

Abstract

Nanoparticles have the potential to contribute to new modalities in molecular imaging and sensing as well as in therapeutic interventions. In this Nano Focus article, we identify some of the current challenges and knowledge gaps that need to be confronted to accelerate the developments of various applications. Using specific examples, we journey from the characterization of these complex hybrid nanomaterials; continue with surface design and (bio)physicochemical properties, their fate in biological media and cells, and their potential for cancer treatment; and finally reflect on the role of animal models to predict their behavior in humans.
On this journey from nanoparticle (NP) synthesis and characterization to animal models, a number of common themes emerge: how do we evaluate the potential for translation of new materials? What should be our standards for NP characterization and for their biological properties? How do we improve the robustness and reproducibility of our devices, measurements, and conclusions? These questions will need to be addressed to progress more effectively and safely toward biomedical applications. Translation toward the clinic is already happening, yet great challenges remain before these approaches will make a significant difference to a large number of patients.
The diversity of nanomaterials available provides a formidable toolbox, but one that is now so vast that we can only use it effectively if we have a clear understanding of what each tool is and of what it can do. In that context, standards and careful characterization of these nanomaterials and their surfaces is essential. Sometimes the tools exist but are not used systematically, while in other cases, new methods are required. Nanoparticles also need to be characterized at different points in their life cycle as they are generally dynamic systems. A question as basic (and fundamentally important) as “do NPs go through biological membrane barriers?” remains open and requires further experimentation. There is also a large knowledge gap regarding the interactions of NPs at the whole animal level that reflect their performance in cancer imaging, sensing, and therapy; their metabolization and expulsion from the body; their effects on the immune system; and the choices of animal models that are effective at predicting outcomes in humans. Better materials characterization, better experimental design, new measurement techniques that allow for real-time molecular-level information at the NP–biological interface, advanced in vitro testing (three-dimensional cocultures) and computer models, systematic sharing of data, more effective interdisciplinary collaborations, publication of both positive and negative results, and critical discussion of published evidence are just a few suggested steps toward a more robust scientific enterprise.
Nonetheless, the societal impact of nanotechnologies, if properly deployed, is enormous. In addition to NPs for human health, NPs are components of next-generation technologies for cleaner water, cleaner air, and cleaner energy. A thorough understanding of the issues raised on the biomedical application side will also help other NP industries produce their technologies in a safe and sustainable manner.
Core–Shell Nanoparticles: The Core Matters
Researchers are generating an immense collection of new nanomaterials, which differ by the composition of their (inorganic) cores and (organic) ligand shells. While many are proposed as potential candidates for biomedical applications, only a few are, or will be, really translated. In this first section, we use iron oxide NPs as a case study to illustrate the need for detailed characterization of the inorganic core. These NPs combine magnetic properties with low toxicity and biodegradability. Translation has started with current applications in sensing, imaging and therapy.
Fundamental changes in the magnetic structure of macroscopic magnetically ordered materials occur when the physical size is reduced. A bulk ferromagnet spontaneously subdivides into a multidomain structure to reduce the magnetostatic energy associated with a large stray field. A critical size exists—which is both material and shape dependent, although typically on the order of 100 nm—at which only a single domain structure can be supported. Further reduction in size leads to the superparamagnetic state. In this state, thermal agitation results in the instability of the magnetic moment, which flips between easy axes on a time scale on the order of nanoseconds.1 Although the spin structure of the superparamagnetic and single domains is identical, the two states differ in the nature and the dynamics of their magnetic response. The magnetic behavior of the superparamagnetic state is analogous to the Langevin model of atomic paramagnetism. However, instead of dealing with the magnetic moment of a single atom, in the superparamagnetic state of a magnetic nanocrystal, there are on the order of 105 atoms that are magnetically coupled by the exchange interaction.2 The reader is referred to the reviews by Pankhurt3,4 for an overview of the underlying physics that governs these changes in magnetic behavior, with Cullity5 recommended for a more detailed discussion.
Bioapplications of magnetic NPs have been explored for several decades6,7 and now extend to magnetic separation, delivery (actuation), hyperthermia treatment, and contrast enhancement agents for magnetic resonance imaging (MRI). In this broad interdisciplinary field, the terms “single domain” and “superparamagnetic” particles are often used interchangeably; the subtle differences in the dynamics of the magnetic response mean that it is important to make the distinction between these two different regimes.
Iron oxide can exist in different chemical compositions such as magnetite (Fe3O4) or maghemite (γ-Fe2O3) or, most likely, a nonstoichiometric combination of the two. Understanding of the long-term biotransformation, and consequently toxicity, requires a precise knowledge of the injected NP.8 As Fe2+ ions play a critical role in biochemical processes that generate free radicals, in vivo applications of superparamagnetic NPs require accurate and precise quantification of the amount of Fe2+. Fe3+ ions are significantly less reactive and are the form used for iron storage within the body.
Although the magnetic response is primarily governed by the object size, it is also affected by the physical shape, the crystallographic structure, and microstructural quality. The contributions to the total energy of the ferromagnetic system are not isotropic, with anisotropic contributions associated with both the crystal structure (magnetocrystalline anisotropy) and the shape (magnetostatics) of the particle. While it is necessary to perform thorough characterization of a range of properties of magnetic NPs, such characterizations should also be done under conditions that mimic the physical environment of the intended application. Though this is by no means trivial, it provides a sound basis for comparison of materials, interpretation of performances, as well as clues for the rational design of the next generation of particles. Key techniques to analyze magnetic cores and the in vitro/in vivo fate are described briefly here.
Structural and Chemical Characterization
Structural characterization can be achieved using a combination of X-ray diffraction (XRD) and electron microscopy—including both standard and high-resolution transmission electron microscopy (TEM, HR-TEM).
Diffraction techniques may be preferred over imaging techniques such as electron microscopy (EM) since they are limited by the small amount of material that can be imaged. The full width at half-maximum (FWHM) of diffraction peaks can be fitted using the Scherrer equation and provides information on microstrain as well as the NP size distribution. For iron oxides, the interpretation of the diffraction peak positions is complicated by the fact that the lattice parameters of magnetite and maghemite are extremely similar (amagnetite = 8.396 Å; amaghemite = 8.346 Å). Structural studies should always be complemented with spectroscopy that probes the chemical environment such as Mössbauer, electron energy loss spectroscopy (EELS), or X-ray photoelectron spectroscopy (XPS). It is only with such spectroscopies that the Fe2+/Fe3+ ratio can be quantified. However, XPS probes the chemical environment at the surface, while Mössbauer probes the mean iron oxidation degree within the whole volume of the NP. On the other hand, EELS is more qualitative and can only be performed on one particle at a time. Therefore, pure magnetite, pure maghemite, core–shell magnetite–maghemite, or intermediate composition can be distinguished by using a combination of these techniques.
Electron microscopy does, however, provide invaluable insights into the range of shapes present and, when combined with electron diffraction, offers a powerful way with which to probe the evolution of crystallographic quality. In some recent work,9,10 the transformation of iron oxide NPs has been observed by following the crystallographic structure evolution of superparamagnetic maghemite into an amorphous, poorly magnetic iron species over the course of three months in vivo. This example nicely illustrates the benefit of complete characterization since it enables demonstration of the association of a loss of magnetic properties with the biotransformation of the material into poorly magnetic species (rather than a disappearance of the material).
Magnetic Characterization
By definition, the superparamagnetic state is characterized by two features: the lack of remanence and the temperature dependence of magnetization curves (magnetization curves normalized by temperature superimpose, i.e., a plot of M/TvsH/T).2 Static magnetization measurements can be used to confirm the superparamagnetic state and can be performed using vibrating sample magnetometry (VSM) or superparamagnetic quantum interference device (SQUID) magnetometry.
By ensuring that all magnetometry measurements are performed on noninteracting systems, analysis of room-temperature magnetization curves can clearly and unambiguously provide a wealth of information concerning the particle size distribution of the starting material.11−14 The initial susceptibility is sensitive to larger particles, while the approach to saturation is governed by smaller particles. The value of magnetization at saturation (MSB) is significantly different for bulk magnetite and maghemite (at 297 K, MSB[magnetite] ∼ 480 emu/cm3 while MSB[maghemite] ∼ 300 emu/cm3). In addition, the large surface-to-volume ratio of NPs results in saturation magnetization of the particles, MSP, that is typically lower than that of the bulk material (due to the higher number of surface defects). This saturation magnetization can be further reduced by poor crystal structure. Thus, to avoid misinterpreting a strongly magnetic maghemite sample with a weakly magnetic magnetite sample, magnetization measurements should always be performed in conjunction with structural and chemical characterization so that the correct phase of iron oxide is identified.
The articles by Chen et al.(12) and El-Hilo13 are two excellent examples that discuss how to extract the magnetic size distributions of superparamagnetic NPs from magnetization curves, which can be achieved by fitting experimentally measured curves using the simple Langevin expression modified by a log-normal distribution. Such articles highlight the need to move beyond working in terms of a single particle size and magnetic response, when instead there is always a distribution of sizes and shapes. Small changes in the shape cannot be neglected when dealing with magnetic species. Changes in shape can cause magnetostatic interactions, which may affect the colloidal stability; those may be exacerbated during their intended applications, as briefly discussed here.
Any application that requires magnetic NPs to be internalized within cells should also characterize the magnetization measurements after uptake in order to determine the effect of intracellular confinement on the magnetic response. The mechanism of uptake of NPs by cells is further discussed below, but it is worth noting here that particles typically end up sequestered in large concentrations within intracellular vesicles. Close confinement can cause dipole–dipole interactions,15,16 and indeed, some groups have reported that MRI relaxivities of superparamagnetic iron oxide NPs are often significantly reduced upon internalization by cells.17,18
Understanding the nature of magnetization reversal dynamics is important for magnetic hyperthermia applications. When subjected to alternating current (AC) magnetic fields, magnetic NPs cause heating of the local area due to losses during magnetization reversal. There are three different mechanisms by which the particles dissipate heat—hysteretic losses, relaxation (Néel or Brownian), and viscous (stirring) heating.19−21 The physical basis of the heating of superparamagnetic particles by AC magnetic fields has been reviewed by Rosensweig.22 Briefly, for small field amplitudes, and assuming minimal interactions between the constituent superparamagnetic particles, the response to an AC field can be described in terms of its complex susceptibility. This results in the generation of heat,22 which can be interpreted physically as meaning that if the magnetization lags the applied field there is a positive conversion of magnetic energy into internal energy. The dominant heat loss mechanism is determined by the shape of the magnetization curve, although these vary in different manners on the amplitude and frequency of the AC magnetic field. Nonetheless, they are all strongly dependent on the mean and width of the size distribution, particle shape, and crystallinity.19,23 These properties can be further affected by both temperature and viscosity of the physiological conditions.
Unfortunately, most publications on the development of magnetic NPs for bioapplications do not report such extensive structural, chemical, and magnetic characterization. Instead these approaches are often restricted to investigators primarily interested in nanophysics. Such physical properties are, however, of direct use to interpret and to understand how the biological environment affects and modifies the NPs’ magnetic properties. Consequently, the current literature is based on a range of materials of various magnetic qualities that have been irregularly characterized, which makes final observed properties difficult to interpret and to compare.
Complexity and Biocompatibility of Nanoparticle Surfaces
As-made colloidal NPs bear adsorbed ions, ligands, or other molecules on their surfaces. Generally speaking, these “native species” on the NP surface are not the final desired surface coatings. As-made NPs may be too hydrophobic for biological applications, or toxicity concerns of the ligands may arise. Therefore, many workers in the field engage in surface bioconjugation of NPs, usually with the goals of making nanomaterials that (i) are biocompatible and (ii) would bind to a particular biological target in or on a living cell. The most common chemical linkages between inorganic NP surfaces and their designer molecular coats are shown in Figure 1.
Figure 1.
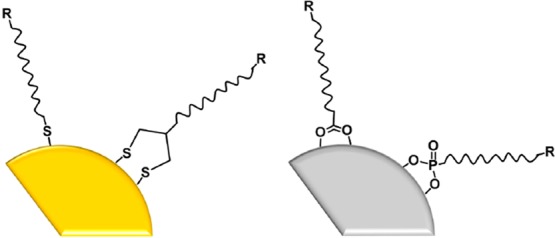
Schematic of (left) a noble metal nanoparticle (NP) with thiol and disulfide surface coatings and (right) a metal oxide NP with carboxylate and phosphonate coatings. R represents a functional group or recognition unit. The left cartoon implies metal–sulfur bonding; the right implies metal cation–anion bonding.
Even for a monolayer of identical molecules on a nanoparticle surface, variations in surface coverage or ligand orientation (e.g., lying down or standing up on the surface) can occur. The situation becomes even more complex when mixtures of surface ligands are coordinated to the NP surface: do the ligands self-assemble into domains, reminiscent of the lipid raft hypothesis for cell membranes? Or are the ligands randomly distributed on the surface (Figure 2)? Simulation tools are valuable guides to the engineering of patchy nanoparticles.24−26 Experimental evidence of “patchiness” relies on either direct molecular-scale imaging, which is difficult to perform on nanoscale curved surfaces and is subject to possible artifacts, or on less direct methods such as mass spectrometry.27−36
Figure 2.
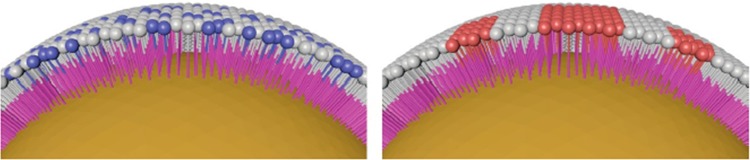
Schematic of randomly arranged molecular adsorbates on a nanoparticle (NP) surface (left), compared to adsorbates arranged in patches (right) on a NP surface.
One grand challenge in the field, then, is a reliable means of measuring the spatial location of molecules on nanoscale curved surfaces that provides chemical information as to what the molecules actually are.
The importance of molecular display on nanoscale curved surfaces becomes apparent when the size of a typical NP (5–200 nm) is compared to that of a small molecule (1 nm), a typical protein (10 nm), and a cell (1000–100 000 nm). Receptor clustering is a phenomenon in which proteins or other molecules cluster together on the surface of a cell an event that is correlated with intracellular signaling. The size of the average NP is beautifully suited to anchor onto a cluster of such molecules and perhaps even to multiple different clusters at once. In other words, even though the “lock and key” idea of chemical binding is long past its prime, it is tempting to imagine that multiple molecular keys on a NP surface might open multiple cellular locks in a coordinated way—if the spatial locations were well-matched.
Even if the presence of a patchy designer coat on a NP surface is not needed for a particular application, NP bioconjugation experiments frequently proceed with multiple ligands, one or more for targeting and one or more for biocompatibility.
The selection of targeting ligands is, for the most part, driven by existing biological recognition partners (antibody/antigen; aptamer/small molecule; receptor/ligand, cyclic RGD/integrin, etc.). While a good idea in principle and in vitro, in practice, the ability to reach their engineered target in vivo is questionable, and may rely more on the enhanced permeability and retention effect than surface chemistry (see below). Another challenge in the field, then, is to measure the percent yield of designer-coated NPs binding to their target and understand the mechanisms by which this event fails.
To improve NP biocompatibility, the most popular coligand is poly(ethylene) glycol (PEG; Figure 3). Although PEG is often considered to be the current best antibiofouling surface ligand, carboxy- and sulfobetaines have been explored as possible alternatives (Figure 3).37−42
Figure 3.
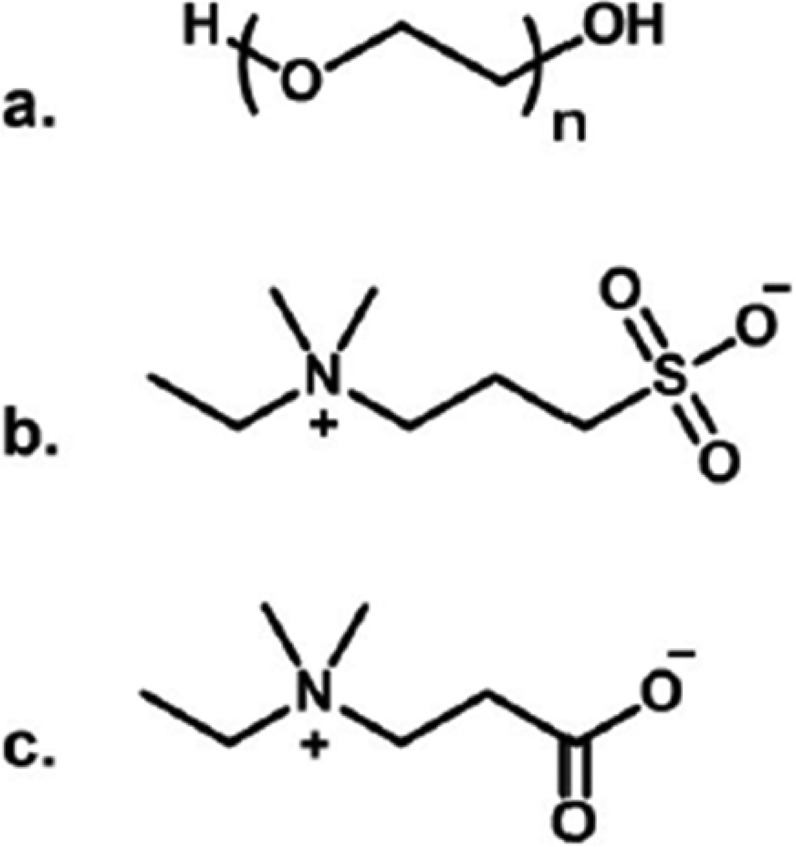
Chemical structures of (a) poly(ethylene) glycol (PEG), (b) sulfobetaine, and (c) carboxybetaine.
Both PEG and the betaine ligands are overall neutral in aqueous solution and very soluble in water; PEG is known to be well-hydrated. Thus, surfaces coated with PEG, or betaines, resist adventitious protein adsorption because (i) there are no charges to make favorable electrostatic contacts with proteins, and (ii) the hydration shell effectively looks like water to the proteins, and so there is little driving force for protein–surface binding. As a result of these ideas, PEG especially is considered “biocompatible” when really what is meant is “resistant to protein (or cellular) adsorption”. The degree of resistivity to protein adsorption is by no means 100%; early studies showed reduction in protein adsorption by a factor of 4 or so.41
Recently, questions about PEG’s biocompatibility have been raised. Although PEG is Generally Regarded as Safe (GRAS) by the U.S. Food and Drug Administration, and its use as a drug modifier is approved in some commercial products, PEG does have limitations in its postuptake chemistry38,39 and also different degrees of organ accumulation as a function of molecular weight.38 Poly(ethylene) glycol properties such as molecular weight and surface density control when bound to a NP do influence NP stability.39,43 There are increasing reports of PEGylated objects inducing complement activation, which is part of the immune cascade;44−48 these reports underscore the need to have a diversity of options when it comes to “biocompatible surfaces”. Understanding and predicting these chemical processes at the NP surface, and extending the molecular level to the cellular or organismal level, is a key challenge for the field.
Utilizing Nanocrystals in Biomedicine: Particle Surface Chemistry and Biological Effects
Nanoparticle High Surface Energy
Nanocrystals, especially those in the smallest size range (below 30 nm), may be rather unstable and short-lived. Energy is required to form NPs, either from the bulk or from molecular precursors, and to create a solid–liquid/air interface. The resulting NPs are reactive and tend to either grow or dissolve (Figure 4). Interfacial energy can also be released via aggregation and interaction with proteins, both of which are of special relevance in the nanobiotechnology context.
Figure 4.

Nanoparticle (NP) cycle. To generate NPs through bottom-up or top-down fabrication requires energy that the formed NPs tend to dissipate either by aggregation or by chemical transformation.
Nanoparticle Aggregation
Although the stability of NPs and conjugates at physiological conditions has been the subject of intensive studies,49−51 the loss of colloidal stability is often underestimated. This problem is particularly acute at high concentrations of NPs, which are required to reach therapeutic doses and for in vivo studies. Since the volume that one can inject is limited, not more than ∼200 μL in mice, for example, concentration needs to be high to achieve reasonable doses. Thus, in many cases, the loss of colloidal stability is behind the lack of (or unexpected) biological effects.52,53 Indeed, surface modification of NPs is always required for biomedical applications because the environment where the NPs are produced and the physiological environment where they should perform are different and usually incompatible. This is especially important in environments where solutions with high ionic strength, such as biological fluids (e.g., blood, lymph, and urine), induce compression and screening of the ionic double layer that forms around the NPs in aqueous solutions, allowing them to get close together, leading to aggregation if a steric stabilizing agent is not present.54
Since small size is a key functional feature of these systems, any induced aggregation will dramatically affect cellular and molecular responses. Additionally, NPs may aggregate to sizes large enough to induce frustrated phagocytosis resulting in chronic inflammation and consequent risk of promoting cancer or neurodegeneration.55
Nanoparticle Protein Corona
Usually, proteins present in biological media play the role of stabilizing agent by forming a protective protein corona on the surface of the NPs as soon as they come into contact with serum, thus preventing aggregation.50,56 However, under certain conditions, this is not enough to prevent aggregation, for example, when working with high concentration of NPs in cell culture media or animal models (>1012 NC/mL).57
Nanoparticles, in general, strongly interact with macromolecules, a special case being interactions with proteins, their equivalent-sized biological counterparts. The interaction of NPs with proteins and other molecules may alter the metabolism of biological molecules, and the conjugates can be detected by the immune system, evoking a pro-inflammatory response. The protein corona also influences the interactions of NPs with the cell membrane.58 Adsorption of proteins also modifies the surface charge50 of the NP and therefore determines how it interacts with biological processes.59,60 In addition, once the protein corona is formed, opsonins and proteins of the complementary system may be able to recognize proteins adsorbed on the surface of the NP which would trigger an immune response.61,62 Additionally, conformational changes due to adsorption of proteins on the particle surface may cause lack of recognition by the organisms or may cause autoimmunity.63
Adsorbed proteins may also influence signaling, when extracellular proteins internalize into cells carried by NPs or when binding of NPs to cells changes structure or association patterns of self-proteins.64 The interaction of proteins with NPs may modify protein activity and structure and interfere with their natural fate. Therefore, the effect of different corona compositions on intracellular detection mechanisms needs to be investigated. In some cases, the adsorbed proteins can induce signaling by exposing new epitopes upon binding to the NP surface,65 thus interacting with specific receptors on the cell membrane, such as receptors involved in lipoprotein trafficking, complement receptors, and pattern recognition receptors as the toll-like receptor family.66 Additionally, modification of the protein structure or environment due to conjugation may have other important effects, as induction of exposure of hydrophobic residues and consequent aggregation and protein denaturalization, or modification in how proteins are recognized, employed, and processed.
Different modes of surface functionalization can also modify the way proteins absorb. This determines the fate of the NPs inside the body, varying from innocuous and eliminated through the urine in minutes67,68 to provoking strong and aggressive immune (anaphylactic) response.69,70
It is important to note that the immune system is especially sensitive to natural nano-objects, such as misfolded or non-self-proteins, protein aggregates, and protein patterns (e.g., virus surfaces). This is why the humoral and cellular immune defense response to engineered NPs is also critical: NPs have dimensions similar to those of proteins, protein aggregates, and viruses and are recognized by immune cells, so they are subject to efficient protective strategies that prevent their uncontrolled access into the body. Nanoparticles could induce immunogenic epitope generation, that is, cause deformation of protein tertiary structure (promoted by the interaction with the curved NP surface) that can induce protein aggregation or make self-proteins immunogenic, thereby inducing autoimmune reactions, as antigens absorbed onto NPs could become allergenic. Additionally, it is known that epitope concentration, repetition, and patterning induced by association of proteins or peptides to NPs can be observed to trigger an immune response.66 Given the above considerations, it is clear that the interaction between NPs and other biological molecules may cause a modification of both, with consequences for human health that are not yet easy to predict. In this context, previous NP albuminization to saturate the NP surface with proteins seems a simple and effective strategy to biocompatibilize and to avoid NP–protein interaction; PEGylation is also used, as discussed above.
Extensive investigations into the formation of the protein corona have shown that hydrophobic NPs get rapidly coated by the fraction of the serum proteins dedicated to detecting foreign objects, as lipopolysaccharide (LPS) or misfolded protein and protein aggregates, basically immunoglobulins and apolipoproteins. Additionally, hydrophilic surface NPs get progressively coated by serum albumin—which counts for 50% of all proteins in the serum—in a process that may take minutes to hours and results in a permanent protein corona enveloping the NPs. Surfactant molecules present on the particle surface may effectively interfere with the absorption process, succeeding in completely avoiding protein absorption in some cases, as with PEGylation.71
Mechanism of Cell Entry and Exit
In a previous Nano Focus article,72 authors stated that “surface-modified NPs can directly reach the cytosols of living cells”. Indeed, this impression seems to be relatively widely shared in the nanoscience and chemistry literature, with many authors making the case for the preparation of new materials on the basis of the special capacity of NPs to cross cell membranes. Vincent Rotello, however, speaking at the E-MRS symposium Q (spring 2013, Strasbourg), noted that “amateurs worry about how NPs get into cells; experts worry about how they get out of the endosome.” A critical look at the literature shows that reaching the cytosol remains a major challenge and that we are far from understanding, let alone controlling, the interactions of NPs with cells or even with model membranes. Before focusing on the recent literature on NP/membrane interactions, it is perhaps useful to reflect on the history of a related field—cell-penetrating peptides (CPPs)—and on some basic biology and thermodynamics arguments.
The CPP field has now entered its third decade. Initial reports on CPPs concluded that there was rapid passive temperature-independent transport of the CPPs and of their cargoes through the cell membrane with low toxicity. These highly exciting properties contributed to the massive growth of this field of research, but it was later found that fixation prior to microscopy observation induced a redistribution of the molecules. In live cells, punctate patterns characteristic of endocytosis were observed. For several years, endocytosis became the generally accepted mode of transport for CPPs, details of molecular pathways were elucidated, and the direct involvement of various cell surface proteins and glycosaminoglycans were demonstrated. In recent years, experiments on model membranes (e.g., giant unilamellar vesicles and cells) have indicated that while a range of endocytotic mechanisms are indeed the main contributors in live cells, in some cases, passive diffusion through the membrane does occur depending on lipid composition, cargoes, and membrane potential.73,74 In spite of over 20 years of research and more than 20 000 publications, many basic questions remain to be clarified in the field of CPPs, and what is emerging is a highly complex set of scenarios resulting in increased uptake through various internalization pathways working in parallel that depend on cell state and peptide sequence; CPPs are not a magical route to the cytosol.73,74
The self-assembly of small molecules into a membrane that separates a living entity from its environment is probably one of the earliest events in the history of life. The plasma membrane of eukaryotic cells tightly controls what does and what does not enter the interior of cells. Polar molecules such as ions, peptides, proteins, oligosaccharides, etc. do not cross the membrane by diffusion: transport is either facilitated by pore/pump proteins or relies on the endocytotic processes. In response, viruses and bacteria have evolved advanced molecular machineries to gain access to the interior of cells through specific interactions with the host. Viruses have similar sizes to NPs, yet no virus relies on simple passive diffusion through the membrane to gain access to the cytosol. The reason is probably simply that the energy barrier for the passive diffusion of a polar particle of several nanometers through the cell membrane is much higher than the thermal energy.
Recent literature on NPs and cells/membranes can be classified in three categories: (1) experimental studies with living cells, (2) theoretical studies (necessarily of model systems), and (3) experimental studies on model membrane systems. The first category has been critically reviewed recently by Iversen et al., who caution against several experimental and interpretation pitfalls and make recommendations for future studies.75 We certainly second their concluding remark that “The complexity, the combination of advanced chemistry and cell biology, makes it important that future research on NPs is performed as a close collaboration between scientists with different backgrounds. This is important to prevent misleading/wrong interpretations and thus aid in bringing NPs faster into clinical use.” In addition to interdisciplinary collaborations, more open and robust discussion of published evidence (taking inspiration from the history of the CPPs field) is required. One example of reported passive diffusion of NPs through cell membranes is the case of the “stripy” NPs;76 however, both the structure and the intracellular localization have been questioned.27,29,36 Two recent studies on lipid NPs for the delivery of siRNA further confirm that reaching the cytosol remains a significant challenge.77,78 Both studies show that internalization occurs via endocytosis and escape to the cytosol is a rare event. Gilleron et al. used gold NPs and with extensive electron microscopy analysis demonstrated that only a minor fraction (1–2%) of siRNAs were released from endosomes.77,79
The second category (i.e., theoretical studies) has been expanding rapidly in the past few years but is unfortunately not yet matched by a growth in the third category of carefully controlled experiments on model membranes. Among theoretical articles, we note that Li et al. offer support for favored transport of NPs with striated ligand patterns across membranes compared with random configuration of ligands.80 On the contrary, Gkeka et al. show that a ligand configuration that avoids hydrophobic patches as much as possible minimizes the energy trap.81 It is worth noting that, in both cases, there is an energy trap of over 10 kBT for these particles that have a significant proportion of hydrophobic ligands with favorable interactions with the interior of the membrane. Incorporation of hydrophobic NPs into lipid membranes is relatively well understood,82 and the use of NPs/colloids as surface active materials to stabilize emulsions is a mature industrial field.83 Clear experimental evidence of passive transport of NPs across model membranes, however, remains elusive.
Inorganic Nanoparticles for Cancer: Hyperthermia and Bimodal Therapy
The use of heat to kill tumors has been known since the ancient Greek times and is called hyperthermia.84 It consists of an increase in temperature just above the physiological temperature to induce cytotoxic effects on cancer cells because, in tumor mass, tumor cells show a disorganized and compact vascular structure, and heat dissipation is hindered in comparison to healthy tissues.85 To increase the temperature, a variety of clinical methods have been employed, including the application of microwaves and ultrasound. However, such methods have limited spatial and temporal control, and therefore, burns to surrounding healthy tissues are often inevitable.
Magnetic hyperthermia, whereby magnetic NPs are used for the controlled generation of heat to kill cancer cells, while limiting damage to the surrounding tissue, was first developed in 1957 (Figure 5).6 Heat generation by means of plasmonic gold or silver NPs under near-infrared (NIR) laser irradiation has also been previously reported.86−88 The traditional understanding is that a sustained change in temperature above a threshold of ∼42 °C results in destruction of malignant tissue, although recent work suggests that toxicity may be achieved without bulk temperature change, possibly through damage to intracellular organelles leading to lysosomal content release.89,90
Figure 5.

Schematic illustrating magnetically induced hyperthermia and drug release. (A) Magnetic nanoparticles (NPs) at a defined concentration, when exposed to an alternating magnetic field, can induce a temperature rise as a consequence of their magnetic vibrations. (B) Heat produced locally by the magnetic NP can be used to release drugs associated to the NP surface via thermosensitive linkers.91−93 In this case, drug release could occur even if the global temperature of the system does not change macroscopically; a local temperature increase is responsible for such release.91,92,94
The heating ability of magnetic NPs is defined as the specific absorption rate (SAR) (sometimes called specific loss power, SLP), which, in turn, is determined by the particle material, size, and shape. The heat generated by NPs can only be understood when the magnetic, physical, and hydrodynamic size distributions are accurately known (see refs (19, 23), and (95) and discussion on core characterization in the previous section). Gonzalo Vallejo-Fernandez et al. argue that SAR can only be reliably measured when the particles are embedded within a solid matrix, thus simulating the NP localization at the tumor.23 It is also important to remark that the field and frequency have to be kept below a certain threshold in order to be clinically applied.95,96 This has not always been the case for many proof-of-concept studies.94,97
Even with high SAR particles, in vivo implementation is constrained by the need to achieve high local concentration of nanomaterials at the tumor site. Superparamagnetic NPs that can circulate long enough into the blood to lead to the required tumor accumulation and that present the optimal therapeutic dose for the heat treatment while avoiding systemic toxicity are still missing. It is unclear whether further improvements to the surface chemistry and colloidal stability of the NPs combined with active targeting will provide a widely applicable strategy since therapies based on systemic injections are fundamentally limited by different physiological barriers: (i) the extravasation first from the vascular system to the tumor, (ii) the distribution among the tumor regions, and finally (iii) the targeting toward different subsets of tumor cell populations (i.e., cancer stem cells (CSCs) versus non-CSCs).
Nonetheless, magnetic NPs have been approved for medical devices, and MagForce (Berlin, Germany) has translated magnetic hyperthermia for the treatment of glioblastoma multiforme. Their treatment does not rely on systemic injection. Instead, it consists of injecting 15 nm aminosilane-coated SPIONs directly into the solid tumor with a specially designed surface to ensure that NPs remain at the site of injection, thus allowing for multiple magnetic exposure.98,99
Since chemo- and radiotherapy are the most common treatments for cancer, NPs that combine the functions of heat mediator foci with the capability to deliver and to release drugs may broaden the scope of biomedical applications. Attempts have been made to exploit the heat produced by the NP as a trigger mechanism enabling spatial and temporal control of drug release. Mechanisms include breaking thermosensitive linkers,91−93 the opening of “polymer gates” from silica-functionalized magnetic NPs,100 or the disruption of molecular assemblies.101
In a couple of recent examples, dehybridization of oligonucleotides was used as a tool to engineer a temperature-sensitive gate for drug release.93,94,102 Ruiz-Ernàndez et al. encapsulated a model drug into silica nanopores blocked by DNA hybridized with iron oxide NPs bearing the complementary DNA strand. In this study, a macroscopic increase in temperature, above 42–45 °C, resulted in melting and subsequent release of the drug, thus combining hyperthermia with drug release. More recently, drug release has been achieved without the need to reach a macroscopic temperature change. This effect relies on the quite significant heat profile generated at the NP surface, upon alternating magnetic field (AMF) exposure, within a contour of only a few nanometers.91,92,94 As such local changes are essentially independent of NP concentration, some of the limitations of systemic injections noted above may be relaxed. Achieving the very high drug loading required to achieve a local therapeutic dose might then become the next challenge.
New opportunities might come from hollow iron oxide NPs, which can be both magnetic hyperthermia agents and nanocontainers for drug molecules.103−105 The thin shell structure can either be broken via sonication104 or similar treatments such as by using focused ultrasound treatment106 or electroshock wave lithotripsy, which is typically applied for the treatment of renal stones. In the case of hollow nanostructures containing platinum, such as iron–platinum NPs or FePt@Fe2O3 yolk–shell NPs, the degradation of the fragile hollow shell under acidic pH in cells also allows the gradual release of cytotoxic ions, the Pt2+ ions, thus generating cytotoxicity effects toward the cells that have internalized the nanostructures.107,108
To introduce new therapeutic agents, the net beneficial effects have to be significant in comparison with the present treatment applied. To reach this point, a careful investigation of potential new nanomedicines must include in vitro characterization of their effects on living cells, proof of principle in vivo (in a suitable model, see discussion in the next section), as well as studies of efficacy, biodistribution, in vivo accumulation, and degradation to elucidate the fate of the materials, their potential long-term toxicity, and their body elimination at the dose needed for the treatment. This approach has already been applied in the case of iron oxide NPs.109 To move ahead, a continuous feedback is needed between design, characterization, and evaluation to optimize the therapeutic performance while minimizing the side effects.
Predicting Nanoparticle Behavior in Humans: How Good Are the Current Cellular and Animal Models?
Profound understanding of how NPs that are made from different materials interact with various organs and cells is important for optimizing novel sensing, imaging, and drug-delivery systems. To achieve this goal, the use of relevant biological models is needed. At the cellular level, it is understandable that cells will vary in their interaction with a particular NP since cells from different organs and origin have different biological functions, membrane compositions, and structures. Thus, both the interaction at the membrane level and the intracellular fate of the NP (trafficking, signaling, and disposition of the NP and its cargo) arefferent in different cell types and within different organs.
As an example, we discuss here the well-studied lipid-based nanoparticles (LNPs) to show how these materials can interact with subsets of cells from the immune system.110,111 Lipid-based NPs (e.g., liposomes, lipoplexes, micelles, etc.) are NPs self-assembled from a variety of naturally occurring and synthetic lipids creating large panels of structures with unique architectures and properties. However, despite the advantages of increased stability, facilitated delivery, and cargo protection, following systemic administration, LNPs may trigger the innate immune arm of the immune system, especially the complement cascade and macrophage/dendritic cells clearance mechanisms (Figure 6).112,113
Figure 6.
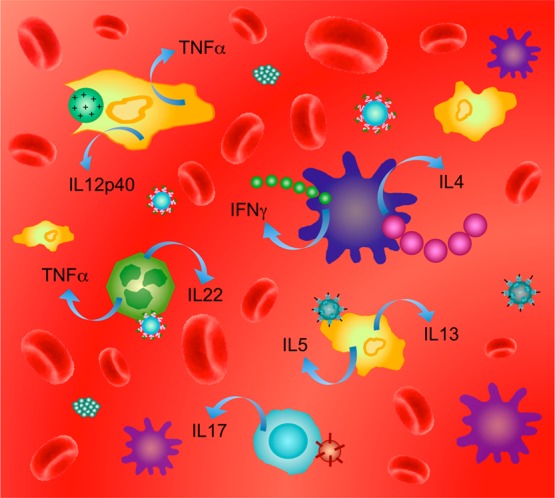
Iteraction of different lipid-based nanoparticles (LNPs) with subsets of leukocytes can suppress or activate the immune response. The first line of defense by the innate immune arm includes different pattern recognition receptors such as membrane-bound toll-like receptors (TLRs), cytoplasmic NOD-like receptors (NLRs), and scavenger receptors on innate immune cells such as monocytes, macrophages, and dendritic cells. The second line of defense includes the adaptive immune arm with several important T helper subsets such as TH1, TH2, TH17, Tregs, TH9, and TH22 cells. Each subset of leukocytes can interact differently with different types of nanoparticles made from different materials and with different sizes, geometries, and surface charges. Adapted with permission from ref (114). Copyright 2012 Elsevier B.V.
Like other circulating particles, LNPs such as liposomes are often first taken up by phagocytic cells (such as blood monocytes and specialized macrophages of the liver [Kupffer cells, also known as stellate macrophages], spleen, and bone marrow).115 There might be undesirable interactions between the liposomes and the immune system, such as immunostimulation or immunosuppression. Liposomes have been well-documented as agonists of toll-like receptors (TLRs)116,117 and can also be internalized into macrophages by scavenger receptors, but there is a lack of immunological knowledge about the interaction of liposomes and other LNPs with nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) and specifically with the inflammasome. Liposomes can also activate, complement, or induce an “educational” event with the adaptive immune system. Interactions of LNPs with TH and B-lymphocytes are less well characterized and have tremendous potential for exploring new avenues in the adaptive immune system.112,118,119
The inadvertent recognition of liposomes as foreign entities by leukocytes may result in a multilevel immune response against the liposomes and eventually lead to toxicity in the host and/or lack of therapeutic benefit.115,120 A harmful activation of the complement cascade may occur in some types of particles. This event may lead to hypersensitivity reactions and anaphylaxis.121 Szebeni et al. showed that intravenous injection of LNPs could cause acute hypersensitivity reactions (HSRs) in up to 45% of patients, with hemodynamic, respiratory, and cutaneous manifestations. The phenomenon can be explained with activation of the complement system on the surface of lipid particles, leading to anaphylatoxin (C5a and C3a) liberation and subsequent release reactions of mast cells, basophiles, and possibly other inflammatory cells in the blood.122
Lipid-based nanoparticles decorated with poly(ethylene) glycol (PEG) and entrapping doxorubicin (Doxil) also activate the complement system. The reported frequency of HSRs to Doxil is up to 25% of all the treated patients. Unlike an IgE-mediated (type I) allergy, these reactions occur mostly at the first exposure to the formulation without prior sensitization.123 An additional example is a harmful activation of the complement system at tumor sites, which may stimulate tumor-associated immune cells and promote their conversion into a tumor-supportive phenotype, thereby stimulating cancer progression.113,121,124
Not only are NP interactions with different cell types still not well-documented but there is also an even larger knowledge gap at the whole animal level. Adequate animal models to study the biological properties of NPs with specific organs in a whole animal are necessary. In that context, NPs that are also imaging contrast agents are advantageous. The majority of antitumor studies, for example, are conducted in immunodeficient mice (e.g., severe combined immunodeficiency, SCID, Rag-deficient mice) to allow investigations on human samples; however, these animals possess a compromised adaptive immune system that reduces the effect of antigen-presenting cell-mediated NP clearance. Nanoparticle pharmacokinetic studies should be conducted in immune-competent animals, both on those that are healthy and others that are diseased. The rapidly induced animal tumors that potentiate the enhanced permeability and retention (EPR) effect may not be representative of all human tumors; therefore, caution must be observed in predicting the translational capability of certain NPs evaluated in these models.
Since it was first reported by Matsumura and Maeda in 1986,125 the EPR effect has been utilized by different macromolecules and, among them, nanotechnology-based platforms to deliver drugs to solid tumors. The defective architecture of tumor neovasculature allows the extravasation of macromolecules over 40 kDa to the tumor interstitial space, while the ineffective lymphatic drainage allows them to remain there (Figure 7).
Figure 7.

Schematic representation of different mechanisms by which nanocarriers can deliver drugs to tumors. Multifunctional lipid-based nanoparticles (LNPs) coencapsulated with chemotherapeutic drug (orange dots) and siRNA are shown as representative nanocarriers. Passive tissue targeting is achieved by extravasation of nanoparticles (NPs) through increased permeability of the tumor vasculature and ineffective lymphatic drainage (EPR effect). Active cellular targeting (inset) can be achieved by decorating the surface of NPs with multiple targeting moieties that promote cell-specific recognition and binding. The NPs can reach different tumor subpopulations concomitantly (i.e., tumor cells and tumor “nurse-like” macrophages) to ensure maximal therapeutic effect and release their contents in close proximity to the target cells, attach to the membrane of the cell, and act as an extracellular sustained-release drug depot or internalize into the cell, introducing their payload to cell cytoplasm.
However, although several EPR-based nanomedicines are in clinical use (e.g., Doxil, Abraxane, and Marqibo), the potential therapeutic efficacy of EPR-based nanomedicines has been hampered by the heterogeneity of the EPR effect within and between different tumors and by limited experimental data from patients on the effectiveness of this mechanism as related to drug accumulation in the tumor site that is translated into real efficacy. The heterogeneity in EPR may be a major contributing factor to the limited success of nanomedicines with reductions in toxicity accompanied by the limited gains in overall survival as compared with traditional chemotherapy and small-molecule anticancer agents.126 Several methods have been proposed to augment the EPR effect, including (1) increasing blood pressure during infusion of a nanomedicine using angiotensin-II and (2) using vascular mediators such as nitroglycerin, angiotensin-converting enzyme inhibitor, or nitric oxide.127
One of the fundamental limitations in evaluating EPR is the lack of knowledge regarding which preclinical tumor models recapitulate patients with solid tumors. Several main factors affect the delivery of nanomedicines to tumors in preclinical models, such as the rate of tumor growth, vasculature, tumor environment, functional mononuclear phagocyte system (MPS), etc., and appear to vary based on the tumor model (e.g., subcutaneous xenograft, orthotopic xenograft, genetically engineered mouse model) used. The majority of antitumor studies are conducted in immune-compromised mice to enable investigations on human xenografts and recently also on a patient’s own tumor, known as tumorgraft. Nevertheless, these animals possess a compromised MPS, which could have a distinct effect on nanomedicine pharmacokinetic studies and thus pharmacokinetic studies should be conducted in immune-competent animals in order to reflect more accurately on human cancers.128 Furthermore, commonly used subcutaneous tumor xenografts possess vasculature found in very high EPR tumors independent of tumor type and thus could provide a false impression regarding the benefit of nanomedicines in most tumor settings when relying on the EPR effect.126
Future studies will need to evaluate these factors systemically in preclinical models and in patients with various solid tumors and match the most suitable preclinical model to each human tumor accordingly. Moreover, further investigations are required to understand how to assess drugs relying on the EPR effect for efficacy in preclinical tumor models and to understand how they reflect the heterogeneity seen in human disease. A promising venue for better understanding and predictability of EPR function in humans comes from the use of clinical imaging studies, which may help the development of more effective nanomedicines. In one such study, pharmacokinetics and biodistribution of 111In-labeled PEGylated liposomes were evaluated in patients with different advanced tumors. Although positive tumor images were obtained in 15 of 17 studies, the levels of tumor liposome uptake varied significantly both between and within tumor types, emphasizing the need for further comprehensive studies of EPR-based nanomedicines in patients.129
At the same time, more selective delivery strategies are being developed,130−133 utilizing active cellular targeting upon reaching the tumor vicinity (Figure 7). It is likely that these strategies will govern the new therapeutic modality for both diagnostics (molecular imaging), therapeutics (targeted, cell-specific delivery), and perhaps also theranostics.134
Acknowledgments
L.B. and R.L. thank EPSRC (EP/H046143/1) for funding. G.P. thanks the European Union (ERDF, European Regional Development Fund) via the INTERREG Upper Rhine program (Nanomatrix). C.M. acknowledges the National Science Foundation (CHE-1240151: Centre for Sustainable Nanotechnology). T.P. acknowledges the financial support of the European project Magnifyco (Contract NMP4-SL-2009-228622). D.P. acknowledges support from INNI, FTA: Nanomedicine for Personalized Theranostics, and from The Leona M. and Harry B. Helmsley Nanotechnology Research Fund. All authors thank the E-MRS for supporting Symposium Q at the Spring conference 2013 (Strasbourg, France) where the idea of this Nano Focus article was conceived.
Views expressed in this Nano Focus are those of the authors and not necessarily the views of the ACS.
The authors declare the following competing financial interest(s): D.P. has financial interests in Quiet Therapeutics.
References
- Neel L. Les Proprietes Magnetiques du Sesquioxyde de Fer Rhomboedrique. C. R. Hebd. Seances Acad. Sci. 1949, 228, 64–66. [Google Scholar]
- Bean C. P.; Livingston J. D. Superparamagnetism. J. Appl. Phys. 1959, 30, 120S–129S. [Google Scholar]
- Pankhurst Q. A.; Connolly J.; Jones S. K.; Dobson J. Applications of Magnetic Nanoparticles in Biomedicine. J. Phys. D: Appl. Phys. 2003, 36, R167–R181. [Google Scholar]
- Pankhurst Q. A.; Thanh N. T. K.; Jones S. K.; Dobson J. Progress in Applications of Magnetic Nanoparticles in Biomedicine. J. Phys. D: Appl. Phys. 2009, 42, 224001. [Google Scholar]
- Cullity B. D.Introduction to Magnetic Materials; Addison-Wesley: Reading, MA, 1972. [Google Scholar]
- Gilchrist R. K.; Medal R.; Shorey W. D.; Hanselman R. C.; Parrott J. C.; Taylor C. B. Selective Inductive Heating of Lymph Nodes. Ann. Surg. 1957, 146, 596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon R. T.; Hines J. R.; Gordon D. Intracellular Hyperthermia—Biophysical Approach to Cancer Treatment via Intracellular Temperature and Biophysical AlterationsS. Med. Hypotheses 1979, 5, 83–102. [DOI] [PubMed] [Google Scholar]
- Mejias R.; Gutierrez L.; Salas G.; Perez-Yague S.; Zotes T. M.; Lazaro F. J.; Morales M. P.; Barber D. F. Long Term Biotransformation and Toxicity of Dimercaptosuccinic Acid-Coated Magnetic Nanoparticles Support Their Use in Biomedical Applications. J. Controlled Release 2013, 171, 225–233. [DOI] [PubMed] [Google Scholar]
- Levy M.; Lagarde F.; Maraloiu V.-A.; Blanchin M.-G.; Gendron F.; Wilhelm C.; Gazeau F. Degradability of Superparamagnetic Nanoparticles in a Model of Intracellular Environment: Follow-Up of Magnetic, Structural and Chemical Properties. Nanotechnology 2010, 21, 395103. [DOI] [PubMed] [Google Scholar]
- Levy M.; Luciani N.; Alloyeau D.; Elgrabli D.; Deveaux V.; Pechoux C.; Chat S.; Wang G.; Vats N.; Gendron F.; et al. Long Term In Vivo Biotransformation of Iron Oxide Nanoparticles. Biomaterials 2011, 32, 3988–3999. [DOI] [PubMed] [Google Scholar]
- Chantrell R. W.; Popplewell J.; Charles S. W. Measurements of Particle-Size Distribution Parameters in Ferrofluids. IEEE Trans. Magn. 1978, 14, 975–977. [Google Scholar]
- Chen D. X.; Sanchez A.; Taboada E.; Roig A.; Sun N.; Gu H. C. Size Determination of Superparamagnetic Nanoparticles from Magnetization Curve. J. Appl. Phys. 2009, 105, 083924. [Google Scholar]
- El-Hilo M. Nano-Particle Magnetism with a Dispersion of Particle Sizes. J. Appl. Phys. 2012, 112, 103915. [Google Scholar]
- Elmore W. C. The Magnetization of Ferromagnetic Colloids. Phys. Rev. 1938, 54, 1092–1095. [Google Scholar]
- Levy M.; Gazeau F.; Bacri J.-C.; Wilhelm C.; Devaud M. Modeling Magnetic Nanoparticle Dipole–Dipole Interactions Inside Living Cells. Phys. Rev. B 2011, 84, 075480. [Google Scholar]
- Levy M.; Wilhelm C.; Luciani N.; Deveaux V.; Gendron F.; Luciani A.; Devaud M.; Gazeau F. Nanomagnetism Reveals the Intracellular Clustering of Iron Oxide Nanoparticles in the Organism. Nanoscale 2011, 3, 4402–4410. [DOI] [PubMed] [Google Scholar]
- Gossuin Y.; Gillis P.; Hocq A.; Vuong Q. L.; Roch A. Magnetic Resonance Relaxation Properties of Superparamagnetic Particles. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol. 2009, 1, 299–310. [DOI] [PubMed] [Google Scholar]
- Klug G.; Kampf T.; Bloemer S.; Bremicker J.; Ziener C. H.; Heymer A.; Gbureck U.; Rommel E.; Noeth U.; Schenk W. A.; et al. Intracellular and Extracellular T-1 and T-2 Relaxivities of Magneto-Optical Nanoparticles at Experimental High Fields. Magn. Reson. Med. 2010, 64, 1607–1615. [DOI] [PubMed] [Google Scholar]
- Hergt R.; Dutz S.; Muller R.; Zeisberger M. Magnetic Particle Hyperthermia: Nanoparticle Magnetism and Materials Development for Cancer Therapy. J. Phys.: Condens. Matter 2006, 18, S2919–S2934. [Google Scholar]
- Dennis C. L.; Ivkov R. Physics of Heat Generation Using Magnetic Nanoparticles for Hyperthermia. Int. J. Hyperthermia 2013, 29, 715–729. [DOI] [PubMed] [Google Scholar]
- Dutz S.; Hergt R. Magnetic Nanoparticle Heating and Heat Transfer on a Microscale: Basic Principles, Realities and Physical Limitations of Hyperthermia for Tumour Therapy. Int. J. Hyperthermia 2013, 29, 790–800. [DOI] [PubMed] [Google Scholar]
- Rosensweig R. E. Heating Magnetic Fluid with Alternating Magnetic Field. J. Magn. Magn. Mater. 2002, 252, 370–374. [Google Scholar]
- Vallejo-Fernandez G.; Whear O.; Roca A. G.; Hussain S.; Timmis J.; Patel V.; O’Grady K. Mechanisms of Hyperthermia in Magnetic Nanoparticles. J. Phys. D: Appl. Phys. 2013, 46, 312001. [Google Scholar]
- Carney R. P.; DeVries G. A.; Dubois C.; Kim H.; Kim J. Y.; Singh C.; Ghorai P. K.; Tracy J. B.; Stiles R. L.; Murray R. W.; et al. Size Limitations for the Formation of Ordered Striped Nanoparticles. J. Am. Chem. Soc. 2008, 130, 798–799. [DOI] [PubMed] [Google Scholar]
- Pons-Siepermann I. C.; Glotzer S. C. Design of Patchy Particles Using Quaternary Self-Assembled Monolayers. ACS Nano 2012, 6, 3919–3924. [DOI] [PubMed] [Google Scholar]
- Zhang Z. L.; Glotzer S. C. Self-Assembly of Patchy Particles. Nano Lett. 2004, 4, 1407–1413. [DOI] [PubMed] [Google Scholar]
- Stirling J.; Woolley R. A. J.; Moriarty P. Scanning Probe Image Wizard: A Toolbox for Automated Scanning Probe Microscopy Data Analysis. Rev. Sci. Instrum. 2013, 84, 113701. [DOI] [PubMed] [Google Scholar]
- Centrone A.; Penzo E.; Sharma M.; Myerson J. W.; Jackson A. M.; Marzari N.; Stellacci F. The Role of Nanostructure in the Wetting Behavior of Mixed-Monolayer-Protected Metal Nanoparticles. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 9886–9891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesbron Y.; Shaw C. P.; Birchall J. P.; Free P.; Levy R. Stripy Nanoparticles Revisited. Small 2012, 8, 3714–3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishler R.; Artzy-Schnirman A.; Peer E.; Wolchinsky R.; Brener R.; Waks T.; Eshhar Z.; Reiter Y.; Sivan U. Mixed Alkanethiol Monolayers on Submicrometric Gold Patterns: A Controlled Platform for Studying Cell–Ligand Interactions. Nano Lett. 2012, 12, 4992–4996. [DOI] [PubMed] [Google Scholar]
- Harkness K. M.; Balinski A.; McLean J. A.; Cliffel D. E. Nanoscale Phase Segregation of Mixed Thiolates on Gold Nanoparticles. Angew. Chem., Int. Ed. 2011, 50, 10554–10559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson A. M.; Myerson J. W.; Stellacci F. Spontaneous Assembly of Subnanometre-Ordered Domains in the Ligand Shell of Monolayer-Protected Nanoparticles. Nat. Mater. 2004, 3, 330–336. [DOI] [PubMed] [Google Scholar]
- Jiang S.; Chen Q.; Tripathy M.; Luijten E.; Schweizer K. S.; Granick S. Janus Particle Synthesis and Assembly. Adv. Mater. 2010, 22, 1060–1071. [DOI] [PubMed] [Google Scholar]
- Verma A.; Uzun O.; Hu Y.; Hu Y.; Han H.-S.; Watson N.; Chen S.; Irvine D. J.; Stellacci F. Surface-Structure-Regulated Cell-Membrane Penetration by Monolayer-Protected Nanoparticles. Nat. Mater. 2008, 7, 588–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. A.; Murphy C. J. Evidence for Patchy Lipid Layers on Gold Nanoparticle Surfaces. Langmuir 2012, 28, 5404–5416. [DOI] [PubMed] [Google Scholar]
- Yu M.; Stellacci F. Response to “Stripy Nanoparticles Revisited”. Small 2012, 8, 3720–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abraham S.; Unsworth L. D. Multi-functional Initiator and Poly(carboxybetaine methacrylamides) for Building Biocompatible Surfaces Using “Nitroxide Mediated Free Radical Polymerization” Strategies. J. Polym. Sci., Polym. Chem. 2011, 49, 1051–1060. [Google Scholar]
- Alconcel S. N. S.; Baas A. S.; Maynard H. D. FDA-Approved Poly(ethylene glycol)–Protein Conjugate Drugs. Polym. Chem. 2011, 2, 1442–1448. [Google Scholar]
- Gref R.; Luck M.; Quellec P.; Marchand M.; Dellacherie E.; Harnisch S.; Blunk T.; Muller R. H. ‘Stealth’ Corona-Core Nanoparticles Surface Modified by Polyethylene Glycol (PEG): Influences of the Corona (PEG Chain Length and Surface Density) and of the Core Composition on Phagocytic Uptake and Plasma Protein Adsorption. Colloids Surf., B 2000, 18, 301–313. [DOI] [PubMed] [Google Scholar]
- Muro E.; Pons T.; Lequeux N.; Fragola A.; Sanson N.; Lenkei Z.; Dubertret B. Small and Stable Sulfobetaine Zwitterionic Quantum Dots for Functional Live-Cell Imaging. J. Am. Chem. Soc. 2010, 132, 4556–4557. [DOI] [PubMed] [Google Scholar]
- Sawhney A. S.; Hubbell J. A. Poly(ethylene oxide)-Graft-Poly(l-lysine) Copolymers To Enhance the Biocompatibility of Poly(l-lysine)-Alginate Microcapsule Membranes. Biomaterials 1992, 13, 863–870. [DOI] [PubMed] [Google Scholar]
- Zhang L.; Cao Z.; Bai T.; Carr L.; Ella-Menye J.-R.; Irvin C.; Ratner B. D.; Jiang S. Zwitterionic Hydrogels Implanted in Mice Resist the Foreign-Body Reaction. Nat. Biotechnol. 2013, 31, 553–556. [DOI] [PubMed] [Google Scholar]
- Wang W.; Wei Q.-Q.; Wang J.; Wang B.-C.; Zhang S.-h.; Yuan Z. Role of Thiol-Containing Polyethylene Glycol (Thiol-PEG) in the Modification Process of Gold Nanoparticles (AuNPs): Stabilizer or Coagulant?. J. Colloid Interface Sci. 2013, 404, 223–229. [DOI] [PubMed] [Google Scholar]
- Andersen A. J.; Windschiegl B.; Ilbasmis-Tamer S.; Degim I. T.; Hunter A. C.; Andresen T. L.; Moghimi S. M. Complement Activation by PEG-Functionalized Multi-walled Carbon Nanotubes Is Independent of PEG Molecular Mass and Surface Density. Nanomedicine 2013, 9, 469–473. [DOI] [PubMed] [Google Scholar]
- Bradley A. J.; Test S. T.; Murad K. L.; Mitsuyoshi J.; Scott M. D. Interactions of IgM ABO Antibodies and Complement with Methoxy-PEG-Modified Human RBCs. Transfusion 2001, 41, 1225–1233. [DOI] [PubMed] [Google Scholar]
- Sroda K.; Rydlewski J.; Langner M.; Kozubek A.; Grzybek M.; Sikorski A. F. Repeated Injections of PEG-PE Liposomes Generate Anti-PEG Antibodies. Cell. Mol. Biol. Lett. 2005, 10, 37–47. [PubMed] [Google Scholar]
- Szebeni J.; Baranyi L.; Savay S.; Milosevits J.; Bunger R.; Laverman P.; Metselaar J. M.; Storm G.; Chanan-Khan A.; Liebes L.; et al. Role of Complement Activation in Hypersensitivity Reactions to Doxil and HYNICPEG Liposomes: Experimental and Clinical Studies. J. Liposome Res. 2002, 12, 165–172. [DOI] [PubMed] [Google Scholar]
- van den Hoven J. M.; Nemes R.; Metselaar J. M.; Nuijen B.; Beijnen J. H.; Storm G.; Szebeni J. Complement Activation by PEGylated Liposomes Containing Prednisolone. Eur. J. Pharm. Sci. 2013, 49, 265–271. [DOI] [PubMed] [Google Scholar]
- Aggarwal P.; Hall J. B.; McLeland C. B.; Dobrovolskaia M. A.; McNeil S. E. Nanoparticle Interaction with Plasma Proteins as It Relates to Particle Biodistribution, Biocompatibility and Therapeutic Efficacy. Adv. Drug Delivery Rev. 2009, 61, 428–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casals E.; Pfaller T.; Duschl A.; Oostingh G. J.; Puntes V. Time Evolution of the Nanoparticle Protein Corona. ACS Nano 2010, 4, 3623–3632. [DOI] [PubMed] [Google Scholar]
- Faunce T. A.; White J.; Matthael K. I. Integrated Research into the Nanoparticle–Protein Corona: A New Focus for Safe, Sustainable and Equitable Development of Nanomedicines. Nanomedicine 2008, 3, 859–866. [DOI] [PubMed] [Google Scholar]
- Cho E. C.; Zhang Q.; Xia Y. N. The Effect of Sedimentation and Diffusion on Cellular Uptake of Gold Nanoparticles. Nat. Nanotechnol. 2011, 6, 385–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oostingh G. J.; Casals E.; Italiani P.; Colognato R.; Stritzinger R.; Ponti J.; Pfaller T.; Kohl Y.; Ooms D.; Favilli F.; et al. Problems and Challenges in the Development and Validation of Human Cell-Based Assays To Determine Nanoparticle-Induced Immunomodulatory Effects. Part. Fibre Toxicol. 2011, 8, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casals E.; Vazquez-Campos S.; Bastus N. G.; Puntes V. Distribution and Potential Toxicity of Engineered Inorganic Nanoparticles and Carbon Nanostructures in Biological Systems. TrAC, Trends Anal. Chem. 2008, 27, 672–683. [Google Scholar]
- Poland C. A.; Duffin R.; Kinloch I.; Maynard A.; Wallace W. A. H.; Seaton A.; Stone V.; Brown S.; MacNee W.; Donaldson K. Carbon Nanotubes Introduced into the Abdominal Cavity of Mice Show Asbestos-like Pathogenicity in a Pilot Study. Nat. Nanotechnol. 2008, 3, 423–428. [DOI] [PubMed] [Google Scholar]
- Goy-Lopez S.; Juarez J.; Alatorre-Meda M.; Casals E.; Puntes V. F.; Taboada P.; Mosquera V. Physicochemical Characteristics of Protein-NP Bioconjugates: The Role of Particle Curvature and Solution Conditions on Human Serum Albumin Conformation and Fibrillogenesis Inhibition. Langmuir 2012, 28, 9113–9126. [DOI] [PubMed] [Google Scholar]
- Comenge J.; Sotelo C.; Romero F.; Gallego O.; Barnadas A.; Parada T. G. C.; Dominguez F.; Puntes V. F. Detoxifying Antitumoral Drugs via Nanoconjugation: The Case of Gold Nanoparticles and Cisplatin. PLoS One 2012, 7, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenzer S.; Docter D.; Kuharev J.; Musyanovych A.; Fetz V.; Hecht R.; Schlenk F.; Fischer D.; Kiouptsi K.; Reinhardt C.; et al. Rapid Formation of Plasma Protein Corona Critically Affects Nanoparticle Pathophysiology. Nat. Nanotechnol. 2013, 8, 772–781. [DOI] [PubMed] [Google Scholar]
- Salvati A.; Pitek A. S.; Monopoli M. P.; Prapainop K.; Bombelli F. B.; Hristov D. R.; Kelly P. M.; Aberg C.; Mahon E.; Dawson K. A. Transferrin-Functionalized Nanoparticles Lose Their Targeting Capabilities When a Biomolecule Corona Adsorbs on the Surface. Nat. Nanotechnol. 2013, 8, 137–143. [DOI] [PubMed] [Google Scholar]
- Maiorano G.; Sabella S.; Sorce B.; Brunetti V.; Malvindi M. A.; Cingolani R.; Pompa P. P. Effects of Cell Culture Media on the Dynamic Formation of Protein-Nanoparticle Complexes and Influence on the Cellular Response. ACS Nano 2010, 4, 7481–7491. [DOI] [PubMed] [Google Scholar]
- Hamad I.; Al-Hanbali O.; Hunter A. C.; Rutt K. J.; Andresen T. L.; Moghimi S. M. Distinct Polymer Architecture Mediates Switching of Complement Activation Pathways at the Nanosphere–Serum Interface: Implications for Stealth Nanoparticle Engineering. ACS Nano 2010, 4, 6629–6638. [DOI] [PubMed] [Google Scholar]
- Sim R. B.; Wallis R. Surface Properties: Immune Attack on Nanoparticles. Nat. Nanotechnol. 2011, 6, 80–81. [DOI] [PubMed] [Google Scholar]
- Nel A. E.; Madler L.; Velegol D.; Xia T.; Hoek E. M. V.; Somasundaran P.; Klaessig F.; Castranova V.; Thompson M. Understanding Biophysicochemical Interactions at the Nano–Bio Interface. Nat. Mater. 2009, 8, 543–557. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S.; Bhattacharya R.; Curley S.; McNiven M. A.; Mukherjee P. Nanoconjugation Modulates the Trafficking and Mechanism of Antibody Induced Receptor Endocytosis. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 14541–14546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z. J.; Liang M. T.; Monteiro M.; Toth I.; Minchin R. F. Nanoparticle-Induced Unfolding of Fibrinogen Promotes Mac-1 Receptor Activation and Inflammation. Nat. Nanotechnol. 2011, 6, 39–44. [DOI] [PubMed] [Google Scholar]
- Bastus N. G.; Sanchez-Tillo E.; Pujals S.; Farrera C.; Lopez C.; Giralt E.; Celada A.; Lloberas J.; Puntes V. Homogeneous Conjugation of Peptides onto Gold Nanoparticles Enhances Macrophage Response. ACS Nano 2009, 3, 1335–1344. [DOI] [PubMed] [Google Scholar]
- Choi H. S.; Liu W.; Misra P.; Tanaka E.; Zimmer J. P.; Ipe B. I.; Bawendi M. G.; Frangioni J. V. Renal Clearance of Quantum Dots. Nat. Biotechnol. 2007, 25, 1165–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H. S.; Liu W. H.; Liu F. B.; Nasr K.; Misra P.; Bawendi M. G.; Frangioni J. V. Design Considerations for Tumour-Targeted Nanoparticles. Nat. Nanotechnol. 2010, 5, 42–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanan-Khan A.; Szebeni J.; Savay S.; Liebes L.; Rafique N. M.; Alving C. R.; Muggia F. M. Complement Activation Following First Exposure to PEGylated Liposomal Doxorubicin (Doxil): Possible Role in Hypersensitivity Reactions. Ann. Oncol. 2003, 14, 1430–1437. [DOI] [PubMed] [Google Scholar]
- Szebeni J. Complement Activation-Related Pseudoallergy: A New Class of Drug-Induced Acute Immune Toxicity. Toxicology 2005, 216, 106–121. [DOI] [PubMed] [Google Scholar]
- Knop K.; Hoogenboom R.; Fischer D.; Schubert U. S. Poly(ethylene glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives. Angew. Chem., Int. Ed. 2010, 49, 6288–6308. [DOI] [PubMed] [Google Scholar]
- Pelaz B.; Jaber S.; de Aberasturi D. J.; Wulf V.; Aida T.; de la Fuente J. M.; Feldmann J.; Gaub H. E.; Josephson L.; Kagan C. R.; et al. The State of Nanoparticle-Based Nanoscience and Biotechnology: Progress, Promises, and Challenges. ACS Nano 2012, 6, 8468–8483. [DOI] [PubMed] [Google Scholar]
- Lee S. H.; Castagner B.; Leroux J. C. Is There a Future for Cell-Penetrating Peptides in Oligonucleotide Delivery?. Eur. J. Pharm. Biopharm. 2013, 85, 5–11. [DOI] [PubMed] [Google Scholar]
- Bechara C.; Sagan S. Cell-Penetrating Peptides: 20 Years Later, Where Do We Stand?. FEBS Lett. 2013, 587, 1693–1702. [DOI] [PubMed] [Google Scholar]
- Iversen T.-G.; Skotland T.; Sandvig K. Endocytosis and Intracellular Transport of Nanoparticles: Present Knowledge and Need for Future Studies. Nano Today 2011, 6, 176–185. [Google Scholar]
- Verma A.; Uzun O.; Hu Y.; Hu Y.; Han H.-S.; Watson N.; Chen S.; Irvine D. J.; Stellacci F. Surface-Structure-Regulated Cell-Membrane Penetration by Monolayer-Protected Nanoparticles. Nat. Mater. 2013, 12, 376–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilleron J.; Querbes W.; Zeigerer A.; Borodovsky A.; Marsico G.; Schubert U.; Manygoats K.; Seifert S.; Andree C.; Stoeter M.; et al. Image-Based Analysis of Lipid Nanoparticle-Mediated siRNA Delivery, Intracellular Trafficking and Endosomal Escape. Nat. Biotechnol. 2013, 31, 638–646. [DOI] [PubMed] [Google Scholar]
- Sahay G.; Querbes W.; Alabi C.; Eltoukhy A.; Sarkar S.; Zurenko C.; Karagiannis E.; Love K.; Chen D.; Zoncu R.; et al. Efficiency of siRNA Delivery by Lipid Nanoparticles Is Limited by Endocytic Recycling. Nat. Biotechnol. 2013, 31, 653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsico G.Response to comment on Gilleron et al. In PubPeer, http://pubpeer.com/publications/BECD9A153DB5564060F49D684588B4/comments/2063, n.d., Retrieved October 8, 2013. [Google Scholar]
- Li Y.; Li X.; Li Z.; Gao H. Surface-Structure-Regulated Penetration of Nanoparticles Across a Cell Membrane. Nanoscale 2012, 4, 3768–3775. [DOI] [PubMed] [Google Scholar]
- Gkeka P.; Sarkisov L.; Angelikopoulos P. Homogeneous Hydrophobic–Hydrophilic Surface Patterns Enhance Permeation of Nanoparticles through Lipid Membranes. J. Phys. Chem. Lett. 2013, 4, 1907–1912. [DOI] [PubMed] [Google Scholar]
- Schulz M.; Olubummo A.; Binder W. H. Beyond the Lipid-Bilayer: Interaction of Polymers and Nanoparticles with Membranes. Soft Matter 2012, 8, 4849–4864. [Google Scholar]
- Binks B. P. Particles as Surfactants—Similarities and Differences. Curr. Opin. Colloid Interface Sci. 2002, 7, 21–41. [Google Scholar]
- Baronzio G. F.; Hager E. D.. Hyperthermia in Cancer Treatment: A Primer; Landes Bioscience and Springer Science+Business Media, LLC: New York, 2006. [Google Scholar]
- Song C. W. Effect of Local Hyperthermia on Blood Flow and Microenvironment. A Review. Cancer Res. 1984, 44, 4721S–4730S. [PubMed] [Google Scholar]
- Huang X. H.; El-Sayed I. H.; Qian W.; El-Sayed M. A. Cancer Cell Imaging and Photothermal Therapy in the Near-Infrared Region by Using Gold Nanorods. J. Am. Chem. Soc. 2006, 128, 2115–2120. [DOI] [PubMed] [Google Scholar]
- Hirsch L. R.; Stafford R. J.; Bankson J. A.; Sershen S. R.; Rivera B.; Price R. E.; Hazle J. D.; Halas N. J.; West J. L. Nanoshell-Mediated Near-Infrared Thermal Therapy of Tumors under Magnetic Resonance Guidance. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 13549–13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardhan R.; Lal S.; Joshi A.; Halas N. J. Theranostic Nanoshells: From Probe Design to Imaging and Treatment of Cancer. Acc. Chem. Res. 2011, 44, 936–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goya G. F.; Asin L.; Ibarra M. R. Cell Death Induced by AC Magnetic Fields and Magnetic Nanoparticles: Current State and Perspectives. Int. J. Hyperthermia 2013, 29, 810–818. [DOI] [PubMed] [Google Scholar]
- Creixell M.; Bohorquez A. C.; Torres-Lugo M.; Rinaldi C. EGFR-Targeted Magnetic Nanoparticle Heaters Kill Cancer Cells without a Perceptible Temperature Rise. ACS Nano 2011, 5, 7124–7129. [DOI] [PubMed] [Google Scholar]
- N’Guyen T. T. T.; Duong H. T. T.; Basuki J.; Montembault V.; Pascual S.; Guibert C.; Fresnais J.; Boyer C.; Whittaker M. R.; Davis T. P.; et al. Functional Iron Oxide Magnetic Nanoparticles with Hyperthermia-Induced Drug Release Ability by Using a Combination of Orthogonal Click Reactions. Angew. Chem., Int. Ed. 2013, 52, 14152–14156. [DOI] [PubMed] [Google Scholar]
- Riedinger A.; Guardia P.; Curcio A.; Garcia M. A.; Cingolani R.; Manna L.; Pellegrino T. Subnanometer Local Temperature Probing and Remotely Controlled Drug Release Based on Azo-Functionalized Iron Oxide Nanoparticles. Nano Lett. 2013, 13, 2399–2406. [DOI] [PubMed] [Google Scholar]
- Yoo D.; Jeong H.; Noh S. H.; Lee J. H.; Cheon J. Magnetically Triggered Dual Functional Nanoparticles for Resistance-Free Apoptotic Hyperthermia. Angew. Chem., Int. Ed. 2013, 52, 13047–13051. [DOI] [PubMed] [Google Scholar]
- Dias J. T.; Moros M.; Del Pino P.; Rivera S.; Grazu V.; de la Fuente J. M. DNA as a Molecular Local Thermal Probe for the Analysis of Magnetic Hyperthermia. Angew. Chem., Int. Ed. 2013, 52, 11526–11529. [DOI] [PubMed] [Google Scholar]
- Hergt R.; Dutz S. Magnetic Particle Hyperthermia: Biophysical Limitations of a Visionary Tumour Therapy. J. Magn. Magn. Mater. 2007, 311, 187–192. [Google Scholar]
- Li T. J.; Huang C. C.; Ruan P. W.; Chuang K. Y.; Huang K. J.; Shieh D. B.; Yeh C. S. In Vivo Anti-cancer Efficacy of Magnetite Nanocrystal-Based System Using Locoregional Hyperthermia Combined with 5-Fluorouracil Chemotherapy. Biomaterials 2013, 34, 7873–7883. [DOI] [PubMed] [Google Scholar]
- Derfus A. M.; von Maltzahn G.; Harris T. J.; Duza T.; Vecchio K. S.; Ruoslahti E.; Bhatia S. N. Remotely Triggered Release from Magnetic Nanoparticles. Adv. Mater. 2007, 19, 3932–3936. [Google Scholar]
- Maier-Hauff K.; Rothe R.; Scholz R.; Gneveckow U.; Wust P.; Thiesen B.; Feussner A.; von Deimling A.; Waldoefner N.; Felix R.; et al. Intracranial Thermotherapy Using Magnetic Nanoparticles Combined with External Beam Radiotherapy: Results of a Feasibility Study on Patients with Glioblastoma Multiforme. J. Neuro-Oncol. 2007, 81, 53–60. [DOI] [PubMed] [Google Scholar]
- Thiesen B.; Jordan A. Clinical Applications of Magnetic Nanoparticles for Hyperthermia. Int. J. Hyperthermia 2008, 24, 467–474. [DOI] [PubMed] [Google Scholar]
- Liu J.; Detrembleur C.; De Pauw-Gillet M. C.; Mornet S.; Vander Elst L.; Laurent S.; Jerome C.; Duguet E. Heat-Triggered Drug Release Systems Based on Mesoporous Silica Nanoparticles Filled with a Maghemite Core and Phase-Change Molecules as Gatekeepers. J. Mater. Chem. B 2014, 2, 59–70. [DOI] [PubMed] [Google Scholar]
- Lee J. H.; Chen K. J.; Noh S. H.; Garcia M. A.; Wang H.; Lin W. Y.; Jeong H.; Kong B. J.; Stout D. B.; Cheon J.; et al. On-Demand Drug Release System for In Vivo Cancer Treatment through Self-Assembled Magnetic Nanoparticles. Angew. Chem., Int. Ed. 2013, 52, 4384–4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Hernandez E.; Baeza A.; Vallet-Regi M. Smart Drug Delivery through DNA/Magnetic Nanoparticle Gates. ACS Nano 2011, 5, 1259–1266. [DOI] [PubMed] [Google Scholar]
- Cheng K.; Peng S.; Xu C. J.; Sun S. H. Porous Hollow Fe3O4 Nanoparticles for Targeted Delivery and Controlled Release of Cisplatin. J. Am. Chem. Soc. 2009, 131, 10637–10644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George C.; Dorfs D.; Bertoni G.; Falqui A.; Genovese A.; Pellegrino T.; Roig A.; Quarta A.; Comparelli R.; Curri M. L.; et al. A Cast-Mold Approach to Iron Oxide and Pt/Iron Oxide Nanocontainers and Nanoparticles with a Reactive Concave Surface. J. Am. Chem. Soc. 2011, 133, 2205–2217. [DOI] [PubMed] [Google Scholar]
- Shin J. M.; Anisur R. M.; Ko M. K.; Im G. H.; Lee J. H.; Lee I. S. Hollow Manganese Oxide Nanoparticles as Multifunctional Agents for Magnetic Resonance Imaging and Drug Delivery. Angew. Chem., Int. Ed. 2009, 48, 321–324. [DOI] [PubMed] [Google Scholar]
- Liu H. L.; Hua M. Y.; Yang H. W.; Huang C. Y.; Chu P. C.; Wu J. S.; Tseng I. C.; Wang J. J.; Yen T. C.; Chen P. Y.; et al. Magnetic Resonance Monitoring of Focused Ultrasound/Magnetic Nanoparticle Targeting Delivery of Therapeutic Agents to the Brain. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 15205–15210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J. H.; Liang G. L.; Cheung J. S.; Pan Y.; Kuang Y.; Zhao F.; Zhang B.; Zhang X. X.; Wu E. X.; Xu B. Multifunctional Yolk–Shell Nanoparticles: A Potential MRI Contrast and Anticancer Agent. J. Am. Chem. Soc. 2008, 130, 11828–11833. [DOI] [PubMed] [Google Scholar]
- Gao J. H.; Liang G. L.; Zhang B.; Kuang Y.; Zhang X. X.; Xu B. FePt@CoS2 Yolk–Shell Nanocrystals as a Potent Agent To Kill HeLa Cells. J. Am. Chem. Soc. 2007, 129, 1428–1433. [DOI] [PubMed] [Google Scholar]
- Lartigue L.; Alloyeau D.; Kolosnjaj-Tabi J.; Javed Y.; Guardia P.; Riedinger A.; Pechoux C.; Pellegrino T.; Wilhelm C.; Gazeaut F. Biodegradation of Iron Oxide Nanocubes: High-Resolution In Situ Monitoring. ACS Nano 2013, 7, 3939–3952. [DOI] [PubMed] [Google Scholar]
- Landesman-Milo D.; Peer D. Altering the Immune Response with Lipid-Based Nanoparticles. J. Controlled Release 2012, 161, 600–608. [DOI] [PubMed] [Google Scholar]
- Peer D. Immunotoxicity Derived from Manipulating Leukocytes with Lipid-Based Nanoparticles. Adv. Drug Delivery Rev. 2012, 64, 1738–1748. [DOI] [PubMed] [Google Scholar]
- Szebeni J.; Moghimi S. M. Liposome Triggering of Innate Immune Responses: A Perspective on Benefits and Adverse Reactions. J. Liposome Res. 2009, 19, 85–90. [DOI] [PubMed] [Google Scholar]
- Szebeni J.; Muggia F.; Gabizon A.; Barenholz Y. Activation of Complement by Therapeutic Liposomes and Other Lipid Excipient-Based Therapeutic Products: Prediction and Prevention. Adv. Drug Delivery Rev. 2011, 63, 1020–1030. [DOI] [PubMed] [Google Scholar]
- Landesman-Milo D.; Peer D. Altering the Immune Response with Lipid-Based Nanoparticles. J. Controlled Release 2012, 161, 600–608. [DOI] [PubMed] [Google Scholar]
- Haber E.; Afergan E.; Epstein H.; Gutman D.; Koroukhov N.; Ben-David M.; Schachter M.; Golomb G. Route of Administration-Dependent Anti-inflammatory Effect of Liposomal Alendronate. J. Controlled Release 2010, 148, 226–233. [DOI] [PubMed] [Google Scholar]
- Kedmi R.; Ben-Arie N.; Peer D. The Systemic Toxicity of Positively Charged Lipid Nanoparticles and the Role of Toll-like Receptor 4 in Immune Activation. Biomaterials 2010, 31, 6867–6875. [DOI] [PubMed] [Google Scholar]
- Lonez C.; Lensink M. F.; Vandenbranden M.; Ruysschaert J. M. Cationic Lipids Activate Cellular Cascades. Which Receptors Are Involved?. Biochim. Biophys. Acta 2009, 1790, 425–430. [DOI] [PubMed] [Google Scholar]
- Moghimi S. M.; Peer D.; Langer R. Reshaping the Future of Nanopharmaceuticals ad Ludicium. ACS Nano 2011, 5, 8454–8458. [DOI] [PubMed] [Google Scholar]
- Moghimi S. M.; Hunter A. C. Recognition by Macrophages and Liver Cells of Opsonized Phospholipid Vesicles and Phospholipid Headgroups. Pharm. Res. 2001, 18, 1–8. [DOI] [PubMed] [Google Scholar]
- Goldsmith M.; Mizrahy S.; Peer D. Grand Challenges in Modulating the Immune Response with RNAi Nanomedicines. Nanomedicine 2011, 6, 1771–1785. [DOI] [PubMed] [Google Scholar]
- Zolnik B. S.; Gonzalez-Fernandez A.; Sadrieh N.; Dobrovolskaia M. A. Nanoparticles and the Immune System. Endocrinology 2010, 151, 458–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szebeni J.; Alving C. R.; Rosivall L.; Bunger R.; Baranyi L.; Bedocs P.; Toth M.; Barenholz Y. Animal Models of Complement-Mediated Hypersensitivity Reactions to Liposomes and Other Lipid-Based Nanoparticles. J. Liposome Res. 2007, 17, 107–117. [DOI] [PubMed] [Google Scholar]
- Chanan-Khan A.; Szebeni J.; Savay S.; Liebes L.; Rafique N. M.; Alving C. R.; Muggia F. M. Complement Activation Following First Exposure to PEGylated Liposomal Doxorubicin (Doxil): Possible Role in Hypersensitivity Reactions. Ann. Oncol. 2003, 14, 1430–1437. [DOI] [PubMed] [Google Scholar]
- Szebeni J.; Baranyi L.; Savay S.; Milosevits J.; Bunger R.; Laverman P.; Metselaar J. M.; Storm G.; Chanan-Khan A.; Liebes L.; et al. Role of Complement Activation in Hypersensitivity Reactions to Doxil and Hynic PEG Liposomes: Experimental and Clinical Studies. J. Liposome Res. 2002, 12, 165–172. [DOI] [PubMed] [Google Scholar]
- Matsumura Y.; Maeda H. A New Concept for Macromolecular Therapeutics in Cancer Chemotherapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986, 46, 6387–6392. [PubMed] [Google Scholar]
- Prabhakar U.; Maeda H.; Jain R. K.; Sevick-Muraca E. M.; Zamboni W.; Farokhzad O. C.; Barry S. T.; Gabizon A.; Grodzinski P.; Blakey D. C. Challenges and Key Considerations of the Enhanced Permeability and Retention Effect for Nanomedicine Drug Delivery in Oncology. Cancer Res. 2013, 73, 2412–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda H.; Nakamura H.; Fang J. The EPR Effect for Macromolecular Drug Delivery to Solid Tumors: Improvement of Tumor Uptake, Lowering of Systemic Toxicity, and Distinct Tumor Imaging in Vivo. Adv. Drug Delivery Rev. 2013, 65, 71–79. [DOI] [PubMed] [Google Scholar]
- Howard K. A.; Peer D. Providing the Full Picture: A Mandate for Standardizing Nanoparticle-Based Drug Delivery. Nanomedicine 2013, 8, 1031–1033. [DOI] [PubMed] [Google Scholar]
- Harrington K. J.; Mohammadtaghi S.; Uster P. S.; Glass D.; Peters A. M.; Vile R. G.; Stewart J. S. Effective Targeting of Solid Tumors in Patients with Locally Advanced Cancers by Radiolabeled PEGylated Liposomes. Clin. Cancer Res. 2001, 7, 243–254. [PubMed] [Google Scholar]
- Peer D.; Karp J. M.; Hong S.; FaroKhzad O. C.; Margalit R.; Langer R. Nanocarriers as an Emerging Platform for Cancer Therapy. Nat. Nanotechnol. 2007, 2, 751–760. [DOI] [PubMed] [Google Scholar]
- Davis M. E.; Zuckerman J. E.; Choi C. H.; Seligson D.; Tolcher A.; Alabi C. A.; Yen Y.; Heidel J. D.; Ribas A. Evidence of RNAi in Humans from Systemically Administered siRNA via Targeted Nanoparticles. Nature 2010, 464, 1067–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrkach J.; Von Hoff D.; Mukkaram Ali M.; Andrianova E.; Auer J.; Campbell T.; De Witt D.; Figa M.; Figueiredo M.; Horhota A.; et al. Preclinical Development and Clinical Translation of a PSMA-Targeted Docetaxel Nanoparticle with a Differentiated Pharmacological Profile. Sci. Transl. Med. 2012, 4, 128ra39. [DOI] [PubMed] [Google Scholar]
- Peer D.; Park E. J.; Morishita Y.; Carman C. V.; Shimaoka M. Systemic Leukocyte-Directed siRNA Delivery Revealing Cyclin D1 as an Anti-inflammatory Target. Science 2008, 319, 627–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinav E.; Peer D. Harnessing Nanomedicine for Mucosal Theranostics—A Silver Bullet at Last?. ACS Nano 2013, 7, 2883–2890. [DOI] [PubMed] [Google Scholar]