

Summary
N-hydroxyaristolactams, stable products of the partial reduction of aristolochic acids, serve as substrates for several sulfotransferases. In turn, the sulfated intermediate reacts with DNA to form aristolactam–DNA adducts. NAD(P)H:quinone oxidoreductase-1-mediated reduction of aristolochic acids is facilitated by SULT1B1.
Abstract
Aristolochic acids are potent human carcinogens; the role of phase II metabolism in their bioactivation is unclear. Accordingly, we tested the ability of the partially reduced metabolites, N-hydroxyaristolactams (AL-NOHs), and their N-O-sulfonated and N-O-acetylated derivatives to react with DNA to form aristolactam–DNA adducts. AL-NOHs displayed little or no activity in this regard while the sulfo- and acetyl compounds readily form DNA adducts, as detected by 32P-post-labeling analysis. Mouse hepatic and renal cytosols stimulated binding of AL-NOHs to DNA in the presence of adenosine 3'-phosphate 5'-phosphosulfate (PAPS) but not of acetyl-CoA. Using Time of Flight liquid chromatography/mass spectrometry, N-hydroxyaristolactam I formed the sulfated compound in the presence of PAPS and certain human sulfotransferases, SULT1B1 >>> SULT1A2 > SULT1A1 >>> SULT1A3. The same pattern of SULT reactivity was observed when N-hydroxyaristolactam I was incubated with these enzymes and PAPS and the reaction was monitored by formation of aristolactam–DNA adducts. In the presence of human NAD(P)H:quinone oxidoreductase, the ability of aristolochic acid I to bind DNA covalently was increased significantly by addition of PAPS and SULT1B1. We conclude from these studies that AL-NOHs, formed following partial nitroreduction of aristolochic acids, serve as substrates for SULT1B1, producing N-sulfated esters, which, in turn, are converted to highly active species that react with DNA and, potentially, cellular proteins, resulting in the genotoxicity and nephrotoxicity associated with ingestion of aristolochic acids by humans.
Introduction
Aristolochic acids (AAs) are naturally occurring nitropolyaromatic compounds found in Aristolochia plants, which are used as herbal remedies in countries throughout the world (1). Serious toxic effects including progressive renal fibrosis and cancer have been associated with prolonged intake of Aristolochia herbs (2). A similar nephropathy affects residents of rural villages in the Danube river basin where ingestion of bread prepared with flour contaminated with Aristolochia clematitis proved to be the causative agent of the so-called Balkan endemic nephropathy (3,4). Both syndromes are termed aristolochic acid nephropathy, which now is recognized as a global disease (5). A striking feature of long-term exposure to AAs is the development of otherwise rare carcinomas of the upper urinary tract in approximately half of the cases of Balkan endemic nephropathy (6). The principal toxic components of Aristolochia species are aristolochic acid I, AA-I, and its 8-demethoxylated form, AA-II (Figure 1) (7). Both compounds are carcinogenic; however, in rodents, only AA-I induces nephrotoxicity (8,9).
Fig. 1.
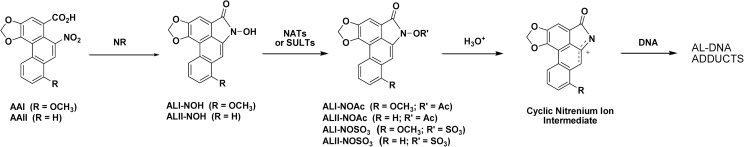
Proposed route for bioactivation of AAs. AA-I and AA-II undergo four electron nitroreduction to form AL-I-NOH and AL-II-NOH followed by N-acetylation or N-O-sulfonation catalyzed by NATs and SULTs, respectively. AL-N-oxyesters (AL-I-N-OAc, AL-II-N-OAc, AL-I-N-OSO3H and AL-II-N-OSO3H) solvolize in aqueous solution, leading to cyclic nitrenium ion formation, which, in turn, reacts with DNA to form AL-DNA adducts.
Following metabolic activation, AA-I and AA-II react with DNA to form covalent aristolactam (AL)-DNA adducts (10). The deoxyguanosine and deoxyadenosine adducts, dG-AL and dA-AL, are mutagenic and block DNA replication (11,12). In human tumors, 7-(deoxyadenosin-N6-yl)-aristolactam I (dA-AL-I) adducts induce A:T transversions on the non-transcribed strand of the TP53 gene, thereby serving as biomarkers of exposure to AAs and reflecting their role in the carcinogenicity of AAs (4,13,14).
Nitroreduction is necessary for the formation of reactive intermediates of AAs (Figure 1) (15). It has been proposed that an intermediate containing the reactive, delocalized nitrenium ion (Figure 1) is the direct precursor of AL-adducts in DNA (15). In the case of analogous nitroaromatic compounds, such as 3-nitrobenzanthrone and its derivatives, acetylation or sulfonation of reduced metabolites increases their electrophilic properties and reactivity with cellular nucleophiles (16,17). The cyclic aristolactam–nitrenium-ion intermediate is proposed to arise from a reduced metabolite of AA, N-hydroxyaristolactam (AL-NOH), the aristolactam-N-acetoxy ester (AL-N-OAc) or aristolactam-N-sulfate (AL-N-OSO3H) (Figure 1).
Several mammalian enzymes capable of nitroreduction are reported to be associated, some only under hypoxic conditions, with the formation of DNA adducts in vitro. These include NAD(P)H:quinone oxidoreductase 1 (NQO1), xanthine oxidase, prostaglandin H synthase, NADPH:CYP reductase and CYP1A1/2 (18). Recently, AA-I was shown to increase expression of mouse NQO1 protein in liver and kidney (19). Mice treated with dicoumarol, an inhibitor of NQO1, exhibited attenuated nephrotoxocity and higher levels of the non-toxic demethoxylated metabolite, AA-Ia, compared with other reduced metabolites in urine (20). Thus, NQO1, with other enzymes as backup, has been considered to be the main cytosolic enzyme involved in AA-I bioactivation.
Formation of aristolactam-N-oxyesters represents a plausible pathway to increase reactivity of nitroreduction intermediates of AAs (Figure 1). However, published data regarding the potential involvement of phase II metabolites in AAs toxicity have been conflicting. In humans, 13 sulfotransferases (SULTs) (21,22) and two N-acetyltransferases (NAT1 and NAT2) have been described(23). These enzymes catalyze the transfer of sulfo and acetoxy groups from 3'-phosphoadenosine-5'-phosphosulfate (PAPS) and acetyl-CoA, respectively. The increased mutagenicity of AA-I has been described in bacterial and mammalian cells harboring human SULTs, SULT1A1 or SULT1B1 (24). In contrast to these findings, Stiborova et al., using somewhat different methods, reported that SULT1A enzymes do ‘not’ stimulate reactivity of AAs with DNA in the presence of NQO1 (25).
Our studies are designed to resolve this apparent discrepancy. We provide evidence of the direct involvement of SULTs in converting AL-NOHs into forms that bind efficiently to DNA. In addition, these studies demonstrate that sulfonation following nitroreduction further increases the mutagenic and cytotoxic potential of AAs.
Materials and methods
Ethics statement
Animal protocols were reviewed and approved by the Stony Brook Institutional Animal Care and Use Committee.
Chemicals
γ-32P-ATP (6000 Ci/mmol) was obtained from PerkinElmer (Boston, MA). liquid chromatography/mass spectrometry (LC/MS) grade acetonitrile and aqueous ammonium hydroxide (28%) were purchased from Fisher Scientific. AA-I, AA-II, N-hydroxyaristolactam I (AL-I-NOH) and N-hydroxyaristolactam II (AL-II-NOH) and its sulfated and acetylated analogs, with a purity of >97%, were synthesized in our laboratory (Attaluri, unpublished data). AAs and their metabolites were dissolved, at 5–40mM, in dimethyl sulfoxide and stored at −20°C. Concentrations of AAs were established by UV absorption at 250 nm (26). Enzymes used for 32P-post-labeling analysis were obtained from Worthington (Newark, NJ) and Sigma–Aldrich (St Louis, MO). Zinc powder, dimethyl sulfoxide, salmon sperm DNA (ssDNA), PAPS (>60% purity) and acetyl-CoA were purchased from Sigma–Aldrich. 7-(deoxyguanosin-N2-yl)-aristolactam II (dG-AL-II) and dA-AL-II containing oligonucleotides were synthesized as described earlier (12).
Human histidine-tagged SULT1A1, SULT1A2 and SULT1A3, expressed in Escherichia coli and purified with the specific activity of 15 pmoles/min/μg, as defined by transfer of sulfonate groups from PAPS to 1-naphtol, were purchased from US Biological (Swampscott, MA). Recombinant human SULT1B1 was purchased from MyBioSource (San Diego, CA). Cytosols from insect cells infected with NAT1 and NAT2 baculovirus expressing vectors were obtained from BD Biosciences (Woburn, MA). Human NQO1 was purchased from Sigma–Aldrich.
Stability of AA-I metabolites
AL-I-NOH, aristolactam-I-N-acetoxy ester (AL-I-N-OAc) and aristolactam-I-N-sulfate (AL-I-N-OSO3H) (50 μM) were prepared in 50mM Tris-HCl buffer (pH 7.5) and incubated for various periods of time at 37°C. Aliquots (100 µL) were withdrawn from the reaction at intervals for analysis on a Waters high-performance liquid chromatography (HPLC) system equipped with an XTerra™ MS C18 column (5 µm, 4.6 × 250mm), a 2996 Photodiode Array detector and Waters Empower software. Samples were eluted over 40min at 1ml/min with a linear gradient starting at 20% solvent B and 80% solvent A and ending with 100% solvent B. Solvent A was 5mM ammonium acetate dissolved in a 1:9 acetonitrile/water mixture; solvent B was 5mM ammonium acetate in 9:1 acetonitrile/water. A chromatogram for each injection was produced by monitoring UV absorption at 254nm, and peak areas used to determine the concentration of each compound.
Cell culture and chemical exposures
The GM00637 human fibroblast cell line was obtained from the Coriell Institute for Medical Research in Camden, NJ. Authenticated cells were grown in 75cm2 flasks as described previously (27). Briefly, cells were passed in a 1:6 ratio and 0.5ml of the culture media maintained on 24-well plates for cytotoxicity assays. Also, 5ml of the culture media was grown to confluence on 60mm plates. Before treatment with the DNA-damaging agents, cultures were washed thoroughly with Dulbecco’s phosphate buffered saline without CaCl2 and MgCl2. Then, 1–50 µM of AAs or their derivatives, diluted in Dulbecco's modified Eagle's medium without the aforementioned supplements, were added and the cell culture incubated under standard conditions. Cells were treated with the chemicals for 24 h for adduct analyses and for 48 h for cytotoxicity studies.
Cell viability assay
Cell viability assay were performed as described previously (27). Briefly, cells, distributed in 24-well plates, were treated with various compounds for 48h, then washed with phosphate buffered saline and lysed. Cytotoxicity was defined as the ratio of adenosine triphosphate in treated cells to adenosine triphosphate in the untreated control. Three different wells were used for each exposure.
Mouse hepatic and renal cytosolic extracts
Four, 8-week-old male C3H/HeJ mice were killed by CO2 asphyxiation. Liver tissues and cortices from both kidneys, total wet weight for each tissue, 933mg, were homogenized in a Dounce homogenizer with 10 strokes of pestle A and 20 strokes of pestle B in 3ml of ice-cold 50mM Tris-HCl (pH 7.6). The homogenates were centrifuged at 150 000g for 40min. Cytosolic preparations were aliquoted and stored at −80°C. The protein content was analyzed by Bradford assay (28), using bovine serum albumin as the standard.
Incubations of AAs and metabolites with DNA
ssDNA (30 μg) in a final volume of 200 μl was incubated with 2 μM of each of the following: AA-I, AA-II, AL-I-NOH, AL-II-NOH and AL-N-oxyesters, dissolved in 50mM KPi buffer, pH 5.8, in the presence or absence of 1mg of zinc powder included in the reaction mixtures as a reducing agent. For dose response studies, incubations were for 2h with 0.1–20 μM of various ALs. Following the reaction, DNA was precipitated by 70% ethanol, resuspended in 0.1×TE buffer and stored at −20°C prior to adduct analysis (see below). In experiments with AAs, 5 μg DNA was subjected to analysis while 1 μg DNA was used for AL-N-oxyesters.
Cytosolic incubations
Incubations with cytosols, in a final volume of 500 μl, consisted of 50mM Tris-HCl pH 7.5, 0.2% Tween 20, 0.2mM PAPS or 1mM acetyl-CoA as cofactors for cytosolic SULTs or NATs, respectively, 0.4mM AL-I-NOH or AL-II-NOH, 0.5mg of mouse hepatic or renal cortex cytosolic proteins and 0.4mg of ssDNA. Reactions were initiated by adding the cofactors. Incubations were carried out at 37°C for 1–8h. Under these conditions, DNA adduct formation was linear up to 6 h. Control incubations were conducted without cytosols, cofactors and AL-NOHs. Samples (80–100 μl) were collected at each time point, mixed with 200 μl water and extracted three times with 300 μl phenol-chloroform-isoamyl alcohol mixture (Sigma). Following the extraction step, DNA was precipitated with three volumes of ethanol and diluted in 150 ul 0.1×TE buffer. DNA was stored at −20°C prior to adduct analysis. Twenty micrograms of DNA was analyzed for AL-DNA adducts, as described below.
Activation of AL-NOHs by SULTs and NATs
Incubation mixtures, in a final volume of 500 μl, consisted of 0.4mg of ssDNA in 50mM Tris-HCl pH 7.5, 0.2% Tween 20, 0.1mM AL-I-NOH or AL-II-NOH, 10–80nM of SULTs or 40 μg of cytosolic extracts from NAT1- or NAT2-infected insect cells. Reactions, initiated by adding 0.2mM PAPS or 1mM acetyl-CoA, were incubated at 37°C for 1–8h. Control incubations were conducted without AL-NOHs, without cofactors, or without enzyme. DNA was collected and stored as described above. Five micrograms of DNA were used for adduct analysis.
Time of Flight LC/MS kinetic study of SULTs
AL-I-NOH (0.5–100 µM) was incubated in 250 µL Tris/Tween buffer (pH 7.5) at 37°C with 0.2mM PAPS and either 30nM SULT1B1, 160nM SULT1A1, 160nM SULT1A2 or 160nM SULT1A3. Reactions were initiated by the addition of PAPS. Aliquots (5 µL) of the mixture were analyzed at various times for AL-I-N-OSO3H formation using an Agilent Technologies 6224 Time of Flight LC/MS system with an electrospray ion source interfaced to a Series 1200 HPLC system. An Agilent Extend C18 column (1.8 µm, 2.1 × 50mm) was utilized for LC with an isocratic mobile phase (200 µL/min) consisting of 1:1 acetonitrile: water containing 0.01% NH4OH. Data were acquired in the negative ion mode over the mass range of 150–1500Da. Mass chromatograms were created for the mass range 387.8–388.2Da and integrated for quantitative analysis. Peak areas were converted to concentrations using a calibration curve generated under similar conditions. Standards for the calibration curve were prepared by including pure, synthetic AL-I-N-sulfate to the reaction mixture at various concentrations, omitting the addition of AL-I-NOH. Reactions were repeated at least three times for each substrate concentration. Product appearance was linear up to 20min. Area under the peak, corresponding to the standard solution, was integrated and the amount of AL-I-N-sulfate formed was estimated. Kinetic parameters were estimated by fitting curves to the Michaelis–Menten equation in Sigma Plot.
Incubations of AAs or 3-nitrobenzanthrone with DNA and NQO1 in the presence of SULT1 enzymes
Reaction mixtures (400 μl) consisted of 0.1mM AA-I, AA-II or 3-nitrobenzanthrone, 0.3mg of calf thymus DNA, 50mM Tris-HCl pH 7.5, 0.2% Tween, 1mM NADPH, 0.2mM PAPS and 500nM NQO1 with or without 500nM SULT1 enzymes. Incubations were carried out at 37°C for 1–6h. Each time point was collected and DNA was extracted as described above. DNA (20 μg) was analyzed for presence of adducts.
32P-post-labeling adduct analysis
DNA adduct levels were measured as described previously (27,29). For each of the following 24mer oligonucleotides, 30–120fmol, corresponding to 2–8 adduct/106 nucleotides in 5 μg DNA, was used as standards.
5'-TCT TCT TCT GTG CXC TCT TCT TCT-3' X = dA-AL-II
5'-TCT TCT TCT GTX CAC TCT TCT TCT-3' X = dG-AL-II
Briefly, DNA (1–20 μg) was digested and the concentration of adducts enriched by butanol extraction (27). AL-DNA adducts were post-labeled with γ-32P-ATP, then loaded on 30% non-denaturating acrylamide gels. After 4 or 12 h, the gel was visualized by phosphorimaging. An Image QuaNT v5.2 (Molecular Dynamics) program was used to estimate the amount of adducts present.
Data analysis
Apparent K m and V max values and the initial velocities of AL-DNA adduct formation were determined using Sigma Plot v8.0 (SPSS).
Results
Comparison of AA-I, AL-I-NOH, AL-I-N-OAc and AL-I-N-OSO3H as precursors of AL-DNA adducts
We monitored the dose response and time course of AL-DNA adduct formation in reactions of AA-I, AL-I-NOH, AL-I-N-OAc and AL-I-N-OSO3H with ssDNA. Following incubation at 37°C, DNA was digested to single nucleotides and/or monoadducts, labeled with γ-32P-ATP and analyzed by polyacrylamide gel electrophoresis. In parallel, oligonucleotides containing dA-AL-II and dG-AL-II were processed as standards. AA-I formed dG-AL-I and dA-AL-I adducts when zinc dust was present in the reaction (Figure 2A, lanes 3–10); however, AL-I-NOH did not form dA or dG adducts in the presence or absence of this reducing agent (Figure 2A, lanes 1 and 2, respectively). In contrast, AL-I-N-acetoxy and AL-I-N-sulfate formed dA and dG adducts at levels more than an order of magnitude greater than AA-I, even in the absence of zinc (Figure 2A, lanes 11–18 and 19–26). Moreover, AL-I adduct levels reached saturation within 15 min (Figure 2B), by which time at least one in 104 nucleotides was modified, corresponding, for 30 μg DNA, to 10 pmol of AL-I adducted nucleosides. The efficiency of AL-DNA adduct formation at saturation, for 400 pmol of activated chemicals, was ~2.5%.
Fig. 2.

Reactivity and toxicity of AA-I and its analogs. (A –C) AA-I, AL-I-NOH, AL-I-N-OAc and AL-I-N-OSO3H were incubated with ssDNA in the presence and absence of zinc, 2mg per reaction, as described in Materials and methods. DNA (2–5 μg) was analyzed for the presence of adducts by 32P-post-labeling analysis. (A) Fragment of a 30% polyacrylamide gel after 32P-labeling of DNA adduct nucleosides. St, mixture of 24mer oligonucleotides (15fmol) containing a single dG-AL-II or dA-AL-II, represented by the upper and lower band, respectively. Each band corresponds to 1 adduct/106 nucleotides for 5 µg DNA. For each DNA digestion, at least three standard mixtures were used. Lanes 1–2, AL-I-NOH and DNA incubated for 6h with and without zinc, respectively; Lanes 3–10, AA-I (2 μM) and DNA, incubated for 1, 2, 4, 6h, respectively. All reactions were conducted in duplicate, digested separately and loaded in wells adjacent to each other. Lanes 11–18, AL-I-N-OAc (2 μM) incubated with DNA for 1–6h in the absence of zinc; Lanes 19–16, AL-I-N-OSO3H (2 μM) incubated for 1–6h in the absence of zinc. (B) Time course of AL-I-DNA adduct formation. All analogs were present at 2 μM. (C) Dose response of AL-I-DNA adduct formation following 2h incubations. Filled circles indicate AL-I-DNA adducts in the presence AA-I and zinc, open circles represent AL-I-DNA adducts in the presence of AL-I-N-OAc and the absence of zinc, filled triangles indicate AL-DNA adducts in the presence of AL-I- N-OSO3H and the absence of zinc. Each point corresponds to mean ± standard deviation and presents at least two independent experiments. (D –E) The GM00637 human fibroblast cell line was treated with various aristolactam analogs, at the concentrations shown, for 48 and 24h, then analyzed for cytotoxicity and genotoxicity, respectively. Results are shown as mean values for two independent experiments; standard deviations are <30%. (D) Cytotoxicity, estimated by measuring adenosine triphosphate content, was determined in human fibroblasts treated with AA-I (filled circle), AL-I-NOH (open circle), AL-I-N-OAc (open triangle) and AL-I-N-OSO3H (filled triangle). (E) AL-I-DNA adduct levels in cells treated with 2 and 5 μM of the compounds.
Similar effects were observed when DNA was permitted to react for 2 h with various ALs (Figure 2C). Acetoxy and sulfate derivatives of AL-I-NOH produced higher adduct levels than AA-I in the presence of zinc, and AL-I-DNA adduct reached saturation when these precursors were present at 10 μM.
A similar reactivity pattern was observed for AA-II, AL-II-NOH and aristolactam-II-N-acetoxy ester (AL-II-N-OAc) incubated with DNA under identical experimental conditions (Supplementary Figure S1, available at Carcinogenesis online). Overall, in reactions with DNA, AA-II produced more DNA adducts than AA-I, whereas the N-substituted AL-I derivatives formed more adducts than the N-acetoxy AL-II derivative.
To assess the relative toxicity of AAs and analogs for cells in culture, human fibroblasts were treated with the compounds. The GM00637 fibroblast cell line has been used previously in our study of pathways engaged in the repair of AL-DNA adducts (27). Based on structure–function studies and our unpublished observations, we postulate that renal cytotoxicity reflects two independent cellular mechanisms, one of which is independent of DNA damage. Thus, GM00637 fibroblasts serve as a general model for overall cytotoxicity of AAs. Bioactivated products of nitroreduction, including N-O- conjugates, were expected to be more cytotoxic and to form more AL-DNA adducts in comparison with simple enzymatic nitroreduction of AA. AL-NOHs and the corresponding AL-N-oxyesters displayed high levels of cytotoxicity as determined by cell survival (Figure 2D and Supplementary Figure S2A, available at Carcinogenesis online). The IC 50 for AA-I was 30 μM, 6-fold greater than that for the AL compounds. Following 48h exposure, AA-II was not cytotoxic at the highest concentration used (50 μM). In contrast, under similar experimental conditions, AL-II-NOH and AL-II-N-OAc exhibited significant cellular toxicity (Supplementary Figure S2A, available at Carcinogenesis online).
AL-I-DNA adduct levels in cells were used as a measure of genotoxicity (Figure 2E). A 24h exposure was chosen to avoid depletion of cells containing high levels of adducts. The highest level of AL-I-DNA adducts, more than two orders of magnitudes greater than for AA-I-treated cells, was observed in cells treated with AL-I-N-OSO3H. AL-I-NOH and AL-I-N-OAc formed similar quantities of adducts, but at lower levels than AL-I-N-OSO3H. Similar results were obtained for AA-II, AL-II-NOH and AL-II-N-OAc treated cells (Supplementary Figure S2B, available at Carcinogenesis online). Overall, AA-I and its metabolic intermediates caused more toxicity and generated higher levels of DNA adducts in fibroblasts in cell culture than did AA-II. These observations support the importance of phase II metabolism in AAs induced toxicity.
Stability of AA-I metabolites
The stability of AL-I-NOH, AL-I-N-OAc and AL-I-N-OSO3H was assessed by incubating each compound in water or Tris-HCl buffer (pH 7.5) at 37°C and analyzing aliquots of the solution by HPLC at various times. Under these conditions, AL-I-NOH and AL-I-N-OAc remained stable over the time period of the experiment (Supplementary Figure S3, available at Carcinogenesis online); however, AL-I-N-OSO3H decomposed rapidly in water and in buffer with a half-life of 15–20min. The major decomposition products were AL-I-NOH and aristolactam-I, as established by electron ionization and electrospray ionization mass spectrometer analysis (data not shown).
Activation of AL-NOHs by mouse renal and hepatic cytosols
To investigate further the potential activation of AL-NOHs by cellular SULTs and/or NATs, cytosolic fractions prepared from mouse renal cortex or liver were incubated with ssDNA, AL-I-NOH or AL-II-NOH and either PAPS or acetyl-CoA. Figure 3A shows the time course of AL-I-DNA adduct formation following the reaction of AL-I-NOH with ssDNA in the presence of cytosolic fractions and PAPS. DNA adducts were not formed in the absence of cofactors or cytosols (Figure 3A, lanes 1–6). AL-I-adducts were formed in a time dependent manner when DNA, PAPS, one of the cytosolic extracts and AL-I-NOH were present in the reaction mixture (Figure 3A, lanes 7–14). AL-I-NOH and AL-II-NOH stimulated adduct formation over a period of 6h (Figure 3B). The liver cytosolic fraction was at least two orders of magnitudes more proficient than the kidney cytosol in forming adducts from both N-hydroxylactams. Both cytosolic enzymes produced greater levels of adducts from AL-II-NOH than from AL-I-NOH.
Fig. 3.

Murine renal and hepatic cytosols activate AL-I-NOH and AL-II-NOH in the presence of PAPS, leading to AL-DNA adduct formation. 0.8mg/ml of ssDNA was incubated with 300 μM of AL-I-NOH or AL-II-NOH and 1mg/ml of mouse cytosolic extracts in the presence of PAPS in a total volume of 500 μl. DNA was extracted and 20 μg of used for the adduct analysis. (A) Fragment of polyacrylamide gel showing results of 32P-post-labeling analysis; St—Mixture of 24mer oligonucleotides (30fmol) containing a single dG-AL-II or dA-AL-II, represented by the upper and lower band, respectively. Lanes 1–6, the following were incubated for 6h in a reaction buffer; 1—DNA; 2—DNA and AL-I-NOH; 3—DNA, AL-I-NOH and PAPS; 4—DNA, AL-I-NOH and acetyl-CoA; 5—DNA, AL-I-NOH and renal cytosol; 6—DNA, AL-I-NOH and hepatic cytosol; Lanes 7–10, DNA, AL-I-NOH, PAPS and renal cytosol, incubated for 2, 4, 6 and 8h, respectively; Lanes 11–14, DNA, AL-I-NOH, PAPS and hepatic cytosol, incubated for 2, 4, 6 and 8h, respectively. (B) Time dependence of AL-DNA adduct formation. Filled and open circles correspond to renal and hepatic SULTs activities towards AL-I-NOH. Filled and open squares correspond to renal and hepatic SULTs activities towards AL-II-NOH. Results are shown as mean values for three independent experiments; standard deviations are <30%.
Importantly, when PAPS was replaced in the reaction mixture by acetyl-CoA, adducts were not detected in incubations of renal cortex or hepatic cytosol with either AL-I-NOH or AL-II-NOH (data not shown). Also, consistent with previous studies (19), AL-DNA adducts were not found in incubations containing mouse kidney cortex cytosol extracts and AA-I or AA-II with NADPH. Incubation of hepatic cytosols with DNA, NADPH and AAs formed only very small amounts of AL-DNA adducts (data not shown). Addition of PAPS to this reaction did not stimulate DNA adduct formation significantly.
Activation of AL-NOHs by human SULTs
Human SULT1A1, SULT1A2, SULT1A3 or SULT1B1 were incubated with AL-I-NOH or AL-II-NOH in the presence of PAPS and ssDNA, followed by DNA adduct analysis. AL-I-NOH and AL-II-NOH concentrations were set at 100 μM in order to remain above the K M reported for these enzymes. Among the SULTs studied, the most active was SULT1B1. Figure 4A and B show the time course for AL-I- and AL-II-adduct formation for different doses of SULT1B1. Only background levels of DNA adducts were found in control reactions containing AL-NOHs and PAPS, or AL-NOHs and SULTs in the absence of PAPS (data not shown). The time course of AL-adduct appearance for each SULT1B1 dose was fitted to a linear regression and initial rates of adduct formation were calculated (Figure 4C). AL-I was formed more efficiently than the AL-II-adduct for all doses of SULT1B1, with the most pronounced differences observed at the lowest enzyme dose used (10nM). The same approach was used for SULT1A1, SULT1A2 and SULT1A3 (Supplementary Figure S4, available at Carcinogenesis online). Appearance of AL-DNA adducts was monitored over time, and the initial rates were compared with those for the SULT1B1 reaction. Supplementary Table S1, available at Carcinogenesis online, shows that mean values for SULT activities across all doses with an AL-I-NOH substrate were at least an order of magnitude less efficient in comparison with SULT1B1. In contrast to AL-I-NOH, all four SULTs activated AL-II-NOH with similar efficiency, suggesting the importance of a methoxy group at C8 for enzyme–substrate interactions.
Fig. 4.

SULT1B1 activation of AL-I-NOH and AL-II-NOH. ssDNA was incubated with 100 μM of AL-I-NOH or AL-II-NOH and 40 (filled circles), 20 (open circles) and 10 (filled triangles) nM of SULT1B1 in the presence of PAPS. 2–5 μg DNA was used for the adduct analysis. (A) Time course of AL-I-DNA adduct formation. (B) Time course of AL-II-DNA adduct formation. Results are shown as mean values and standard deviations for three independent experiments. (C) Initial rates of AL-I- and AL-II-DNA-adduct formation for each concentration of enzyme.
Since AL-DNA adducts were formed when AL-N-OAc was incubated with DNA, AL-I-NOH or AL-II-NOH was incubated with human cytosolic NATs, NAT1 and NAT2, in the presence of acetyl-CoA. AL-I and AL-II adducts were detected only in prolonged incubations with high levels of NAT2 (Supplementary Figure S5, available at Carcinogenesis online). Thus, efficient bioactivation of AL-NOHs appears to be specific to SULTs, with SULT1B1 being the most active enzyme examined to date.
Kinetic studies of AL-I-NOH sulfonation
To define the reaction product arising from AL-NOHs in the presence of SULTs, reaction mixtures containing AL-I-NOH, PAPS and one of the SULTs were subjected to LC/MS electrospray ionization-Time of Flight analysis. Pure, synthetic AL-I-N-OSO3H was used as a reference compound. HPLC retention time and negative ion electrospray ionization mass spectroscopy were used to confirm enzymatic formation of N-sulfated compound.
SULTs were incubated with 0.5–10 μM AL-I-NOH for 1–30min to establish optimal conditions for kinetic studies. AL-I-N-OSO3H formation was linear up to 20min; thus, a 10min time point was selected in order to quantify product accumulation in the linear range. Figure 5A–C represents Michaelis–Menten kinetics for SULT1B1, SULT1A1 and SULT1A2. No significant activity was found for SULT1A3, which agrees with the data presented in this paper for adduct formation. Despite reports of substrate inhibition in the literature (30), SULT1A1 showed no significant inhibitory effect with high concentrations of AL-I-NOH. SULT1B1 displayed the lowest K m and the highest k cat values, 0.71 μM and 4.2/min, respectively (Figure 5D). SULT1A1 and SULT1A2 showed similar values for apparent K m, but SULT1A2 had a higher turnover rate. The k cat value for SULT1B1 was at least two orders of magnitude greater than those for other enzymes studied.
Fig. 5.

AL-I-NOH sulfonation by human SULTs. AL-I-NOH (0.5–50 μM) was incubated for 1–30min with each of the following enzymes, (A) SULT1B1, (B) SULT1A1 and (C) SULT1A2 in the presence of PAPS. Time course of AL-I-N-OSO3H formation was monitored by Time of Flight LC/MS. Initial rates were calculated using linear regression analysis in Sigma Plot and plotted against dose of AL-I-NOH. Product formation was linear up to 20min. Results are shown as mean values with standard deviations for at least three independent experiments. (D) Kinetic parameters of human SULTs with AL-I-NOH as a substrate. Apparent kinetic parameters were obtained by fitting curves to Michaelis–Menten equation.
Formation of AL-I-DNA adduct in a reaction containing AA-I, NQO1 and SULT1B1
AA-I was incubated with DNA, NADPH, NQO1, PAPS and SULT1B1, and the time dependence of AL-I-adduct formation was monitored. Figure 6A shows the post-labeling gel, where lanes 1–5 represent adduct formation in the presence of NQO1 and lanes 6–10 represent adduct formation in the presence of NQO1 and SULT1B1 at six time points. For a negative control, we replaced SULT1B1 by SULT1A2, which was shown to have no effect on formation of AL-I-DNA adducts in the presence of NQO1 (25). As expected, SULT1A2 did not alter the rate of AL-I-DNA adduct formation in comparison with NQO1 (Figure 6B). However, incorporation of SULT1B1 significantly stimulated formation of AL-I-adducts (Figure 6B). In contrast, for the structurally related carcinogen, 3-nitrobenzanthrone, DNA adduct formation was stimulated by SULT1A2 but not SULT1B1 (Figure 6C). In the case of AA-II, only a 1.5-fold increase of AL-II-adduct accumulation was monitored in incubations of AA-II with DNA, NQO1 and SULT1B1, compared with NQO1 incubations only (Supplementary Figure S6A and B, available at Carcinogenesis online). In the presence of SULT1A2, slight inhibition of AL-II-adduct formation was found (data not shown), consistent with the literature data (31).
Fig. 6.

SULT1B1 stimulates AA-I reactivity with DNA in the presence of NQO1. AA-I or 3-nitrobenzanthrone (100 μM) were incubated with DNA, PAPS, NADPH, 500nM of SULT1 enzymes and/or NQO1. Twenty micrograms of DNA was used for the adduct analysis. (A) Fragment of a 30% polyacrylamide gel following 32P-labeling. dG-AL-II or dA-AL-II (upper and lower band, respectively). Lanes 1–5, 2, 3, 4, 5, 6h incubations of AA-I, NQO1 and DNA, respectively; Lanes 6–10, AA-I, NQO1 and SULT1B1. (B) Time dependence for AL-I-DNA adducts formation. (C) The same experiment using 3-nitrobenzanthrone. Filled circles DNA adducts in the presence of NQO1, open circles represent DNA adducts in the presence of SULT1A2 and NQO1, filled triangles represent DNA adducts in the presence of SULT1B1 and NQO1. Results shown as mean values for at least two independent experiments.
Discussion
In this paper, we investigated the contribution of phase II metabolism to the bioactivation of AAs prior to their reaction with DNA to form mutagenic adducts. Novel findings in this paper include the (i) high reactivity of sulfated and acetylated AL-NOHs with DNA in the absence of enzymes or reducing agents; (ii) conversion of AL-NOHs to DNA-reactive metabolites, catalyzed by human SULTs; and (iii) accelerated formation of DNA adducts catalyzed by SULT1B1, following NQO1-mediated bioactivation of AAs.
Many nitroaromatic compounds share a common metabolic pathway leading to reactive intermediates that form mutagenic adducts with DNA (32). Reduction of the nitro group is the essential first step in the generation of carcinogenic intermediates. O-sulfonylation (33) and O-acetylation (34) increase the reactivity of N-hydroxy metabolites, with solvolytic cleavage generating the reactive species. AL-NOHs have been found in the urine of rodents exposed to AAs (35). For AA-I and AA-II, the N-hydroxy intermediate, formed during reduction to the N-amino-compound, is believed to undergo heterolytic cleavage of the N-O bond, forming a nitrenium/carbenium ion that subsequently reacts with DNA to form covalent adducts (10,15). The charge on the nitrenium ion in ALs often is delocalized and when the charge resides principally on the nitrogen atom, the nitrenium ion will react with carbon atoms in the nucleobases (most often at C8 of purines). When the charge resides principally on carbon, the carbenium ion will preferentially react with exocyclic amino groups. In theory, simple reduction of AAs is capable of producing the corresponding N-hydroxyaristolactams. However, we find these compounds to be stable both as solids and in solution and were unable to generate DNA adducts efficiently in their presence. In the case of AAs, further activation by N-O-sulfonation or N-O-acetylation is required prior to nitrenium ion formation. Therefore, this result requires refinement of the currently proposed activation mechanism for AAs.
Treatments of fibroblasts in culture with AL-NOHs and AL-N-oxyesters led to a significant increase in adduct levels and cellular toxicity, suggesting the importance of nitroreduction and further conjugation. In accord with these results, SULT messenger RNA transcripts have been found in human fibroblasts in culture, with SULT1A1 and SULT1A3 being expressed ubiquitously across epithelial tissues and cell lines (36).
Liver and kidney are target tissues for AAs, as well as being the site of the majority of the metabolic reactions involving AAs. Biotransformation enzymes found in cytosols of these tissues provide insight into the fate of the N-hydroxyaristolactams (37,38). In humans, SULT1A1 proved to be the principal SULT found in hepatic cytosols (39). Significant amounts of SULT1B1, although less than the amount of SULT1A1, were demonstrated in hepatic cytosols( 39). Human kidney predominantly expresses SULT1A1, together with minor quantities of SULT1B1 and SULT1A3 (24,39,40). It appears that only the SULT1A2 gene is transcribed, although the protein formed was difficult to detect in any tissue (36,40). Although tissues of humans and mice differ in the distribution of SULT isoforms, renal and hepatic cytosols from mice were used as the source of enzymes for this preliminary assessment of the activation of AL-NOHs. Recently, we established the mouse as a robust model for studying the nephrotoxic and carcinogenic effects of AA (8). In the presence of PAPS, mouse cytosol fractions catalyzed the conversion of AL-NOHs to DNA-reactive intermediates. These results prompted us to test the ability of selected human SULT isoforms, SULT1A1, SULT1A2, SULT1A3 and SULT1B1, to activate AL-NOHs. Results of 32P-post-labeling DNA adduct analysis and Time of Flight LC/MS were consistent, revealing that SULT1B1 displayed the highest level of AL-NOH activation. Moreover, the efficiency of processing of AL-I-NOH by SULT1B1 was much higher than that reported for the proposed endogenous substrate of this enzyme, xanthurenic acid (41).
Activation of heterocyclic amines has been described for the arylamine NATs, NAT1 and NAT2 (34,42). High levels of both enzymes are found in mouse and human kidney and liver (43,44). However, despite the observed reactivity of synthetic AL-N-O-acetyl compounds with duplex DNA, only minor DNA adduction was observed when AL-NOHs were incubated with DNA, acetyl-CoA and human NAT2. Moreover, adducts were not formed when NAT1 or mouse hepatic and renal cortex cytosols were employed in a similar reaction. Thus, we conclude that arylamine N-acetyltransferases, NAT1 and NAT2, do not play a significant role in AL-DNA adduct formation.
In vitro experiments in which nitroreduction of AAs, catalyzed by NQO1 is coupled with sulfonation, catalyzed by SULT1B1, revealed a significant increase in AL-I–DNA-adduct formation in comparison with nitroreduction of AA-I alone. The increase in AL-DNA adduct levels was much higher for AA-I than for AA-II, consistent with the preferential activation of AL-I-NOH by SULT1B1. This result could not be duplicated in reactions with SULT1A2, nor was there an increase in adduct formation in the presence of NQO1 and SULT1A isoforms, as reported by Stiborova et al. (25). However, many xenobiotics are selectively activated by one or more SULTs (33,45). The lack of specific SULTs in mouse tissues might account for the lower AL-DNA adduct levels observed after addition of PAPS when mouse hepatic cytosols are incubated with AAs, NADPH and DNA.
In humans, AL-DNA adducts generate mutations that play a role in initiating upper urinary tract cancers (4,14,46). SULT1B1 is one of the SULT isoforms detected in human hepatic tissues and is active in transforming AA into a highly reactive species. In principle, the formation of AL-N-O-sulfated metabolites in liver could precede the formation of tumors in the kidney as analogous, relatively unstable compounds can be transported from the liver to the kidney. For example, serum albumin substantially prolongs the half-life of the sulfate ester of the human renal carcinogen, 1-hydroxymethylpyrene (47). Moreover, hepatic secretion of sulfated 1-hydroxymethlpyrene, its transport to kidney and subsequent uptake into proximal tubule cells by human organic anion transporters has been documented, suggesting a pathway that would explain the renal toxicity caused by AAs (48,49). Further studies are required to establish whether the liver or kidney is the site of bioactivation of AAs in mice and in humans and which SULTs are actively involved.
Humans vary in their susceptibility to the toxic effects of AA; consequently, polymorphisms in genes controlling the activities of enzymes may enable the identification of individuals at risk. Polymorphisms of SULT1A1 have been studied with respect to their association with various cancers (50,51). In particular, the SULT1A1*2 allozyme, defined by an Arg213His amino acid substitution, is reported to confer susceptibility to upper urinary tract tumors (52). However, in contrast to SULT1A1*1, this amino acid change results in a decrease of activity towards polyphenols (53). Thus, this polymorphism, in principle, could decrease sulfonation of AA-1a but would not be expected to increase the rate of bioactivation (35,54). However, 17 polymorphisms, including 2 non-synonymous and 12 non-coding nucleotide substitutions with unknown consequences for gene and/or protein function, have been described in the SULT1B1 gene (55). Such polymorphisms could result in variable sensitivity to AA among Taiwanese patients that develop upper urinary tract cancer (14,46).
In conclusion, we have shown that AL-NOHs, stable intermediates produced by the partial enzymatic reduction of AA-I and AA-II, serve as substrates for several SULTs, leading to the preferential activation of AA-I and AA-II by SULT1B1. In turn, these sulfated compounds react with DNA in vitro and in human cells to form mutagenic AL-dA adducts. NQO1-mediated reduction of AAs is facilitated by SULT1B1 but not by SULT1A2. Taken together, these studies indicate that sulfonation following nitroreduction increases significantly the mutagenic and cytotoxic potential of AAs.
Supplementary material
Supplementary Table S1 and Supplementary Figures S1–7 can be found at http://carcin.oxfordjournals.org/
Funding
National Institutes of Health (ES004068 to A.P.G.); Henry Laufer and Marsha Laufer; Zickler Family Foundation (Translational Research Scholar award to K.D.).
Conflict of Interest Statement: None declared.
Supplementary Material
Glossary
Abbreviations:
- AA-I
aristolochic acid I
- AA-II
aristolochic acid II
- AAs
collective term describing both AA-I and AA-II
- AL
aristolactam
- AL-DNA
aristolactam–DNA
- AL-II-N-OAc
aristolactam-II-N-acetoxy ester
- AL-II-NOH
N-hydroxyaristolactam II
- AL-I-N-OAc
aristolactam-I-N-acetoxy ester
- AL-I-NOH
N-hydroxyaristolactam I
- AL-I-N-OSO3H
aristolactam-I-N-sulfate
- AL-NOHs
collective term describing both, N-hydroxyaristolactam-I and N-hydroxyaristolactam-II
- dA-AL-I
7-(deoxyadenosin-N6-yl)-aristolactam I
- dA-AL-II
7-(deoxyadenosin-N6-yl)-aristolactam II
- dG-AL-I
7-(deoxyguanosin-N2-yl)-aristolactam I
- dG-AL-II
7-(deoxyguanosin-N2-yl)-aristolactam II
- HPLC
high-performance liquid chromatography
- LC/MS
liquid chromatography/mass spectrometry
- NATs
N-acetyltransferases NAT1 and NAT2
- NQO1
NAD(P)H:quinone oxidoreductase 1
- PAPS
3'-phosphoadenosine-5'-phosphosulfate
- ssDNA
salmon sperm DNA
- SULT
sulfotransferase.
References
- 1. Debelle F.D., et al. (2008). Aristolochic acid nephropathy: a worldwide problem. Kidney Int., 74, 158–169 [DOI] [PubMed] [Google Scholar]
- 2. Vanherweghem J.L., et al. (1993). Rapidly progressive interstitial renal fibrosis in young women: association with slimming regimen including Chinese herbs. Lancet, 341, 387–391 [DOI] [PubMed] [Google Scholar]
- 3. Jelaković B., et al. (2012). Aristolactam-DNA adducts are a biomarker of environmental exposure to aristolochic acid. Kidney Int., 81, 559–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Grollman A.P., et al. (2007). Aristolochic acid and the etiology of endemic (Balkan) nephropathy. Proc. Natl. Acad. Sci. U. S. A., 104, 12129–12134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. National Toxicology Program (2011) Aristolochic acids. Rep Carcinog., 12, 45–49 [PubMed] [Google Scholar]
- 6. Nortier J.L., et al. (2000). Urothelial carcinoma associated with the use of a Chinese herb (Aristolochia fangchi). N. Engl. J. Med., 342, 1686–1692 [DOI] [PubMed] [Google Scholar]
- 7. Kumar V., et al. (2003). Naturally occurring aristolactams, aristolochic acids and dioxoaporphines and their biological activities. Nat. Prod. Rep., 20, 565–583 [DOI] [PubMed] [Google Scholar]
- 8. Shibutani S., et al. (2007). Selective toxicity of aristolochic acids I and II. Drug Metab. Dispos., 35, 1217–1222 [DOI] [PubMed] [Google Scholar]
- 9. Sato N., et al. (2004). Acute nephrotoxicity of aristolochic acids in mice. J. Pharm. Pharmacol., 56, 221–229 [DOI] [PubMed] [Google Scholar]
- 10. Pfau W., et al. (1990). Aristolochic acid binds covalently to the exocyclic amino group of purine nucleotides in DNA. Carcinogenesis, 11, 313–319 [DOI] [PubMed] [Google Scholar]
- 11. Schmeiser H.H., et al. (1991). Activating mutations at codon 61 of the c-Ha-ras gene in thin-tissue sections of tumors induced by aristolochic acid in rats and mice. Cancer Lett., 59, 139–143 [DOI] [PubMed] [Google Scholar]
- 12. Attaluri S., et al. (2010). DNA adducts of aristolochic acid II: total synthesis and site-specific mutagenesis studies in mammalian cells. Nucleic Acids Res., 38, 339–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schmeiser H.H., et al. (1996). Detection of DNA adducts formed by aristolochic acid in renal tissue from patients with Chinese herbs nephropathy. Cancer Res., 56, 2025–2028 [PubMed] [Google Scholar]
- 14. Chen C.H., et al. (2012). Aristolochic acid-associated urothelial cancer in Taiwan. Proc. Natl. Acad. Sci. U. S. A., 109, 8241–8246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pfau W., et al. (1990). The structural basis for the mutagenicity of aristolochic acid. Cancer Lett., 55, 7–11 [DOI] [PubMed] [Google Scholar]
- 16. Purohit V., et al. (2000). Mutagenicity of nitroaromatic compounds. Chem. Res. Toxicol., 13, 673–692 [DOI] [PubMed] [Google Scholar]
- 17. Arlt V.M., et al. (2005). Environmental pollutant and potent mutagen 3-nitrobenzanthrone forms DNA adducts after reduction by NAD(P)H:quinone oxidoreductase and conjugation by acetyltransferases and sulfotransferases in human hepatic cytosols. Cancer Res., 65, 2644–2652 [DOI] [PubMed] [Google Scholar]
- 18. Stiborova M., et al. (2009). The role of biotransformation enzymes in the development of renal injury and urothelial cancer caused by aristolochic acid: urgent questions and difficult answers. Biomed. Pap. Med. Fac. Univ. Palacky. Olomouc. Czech. Repub., 153, 5–11 [DOI] [PubMed] [Google Scholar]
- 19. Levova K., et al. (2012). NAD(P)H:quinone oxidoreductase expression in Cyp1a-knockout and CYP1A-humanized mouse lines and its effect on bioactivation of the carcinogen aristolochic acid I. Toxicol. Appl. Pharmacol., 265, 360–367 [DOI] [PubMed] [Google Scholar]
- 20. Chen M., et al. (2011). Inhibition of renal NQO1 activity by dicoumarol suppresses nitroreduction of aristolochic acid I and attenuates its nephrotoxicity. Toxicol. Sci., 122, 288–296 [DOI] [PubMed] [Google Scholar]
- 21. Blanchard R.L., et al. (2004). A proposed nomenclature system for the cytosolic sulfotransferase (SULT) superfamily. Pharmacogenetics, 14, 199–211 [DOI] [PubMed] [Google Scholar]
- 22. Freimuth R.R., et al. (2004). Human cytosolic sulfotransferase database mining: identification of seven novel genes and pseudogenes. Pharmacogenomics J., 4, 54–65 [DOI] [PubMed] [Google Scholar]
- 23. Blum M., et al. (1990). Human arylamine N-acetyltransferase genes: isolation, chromosomal localization, and functional expression. DNA Cell Biol., 9, 193–203 [DOI] [PubMed] [Google Scholar]
- 24. Meinl W., et al. (2006). Human sulphotransferases are involved in the activation of aristolochic acids and are expressed in renal target tissue. Int. J. Cancer, 118, 1090–1097 [DOI] [PubMed] [Google Scholar]
- 25. Stiborová M., et al. (2011). The human carcinogen aristolochic acid i is activated to form DNA adducts by human NAD(P)H:quinone oxidoreductase without the contribution of acetyltransferases or sulfotransferases. Environ. Mol. Mutagen., 52, 448–459 [DOI] [PubMed] [Google Scholar]
- 26. O’Neil M.J., et al. (2006). An Encyclopedia of Chemicals, Drugs and Biologicals. Merck & Co, Whitehouse Station, NJ [Google Scholar]
- 27. Sidorenko V.S., et al. (2012). Lack of recognition by global-genome nucleotide excision repair accounts for the high mutagenicity and persistence of aristolactam-DNA adducts. Nucleic Acids Res., 40, 2494–2505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bradford M.M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem., 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 29. Shibutani S., et al. (2006). 32P-postlabeling DNA damage assays: PAGE, TLC, and HPLC. Methods Mol. Biol., 314, 307–321 [DOI] [PubMed] [Google Scholar]
- 30. Gamage N.U., et al. (2003). Structure of a human carcinogen-converting enzyme, SULT1A1. Structural and kinetic implications of substrate inhibition. J. Biol. Chem., 278, 7655–7662 [DOI] [PubMed] [Google Scholar]
- 31. Martinek V., et al. (2011). Comparison of activation of aristolochic acid I and II with NADPH:quinone oxidoreductase, sulphotransferases and N-acetyltranferases. Neuro Endocrinol. Lett., 32 (suppl 1), 57–70 [PubMed] [Google Scholar]
- 32. Boelsterli U.A., et al. (2006). Bioactivation and hepatotoxicity of nitroaromatic drugs. Curr. Drug Metab., 7, 715–727 [DOI] [PubMed] [Google Scholar]
- 33. Glatt H. (2000). Sulfotransferases in the bioactivation of xenobiotics. Chem. Biol. Interact., 129, 141–170 [DOI] [PubMed] [Google Scholar]
- 34. Hein D.W., et al. (1993). Metabolic activation and deactivation of arylamine carcinogens by recombinant human NAT1 and polymorphic NAT2 acetyltransferases. Carcinogenesis, 14, 1633–1638 [DOI] [PubMed] [Google Scholar]
- 35. Chan W., et al. (2007). Investigation of the metabolism and reductive activation of carcinogenic aristolochic acids in rats. Drug Metab. Dispos., 35, 866–874 [DOI] [PubMed] [Google Scholar]
- 36. Dooley T.P., et al. (2000). Expression profiling of human sulfotransferase and sulfatase gene superfamilies in epithelial tissues and cultured cells. Biochem. Biophys. Res. Commun., 277, 236–245 [DOI] [PubMed] [Google Scholar]
- 37. Krumbiegel G., et al. (1987). Studies on the metabolism of aristolochic acids I and II. Xenobiotica., 17, 981–991 [DOI] [PubMed] [Google Scholar]
- 38. Rosenquist T.A., et al. (2010). Cytochrome P450 1A2 detoxicates aristolochic acid in the mouse. Drug Metab. Dispos., 38, 761–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Riches Z., et al. (2009). Quantitative evaluation of the expression and activity of five major sulfotransferases (SULTs) in human tissues: the SULT ‘pie’. Drug Metab. Dispos., 37, 2255–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Teubner W., et al. (2007). Identification and localization of soluble sulfotransferases in the human gastrointestinal tract. Biochem. J., 404, 207–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Senggunprai L., et al. (2008). Involvement of ST1B subfamily of cytosolic sulfotransferase in kynurenine metabolism to form natriuretic xanthurenic acid sulfate. J. Pharmacol. Exp. Ther., 327, 789–798 [DOI] [PubMed] [Google Scholar]
- 42. Hein D.W. (1988). Acetylator genotype and arylamine-induced carcinogenesis. Biochim. Biophys. Acta, 948, 37–66 [DOI] [PubMed] [Google Scholar]
- 43. Husain A., et al. (2007). Identification of N-acetyltransferase 2 (NAT2) transcription start sites and quantitation of NAT2-specific mRNA in human tissues. Drug Metab. Dispos., 35, 721–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McQueen C.A., et al. (2003). Neonatal ontogeny of murine arylamine N-acetyltransferases: implications for arylamine genotoxicity. Toxicol. Sci., 73, 279–286 [DOI] [PubMed] [Google Scholar]
- 45. Oda Y., et al. (2012). Roles of human sulfotransferases in genotoxicity of carcinogens using genetically engineered umu test strains. Environ. Mol. Mutagen., 53, 152–164 [DOI] [PubMed] [Google Scholar]
- 46. Hoang M.L., et al. (2013). Mutational signature of aristolochic acid exposure as revealed by whole-exome sequencing. Sci. Transl. Med., 5, 197ra102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ma L., et al. (2003). Albumin strongly prolongs the lifetime of chemically reactive sulphuric esters and affects their biological activities in the rat. Nova Acta Leopoldina, 329, 265–272 [Google Scholar]
- 48. Monien B.H., et al. (2009). Probenecid, an inhibitor of transmembrane organic anion transporters, alters tissue distribution of DNA adducts in 1-hydroxymethylpyrene-treated rats. Toxicology, 262, 80–85 [DOI] [PubMed] [Google Scholar]
- 49. Bakhiya N., et al. (2006). Uptake of chemically reactive, DNA-damaging sulfuric acid esters into renal cells by human organic anion transporters. J. Am. Soc. Nephrol., 17, 1414–1421 [DOI] [PubMed] [Google Scholar]
- 50. Jiang Y., et al. (2010). Association of sulfotransferase SULT1A1 with breast cancer risk: a meta-analysis of case-control studies with subgroups of ethnic and menopausal statue. J. Exp. Clin. Cancer Res., 29, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hirata H., et al. (2008). CYP1A1, SULT1A1, and SULT1E1 polymorphisms are risk factors for endometrial cancer susceptibility. Cancer, 112, 1964–1973 [DOI] [PubMed] [Google Scholar]
- 52. Rouprêt M., et al. (2007). Phenol sulfotransferase SULT1A1*2 allele and enhanced risk of upper urinary tract urothelial cell carcinoma. Cancer Epidemiol. Biomarkers Prev., 16, 2500–2503 [DOI] [PubMed] [Google Scholar]
- 53. Ung D., et al. (2007). Variable sulfation of dietary polyphenols by recombinant human sulfotransferase (SULT) 1A1 genetic variants and SULT1E1. Drug Metab. Dispos., 35, 740–746 [DOI] [PubMed] [Google Scholar]
- 54. Priestap H.A., et al. (2012). Aristolochic acid I metabolism in the isolated perfused rat kidney. Chem. Res. Toxicol., 25, 130–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hildebrandt M.A., et al. (2007). Genetic diversity and function in the human cytosolic sulfotransferases. Pharmacogenomics J., 7, 133–143 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.