Pressed pellets of yttrium metal (200 mg, 99.99% purity; American Elements, USA) were irradiated with a proton beam of 15 MeV and a current of 20 μA for 2–4 h on a GE PETtrace cyclotron. 89Zr was separated from the yttrium target material by the use of hydroxamate resin as described by Holland et al.[22] Briefly, the target material was dissolved in 4 × 0.5 mL fraction of 6M HCl. After 1 h, the undissolved solid residue was separated by filtration, the resulting solution diluted to 5 mL with de-ionized water and loaded onto the hydroxamate resin column. The column was then washed with 4 × 2.5 mL of 2M HCl and 4 × 2.5 mL de-ionized water. After the solution was removed from the column, the 89Zr was eluted with successive portions of 1M oxalic acid. The first 0.4 mL fraction was discarded and the next 0.7 mL fraction collected for further use.
Complexation study with 89Zr
All solutions described below were prepared with de-ionized water purified through a chelex column prior to use. Stock solutions of 89Zr at pH 7 were prepared as follow: To 450 μL of the 89Zr solution in oxalic acid were added 450 μL of a 1M Na2CO3 solution and the pH was adjusted to 7 by addition of small aliquots of a 0.1 M Na2CO3 or 0.1 M HCl. 450 μL of water containing 3 % bovine serum albumin were then added. To 45 μL of stock solution (∼ 1.3 MBq of 89Zr in a typical experiment) were added 7.5 nmol (5 μL) of L5, L6, L7, C5, C6 or C7 in DMSO or desferrioxamine B (DFB) mesylate in de-ionized water. These solutions were incubated at desired temperatures and time of reaction and analyzed by ITLC-SG using a 50 mM EDTA (ethylenediaminetetraacetic acid) solution adjusted to pH = 7 in de-ionized water as eluant, and analyzed with a Typhoon 8600 scanner (GE Healthcare) in phosphorimaging mode. The percentage of the activity bound to the ligand after TLC was calculated by converting the TLC scan into a chromatogram and integrating the peak corresponding to the spot at the bottom of the TLC. Activity ratios were averaged out of a minimum of 2 TLCs for each condition. A selection of representative TLCs is displayed in supporting information. For stability studies described below, higher activities were used with ∼ 50 MBq 89Zr reacted with 38 nmol of ligand. The specific activity achieved was 0.96 MBq/nmol with L5, 0.30 MBq/nmol with C5, 1.26 MBq/nmol with L6, 1.21 MBq/nmol with C6, and 1.31 MBq/nmol with L7, C7, and DFB.
In vitro stability of the [89Zr]-zirconium(IV) complexes
10 μL of each 89Zr complex solution prepared as described above were incubated for 7 days in 40 μL Phosphate-Buffered Saline (pH 7.4-10% DMSO) at 20 °C, 40 μL sodium phosphate buffer (0.1 M, pH 6.5- 10% DMSO) at 37 °C, 40 μL human serum at 37°C, or 40 μL 50 mM EDTA (pH 7, 10% DMSO) at 37 °C, respectively, and analyzed using the chromatographic system described in the above section after 1 h, 3 h, 1, 2, 3, 4, 5, 6 and 7 days.
Quantum Chemical Study
The X-ray structure of PuIV complexed with DFE reported previously[21] was used as a template to build the ZrIV-C7 complex. After the Zr subsitution for Pu, each amide group in the ring was converted to (-CH2CH2-) and then a fourth hydroxamate group with a 9-carbon alkyl chain on each end was inserted. After the geometry optimization at the level of B3LYP/LanL2DZ, a CH2CH2 moiety in each ring were deleted, and the resulting ZrIV-C7 complex was further optimized in the gaseous phase at the level of M06L with the pseudopotential LanL2DZ for the Zr atom and the 6-31+G* basis set for the rest of the atoms. The acyclic ZrIV-L7 complex was constructed by modifying the optimized ZrIV-C7 complex, and then energy minimized. Starting with these two structures, the rest of the cyclic and acyclic complexes were built successively by deleting a CH2 moiety in each ring in their respective ge parenthesis represent the values ate the ΔG and ΔH for complexation reaction, each ligand was constructed from its corresponding geometry optimized complex by deleting ZrIV, and then protonating each of the 4 hydroxamates. The geometries of these ligands were optimized at the level of M06L/6-31+G*. In order to approximate the solvent effect onto the energetics, single point energy calculations were also done in the reaction field of H2O with the PCM model as implemented in Gausssian 09 software.[19] All calculations have utilized the 5-component d functions, and the coordinates of the ligands and complexes are listed in the supporting information.
Supplementary Material
Acknowledgments
This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research and the Center for Information Technology. The quantum chemical study utilized PC/LINUX clusters at the Center for Molecular Modeling of the NIH (http://cit.nih.gov). The authors are grateful to Dr. L.P. Szajek at the PET department for providing 89Zr.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemeurj.org/ or from the author.
References
- 1.For review see: Tredwell M, Gouverneur V. Angew Chem Int Ed. 2012;51:11426–11437. doi: 10.1002/anie.201204687.Li Z, Conti PS. Adv Drug Deliv Rev. 2010;62:1031–1051. doi: 10.1016/j.addr.2010.09.007.
- 2.For review see: Wadas TJ, Wong EH, Weisman GR, Anderson CJ. Chem Rev. 2010;110:2858–2902. doi: 10.1021/cr900325h.Zhou Y, Baidoo KE, Brechbiel MW. Adv Drug Deliv Rev. 2013;65:1098–1111. doi: 10.1016/j.addr.2012.10.012.Nayak TK, Brechbiel MW. Bioconjugate Chem. 2009;20:825–841. doi: 10.1021/bc800299f.
- 3.Fischer G, Seibold U, Schirrmacher R, Wängler B, Wängler C. Molecules. 2013;18:6469–6490. doi: 10.3390/molecules18066469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Codd R. Coord Chem Rev. 2008;252:1387–1408. [Google Scholar]
- 5.Vosjan MJWD, Perk LR, Visser GWM, Budde M, Jurek P, Kiefer GE, van Dongen GAMS. Nat Protoc. 2010;5:739–743. doi: 10.1038/nprot.2010.13. [DOI] [PubMed] [Google Scholar]
- 6.Zeglis BM, Mohindra P, Weissmann GI, Divilov V, Hilderbrand SA, Weissleder R, Lewis JS. Bioconjugate Chem. 2011;22:2048–2059. doi: 10.1021/bc200288d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a Deri MA, Zeglis BM, Francesconi LC, Lewis JS. Nucl Med Biol. 2013;40:3–14. doi: 10.1016/j.nucmedbio.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Holland JP, Evans MJ, Rice SL, Wongvipat J, Sawyers CL, Lewis JS. Nat Med. 2012;18:1586–1591. doi: 10.1038/nm.2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Nayak TK, Garmestani K, Milenic DE, Brechbiel MW. J Nucl Med. 2012;53:113–120. doi: 10.2967/jnumed.111.094169. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Abou DS, Ku T, Smith-Jones PM. Nucl Med Biol. 2011;38:675–681. doi: 10.1016/j.nucmedbio.2010.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perk LR, Vosjan MJWD, Visser GWM, Budde M, Jurek P, Kiefer GE, van Dongen GAMS. Eur J Nucl Med Mol Imaging. 2010;37:250–259. doi: 10.1007/s00259-009-1263-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tinianow JN, Gill HS, Ogasawara A, Flores JE, Vanderbilt AN, Luis E, Vandlen R, Darwish M, Junutula JR, Williams SP, et al. Nucl Med Biol. 2010;37:289–297. doi: 10.1016/j.nucmedbio.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 11.Price EW, Zeglis BM, Lewis JS, Adam MJ, Orvig C. Dalton Trans. 2013;43:119–131. doi: 10.1039/c3dt51940f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guérard F, Lee YS, Tripier R, Szajek LP, Deschamps JR, Brechbiel MW. Chem Commun. 2013;49:1002–1004. doi: 10.1039/c2cc37549d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cram DJ. Angew Chem Int Ed Engl. 1986;25:1039–1057. [Google Scholar]
- 14.Liu S, Edwards DS. Bioconjugate Chem. 2001;12:7–34. doi: 10.1021/bc000070v. [DOI] [PubMed] [Google Scholar]
- 15.Reddy AS, Kumar MS, Reddy GR. Tetrahedron Lett. 2000;41:6285–6288. [Google Scholar]
- 16.Prášilová J, Havlíček J. J Inorg Nucl Chem. 1970;32:953–960. [Google Scholar]
- 17.Zhao Y, Truhlar DG. J Chem Phys. 2006;125:194101–18. doi: 10.1063/1.2370993. 194101. [DOI] [PubMed] [Google Scholar]
- 18.Wadt WR, Hay PJ. J Chem Phys. 1985;82:284–298. [Google Scholar]
- 19.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, revision A.02. Gaussian Inc.; Wallingford CT: 2009. [Google Scholar]
- 20.In this model, Zr(OH)4 can be considered as the predominant form of ZrIV in water at pH 7 in the absence of strong ligand at high dilution, as reported previously. See: Veyland A, Dupont L, Pierrard J, Rimbault J, Aplincourt M. Eur J Inorg Chem. 1998:1765–1770.
- 21.Neu MP, Matonic JH, Ruggiero CE, Scott BL. Angew Chem Int Ed. 2000;39:1442–1444. doi: 10.1002/(sici)1521-3773(20000417)39:8<1442::aid-anie1442>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 22.Holland JP, Sheh Y, Lewis JS. Nucl Med Biol. 2009;36:729–739. doi: 10.1016/j.nucmedbio.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.