

Abstract
The measles virus (MV) vaccine lineage is a promising oncolytic but prior exposure to the measles vaccine or wild-type MV strains limits treatment utility due to the presence of anti-measles antibodies. MV entry can be redirected by displaying a polypeptide ligand on the Hemagglutinin (H) C-terminus. We hypothesized that retargeted MV would escape neutralization by monoclonal antibodies (mAbs) recognizing the H receptor-binding surface and be less susceptible to neutralization by human antisera. Using chimeric H proteins, with and without mutations that ablate MV receptor binding, we show that retargeted MVs escape mAbs that target the H receptor-binding surface by virtue of mutations that ablate infection via SLAM and CD46. However, C-terminally displayed domains do not mediate virus entry in the presence of human antibodies that bind to the underlying H domain. In conclusion, utility of retargeted oncolytic measles viruses does not extend to evasion of human serum neutralization.
Keywords: Measles Virus, virus retargeting, neutralization, monoclonal antibodies, human serum, oncolytic virotherapy, scFv, ligand display, cancer
INTRODUCTION
Oncolytic virotherapy is an emerging treatment modality for cancer, which exploits viruses that preferentially infect and kill cancer cells. These oncolytic viruses include naturally occurring viruses and viruses that have been engineered for tumor selectivity [1–3]. Oncolytic measles virus (MV) vaccine strains, in particular a laboratory adapted strain of Edmonston vaccine lineage (MV-Edm), has demonstrated therapeutic potential against different solid tumors and hematologic malignancies such as hepatocellular carcinoma [4], breast cancer [5, 6], prostate cancer [7, 8], ovarian cancer [9, 10], multiple myeloma [11, 12], lymphoma [13] and glioblastoma multiforme [14] in preclinical studies. MV-Edm is also being tested clinically for the treatment of multiple myeloma (100), ovarian cancer [15][101], glioblastoma multiforme [102] and mesothelioma [103].
MV is an enveloped, negative-strand RNA virus of the family Paramyxoviridae [16]. MV-Edm has a tropism for three cellular receptors: The signaling lymphocyte activating molecule (SLAM), expressed on activated T and B cells and macrophages [17–20]; Nectin-4, a cellular adhesion molecule expressed in the placenta, trachea, oral mucosa, nasopharynx, and lungs [21, 22] and over expressed on several types of cancer [23–25] and CD46 which is a cellular receptor for laboratory-adapted MV strains [26]. CD46 is a regulator of complement activation [26, 27] that is ubiquitously expressed on all human nucleated cells and over expressed on many different cancer cell types making them highly susceptible to MV-Edm infection and its cytopathic effects [28].
MV-Edm can be retargeted to specific tumor cells by linking a single-chain antibody (single chain fragment variable, scFv) or naturally occurring ligand to the virus attachment hemagglutinin (H) glycoprotein displayed on the virus surface. The ablation of receptor CD46 and SLAM binding sites limits virus attachment and entry to cells expressing the receptor for the scFv or ligand linked to H. Retargeted MV-Edm derivatives retain their oncolytic activity against xenografts expressing target receptors [29–37]. A variety of scFv’s have been displayed on H against different receptors: EGFR (epidermal growth factor receptor) [29, 31]; EGFRvIII [29, 32]; HER2/neu (HER2: Human Epidermal Growth Factor Receptor 2) [38], CD20 [36, 37]; folate receptor alpha [33]; CD38 [29]; CEA (carcinoembryonic antigen) [39], prostate-specific membrane antigen (PSMA) [40] and an unidentified receptor over-expressed on multiple myeloma cells that can be targeted by Wue scFv [35]. Ligands linked to H have also successfully redirected entry, for example: amino-terminal fragment of urokinase plasminogen activator (uPA) targeting uPA receptor on breast tumors and tumor stroma [34]; snake venom peptide echistatin, targeting integrins αvβ3 and α5β1 expressed on vascular endothelium [41]; single-chain T-cell receptor (scTCR) targeting a specific peptide/MHC complex [42] and interleukin-13 targeting gliomas [30].
One of the major hurdles for oncolytic virotherapy is pre-existing immunity against the oncolytic virus [43, 44]. Measles oncolytic virotherapy is limited by preexisting immunity due to widespread global vaccination against measles [45]. The hemaggluntinin attachment protein is the major target for neutralizing antibodies [46] that tend to cluster at the receptor binding surface targeting a conserved neutralizing antigenic region [47–51]. Retargeted MV derivatives have two modifications that could potentially destroy or shield epitopes within the receptor-binding surface. The first modification is a set of two (Y481A and R533A) or four (Y481A, R533A, S548L and F549S) mutations that ablate infection via CD46 and SLAM [29]. The second modification is the scFv or ligand linked to the H C-terminus used to retarget MV to specific receptors. This additional polypeptide domain could shield one or more antibody epitopes and protect the virus from neutralization [52]. Should the utility of retargeted oncolytic MVs extend to evasion of serum neutralization it would render them superior to MV derivatives currently tested clinically.
In this study we used chimeric H proteins with and without mutations that ablate MV receptor binding to determine if these mutations protect MV-Edm from mAbs targeting the mutated receptor-binding surface. We investigated if the displayed domain can shield mAb epitope(s) and if the size of the domain determines how well an epitope is protected. We then addressed the question if retargeted MV derivatives evade human serum neutralization, since entry is no longer dependent on H binding MV receptors, but is mediated by a separate polypeptide domain attached to the H C-terminus by a linker. Our data demonstrate that mutations that ablate CD46 and SLAM binding protect retargeted MV from mAbs targeting the receptor binding-surface but not from human serum neutralization. The displayed domain provided no significant additional protection from neutralizing antibodies tested.
MATERIALS AND METHODS
Cell Culture
Retargeted MVs were propagated and titered on Vero Cells (African green monkey kidney cells) stably expressing membrane-anchored single-chain antibody that recognizes a six-histidine peptide (Vero-His), described previously [29, 53]. Vero-His cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) with 5% Fetal Bovine Serum (FBS). Culture media was supplemented with 1% penicillin and streptomycin (P/S) and cells were grown at 37°C in a humidified atmosphere of 5% CO2.
Cloning of MV-H82.αEGFR
First, the H mutant (H82) was generated by engineering escape mutations in the Hemagglutin (H) gene (Accession AB583749) encoded in the pCG plasmid (pCG-H). Mutations in H82 are as follows: L284S, E395K, E398G, E535N, H536A, A537T, Y310C, D416N, S546N, R547A, S550T, F552N, Y553G, P554T, S590N, G592S. Mutations were added sequentially by site-directed mutagenesis using the QuikChange® Lightning Site-Directed Mutagenesis Kit (Agilent Technologies) as per manufacture’s directions. The IEGR-AAQPA-EGFR.scFv-AAA-RGSHHHHHH C-terminal linker and EGFR.scFv domain, was added to H82 by digesting pCG-H82 and pTN-H6-Haa-αEGFR [29] with Pac1 and PvuII-HF (New England biolabs). The H fragment from pCG-H82 replaced the H fragment in pTN-H6-Haa-αEGFR to generate pTN-H82-αEGFR. P+MV-H82.αEGFR was then generated by replacing the H gene in p+MV-eGFP, with H82.αEGFR by digesting p+MVeGFP and pTN-H82-αEGFR with restriction enzymes Pac1 and Spe1 (New England biolabs).
Plasmids, virus rescue and titration
Plasmids encoding full-length MV recombinant derivatives used for virus rescue are as follows: p+MV-eGFP [54], p+MV-H-αWue1, p+MV-HAALS-αWue1, p+MV-H-echistatin [41], p+MV-HAALS-echistatin, p+MV-HAALS.αEGFR and p+MV-HAALS [29]. EGFR.scFv, Wue1.scFv and echistatin were inserted into SfiI/NotI sites of pTNH6-H or pTNH6-HAALS. Then, the PacI/SpeI fragment of all H constructs was inserted into the corresponding sites of p (+)MVeEGFP as described in [29]. MV rescue system used for retargeted MV was described previously in [29, 55]. Titers were determined by 50% tissue culture infective dose (TCID50) on Vero-His cells. MV-eGFP is referred to in the text as MV-Hedm.
Immunoblot Analysis
Viruses (1×105 PFU) were boiled in SDS loading buffer for 5 min at 95°C and separated on a 7.5% Tris-glycine SDS-polyacrylamide gel. Proteins were blotted to nitrocellulose membrane, immunoblotted with primary antibody: anti-rabbit anti- MV H protein (1:6000 dilution) (made by K.W. Peng, Mayo Clinic) and secondary antibody: HRP-conjugated goat anti-rabbit (1:5000 dilution).
MV mAb Neutralization Assay
Due to limited amounts of Monoclonal antibodies (mAbs) the viruses were assayed in duplicate wells and the experiment was repeated three times. The neutralizing concentration for each mAb was first determined by titration. Neutralizing concentrations used for the Neutralization Assay were: 16DE6 (1.5ul ascites/well), 16CD11 (2ul ascites/well), I-41 (1ul ascites/well), I-44 (1.5ul ascites/well), I-29 (6ul ascites/well), BH97 (2.5ug/well), cl48 (1ug/well), 20H6 (50ul hybridoma supernatant). Each mAbs was added to a total volume of 50ul Opti-MEM per well. This was mixed with equal volume of MV-Hedm or retargeted MV derivatives (4000TCID50/ml - which would yield about 30 syncytia/well 44hours post infection in the absence of mAbs). We aim for 30 syncytia/well because based on experience more than 40 syncytia/well following MV-Hedm (control) infection makes it very difficult to count individual infectious foci due to overlap between expanding syncytia. In the case of mAb 20H6, 50ul of hybridoma supernatant was mixed with equal volume of virus. Infection in the absence of monoclonal antibody (no mAb control), was determined by mixing the virus with equal volume of Opti-MEM. The mixture was incubated for 1hour at 37°C before the addition of Vero-His cells (10,000 cells/well in 50ul DMEM 5% FBS). Forty-four hours post infection the number of eGFP expressing syncytia/well in the absence and presence of mAbs were counted under a fluorescent microscope. The average number of infectious foci in the absence of mAb was equated to 100% and the infection in the presence of mAbs was presented as a percentage of infection in the absence of mAb.
Cell-Cell fusion Assay
MV-eGFP (Hedm) and retargeted MV derivatives were diluted to 4000TCID50/ml in Opti-MEM. 50ul of virus was added per well in a 96 well format. Vero-His cells in DMEM 5% FBS were then added to the viruses (10 000 cells/well) and the plates were incubated at 37°C. Twelve to thirteen hours post infection, mAbs (BH97, 20H6, I-41, 16DE6) were diluted as described above in the “MV mAb neutralization assay”. The media with virus was removed from the cells and 50ul of the diluted mAbs was added to the cells followed by 50ul fresh DMEM 5%. Fourty-four hours post infection the average syncytia size was quantified by counting the number of nuclei per syncytium in 20 syncytia per well and allocating a score of +/− (≤ 5 nuclei/cells per syncytium), + (6–20 nuclei/cells per syncytium), ++ (21–50 nuclei/cells per syncytium), or +++ (>50 nuclei/cells per syncytium) [56]. This was done at a magnification of 20x using fluorescence and phase-contrast microscopy. The data is represented as the percentage of syncytia of a particular size in the absence (no mAb) of presence of mAbs targeting the receptor-binding interface.
Structural Modeling
Models of the hemagglutinin protein were generated using crystal structure of measles virus hemagglutinin protein alone (PDB 2ZB5) [47], in complex with CD46 (PDB 3INB) [49] or SLAM (3ALZ) [48]. The N-linked complex-sugar model of MV hemagglutinin globular head was built manually by using the complex-type penta-antennary sugars whose coordinates were obtained from the web site http://www.glycosciences.de [47, 48]. A representative structure of a single chain antibody fragment of the same size as Wue and EGFR scFv with a C-terminal six Histidine tag was added manually using scFv-against-IL-1β coordinates [57]. Crystal structures were analyzed and manipulated using Educational-use-only PyMOL software. (http://pymol.org/educational).
Monoclonal Antibodies
Monoclonal antibody BH97 was a kind gift from Dr. Claude Muller, Laboratoire National de Sante, Luxemburg. Antibodies 16DE6, 16CD11, I-41, I-44, I-29 [58] were gifts from Erling Norrby and Mariethe Ehnlund, Karolinska Institute, Sweden. MAb cl48 [59] was gifted by Guy Griesmann and Mark Federspiel at the Mayo Clinic, USA. Antibody 20H6 [60] was gifted by Ianko Iankov and Eva Galanis, Mayo Clinic, USA.
Plaque Reduction neutralization Test
Human serum used for this study was as follows: Heat-inactivated pooled human serum from ~150 male donors (Human AB serum – sterile filtered, Valley Biomedical, product number HS1017, lot number C80553). Individual serum from five males diagnosed with malignant neoplasms, was provided by the Mayo Clinic Cancer/Normal Serum Bank (IRB Application #: 13-002217). Serum was diluted in 2 or 4 fold serial dilutions in Opti-MEM (Gibco, Invitrogen). 50ul of each serum dilution was added to wells in a 96 well plate in triplicate. Control wells had 50ul of Opti-MEM with no serum. Equal volume of MV recombinant derivatives was mixed with serum dilutions or Opti-MEM control. A standard dilution of virus in Opti-MEM was adjusted to yield 30 infectious foci per well in the absence of serum forty-four hours post infection. The virus/serum mixture was incubated for 2 hours at 37°C before the addition of Vero-His cells in DMEM, 5%FBS (10,000 cells/well). Plates were incubated for 44hours at 37°C and the number of eGFP expressing foci per well were counted using a fluorescent microscope. The neutralization titer of 100% (NT) was the reciprocal of the dilution of human serum where there was no infection.
RESULTS
Mutations ablating infections via receptors CD46 and SLAM are in proximity to mAb epitopes within receptor-binding interface
Retargeted recombinant oncolytic measles virus derivatives used in this study (figure 1) have been characterized previously: MV-H-EGFR.scFv (MV-H6-HAALS-αEGFR) [29]; MV-H-Ech (MV-ERV) [41]; MV-H-Wue.scFv (MV-Wue) [35]. The control virus MV-Hedm will have a similar susceptibility to monoclonal anti-H antibodies and anti-MV antibodies present in human serum as oncolytic MVs tested clinically [100–103], [15] as they differ from one another only by the transgene they encode. Since retargeted H glycoproteins are of a higher molecular weight their incorporation into the virons is confirmed by comparing their size to Hedm by immunoblotting for H (figure 2).
Figure 1. Recombinant oncolytic measles viruses used in this study.
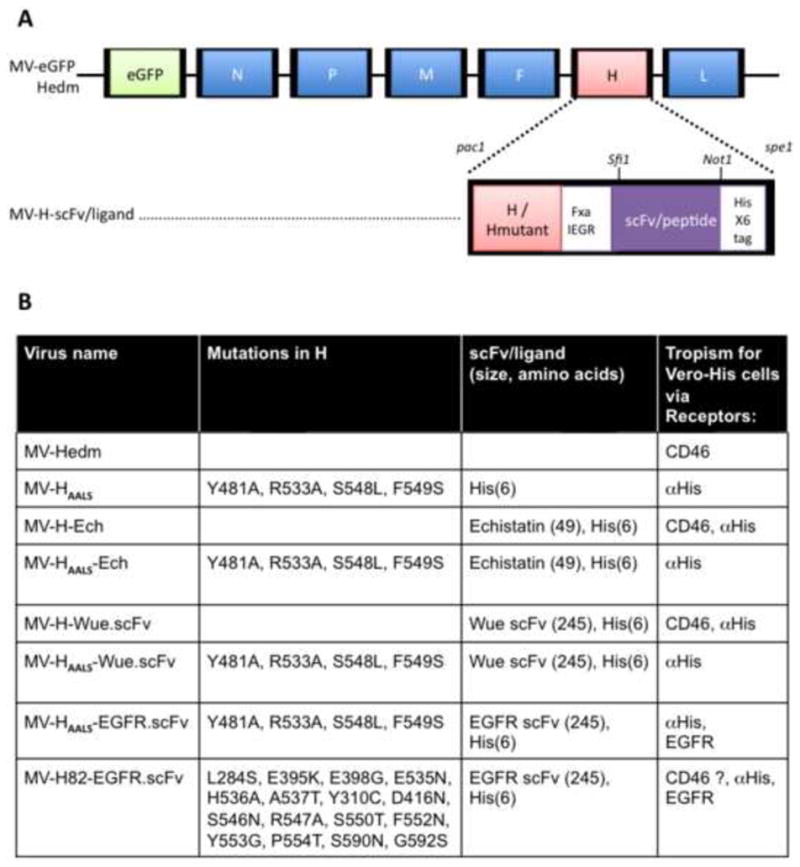
A) Schematic representation of MV.eGFP-edm (edmonston vaccine lineage, edm). The gene encoding enhanced green fluorescent protein (eGFP) is at position 1 followed by genes coding for N nucleoprotein, P phosphoprotein, M matrix, F fusion protein, H hemagglutinin and L large protein. Retargeted viruses are generated by replacing the H gene in MV-eGFP-Hedm with retargeted H using pac1/spe1 restriction enzymes. Retargeted H can be either CD46/SLAM tropic (H) or CD46/SLAM blind due to mutations Y481A, R544A, S548L, F549S (HAALS). Retargeted H or HAALS has a C-terminal scFv/ligand that can be exchanged with restriction enzymes sfi1 and Not1. A 6-histidine peptide (Hisx6) is attached to the C-terminus of scFv/ligand or HAALS. The Hisx6 acts as a ligand for entry and propagation in Vero cells engineered to express anti-Hisx6 scFv (Vero-HIS cells). The factor Xa (IEGR) cleavage site allows for the removal of the scFv/ligand/Hisx6 from H and is used to demonstrate that a blind retargeted MV infects cells via scFv/ligand or Hisx6. B) The table lists the recombinant viruses used in this study; the mutations in H; the scFv or ligand displayed on H and its size in amino acids. All viruses encode eGFP. For simplicity MV-eGFP is referred to as MV-Hedm. MV-H82-EGFRscFv has a H glycoprotein with 16 mutations designed to protect seven epitopes from monoclonal antibodies used in this study and is used as a control.
Figure 2. Chimeric H proteins displayed on the measles virus.
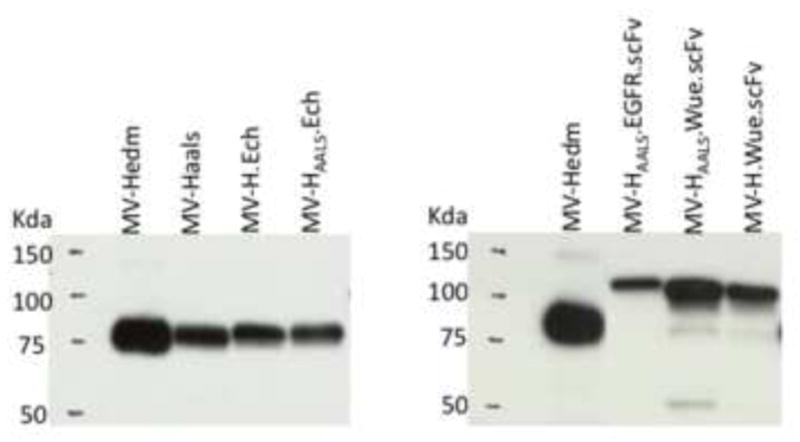
Western immunoblotting of purified MV-Hedm and retargeted MVs. The size difference between the different chimeric H proteins is based on the size of their C-terminal scFv/ligand.
To determine the proximity of mAb epitopes that cluster within and around the receptor-binding surface to mutations in HAALS (Y481A, R533A, S5548L and F549S) that ablate CD46/SLAM binding, we mapped the mutations and mAb epitopes onto the crystal structure of the H protein head domain (figure 3). MV receptors SLAM (figure 3A, yellow), CD46 (figure 3B, orange) and Necitn-4 (not shown) bind to the same side of the H head domain [61]. The receptor ablating mutations in HAALS (figure 3C, marine blue, green) map onto the CD46/SLAM footprint. Monoclonal antibody (mAb) epitopes clustering within and around the receptor-binding interface are mapped in magenta (figure 3C). Residue R533 (green) forms part of the 16DE6 epitope and the R533A mutation in HAALS. MAb epitopes for I-41, 16DE6, cl48, I-44, I-29, 16CD11 have been characterized in previous papers [58, 62]. Epitopes for 20H6 and BH97 were identified by selecting MV-Hedm in the presence of 20H6 or BH97 and by rational design (unpublished data). Figure 3C shows the close proximity of HAALS mutations to mAbs BH97, 20H6 and I-41 and overlap with 16DE6 epitopes. We hypothesized that the scFv/ligand domain of retargeted H could shield one or more mAb epitopes. In order to visualize the relative size of a 250 residue scFv domain compared to H we manually place a 250 residue scFv domain using coordinates from the crystal structure of scFv-against-IL-1β [57]. The location of the scFv/ligand domain relative to H is not known as the last ten C-terminal H residues are missing from this structure and the scFv/ligand domain is separated from H by a 9 residue linker.
Figure 3. HAALS mutations and mAb epitopes map within and around CD46/SLAM binding surface on the Hemagglutinin head domain.

The crystal structure of the H dimer head domain [29] (light gray surface representation) is shown with its native N-linked glycans N168, N187, N200 and N215 (dark gray), which have been built manually using complex-type penta-antennary sugars. The receptor-binding surface is shared by receptors (A) SLAM (yellow) [31] and (B) CD46 (orange) [62]. C) HAALS encodes mutations that disrupt SLAM and CD46 binding: Y481A, R533A, S548L, F549S (boxed/highlighted marine blue). Mutations known to escape monoclonal antibodies (red text) within and around the receptor-binding surface are highlighted in magenta. In green is residue R533, which is mutated in HAALS and forms part of mAb 16DE6 epitope. A representative crystal structure of a 250 amino acid scFv with a C-terminal 6-His peptide domain [57] (blue cartoon representation) is manually placed alongside H to demonstrate its relative size.
Retargeted MVs escape neutralization by mAbs targeting the receptor binding surface but not surrounding areas
Next we investigated if the HAALS mutations protected MV from neutralizing mAbs targeting the receptor-binding surface and if a 49 residue echistatin domain (Ech) or larger 245 residue Wue or EGFR scFv domain protected MV from the mAbs analyzed.
MV derivatives, with and without HAALS mutations and scFv/ligand domains, were incubated with mAbs or media for 1hour at 37°C prior to infection of Vero-His cells. Infection was quantified two days latter by counting the number of eGFP expressing syncytia per well (figure 4). As a positive control we used MV-H82-EGFR.scFv, whose H protein is modified with a C-terminal EGFR scFv/His x6 domain allowing it to be retargeted to EGFR and HIS scFv expressed on Vero-His cells. The H protein in MV-H82-EGFR.scFv is engineered to escape completely or partially mAbs tested in this study by encoding mAb escape mutations and rationally designed N-linked glycosylations sites within or adjacent to mAb epitopes. A glycosylation at these sites would have the potential to cloak the epitope from antibody recognition [62], however we have not determined if the carbohydrates are shielding the epitopes or the potential N-linked glycosylation site engineered into the virus is destroying the epitope. The oncolytic potential of MV-H82-EGFR.scFv has not been tested.
Figure 4. HAALS mutations protect MV from mAbs targeting the receptor binding surface but not surrounding areas.

Measles Viruses were incubated in media (no mAb) or monoclonal antibodies: BH97, cl48, 20H6, I-29, I-44, 16CD11, I-41 and 16DE6 for 1 hour at 37°C, prior to the addition of Vero-His cells. Two days post infection the number of eGFP expressing infected foci/well were counted using a fluorescent microscope and plotted as a percentage of infection in the absence of mAb. This is a representative experiment that was repeated three times. Each data point is an average of duplicate wells.
MV derivatives were challenged with mAbs whose epiotpes were within (BH97, 20H6, I-41, 16DE6) and outside (cl48, I-29, I-44, 16CD11) of the receptor-binding surface. MV derivatives, encoding HAALS mutations evaded neutralization by mAbs 20H6, I-41, 16DE6 and BH97. (figure 4, astrix), whose epitopes all mapped in close proximity to HAALS mutations (figure 3). MAbs whose epitopes mapped outside of the receptor-binding surface neutralized all MVs except for control MV-H82.EGFR.scFv. In the presence of 16CD11 we saw low levels of infection by MVs displaying scFv/ligand domains with and without HAALS mutations. MV-HAALS and MV-Hedm, which do not display a scFv/ligand domain were consistently neutralized. However, following repeat experiments the level of infection was inconsistent between retargeted MVs displaying scFv/ligand, except MV-H82.EGFR.scFv. We can therefore not conclude that a scFv/ligand domain interferes with 16CD11 binding.
Retargeted MV derivatives are neutralized by human serum despite their resistance to monoclonal antibodies recognizing the receptor-binding surface
We then investigated if retargeted MV derivatives are less susceptible to human serum neutralization. Recombinant MV derivatives were incubated for two hours at 37°C in serial dilutions of human serum prior to infection. We analyzed pooled serum collected from ~100–150 male donors at FDA-licensed commercial donor centers within the US (figure 5A) or individual males diagnosed with malignant neoplasms (figure 5B–F). Retargeted MV derivates did not infect cells at higher human serum concentrations than control MV-Hedm (figure 5). The NT (neutralization titer, dilution of serum that neutralized 100% of the virus) differed up to 2-fold between the viruses, which is within the range of experimental variability.
Figure 5. Retargeted MVs do not escape neutralization by human sera.

Control MV-Hedm and retargeted MVs were incubated with decreasing concentrations of heat-inactivated commercially available pooled human sera (A) or sera from five individuals (B–F) diagnosed with malignant neoplasms. Plotted is the reciprocal serum dilution at which there was no infection (NT, neutralization titer). Dotted line marks 1/100 serum dilution.
Retargeted H glycoproteins with no mutations in H can trigger cell-cell fusion in the presence of mAb BH97, 20H6 and I-41 targeting the receptor-binding surface
MV entry into the cell begins with H tethering to the target receptor. H binding to its receptors could induce a change in H head orientation, stalk conformation and/or oligomerization [63, 64]. H stalk cross-linking studies, suggest that the fusion signal is transmitted via the central region of the H stalk to F [65–67], which is subsequently destabilized. F then undergoes a series of conformational changes to induce membrane-membrane fusion [63, 67]. Cells expressing MV H and F glycoproteins interact with MV receptors on neighboring cells and in the same manner can fuse these cells together forming syncytia.
We hypothesized that chimeric H glycoproteins - with no mutations in H – can still bind to the retargeted receptor via their C-terminal ligand and trigger fusion in the presence of a mAb recognizing the SLAM/CD46 receptor-binding surface. To test this hypothesis we performed a cell-cell fusion assay in which mAbs were added to Vero cells after they were infected with MVs and quantified the degree of fusion inhibition (figure 6). Syncytia size in the presence and absence of mAbs was quantified by counting the number of nuclei per syncytium and allocating a score of +/− (≤ 5 nuclei/cells per syncytium), + (6–20 nuclei/cells per syncytium), ++ (21–50 nuclei/cells per syncytium), or +++ (>50 nuclei/cells per syncytium). MAbs BH97, 20H6, and I-41, which bind to the receptor-binding surface, inhibit syncytia formation between cells infected with MV-Hedm (figure 6i), probably because they inhibit H binding to CD46 expressed in Vero cells. mAb 16DE6 does not inhibit fusion of Vero cells as its epitope is in the SLAM binding interface (R533) [29, 48]. MAbs BH97, 20H6, and I-41 did not inhibit fusion between cells infected with retargeted viruses with no mutations in H to the same degree as cells infected with MV-Hedm (figure 6i,iii,v). Since mAbs BH97, 20H6, and I-41 neutralize these viruses (MV-H.Ech, MV-H.WUE, figure 4) they are probably binding to the SLAM/CD46 receptor-binding interface of H proteins displayed on the cell surface. This suggests that fusion is triggered by the H C-terminal ligand binding to the retargeted receptor and that MV-H.Ech and MV-H.Wue are neutralized by these mAbs by a mechanism other than the inhibition of receptor attachment. As expected syncytia formation between cells infected by MV’s encoding HAALS mutations (that escape neutralization by these mAbs) are not inhibited (figure 6ii,iv,vi,vii).
Figure 6. Monoclonal antibodies targeting the receptor binding surface inhibit fusion between cells infected with MV-Hedm but not Retargeted MVs.
Vero-His cells were infected with control MV-Hedm (i) or retargeted MV derivatives (ii–vii) for 13 hours prior to replacing the media with media containing mAbs targeting the receptor binding surface. Average syncytia size was quantified by counting the number of nuclei per syncytium and allocating a score (viii) of +/− (≤ 5 nuclei/cells per syncytium), + (6–20 nuclei/cells per syncytium), ++ (21–50 nuclei/cells per syncytium), or +++ (>50 nuclei/cells per syncytium) (table viii). The data is presented as the % of syncytia of the indicated size per 20 syncytia per well. * astrix marks percentage of syncytia that have > 50 nuclei/cells per syncytium in the presence of mAbs BH97, 20H6, I-41
DISCUSSION
MV is a promising oncolytic in preclinical animal models but anti-measles antibodies limit treatment efficacy in patients. MV H glycoproteins are the major target for neutralizing antibodies. The receptor-binding surface is a major target for neutralizing monoclonal antibodies and is a conserved neutralizing antigenic site [48, 51, 68]. Since, retargeted MV derivatives use a separate domain displayed on H to bind selected receptors and encode mutations ablating native MV receptor binding, it is hypothesized that retargeted MV derivatives may be less susceptible to human serum neutralization. Furthermore, the displayed domain could potentially contribute to serum resistance by shielding one or more antibody epitopes. This hypothesis is supported by work performed by Kneissl et al. [52], who demonstrated that lentiviral vectors pseudotyped with retargeted MV H and F glycoproteins were less susceptible to neutralization by human sera from two selected donors [52].
We therefore studied the susceptibility of recombinant MV derivatives expressing chimeric H proteins to mAbs and human serum. Our major findings are summarized in table 1. We demonstrate that mutations in H (HAALS: Y481A, R533A, S5548L, F549S) that inhibit entry via receptors SLAM and CD46, protect MV from neutralizing mAbs targeting the receptor-binding surface (BH97, 20H6, I-41 and 16DE6), but not surrounding areas. The HAALS mutations do not completely overlap with known epitopes for BH97, 20H6 and I-41 and we do not know the position of the scFv/ligand relative to the H protein because a structure of a retargeted chimeric H protein has not been solved. However based on our results we conclude that the HAALS mutations are responsible for escaping neutralizing mAbs BH97, 20H6, I-41 and 16DE6 for the following reasons: i) Epitope for 16DE6 overlaps with the HAALS mutations R533A. ii) Only retargeted viruses encoding the HAALS mutations escape mAbs targeting the receptor binding surface and these viruses escape neutralization despite the size of the C-terminal extension (i.e., 6 amino acid His peptide, 49 amino acid echistatin domain or 245 amino acid scFv domain). For example these mAbs neutralize MV-H-Wue1/Ech but not MV-HAALS-Wue1/Ech. Iii) N-linked glycosylations native to H do not provide protection against mAbs tested because Hedm shares the same native carbohydrate side chains (N-linked glycosylations) as the retargeted H proteins. Control MV-H82-EGFR.scFv encodes additional N-linked glycosylation sites within or adjacent to mAb epitopes, which protect the virus from being neutralized completely by mAbs tested, but these modifications are not present in the MVs being studied.
Table 1.
Summary of major findings.
| Virus name | Mutations ablating CD46/SLAM receptor binding | Retargeted via C-terminal peptide | mAbs targeting RBS Inhibit infection | mAbs targeting RBS Inhibit fusion via CD46 | Human antibodies inhibit infection |
|---|---|---|---|---|---|
| MV-Hedm | No | No | Yes | Yes | Yes |
| MV-HAALS | Yes | Hisx6 | No | No | Yes |
| MV-H-Ech | No | Ech, Hisx6 | Yes | No | Yes |
| MV-HAALS-Ech | Yes | Ech, Hisx6 | No | No | Yes |
| MV-H-Wue.scFv | No | Wue scFv, Hisx6 | Yes | No | Yes |
| MV-HAALS | Yes | Wue scFv, Hisx6 | No | No | Yes |
| MV-HAALS-EGFR.scFv | Yes | EGFR scFv, Hisx6 | No | No | Yes |
RBS, receptor binding site; scFv, single chain fragment variable; Hisx6, six histidine peptide; Ech, Echistatin; AALS, Y481A, R533A, S548L, F549S; mAbs, monoclonal antibodies.
We cannot conclude that the C-terminal scFv/ligand domain shields the 16CD11 epitope. It is possible that the 49 and 245 residue domains provide marginal protection because most of the viruses that display these domains can infect cells at low levels: MV-H.Ech, MV-HAALS-Ech, MV-HAALS-EGFR, and viruses that do not have these domains, MV-Hedm and MV-HAALS-His, are neutralized. However, we cannot explain why MV-HAALS-Wue1 but not MV-H-Wue1 is neutralized and why the degree of escape by the other viruses varies between experiments. Due to the fact that the receptor-binding surface of the H protein is a hot spot for neutralizing antibodies [47–51] we were surprised that the HAALS mutations protected this antigenic site from mAbs but provided no advantage in escaping neutralization by human sera. We tested pooled human sera available commercially and sera from five individuals diagnosed with malignant neoplasms, as this would be the patient population for oncolytic virotherapy using retargeted MVs.
To escape neutralization by a polyclonal attack by human antibodies, multiple epitopes may need to be destroyed/protected on H. The question is which epitopes and how many should be protected simultaneously. MV-H82-EGFR, in addition to escaping mAbs targeting the receptor binding surface, escaped mAbs cl48/I-44 targeting epitope E3 [62], I-29 targeting epitope E4 [62] also known as epitope vi [69] and partially escaped 16CD11 targeting epitope II [69] (figure 4). Out of the human sera tested in figure 5, two of the individuals had a 4-fold lower NT for MV-H82-EGFR.scFv than MV-Hedm (data not shown). The other three individuals NT was 2-3-fold lower, which we consider to be within the variability range between repeat PRNT experiments. Hence the combination of protecting these epitopes gives this virus a marginal advantage in sera from selected individuals. It would be interesting to protect additional epitopes and study their contribution to human antibody evasion.
Most of the mAbs targeting the receptor-binding surface inhibited cell-cell fusion between MV-Hedm infected cells, probably because they interfere with CD46 receptor binding. We made the interesting observation that MV-H-Wue and MV-H-Ech displaying unmutated H proteins can trigger fusion in the presence of these mAbs (figure 6). However infection is still inhibited (figure 4). If we assume that these mAbs are bound to the CD46/SLAM receptor-binding surface whilst H.scFv/ligand/Hisx6 is attached to His.scFv displayed on Vero-His cells, then it would be interesting to understand how these mAbs inhibit infection if fusion is accomplished in future studies.
CONCLUSIONS
To date engineered oncolytic viruses currently tested in clinical trials have not demonstrated dose-limiting toxicities. However retargeted MV derivatives could potentially be translated into the clinic if they are shown to be less susceptible to neutralization by human anti-measles antibodies or to address any future toxicity concerns associated with higher viral doses or treatment of tumors with low CD46 expression. Here we demonstrate that retargeted MV derivatives escape neutralization by mAbs targeting the receptor-binding surface. However, this gives them no advantage over non-retargeted MVs, in escaping neutralization by human serum. We propose that this is because multiple neutralizing epitopes need to be protected simultaneously.
MAJOR FINDINGS.
Retargeted MVs with CD46/SLAM binding ablated (HAALS), escape neutralization by mAb targeting the receptor binding surface (RBS) but not human antisera
Retargeted MVs with no mutations in H are neutralized by mAbs targeting the RBS. Interestingly, these ligand displaying H proteins can still bind to their retargeted receptors and trigger fusion in a cell-cell fusion assay in the presence of these mAbs. Whilst, non-retargeted Hedm mediated fusion is inhibited.
Displayed ligands do not protect MV from mAbs tested
Research Highlights.
We tested whether retargeted ligand-displaying MVs are resistant to Ab neutralization
MVs with HAALS mutations evade selected neutralizing mAbs targeting RBS*
Displayed ligands do not protect MV from Abs tested, nor from serum neutralization
Acknowledgments
We would like to sincerely thank the following individuals for providing us with reagents and models used in this work: G. Griesmann and Dr. M. Federspiel for sufficient quantities of purified cl48 for our selection studies (originally from T.F Wild). Dr M. Ehnlund for providing us with generous quantities of 16DE6, I-44, I-41 and I-29 (originally from E. Norrby). Dr. Ianko Iankov and Dr. Eva Galanis for mAb 20H6. Dr Takao Hashiguchi for sharing his PDB files and expertise, which aided in our modeling of H modified with N-linked glycans. This work was supported by funds from Mayo Clinic, NIH/NCI (R01 CA129966), and the Richard M Schulze Family Foundation. We would like to acknowledge Ruth Bushnell and John K. Tobin for testing if selected mutations in H82 glycoproteins interfered with the ability of H to induce cell-cell fusion when co-expressed with the fusion glycoprotein in cultured cells. SJR, PLN, PJL & GJT conceived and designed the H82 glycoprotein with masked epitopes.
Footnotes
RBS, receptor-binding surface
Work performed at 1Department of Molecular Medicine, 200 1st Street SW, Guggenheim Building 18, Mayo Clinic, Rochester, MN, USA
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Roland Pappoe, Email: PappRol@bvu.edu.
Takafumi Nakamura, Email: taka@med.tottori-u.ac.jp.
Stephen J. Russell, Email: sjr@mayo.edu.
References
- 1.Eager RM, Nemunaitis J. Clinical development directions in oncolytic viral therapy. Cancer Gene Ther. 18(5):305–17. doi: 10.1038/cgt.2011.7. [DOI] [PubMed] [Google Scholar]
- 2.Donnelly OG, et al. Recent clinical experience with oncolytic viruses. Curr Pharm Biotechnol. 13(9):1834–41. doi: 10.2174/138920112800958904. [DOI] [PubMed] [Google Scholar]
- 3.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 30(7):658–70. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blechacz B, et al. Engineered measles virus as a novel oncolytic viral therapy system for hepatocellular carcinoma. Hepatology. 2006;44(6):1465–77. doi: 10.1002/hep.21437. [DOI] [PubMed] [Google Scholar]
- 5.McDonald CJ, et al. A measles virus vaccine strain derivative as a novel oncolytic agent against breast cancer. Breast Cancer Res Treat. 2006;99(2):177–84. doi: 10.1007/s10549-006-9200-5. [DOI] [PubMed] [Google Scholar]
- 6.Iankov ID, et al. Demonstration of anti-tumor activity of oncolytic measles virus strains in a malignant pleural effusion breast cancer model. Breast Cancer Res Treat. 122(3):745–54. doi: 10.1007/s10549-009-0602-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Msaouel P, et al. Engineered measles virus as a novel oncolytic therapy against prostate cancer. Prostate. 2009;69(1):82–91. doi: 10.1002/pros.20857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Msaouel P, et al. Noninvasive imaging and radiovirotherapy of prostate cancer using an oncolytic measles virus expressing the sodium iodide symporter. Mol Ther. 2009;17(12):2041–8. doi: 10.1038/mt.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peng KW, et al. Intraperitoneal therapy of ovarian cancer using an engineered measles virus. Cancer Res. 2002;62(16):4656–62. [PubMed] [Google Scholar]
- 10.Hasegawa K, et al. Dual therapy of ovarian cancer using measles viruses expressing carcinoembryonic antigen and sodium iodide symporter. Clin Cancer Res. 2006;12(6):1868–75. doi: 10.1158/1078-0432.CCR-05-1803. [DOI] [PubMed] [Google Scholar]
- 11.Peng KW, et al. Systemic therapy of myeloma xenografts by an attenuated measles virus. Blood. 2001;98(7):2002–7. doi: 10.1182/blood.v98.7.2002. [DOI] [PubMed] [Google Scholar]
- 12.Dingli D, et al. Image-guided radiovirotherapy for multiple myeloma using a recombinant measles virus expressing the thyroidal sodium iodide symporter. Blood. 2004;103(5):1641–6. doi: 10.1182/blood-2003-07-2233. [DOI] [PubMed] [Google Scholar]
- 13.Grote D, et al. Live attenuated measles virus induces regression of human lymphoma xenografts in immunodeficient mice. Blood. 2001;97(12):3746–54. doi: 10.1182/blood.v97.12.3746. [DOI] [PubMed] [Google Scholar]
- 14.Phuong LK, et al. Use of a vaccine strain of measles virus genetically engineered to produce carcinoembryonic antigen as a novel therapeutic agent against glioblastoma multiforme. Cancer Res. 2003;63(10):2462–9. [PubMed] [Google Scholar]
- 15.Galanis E, et al. Phase I trial of intraperitoneal administration of an oncolytic measles virus strain engineered to express carcinoembryonic antigen for recurrent ovarian cancer. Cancer Res. 70(3):875–82. doi: 10.1158/0008-5472.CAN-09-2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Griffin D. In: Fields’ Virology. 5. Fields B, Knipe DM, Howley PM, editors. Lippincott Williams and Wilkins; 2007. [Google Scholar]
- 17.Tatsuo H, et al. SLAM (CDw150) is a cellular receptor for measles virus. Nature. 2000;406(6798):893–7. doi: 10.1038/35022579. [DOI] [PubMed] [Google Scholar]
- 18.Hahm B, et al. Measles virus infects and suppresses proliferation of T lymphocytes from transgenic mice bearing human signaling lymphocytic activation molecule. J Virol. 2003;77(6):3505–15. doi: 10.1128/JVI.77.6.3505-3515.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schneider-Schaulies S, et al. Regulation of gene expression in lymphocytes and antigen-presenting cells by measles virus: consequences for immunomodulation. J Mol Med (Berl) 2002;80(2):73–85. doi: 10.1007/s00109-001-0299-x. [DOI] [PubMed] [Google Scholar]
- 20.Schneider-Schaulies S, et al. Measles virus: immunomodulation and cell tropism as pathogenicity determinants. Med Microbiol Immunol. 2002;191(2):83–7. doi: 10.1007/s00430-002-0121-6. [DOI] [PubMed] [Google Scholar]
- 21.Reymond N, et al. Nectin4/PRR4, a new afadin-associated member of the nectin family that trans-interacts with nectin1/PRR1 through V domain interaction. J Biol Chem. 2001;276(46):43205–15. doi: 10.1074/jbc.M103810200. [DOI] [PubMed] [Google Scholar]
- 22.Noyce RS, et al. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog. 7(8):e1002240. doi: 10.1371/journal.ppat.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Derycke MS, et al. Nectin 4 overexpression in ovarian cancer tissues and serum: potential role as a serum biomarker. Am J Clin Pathol. 134(5):835–45. doi: 10.1309/AJCPGXK0FR4MHIHB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takano A, et al. Identification of nectin-4 oncoprotein as a diagnostic and therapeutic target for lung cancer. Cancer Res. 2009;69(16):6694–703. doi: 10.1158/0008-5472.CAN-09-0016. [DOI] [PubMed] [Google Scholar]
- 25.Fabre-Lafay S, et al. Nectin-4, a new serological breast cancer marker, is a substrate for tumor necrosis factor-alpha-converting enzyme (TACE)/ADAM-17. J Biol Chem. 2005;280(20):19543–50. doi: 10.1074/jbc.M410943200. [DOI] [PubMed] [Google Scholar]
- 26.Naniche D, et al. Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus. J Virol. 1993;67(10):6025–32. doi: 10.1128/jvi.67.10.6025-6032.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dorig RE, et al. The human CD46 molecule is a receptor for measles virus (Edmonston strain) Cell. 1993;75(2):295–305. doi: 10.1016/0092-8674(93)80071-l. [DOI] [PubMed] [Google Scholar]
- 28.Anderson BD, et al. High CD46 receptor density determines preferential killing of tumor cells by oncolytic measles virus. Cancer Res. 2004;64(14):4919–26. doi: 10.1158/0008-5472.CAN-04-0884. [DOI] [PubMed] [Google Scholar]
- 29.Nakamura T, et al. Rescue and propagation of fully retargeted oncolytic measles viruses. Nat Biotechnol. 2005;23(2):209–14. doi: 10.1038/nbt1060. [DOI] [PubMed] [Google Scholar]
- 30.Allen C, et al. Interleukin-13 displaying retargeted oncolytic measles virus strains have significant activity against gliomas with improved specificity. Mol Ther. 2008;16(9):1556–64. doi: 10.1038/mt.2008.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paraskevakou G, et al. Epidermal growth factor receptor (EGFR)-retargeted measles virus strains effectively target EGFR- or EGFRvIII expressing gliomas. Mol Ther. 2007;15(4):677–86. doi: 10.1038/sj.mt.6300105. [DOI] [PubMed] [Google Scholar]
- 32.Allen C, et al. Retargeted oncolytic measles strains entering via the EGFRvIII receptor maintain significant antitumor activity against gliomas with increased tumor specificity. Cancer Res. 2006;66(24):11840–50. doi: 10.1158/0008-5472.CAN-06-1200. [DOI] [PubMed] [Google Scholar]
- 33.Hasegawa K, et al. The use of a tropism-modified measles virus in folate receptor-targeted virotherapy of ovarian cancer. Clin Cancer Res. 2006;12(20 Pt 1):6170–8. doi: 10.1158/1078-0432.CCR-06-0992. [DOI] [PubMed] [Google Scholar]
- 34.Jing Y, et al. Tumor and vascular targeting of a novel oncolytic measles virus retargeted against the urokinase receptor. Cancer Res. 2009;69(4):1459–68. doi: 10.1158/0008-5472.CAN-08-2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hummel HD, et al. Genetically engineered attenuated measles virus specifically infects and kills primary multiple myeloma cells. J Gen Virol. 2009;90(Pt 3):693–701. doi: 10.1099/vir.0.007302-0. [DOI] [PubMed] [Google Scholar]
- 36.Ungerechts G, et al. Lymphoma chemovirotherapy: CD20-targeted and convertase-armed measles virus can synergize with fludarabine. Cancer Res. 2007;67(22):10939–47. doi: 10.1158/0008-5472.CAN-07-1252. [DOI] [PubMed] [Google Scholar]
- 37.Yaiw KC, et al. CD20-targeted measles virus shows high oncolytic specificity in clinical samples from lymphoma patients independent of prior rituximab therapy. Gene Ther. 18(3):313–7. doi: 10.1038/gt.2010.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hasegawa K, et al. Affinity thresholds for membrane fusion triggering by viral glycoproteins. J Virol. 2007;81(23):13149–57. doi: 10.1128/JVI.01415-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ungerechts G, et al. An immunocompetent murine model for oncolysis with an armed and targeted measles virus. Mol Ther. 2007;15(11):1991–7. doi: 10.1038/sj.mt.6300291. [DOI] [PubMed] [Google Scholar]
- 40.Liu C, et al. Prostate-specific membrane antigen retargeted measles virotherapy for the treatment of prostate cancer. Prostate. 2009;69(10):1128–41. doi: 10.1002/pros.20962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hallak LK, et al. Targeted measles virus vector displaying echistatin infects endothelial cells via alpha(v)beta3 and leads to tumor regression. Cancer Res. 2005;65(12):5292–300. doi: 10.1158/0008-5472.CAN-04-2879. [DOI] [PubMed] [Google Scholar]
- 42.Peng KW, et al. Targeting virus entry and membrane fusion through specific peptide/MHC complexes using a high-affinity T-cell receptor. Gene Ther. 2004;11(15):1234–9. doi: 10.1038/sj.gt.3302286. [DOI] [PubMed] [Google Scholar]
- 43.Parato KA, Lichty BD, Bell JC. Diplomatic immunity: turning a foe into an ally. Curr Opin Mol Ther. 2009;11(1):13–21. [PubMed] [Google Scholar]
- 44.Willmon C, et al. Cell carriers for oncolytic viruses: Fed Ex for cancer therapy. Mol Ther. 2009;17(10):1667–76. doi: 10.1038/mt.2009.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Russell SJ, Peng KW. Measles virus for cancer therapy. Curr Top Microbiol Immunol. 2009;330:213–41. doi: 10.1007/978-3-540-70617-5_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bouche FB, Ertl OT, Muller CP. Neutralizing B cell response in measles. Viral Immunol. 2002;15(3):451–71. doi: 10.1089/088282402760312331. [DOI] [PubMed] [Google Scholar]
- 47.Hashiguchi T, et al. Crystal structure of measles virus hemagglutinin provides insight into effective vaccines. Proc Natl Acad Sci U S A. 2007;104(49):19535–40. doi: 10.1073/pnas.0707830104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hashiguchi T, et al. Structure of the measles virus hemagglutinin bound to its cellular receptor SLAM. Nat Struct Mol Biol. 18(2):135–41. doi: 10.1038/nsmb.1969. [DOI] [PubMed] [Google Scholar]
- 49.Santiago C, et al. Structure of the measles virus hemagglutinin bound to the CD46 receptor. Nat Struct Mol Biol. 17(1):124–9. doi: 10.1038/nsmb.1726. [DOI] [PubMed] [Google Scholar]
- 50.Ertl OT, et al. Immunodominant domains of the Measles virus hemagglutinin protein eliciting a neutralizing human B cell response. Arch Virol. 2003;148(11):2195–206. doi: 10.1007/s00705-003-0159-9. [DOI] [PubMed] [Google Scholar]
- 51.Tahara M, et al. The receptor-binding site of the measles virus hemagglutinin protein itself constitutes a conserved neutralizing epitope. J Virol. 87(6):3583–6. doi: 10.1128/JVI.03029-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kneissl S, et al. Measles virus glycoprotein-based lentiviral targeting vectors that avoid neutralizing antibodies. PLoS One. 7(10):e46667. doi: 10.1371/journal.pone.0046667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Douglas JT, et al. A system for the propagation of adenoviral vectors with genetically modified receptor specificities. Nat Biotechnol. 1999;17(5):470–5. doi: 10.1038/8647. [DOI] [PubMed] [Google Scholar]
- 54.Duprex WP, et al. Observation of measles virus cell-to-cell spread in astrocytoma cells by using a green fluorescent protein-expressing recombinant virus. J Virol. 1999;73(11):9568–75. doi: 10.1128/jvi.73.11.9568-9575.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Msaouel P, et al. Oncolytic measles virus retargeting by ligand display. Methods Mol Biol. 797:141–62. doi: 10.1007/978-1-61779-340-0_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fielding AK, et al. A hyperfusogenic gibbon ape leukemia envelope glycoprotein: targeting of a cytotoxic gene by ligand display. Hum Gene Ther. 2000;11(6):817–26. doi: 10.1089/10430340050015437. [DOI] [PubMed] [Google Scholar]
- 57.Wilkinson IC, et al. High resolution NMR-based model for the structure of a scFv-IL-1beta complex: potential for NMR as a key tool in therapeutic antibody design and development. J Biol Chem. 2009;284(46):31928–35. doi: 10.1074/jbc.M109.025304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu A, et al. Molecular characterization of epitopes on the measles virus hemagglutinin protein. Virology. 1993;192(1):351–4. doi: 10.1006/viro.1993.1042. [DOI] [PubMed] [Google Scholar]
- 59.Giraudon P, Wild TF. Monoclonal antibodies against measles virus. J Gen Virol. 1981;54(Pt 2):325–32. doi: 10.1099/0022-1317-54-2-325. [DOI] [PubMed] [Google Scholar]
- 60.Iankov ID, et al. Neutralization capacity of measles virus H protein specific IgG determines the balance between antibody-enhanced infectivity and protection in microglial cells. Virus Res. 172(1–2):15–23. doi: 10.1016/j.virusres.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hashiguchi T, Maenaka K, Yanagi Y. Measles virus hemagglutinin: structural insights into cell entry and measles vaccine. Front Microbiol. 2:247. doi: 10.3389/fmicb.2011.00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lech PJ, et al. Epitope dampening monotypic measles virus hemagglutinin glycoprotein results in resistance to cocktail of monoclonal antibodies. PLoS One. 8(1):e52306. doi: 10.1371/journal.pone.0052306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lamb RA, Paterson RG, Jardetzky TS. Paramyxovirus membrane fusion: lessons from the F and HN atomic structures. Virology. 2006;344(1):30–7. doi: 10.1016/j.virol.2005.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yuan P, et al. Structure of the Newcastle disease virus hemagglutinin-neuraminidase (HN) ectodomain reveals a four-helix bundle stalk. Proc Natl Acad Sci U S A. 108(36):14920–5. doi: 10.1073/pnas.1111691108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Navaratnarajah CK, et al. The heads of the measles virus attachment protein move to transmit the fusion-triggering signal. Nat Struct Mol Biol. 18(2):128–34. doi: 10.1038/nsmb.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Navaratnarajah CK, et al. Membrane fusion triggering: three modules with different structure and function in the upper half of the measles virus attachment protein stalk. J Biol Chem. doi: 10.1074/jbc.M112.410563. [DOI] [PMC free article] [PubMed]
- 67.Russell CJ, Luque LE. The structural basis of paramyxovirus invasion. Trends Microbiol. 2006;14(6):243–6. doi: 10.1016/j.tim.2006.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Casasnovas JM, Larvie M, Stehle T. Crystal structure of two CD46 domains reveals an extended measles virus-binding surface. EMBO J. 1999;18(11):2911–22. doi: 10.1093/emboj/18.11.2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tahara M, et al. Functional and structural characterization of neutralizing epitopes of measles virus hemagglutinin protein. J Virol. 87(1):666–75. doi: 10.1128/JVI.02033-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Websites
- 100.Vaccine Therapy With or Without Cyclophosphamide in Treating Patients With Recurrent or Refractory Multiple Myeloma. NCT00450814 http://clinicaltrials.gov/ct2/show/NCT00450814.
- 101.Recombinant Measles Virus Vaccine Therapy and Oncolytic Virus Therapy in Treating Patients With Progressive, Recurrent, or Refractory Ovarian Epithelial Cancer or Primary Peritoneal Cancer. NCT00408590. http://clinicaltrials.gov/ct2/show/NCT00408590?term=measles+virus+cancer&rank=1.
- 102.Phase I Trial of a Measles Virus Derivative Producing CEA (MV-CEA) in Patients with Recurrent Glioblastoma Multiforme (GBM). US-0770. www.gemcris.od.nih.gov/Contents/GC_CLIN_TRIAL_RPT_VIEW.asp?WIN_TYPE=R&CTID=773.
- 103.Intrapleural Measles Virus Therapy in Patients With Malignant Pleural Mesothelioma. NCT01503177. http://clinicaltrials.gov/ct2/show/NCT01503177?term=measles+virus+cancer&rank=5.