

Abstract
Tumor phenotype is a result of the complex interactions between malignant cells and sorrounding stroma. However, the mechanisms by which cancer cells and fibroblasts, the most abundant and active part of the tumor stroma, interact remain to be elucidated. The K303R mutation of estrogen-receptor (ERα) was identified in 50% of invasive breast tumors and associated with poorer survival outcomes. Here, we show that human cancer-associated fibroblasts (CAFs) stimulated proliferation and migration of wild-type (WT) ERα stably transfected breast cancer cells and to a higher extent in cells expressing the K303R ERα hyperactive receptor. We identified, for the first time, leptin, a known cytokine involved in breast cancer development, as a determinant for CAFs tumor-promoting activities in both WT and K303R ERα-expressing cells. Indeed, we found an increase in leptin receptor isoforms expression, and in its signalling activation in K303R-expressing cells compared to WT ERα clones. These data correlated well with the amplified effects of leptin on cell growth, motility and invasiveness in mutant cells. Mutant expression generated a leptin hypersensitive phenotype also in vivo. Lastly, K303R ERα cell-secreted factors stimulated CAFs proliferation and migration and their ability to secrete leptin. We demonstrated that the epidermal growth factor is the paracrine factor by which breast cancer cells affect CAFs phenotype. Thus, our work uncovers a bidirectional cross-talk between breast cancer cells and ‘educated’ CAFs, which leads via leptin signaling to increased tumor progression. The blockade of these intercellular communications might represent an effective strategy for molecular targeted therapies in breast cancer.
Keywords: Breast Cancer, Estrogen Receptor, K303R mutant ERα, CAFs, Leptin Signaling Pathway
Introduction
For the past three decades, cancer research focused predominantly on the characteristics of breast cancer cells. Recently, clinical and experimental studies revealed that both tumor initiation and progression are related to the complex interactions that transpire within the tumor microenvironment. The stromal compartment is composed of mesenchymal cells (fibroblasts/adipocytes/blood cells), extracellular matrix-ECM (lamin/fibronectin/collagen/proteoglicans/etc), and signals from these cells come as soluble secreted factors, ECM-components or direct cell-cell contacts. Growth factors, cytokines, adipokines, proteases, and vascular-stimulating factors are involved in stroma-mediated procancerous activities (1-4). The chemokines CXCL12, CXCL14, and CCL7 stimulated tumor cell proliferation and invasion in vitro and in vivo, increased tumor angiogenesis and macrophage presence at tumor sites (5-7). The interleukin-1 and -8 induced cancer progression by enhancing metastasis and cachexia (8,9). As important adipocyte-derived endocrine and paracrine mediator, the adipokine leptin has been correlated with breast cancer occurrence. Indeed, leptin synthesis and plasma levels increase with obesity, a pandemic condition that influences both risk and prognosis of breast cancers (10).
The processes of heterotypic signalling involve a constant bidirectional cross-talk between stromal cells, and malignant cells. Stromal cells influence tumor invasiveness and malignancy, whereas at the onset and during breast cancer progression, the microenvironment is reorganized by cancer cells (11). Tumors recruit stromal fibroblasts in a process referred to as the desmoplasmic reaction, and these carcinoma-associated fibroblasts (CAFs) are reprogrammed to produce growth factors, cytokines, and ECM-remodeling proteins, that acting in autocrine and paracrine fashion support tumor proliferation and invasion into surrounding tissues (4). Moreover, a variety of these factors may activate estrogen receptor alpha (ERα) (12).
Estrogens and its receptor play a crucial role in regulating breast cancer growth and differentiation. Variant forms of ERα due to alternative splicing or gene mutation have been reported, but their clinical significance is still unresolved (13,14). A naturally-occurring mutation at nucleotide 908, introducing a lysine to arginine transition at residue 303 within the hinge domain of the receptor (K303R ERα), was identified in a third of premalignant breast hyperplasias, and one-half of invasive breast tumors. This mutation correlated with poor outcomes, older age, larger tumor size, and lymph node-positive disease (15,16). Other studies did not detect the mutation in invasive cancers (17-20), but our studies suggest that the detection method used might be insensitive. However, K303R expression was found at low frequency in invasive breast tumors by Conway and colleagues (21). K303R mutation allows ERα to be more highly phosphorylated by different kinases, and it alters the dynamic recruitment of coactivators and corepressors (22-24). Mutant overexpression in MCF-7 breast cancer cells increased sensitivity to subphysiological levels of estrogen, and decreased tamoxifen responsiveness when elevated growth factors signaling was present (15,25). K303R ERα mutation also conferred resistance to the aromatase inhibitor anastrozole (23,26), suggesting a pivotal role for this mutation in more aggressive breast cancers.
The aim of this study was to elucidate the mechanisms underlying tumor/stroma interaction in ERα–positive breast cancer cells. First, we investigated how tumoral microenvironment pressure, exerted by CAFs, impacts breast cancer cell proliferation, migration and invasiveness in relation to the expression of wild-type or the K303R ERα. We then defined the effect that a single factor-leptin has on stroma-mediated breast cancer progression. Finally, we examined the bidirectional interactions between CAFs and breast cancer cells, leading to increased malignancy.
Materials and Methods
Reagents and antibodies
Leptin/17β-estradiol/EGF from Sigma. ICI182,780 by Tocris Bioscience. AG490/AG1478/PD98059/LY294002 by Calbiochem. ERα/ERβ/GAPDH/ObRl/ObRs/Ob/Akt/pAktSer437 antibodies from Santa Cruz Biotechnology. MAPK/JAK2/STAT3/pMAPKThr202/Tyr204/pJAK2Tyr1007/1008/pSTAT3Tyr705/pERαSer118/pERαSer167 from Cell Signaling Technology.
Plasmids
Generation of yellow-fluorescent-protein/YFP-tagged expression constructs, YFP-WT and YFP-K303R-ERα, as described (22). XETL-plasmid, containing an estrogen-responsive-element, by Dr Picard, University Geneva/Switzerland.
Cell culture
MCF-7 and SKBR3 cells were acquired in 2010 from American-Type-Culture-Collection where they were authenticated, stored according to supplier’s instructions, used within four-months after frozen aliquots resuscitations. YFP-WT and YFP-K303R ERα-stable expressing MCF-7 cells, MCF-7 and SKBR3 pools stably transfected with YFP-WT and YFP-K303R ERα generated as described (23,26). Immortalized normal human foreskin-fibroblasts BJ1-hTERT by Dr Lisanti, Jefferson University/Philadelphia/USA. Every four-months, cells were authenticated by single-tandem-repeat-analysis at our sequencing-core, morphology, doubling-times, estrogen-sensitivity, tested for mycoplasma-negativity (MycoAlert/Lonza).
CAFs-isolation
Human breast cancer specimens were collected in 2011 from primary tumors of patients who signed informed-consent. Following tumor excision, small pieces were digested (500IUcollagenase in Hank’s-Balanced-salt-solution, Sigma; 37°C/2h). After differential centrifugation (90g/2min), the surnatant containing cancer-associated fibroblasts (CAFs) was centrifugated (500g/8min), resuspended and cultured in RPMI-1640 medium supplemented with 15% Fetal-Bovine-Serum/FBS, and antibiotics. CAFs between 4-10passages were used, tested by mycoplasma-presence, authenticated by morphology and FAP expression.
Conditioned medium systems
CAFs were incubated with regular-full media (48-72h). Conditioned media were collected, centrifugated to remove cellular debris and used in respective experiments. Alternatively, conditioned media were collected from WT and K303R ERα-expressing MCF-7 cells incubated in 5% charcoal-stripped-FBS (72h).
Expression microarray analysis
Expression profiles were determined with Affymetrix-GeneChip human genome U133plus2.0 arrays. Data quality and statistical analysis as described (23). Microarray study followed MIAME-(Minimum-Information-About-a-Microarray-Experiment) guidelines. All data are available in our previous publication (23).
Immunoblot analysis
Protein extracts were subjected to SDS-PAGE as described (27). Immunoblots show a single representative of three separate experiments.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde, permeabilized with PBS+0.2%TritonX-100 followed by blocking with 5%BSA (1h/room temperature), incubated with anti-ObR antibody (4°C/overnight) and with FITC-conjugated secondary antibody (30min/room temperature). IgG primary antibody as negative control. 4′,6-Diamidino-2-phenylindole (DAPI, Sigma) staining for nuclei detection. Fluorescence was photographed using OLYMPUS BX51 microscope,100Xobjective.
RT-PCR and Real-time RT-PCR assays
FAP/Ob/CyclinD1/pS2/CathepsinD/36B4 gene expression were evaluated by reverse transcription(RT)-PCR method as decribed (27). ObRl/ObRs/CXCR4/IR/IL2RB/IL6R/EGFR/IGF1R/FGFR3/ERα/EGF/IL6/ Insulin gene expression were assessed by Real-time RT-PCR, using SYBR Green Universal PCR Master Mix (Biorad). Each sample was normalized on 18s mRNA content. Relative gene expression levels were calculated as described (28). Primers in Supplementary Table 1.
ERα-transactivation assays
ERα-transactivation as described (29).
Cell proliferation assays
MTT assays
After 4 days of treatments, cell proliferation was assessed using 3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide reagent/MTT (Sigma) and expressed as fold change relative to vehicle-treated cells.
Tripan-blue cell-count assays
After 4 days of treatment, cell numbers were evaluated by trypsin suspension of samples followed by microscopic evaluation using a hemacytometer.
Soft agar growth assays
Anchorage-independent growth as described (26).
Data represent three-independent experiments, performed in triplicate.
Wound-healing scratch assays
Cell monolayers were scraped and treated as indicated. Wound closure was monitored over 24h, cells were fixed and stained with Comassie-Brillant-Blue. Pictures represent one of three-independent experiments (10Xmagnifications, phase-contrast microscopy).
Transmigration assays
Cells treated with/without leptin were placed in Boyden-chamber upper compartments (8micron membranes/Corning Costar). Bottom well contained regular-full media. After 24h, migrated cells were fixed and stained with Comassie-Brillant-Blue. Migration was quantified by viewing five separate fields per membrane at 20Xmagnification and expressed as the mean number of migrated cells. Data represent three-independent experiments, assayed in triplicate.
Invasion assays
Matrigel-based invasion assay was performed by invasion-chambers (8micron membranes) coated with Matrigel (BD Bioscences,0.4μg/ml). Cells treated with/without leptin were seeded into top transwell-chambers, while regular-full medium was used as chemoattractant in lower chambers. After 24h, invaded cells were evaluated as described for transmigration assays. Data represent three-independent experiments, assayed in triplicate.
Tumor Xenografts
In vivo studies performed as described (30).
Leptin measurement by RIA
Leptin was measured by a competitive in-house immunoassay (Chematil) following manufacturer’s protocol. Results are presented as ng/cells.
Leptin-immunodepleted conditioned media
ProteinG-agarose beads were incubated with anti-leptin or IgG antibodies. Antibody-beads complexes were incubated with CAFs-conditioned media and centrifugated. Leptin-immunodepletion was verified by RIA.
Statistical analysis
Data were analyzed for statistical significance using two-tailed student’s Test, GraphPad-Prism4. Standard deviations/S.D. are shown. Survival curves were computed by Kaplan–Meier-method and compared using two-sided log-rank tests.
Results
Tumor/stroma interactions stimulate cell proliferation and motility
Epithelial-stromal interactions support tumor cell proliferation and invasion. Thus, we first investigated the role of tumoral microenvironment in influencing breast cancer phenotype in relation to the expression of wild-type (WT) or K303R ERα mutant receptor.
We used as experimental models for breast cancer ERα-positive MCF-7 cells stably transfected with YFP-WT or YFP-K303R ERα expression vectors. We chose this approach because WT receptor was present along with K303R ERα in invasive breast tumors (16). Stable clones were screened for ERα expression using immunoblot analysis (Fig. 1A). Two clones stably expressing YFP-WT (WT1-2) or YFP-K303R ERα (K303R1-2) are shown along with WT or mutant receptor stable pools (WT P and K303R P). As stromal cells, we employed cancer-associated fibroblasts (CAFs), isolated from biopsies of primary breast tumors. CAFs possessed the basic fibroblast characteristics of long and spindle-shaped morphology, and highly expressed the fibroblast activation protein-FAP (Fig. 1B). To create in vitro conditions that can mimic the complex in vivo microenvironment, we used coculture experiments. Breast cancer cells were incubated with regular-full media (FM), CAFs-derived conditioned media (CM) or normal fibroblasts (NFs)-CM and growth was evaluated by soft agar assays (Fig. 1C). As previously demonstrated (23,26), control basal growth of mutant-expressing cells was elevated compared to WT-expressing cells. CAFs-CM significantly increased colony numbers in both WT and K303R ERα-expressing cells; however, CAFs-CM enhanced K303R-expressing cell growth at a higher extent compared to WT-expressing cells. We then examined the ability of CAFs-CM to promote WT- and K303R-expressing cell movement in wound-healing scratch assays (Fig. 1C). The mutant cells moved the farthest in either direction to close the gap compared to WT-expressing cells. CAFs-CM promoted net movement of WT-expressing cells compared to FM; but, K303R-expressing cells exposed to CAFs-CM moved at higher rate to close the gap in the cell bed. As expected, CAFs possessed a higher ability to enhance both proliferation and motility of breast cancer cells than NFs (Fig. 1C). CAFs-CM-induced cell growth and migratory potential was blocked by inhibition of the classic cytokine JAK2/STAT3 signaling cascade (AG490) and the ERα signaling inhibitor (ICI182,780), although to a higher extent in K303R clones (Fig. 1D). All functional effects described so far are the results of exposure to the total complement of CAFs-secreted proteins. However, it is desirable albeit experimentally difficult to define the contribution of a single factor. Thus, we addressed which CAFs-secreted factor may promote breast cancer cell growth and motility.
Figure 1.
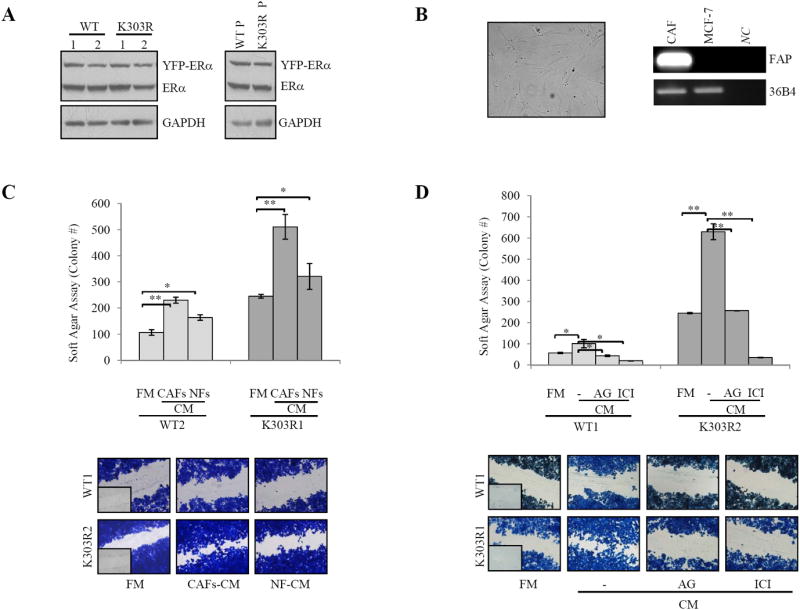
CAFs-induced breast cancer cell growth and motility. A, Immunoblotting for ERα expression in YFP-WT and YFP-K303R ERα stable-expressing MCF-7 cells and WT P and K303R P stable pools. GAPDH, loading control. B, CAFs morphology in monolayer growth using phase-contrast microscopy. RT-PCR for fibroblast activation protein-FAP and 36B4 (internal standard). NC, negative-control. C, Soft agar and scratch assays in cells treated with regular full media (FM), CAFs- or normal fibroblasts (NFs)-derived conditioned media (CM). D, Soft agar and scratch assays in cells treated with FM, CAFs-CM with/without AG490 (AG,10μM) or ICI182,760 (ICI,1μM). *P<0.05, **P<0.005. Small squares, time 0.
Gene transcription patterns of WT and K303R ERα-overexpressing cells
Diffusible growth factors, interleukins, chemokines and adipokines implicated as mediators of stromal-epithelial interactions are involved in breast carcinoma initiation and progression. To determine changes in gene expression for the different receptors of CAFs-secreted factors, that may be responsible of the different sensitivity of WT and mutant clones to CAFs-CM exposure, we performed microarray analysis. Gene expression profile comparing RNA isolated from K303R-expressing with WT-expressing cells are shown in Table 1 and Supplementary Table 2. K303R ERα expression induced several genes potentially involved into tumor/stroma interactions; however, the leptin receptor (ObR) gene was the most highly induced (2.4fold, Table 1). We also observed increased expression of different leptin signaling downstream effectors such as JAK2, the transcription factors fos, STAT, as well as the suppressor of cytokine signaling 3 (Supplementary Table 3). To validate the microarray study, YFP-WT and YFP-K303R ERα-expressing cells were evaluated for a panel of genes using real-time PCR (Fig. 2A). K303R-associated induction could be confirmed for all of them, and, again, the gene encoding the long and short leptin receptor isoforms (ObRl/ObRs) was the most highly upregulated in mutant-expressing cells. However, we did not detect any differences in IGF1R mRNA expression levels between the two cells, although microarray analysis showed a significant decrease of IGF1R. ERα RNA levels were similar between K303R and WT ERα-expressing cells.
Table 1.
Gene expression profile of the different receptors of CAFs-secreted factors among WT and K303R ERα-expressing MCF-7 breast cancer cells.
| Gene Name | Gene Symbol | Parametric P-value | Fold Change in K303R Clones |
|---|---|---|---|
| Leptin receptor | ObR/LepR | <1e-07 | 2.4 |
| Interleukin 28 receptor α | IL28RA | <1e-07 | 1.9 |
| Chemokine (C-X-C motif) receptor 4 | CXCR4 | <1e-07 | 1.9 |
| Insulin receptor | IR | 1.7e-06 | 1.68 |
| Interleukin 17 receptor C | IL17RC | 7e-07 | 1.6 |
| Insulin-like growth factor 2 receptor | IGF2R | 1e-07 | 1.57 |
| Interleukin 15 receptor β | IL15RA | 3e-07 | 1.5 |
| Macrophage stimulating receptor 1 | MSR1 | 9.2e-06 | 1.44 |
| Hepatocyte growth factor receptor | MET | 1.69e-05 | 1.39 |
| Interleukin 1 receptor, type I | IL1R1 | 0.0004 | 1.38 |
| Chemokine (C-C motif) receptor 7 | CCR7 | 0.0001 | 1.3 |
| Chemokine (C-X3-C motif) | CX3CR1 | 7.9e-05 | 1.32 |
| receptor 1 | 1.27 | ||
| Interleukin 2 receptor β | IL2RB | 3.57e-05 | 1.25 |
| Interleukin 9 receptor | IL9R | 0.0002 | 1.23 |
| Interleukin 10 receptor β | IL10RB | 0.0003 | 1.21 |
| Interleukin 6 receptor | IL6R | 0.01 | 1.2 |
| Interleukin 21 receptor | IL21R | 0.0006 | 0.8 |
| Interleukin 4 receptor | IL4R | 0.0007 | 0.7 |
| Epidermal growth factor receptor | EGFR | 7.6e-06 | 0.7 |
| Insulin-like growth factor 1 receptor | IGF1R | 0.0006 | 0.7 |
| Fibroblast growth factor receptor 3 | FGFR3 | 0.0005 | 0.7 |
Representative probesets from pathway analysis showing gene expression changes in the receptor family of CAFs-secreted factors along with the p-value and the fold change of K303R-expressing cells compared to WT cells studied by microarray analysis. In cases where the same genes were deemed significant across multiple probesets, only one is shown.
Figure 2.
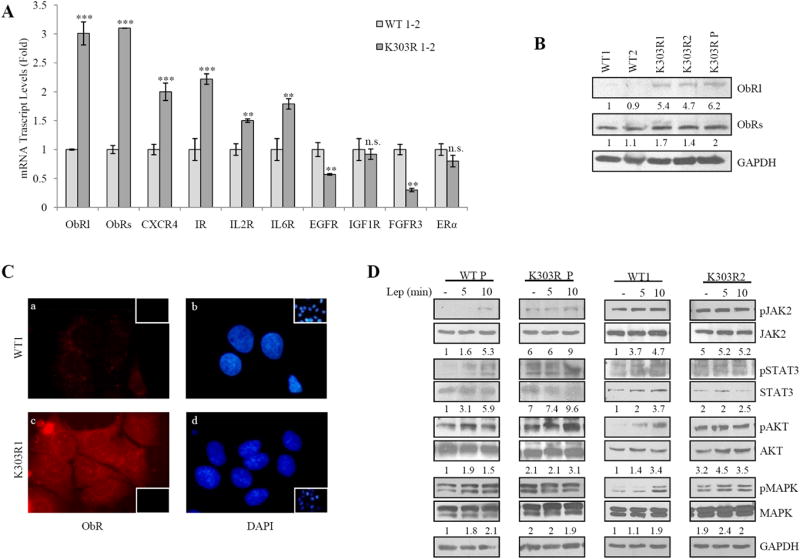
Leptin signaling activation in mutant cells. A, Real-time RT-PCR for different receptors of CAFs-secreted factors. n.s.=nonsignificant, **P<0.01, ***P<0.005. B, Immunoblotting showing leptin receptor long and short isoforms (ObRl/ObRs). GAPDH, loading control. Numbers represent the average fold change in ObRl/GAPDH and ObRs/GAPDH levels. C, Immunofluorescence of ObR (a,c) and DAPI (b,d). Small squares, negative-controls. D, Immunoblotting of phosphorylated pJAK2/pSTAT3/pAKT/pMAPK, and total proteins from cells treated with vehicle(-) or leptin (Lep100ng/ml, 5 and 10min). GAPDH, loading control. Numbers represent the average fold change between phospho/total/GAPDH levels.
The increase in both ObRl and ObRs was then confirmed by evaluating protein levels using immunoblotting analysis (Fig. 2B) and immunofluorescence staining of WT and K303R ERα-expressing cells (red, Fig. 2C).
K303R ERα-overexpressing cells exhibit increased leptin signaling activation
Given the gene expression profile identified in the microarray study, we defined the impact that a single factor-leptin may have on K303R ERα breast cancer cell progression. First, time-course response studies were performed to analyze phosphorylation of leptin downstream effectors using immunoblot analysis (Fig. 2D). WT-expressing cells exhibited low basal levels of phosphorylated JAK2, STAT3, Akt and MAPK that were increased in a time-dependent manner after leptin treatment. In contrast, K303R-expressing cells showed elevated constitutive phosphorylation of these signaling molecules in control-vehicle conditions that was slightly increased after leptin treatment. Thus, the mutant ERα expression was associated with increased leptin signaling activation.
Leptin directly activates ERα in the absence of its own ligand in MCF-7 breast cancer cells (29). As a consequence of the enhanced leptin signaling, we found increased ERα-transcriptional activity and upregulated mRNA levels of the classical ERα-target genes Cyclin D1, pS2 and Cathepsin D in both control and leptin-treated conditions in K303R ERα-expressing cells. In addition, the mutant exhibited elevated pS118 and pS167 YFP-ERα levels (Supplementary Fig. 1).
K303R ERα mutation and leptin hypersensitivity
We next used these stably transfected clones as model systems to study leptin sensitivity, in relation to mutant receptor expression. First, we evaluated leptin effects on growth using anchorage-dependent growth assays (Supplementary Fig. 2). As expected, in both WT and K303R-expressing cells treatment with leptin 100ng/ml increased cell proliferation. However, low leptin treatment (10ng/ml) significantly enhanced cell viability only in K303R-expressing clones. We also evaluated leptin-mediated proliferative effects in anchorage-independent growth assays (Fig. 3A). Leptin treatments at 100 and 1000ng/ml concentrations enhanced colony numbers in all four clones tested, even though to a higher extent in mutant-expressing cells. Again, leptin at 10ng/ml increased anchorage-independent growth only in K303R cells. The increase in colony numbers induced by leptin was reversed by the JAK2/STAT3 inhibitor AG490 (Fig. 3B). We also used the antiestrogen ICI182,780 and found that this treatment suppressed anchorage-independent growth of both cell lines, indicating that ER expression remains important in growth regulation of these cells (Fig. 3B).
Figure 3.
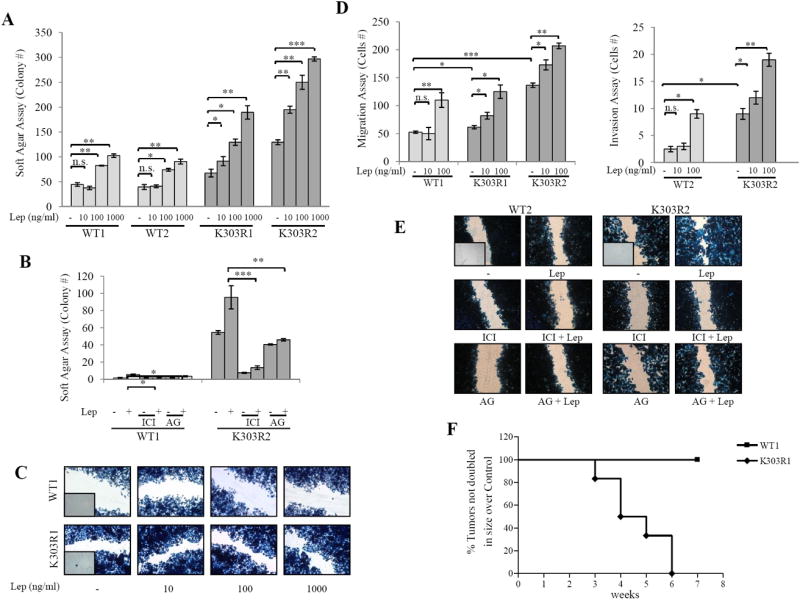
The K303R ERα mutation generates a leptin hypersensitive phenotype. A, Soft agar assay in cells treated with vehicle(-), or Lep10/100/1000ng/ml. B, Soft agar assay in cells treated with vehicle(-) or Lep100ng/ml, with/without ICI182,760 (ICI,1μM) or AG490 (AG,10μM). C, scratch, and D, transmigration and invasion assays in cells treated as indicated. n.s.=nonsignificant, *P<0.05, **P<0.005, ***P<0.001. E, Scratch assay in cells treated with vehicle(-) or Lep100ng/ml, with/without ICI182,760 or AG490. Small squares, time 0. F, WT and K303R ERα-expressing cells were injected into mice (n=6/group) supplemented with E2 (0.72mg/pellet/90-day-release) and 230μg/kg leptin or vehicle (Control). Survival curves (shown as % of mice in which tumors had not doubled in size) are graphed as the time in weeks from treatment to a twofold increase in total tumor volume over baseline (time to tumor doubling).
We next evaluated the ability of increasing doses of leptin to influence cell migration in wound-healing scratch assays (Fig. 3C). Again, the mutant cells moved farthest in either direction to close the gap compared to WT-expressing cells. Leptin treatments at 100 and 1000ng/ml promoted cell motility in both WT and K303R-expressing cells, although to a higher extent in mutant cells. Interestingly, leptin at 10ng/ml stimulated migration only in K303R-expressing cells. Then, the capacity of cells to migrate across uncoated membrane in transmigration assays or to invade an artificial basement membrane-Matrigel in invasion assays was tested in the presence of leptin (Fig. 3D). While WT cells exhibited little motile and no invasive behaviour in vitro, our data clearly demonstrated that mutant receptor expression increased both motility and invasion of cells. High doses of leptin increased the number of migrated and invaded cells in both clones and again low doses of leptin stimulated motility and invasion only of cells expressing the K303R receptor. As expected, treatment with AG490 and ICI182,780 resulted in a clear reduction of both control-untreated and leptin-induced cell motility in wound-healing scratch assays, especially in K303R-expressing cells (Fig. 3E). We recently published that K303R ERα MCF-7 xenograft tumors grew faster than WT ERα tumors (26). In addition, MCF-7 xenograft tumors doubled control value after 13 weeks of leptin exposure (30). Thus, we determined if the mutant receptor-expressing breast cancer cells might exhibit an increased sensitivity to leptin stimulation also in vivo. We found that in mice treated with leptin, all xenografts derived from cells with K303R ERα expression doubled in size within 6 weeks of treatment, while none of xenografts from WT ERα-expressing cells doubled in size during this experiment (Fig. 3F). Thus, expression of the mutant generated a leptin hypersensitive phenotype in vitro and in vivo.
Leptin is responsible for CAFs-induced cell growth and motility
We next assessed the role of leptin in the context of heterotypic signalings working in tumor/stroma interactions. First, RIA measurement in CAFs-CM showed that leptin secretion varied from 10±4.5ng/200.000cells. RT-PCR evidenced Ob mRNA expression in CAFs; CAFs also expressed ObR long isoform, but they did not express ObR short isoform, ERα, or ERβ (Supplementary Fig. 3). Leptin was then immunodepleted from CAFs-CM by leptin specific antibodies and resulting media were tested for the ability to induce anchorage-independent growth and migration of breast cancer cells. Leptin-depletion (CM+LepAb) significantly decreased growth and migration-promoting activities of CAFs-CM, particularly in K303R-expressing cells (Fig. 4A). CM treated with a non specific mouse IgG had not effects, suggesting that the neutralizing effects of leptin antibodies were specific. Our results identify leptin as a main molecular player that mediates CAFs effects on tumor cell growth and migration.
Figure 4.
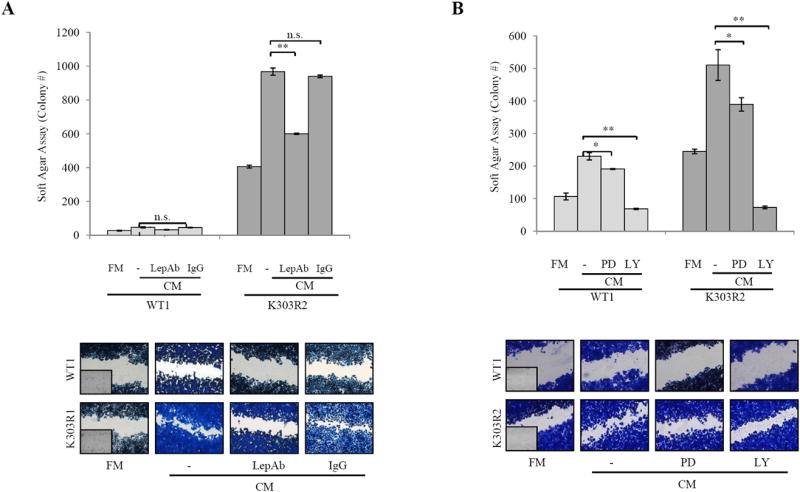
Leptin-immunodepletion reduces CAFs-induced cell growth and migration. A, Soft agar and scratch assays in cells treated with FM, CAFs-CM, or leptin-depleted CM (CM+LepAb). CM treated with a non specific IgG as a control (CM+IgG). B, Soft agar and scratch assays in cells treated with FM, CAFs-CM with/without PD98059 (PD,10μM) or LY294002 (LY,10μM). n.s.=nonsignificant, *P<0.05, **P<0.005. Small squares, time 0.
Leptin activates via JAK2 the MAPK and PI3K/Akt pathways (31). Thus, we investigated the specific signaling involved in the CAFs/breast cancer cells interaction, and found that the PI3K/Akt inhibitor LY294002 was more effective in inhibiting CAFs-induced proliferation and migration than the MEK1 inhibitor PD98059 (Fig. 4B).
Tumor/stroma interactions in SKBR3 breast cancer cells
To extend the results obtained, we generated pools of YFP-WT and YFP-K303R ERα stable transfectants in ERα-negative SKBR3 breast cancer cells (Supplementary Fig. 4). As previously shown for MCF-7 cells, we found a significant increase in both long and short leptin receptor isoforms mRNA in mutant-expressing cells. Again, treatment with leptin at 100 and 1000ng/ml significantly increased colony numbers of WT clones, and to a higher extent of K303R-expressing cells; however 10ng/ml of leptin enhanced anchorage-independent growth only in K303R-expressing clones. Similarly, low leptin promoted migration only in mutant cells. Finally, we tested CAFs-CM for its effects on cell growth and migration. We found a great induction of anchorage-independent growth and motility after treatment with CM, especially in K303R-expressing SKBR3 pools. Leptin-immunodepleted CM strongly reduced CM-proliferative and migratory-promoting activities on K303R cells, confirming that leptin hypersensitive phenotype was associated with K303R ERα expression in different cellular backgrounds.
Effects of breast cancer cell-secreted factors on CAFs phenotype
CAFs and tumor cells cross-talk via different soluble factors, whose effects on both subpopulations determine the final outcome of the tumorogenic process. Thus, as a final step of this study we defined the effects of CM from WT and K303R ERα-expressing breast cancer cells on CAFs phenotype. Treatment with K303R-CM elicited a dramatic alteration in the shape of CAFs in vitro, accompanied by an increased FAP mRNA expression (Fig. 5A). K303R-CM also stimulated CAFs viability and motility compared to WT-CM effects (Fig. 5B), suggesting how soluble K303R ERα cell-secreted factors may generate a more activated CAFs phenotype. Since leptin synthesis is influenced by different humoral factors (32-34), we evaluated the effects of breast cancer-derived CM in modulating leptin secretion from CAFs. Incubation of CAFs with K303R-CM increased leptin mRNA expression and leptin release compared with WT-CM, while no differences were detected in leptin levels among WT and K303R-CM (Fig. 5C). Finally, to investigate the paracrine factor by which breast cancer cells may affect CAFs phenotype, we used microarray analyses to measure the expression of different genes known to be associated with CAFs and/or leptin secretion (11,32-35). Our results showed that the genes encoding for EGF (2,8 fold), IL6 (1,2 fold) and Insulin (1,2 fold) were induced in mutant cells, and realtime PCR confirmed that the EGF gene was the most highly upregulated (Fig. 5D). Thus, we evaluated the role of EGF. First, addition of EGF in WT-CM mimicked the induction of K303R-CM on CAF motility, and the EGFR signaling inhibitor (AG1478) reduced K303R-CM effects (Fig. 5E). Second, treatment with AG1478 reversed K303R-CM stimulated FAP mRNA expression (Fig. 5F). Third, Ob mRNA expression and leptin secretion from CAFs cocultured with K303R-CM was significantly decreased in the presence of AG1478 (Fig. 5F).
Figure 5.
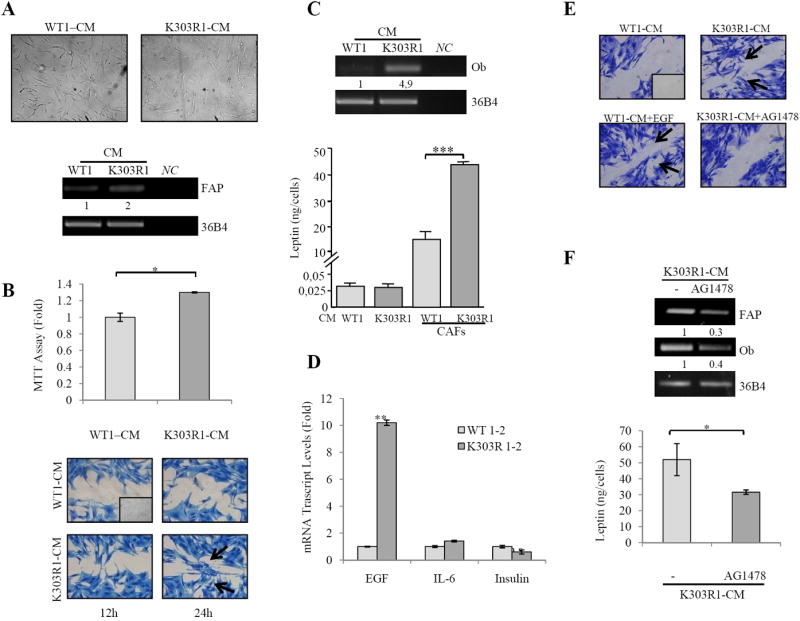
CAFs activated phenotype after K303R ERα cell-CM exposure. A, Phase-contrast microscopy for CAFs morphology and RT-PCR for FAP and 36B4 (internal standard) after treatment with CM from WT or K303R ERα-expressing cells. NC, negative-control. B, MTT growth and scratch assays in CAFs treated with WT-CM and K303R-CM. C, RT-PCR for leptin/Ob and 36B4, and leptin release by RIA. D, Real-time RT-PCR for EGF, IL6 and Insulin. E, Scratch assays in CAFs treated with WT-CM with/without EGF100ng/ml or K303R-CM with/without AG1478 10μM. F, RT-PCR for FAP, leptin/Ob and 36B4, and leptin release from CAFs by RIA. Numbers represent the average fold change of FAP/36B4 and Ob/36B4 levels. n.s.=nonsignificant, *P<0.05, **P<0.005, ***P<0.0001. Small squares, time 0.
Our data show that K303R ERα-expressing breast cancer cells through their soluble secreted factors may take advantage of the plastic nature of reactive surrounding cell populations, as CAFs, to generate a tumor enhancing microenvironment.
Discussion
ERα expression has important implications for breast cancer biology and therapy. Fuqua et al. identified a lysine to arginine transition at residue 303 of ERα (K303R ERα) in 30% of breast hyperplasias, and in 50% of invasive breast cancers (15,16), although using another detection method the mutation was identified in only 6% of tumors (21); thus the frequency is still unresolved. This mutation was associated with older age, larger tumor size, lymph-node positivity, and shorter time to recurrence, all features related with a more aggressive breast cancer phenotype. Because of the recently-recognized importance of tumor/stroma cross-talk in promoting breast cancer progression and metastasis, it is imperative to elucidate the molecular events occurring between cancer cells and adjacent stroma at the site of primary tumors to provide new treatment options for breast cancer.
Here, we elucidated the complex interactions between peritumoral tissue, locally-derived factors and neoplastic cells in dependency of ERα status, with a special focus on leptin effects in influencing the behavior of breast cancer cells bearing the naturally-occurring K303R ERα mutation. We proposed a model in which leptin, secreted from CAFs, binds to its receptor, activates K303R ERα and promotes proliferation, migration and invasiveness of K303R ERα-expressing breast cancer cells. In turn, K303R cells release factors as EGF that ‘educate’ CAFs to enhance secretion of leptin, which, acting back on malignant cells, may establish a positive feedback loop between cancer and stromal cells to further support breast tumor progression (Fig. 6).
Figure 6.
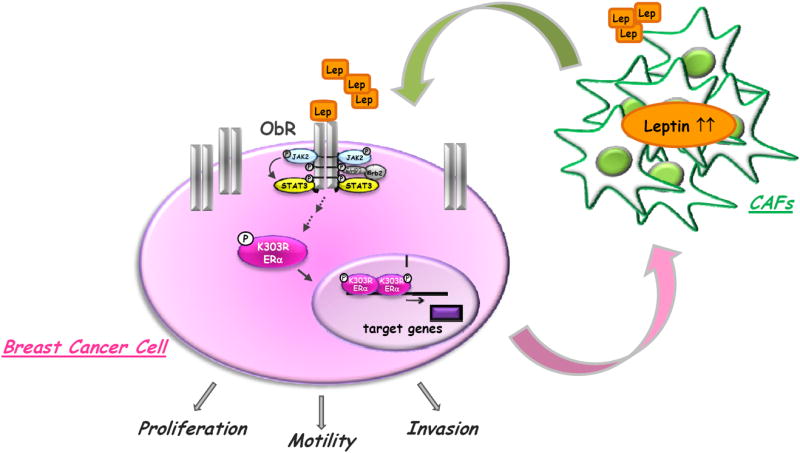
Schematic illustration of tumor/stroma interactions in K303R ERα breast cancer microenvironment. CAFs secrete leptin, which, acting in a paracrine fashion, binds to its cognate receptors (ObR), overexpressed on the surface of K303R ERα breast cancer cells and activates K303R ERα. This results in increased cell proliferation, motility, and invasion. K303R ERα-expressing cells, in turn, secrete factors that stimulate leptin production by adjacent CAFs, thus creating a positive feedback loop between cancer and stromal cells to further promote breast tumor progression.
CAFs promote breast cancer cell malignancy through leptin signaling
The phenotype of malignant cells appears regulated not only by cell autonomous signals, but also is dependent on heterotypic signals coming from surrounding stromal cells, able to create a specific local microenvironment to tightly control breast cancer proliferation and differentiation (36-38).
We defined the molecular interactions between stromal fibroblasts isolated from biopsies of primary breast tumors (CAFs), WT and K303R ERα-expressing MCF-7 breast cancer cells. The initial conditioned media experiments demonstrated that the entire complement of secretory proteins released by CAFs have more profound effects on K303R ERα-expressing cell proliferation and migration than on WT ERα cells. We evidenced an important role for JAK2/STAT3 and ERα signaling pathways in CM-mediated effects. Our microarray study pointed to the regulation of several important transcriptional programs of growth factors and cytokines receptors that, acting as mediators of stromal-epithelial interactions, are potentially involved in carcinoma progression. Among them, the gene encoding for leptin receptor was the most highly induced in K303R-expressing breast cancer cells.
Leptin is primarily synthesized from adipocytes, but is also produced by other cells, including fibroblasts (39-41). We demonstrated, for the first time, Ob mRNA expression and leptin secretion in CAFs. CAFs expressed ObR long isoforms, implying that an autocrine feedback loop may exist. Leptin-immunodepletion from CAFs-CM substantially reduced the growth and migration-promoting activities of CAFs. As one of the leptin downstream effectors (31), we found that the PI3K/Akt inhibitor LY294002 was effective in inhibiting CAFs-induced effects.
Since fibroblasts are the principal cellular component of the stroma, our results suggest that in the breast microenvironment CAFs through leptin signaling may become the main actor in influencing tumor cell behavior, especially in K303R ERα-expressing breast cancer cells.
Crosstalk between leptin and K303R ERα signaling pathways in breast cancer
Leptin, a pleiotropic molecule that regulates food intake, haematopoiesis, inflammation, cell differentiation and proliferation, is also required for mammary gland development and tumorigenesis. Indeed, leptin and its receptor isoforms (ObRs) have been detected in mammary epithelium and breast cancer cell lines, and are overexpressed in cancer tissue compared with healthy epithelium, with a positive correlation between ObR and ERα expression (42,43). Realtime PCR, immunoblotting and immunofluorescence experiments revealed an increase in mRNA and protein expressions of ObR long and short isoforms in K303R ERα-expressing cells. We also demonstrated that the mutant expression was associated with enhanced leptin signaling activation, and increased sensitivity to leptin stimulation on growth, motility, and invasiveness. Moreover, a significant increase in the growth of leptin-treated mutant tumors was observed in vivo.
Leptin is a potent modulator of the estrogen signaling pathway (29,44). On the other hand, estradiol modulates ObR expression in rat brain, through a putative estrogen-responsive-element in its promoter (45,46), and others demonstrated that estradiol induces leptin and ObR expression in MCF-7 breast cancer cells (43). Thus, leptin and estrogen might cooperate in sustaining estrogen-dependent breast carcinoma growth. We showed an increased S167 and S118 phosphorylation of the K303R receptor, an enhanced K303R ERα transactivation, and a more pronounced up-regulation of classical estrogen-regulated genes in K303R-expressing cells. Indeed, the pure antiestrogens ICI182,760 drastically suppressed leptin-stimulated anchorage-independent growth and motility of mutant cells.
These results suggest that the mutation may potentiate ERα’s role as an effector of leptin intracellular signal transduction, which may enhance cell proliferation, migration and invasiveness, contributing to the more aggressive phenotype of K303R-associated breast cancers.
K303R ERα cell-derived factors contribute to CAFs tumor-promoting activities
In the same way as tumor microenvironment plays active roles in shaping the fate of a tumor, cancer cells actively recruit fibroblasts into the tumor mass, in particular, the subpopulation named cancer-associated fibroblasts (CAFs). This cell-type is defined based on morphological characteristics or expression of markers as the fibroblast activating protein-FAP (1-4).
Studies addressing these issues are heterogeneous in terms of cell systems used, tumor cell types, and fibroblast sources. Experimental systems have used different tumor-derived conditioned media to stimulate CAFs, and others have cocultured tumor cells with normal fibroblasts or mesenchymal stem cells and measured chemokines levels in the resulting CM. For instance, fibroblasts growth with tumor cells resulted in increased production of chemokines whose source is in CAFs themselves. Chemokines produced under these “mixed” conditions promoted tumor promalignancy activities (6,9,47). We showed increased leptin mRNA expression and secretion by CAFs in response to soluble K303R ERα cell-secreted factors compared to WT-CM, suggesting that K303R cells have the ability to instruct their surrounding fibroblasts to augment leptin production, thereby enhancing tumor growth. This further indicates that interactions between the two subpopulations are actually bidirectional.
These interactions become more productive when tumor cells have a higher aggressiveness phenotype (47-49). CAFs exposed to K303R cell-derived CM acquired a more activated phenotypic characteristic, as revealed by an altered morphology, an increased FAP mRNA expression, and enhanced proliferative and migratory capabilities. We identified the epidermal growth factor, known to affect CAFs phenotype and leptin secretion (11,32-35), as the factor responsible of the paracrine activation of the surrounding stroma. Thus, a positive feedback loop is established which leads to the development and growth of the tumor.
Conclusions
Our study highlights the functional importance of tumor-host cross-talk in impacting malignant cell behavior, and implies several clinical implications. First, since K303R mutation was identified in breast premalignant hyperplasia, it is tempting to speculate that this specific mutation hypersensitive to leptin signaling may promote or accelerate the development of cancers from premalignant breast lesions, further increasing risk in obese women. Second, understanding the key genes involved differently in relation to ERα status in tumor/stroma interactions may help to identify novel biomarkers for breast cancer. Finally, our findings support the development of new therapeutics targeting stroma signaling components (e.g. leptin) to be implemented in the adjuvant therapy for improving clinical care and reducing mortality from breast cancer.
Supplementary Material
Acknowledgments
We are grateful to Dr F. Romeo for providing the breast cancer specimens.
Grant Support
This work was supported by Reintegration AIRC/Marie Curie International Fellowship in Cancer Research to IB, Lilli Funaro Foundation to IB and LG, European Commission/FSE/Regione Calabria to LG, AIRC Grant (IG11595) to SC, DB, CG and SA, and NCI RO1 CA72038 to SF.
Footnotes
Disclosure of Potential Conflict of Interest: The authors declare they have no conflict of interest.
References
- 1.Bhowmick NA, Neilson EG, Moses HL. Stromal fibroblasts in cancer initiation and progression. Nature. 2004;432:332–37. doi: 10.1038/nature03096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 3.Mueller MM, Fusenig NE. Friends or foes - bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4:839–49. doi: 10.1038/nrc1477. [DOI] [PubMed] [Google Scholar]
- 4.Orimo A, Weinberg RA. Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle. 2006;5:1597–601. doi: 10.4161/cc.5.15.3112. [DOI] [PubMed] [Google Scholar]
- 5.Augsten M, Hagglof C, Olsson E, Stolz C, Tsagozis P, Levchenko T, et al. CXCL14 is an autocrine growth factor for fibroblasts and acts as a multimodal stimulator of prostate tumor growth. Proc Natl Acad Sci. 2009;106:3414–19. doi: 10.1073/pnas.0813144106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jung DW, Che ZM, Kim J, Kim K, Kim KY, Williams D. Tumor-stromal crosstalk in invasion of oral squamous cell carcinoma: a pivotal role of CCL7. Int J Cancer. 2009;127:332–44. doi: 10.1002/ijc.25060. [DOI] [PubMed] [Google Scholar]
- 7.Matsuo Y, Ochi N, Sawai H, Yasuda A, Takahashi H, Funahashi H, et al. CXCL8/IL-8 and CXCL12/SDF-1 co-operatively promote invasiveness and angiogenesis in pancreatic cancer. Int J Cancer. 2009;124:853–61. doi: 10.1002/ijc.24040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolf JS, Chen Z, Dong G, Sunwoo JB, Bancroft CC, Capo DE, et al. IL (interleukin)-1 alpha promotes nuclear factor-kappaB and AP-1-induced IL-8 expression, cell survival, and proliferation in head and neck squamous cell carcinomas. Clin Cancer Res. 2001;7:1812–20. [PubMed] [Google Scholar]
- 9.Kumar S, Kishimoto H, Chua HL, Badve S, Miller KD, Bigsby RM, et al. Interleukin-1 alpha promotes tumor growth and cachexia in MCF-7 xenograft model of breast cancer. Am J Pathol. 2003;163:2531–41. doi: 10.1016/s0002-9440(10)63608-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu MH, Chou YC, Chou WY, Hsu GC, Chu CH, Yu CP, et al. Circulating levels of leptin, adiposity and breast cancer risk. Br J Cancer. 2009;100:578–82. doi: 10.1038/sj.bjc.6604913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schäffler A, Schölmerich J, Buechler C. Mechanisms of disease: adipokines and breast cancer-endocrine and paracrine mechanisms that connect adiposity and breast cancer. Nat Clin Pract Endocrinol Metab. 2007;3:345–54. doi: 10.1038/ncpendmet0456. [DOI] [PubMed] [Google Scholar]
- 12.Osborne CK, Schiff R. Estrogen-receptor biology: continuing progress and therapeutic implications. J Clin Oncol. 2005;23:1616–22. doi: 10.1200/JCO.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 13.Herynk MH, Fuqua SA. Estrogen receptor mutations in human disease. Endocr Rev. 2004;25:869–98. doi: 10.1210/er.2003-0010. [DOI] [PubMed] [Google Scholar]
- 14.Barone I, Brusco L, Fuqua SA. Estrogen receptor mutations and changes in downstream gene expression and signaling. Clin Cancer Res. 2010;16:2702–08. doi: 10.1158/1078-0432.CCR-09-1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fuqua SA, Wiltschke C, Zhang QX, Borg A, Castles CG, Friedrichs WE, et al. A hypersensitive estrogen receptor-alpha mutation in premalignant breast lesions. Cancer Res. 2000;60:4026–29. [PubMed] [Google Scholar]
- 16.Herynk MH, Parra I, Cui Y, Beyer A, Wu MF, Hilsenbeck SG, et al. Association between the estrogen receptor alpha A908G mutation and outcomes in invasive breast cancer. Clin Cancer Res. 2007;13:3235–43. doi: 10.1158/1078-0432.CCR-06-2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tebbit CL, Bentley RC, Olson JA, Marks JR. Estrogen receptor a (ESR1) mutant A908G is not a common feature in benign and malignant proliferations of the breast. Genes Chromosomes Cancer. 2004;40:51–4. doi: 10.1002/gcc.20017. [DOI] [PubMed] [Google Scholar]
- 18.Tokunaga E, Kimura Y, Maehara Y. No hypersensitive estrogen receptor-a mutation (K303R) in Japanese breast carcinomas. Breast Cancer Res Treat. 2004;84:289–92. doi: 10.1023/B:BREA.0000019963.67754.93. [DOI] [PubMed] [Google Scholar]
- 19.Davies MP, O’Neill PA, Innes H, Sibson DR. Hypersensitive K303R oestrogen receptor-a variant not found in invasive carcinomas. Breast Cancer Res. 2005;7:R113–18. doi: 10.1186/bcr965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Z, Yamashita H, Toyama T, Omoto Y, Sugiura H, Hara Y, et al. Estrogen receptor a mutation (A-to-G transition at nucleotide 908) is not found in different types of breast lesions fromJapanese women. Breast Cancer. 2003;10:70–3. doi: 10.1007/BF02967628. [DOI] [PubMed] [Google Scholar]
- 21.Conway K, Parrish E, Edmiston SN, Tolbert D, Tse CK, Geradts J, et al. The estrogen receptor-a A908G (K303R) mutation occurs at a low frequency in invasive breast tumors: results from a population-based study. Breast Cancer Res. 2005;7:R871–80. doi: 10.1186/bcr1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cui Y, Zhang M, Pestell R, Curran EM, Welshons WV, Fuqua SA. Phosphorylation of estrogen receptor alpha blocks its acetylation and regulates estrogen sensitivity. Cancer Res. 2004;64:9199–208. doi: 10.1158/0008-5472.CAN-04-2126. [DOI] [PubMed] [Google Scholar]
- 23.Barone I, Iacopetta D, Covington KR, Cui Y, Tsimelzon A, Beyer A, et al. Phosphorylation of the mutant K303R estrogen receptor alpha at serine 305 affects aromatase inhibitor sensitivity. Oncogene. 2010;29:2404–14. doi: 10.1038/onc.2009.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herynk MH, Hopp T, Cui Y, Niu A, Corona-Rodriguez A, Fuqua SA. A hypersensitive estrogen receptor alpha mutation that alters dynamic protein interactions. Breast Cancer Res Treat. 2010;122:381–93. doi: 10.1007/s10549-009-0580-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giordano C, Cui Y, Barone I, Andò S, Mancini M, Berno V, et al. Growth factor-induced resistance to tamoxifen is associated with a mutation of estrogen receptor α and its phosphorylation at serine 305. Breast Cancer Res Treat. 2010;119:71–81. doi: 10.1007/s10549-009-0334-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barone I, Cui Y, Herynk MH, Corona-Rodriguez A, Giordano C, Selever J, et al. Expression of the K303R estrogen receptor α breast cancer mutation induces resistance to an aromatase inhibitor via addiction to the PI-3K/Akt kinase pathway. Cancer Res. 2009;69:4724–32. doi: 10.1158/0008-5472.CAN-08-4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Catalano S, Malivindi R, Giordano C, Gu G, Panza S, Bonofiglio D, et al. Farnesoid X receptor, through the binding with steroidogenic factor 1-responsive element, inhibits aromatase expression in tumor Leydig cells. J Biol Chem. 2010;285:5581–93. doi: 10.1074/jbc.M109.052670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sirianni R, Chimento A, Malivindi R, Mazzitelli I, Andò S, Pezzi V. Insulin-like growth factor-I, regulating aromatase expression through steroidogenic factor 1, supports estrogen-dependent tumor Leydig cell proliferation. Cancer Res. 2007;67:8368–77. doi: 10.1158/0008-5472.CAN-06-4064. [DOI] [PubMed] [Google Scholar]
- 29.Catalano S, Mauro L, Marsico S, Giordano C, Rizza P, Rago V, et al. Leptin induces, via ERK1/ERK2 signal, functional activation of estrogen receptor alpha in MCF-7 cells. J Biol Chem. 2004;279:19908–15. doi: 10.1074/jbc.M313191200. [DOI] [PubMed] [Google Scholar]
- 30.Mauro L, Catalano S, Bossi G, Pellegrino M, Barone I, Morales S, et al. Evidences that leptin up-regulates E-cadherin expression in breast cancer: effects on tumor growth and progression. Cancer Res. 2007;67:3412–21. doi: 10.1158/0008-5472.CAN-06-2890. [DOI] [PubMed] [Google Scholar]
- 31.Cirillo D, Rachiglio AM, la Montagna R, Giordano A, Normanno N. Leptin signaling in breast cancer: an overview. J Cell Biochem. 2008;105:956–64. doi: 10.1002/jcb.21911. [DOI] [PubMed] [Google Scholar]
- 32.Leroy P, Dessolin S, Villageois P, Moon BC, Friedman JM, Ailhaud G, et al. Expression of ob gene in adipose cells Regulation by insulin. J Biol Chem. 1996;271:2365–68. doi: 10.1074/jbc.271.5.2365. [DOI] [PubMed] [Google Scholar]
- 33.Cascio S, Ferla R, D’Andrea A, Gerbino A, Bazan V, Surmacz E, et al. Expression of angiogenic regulators, VEGF and leptin, is regulated by the EGF/PI3K/STAT3 pathway in colorectal cancer cells. J Cell Physiol. 2009;221:189–94. doi: 10.1002/jcp.21843. [DOI] [PubMed] [Google Scholar]
- 34.Trujillo ME, Sullivan S, Harten I, Schneider SH, Greenberg AS, Fried SK. Interleukin-6 regulates human adipose tissue lipid metabolism and leptin production in vitro. J Clin Endocrinol Metab. 2004;89:5577–82. doi: 10.1210/jc.2004-0603. [DOI] [PubMed] [Google Scholar]
- 35.Iwabu A, Smith K, Allen FD, Lauffenburger DA, Wells A. Epidermal growth factor induces fibroblast contractility and motility via a protein kinase C delta-dependent pathway. J Biol Chem. 2004;279:14551–60. doi: 10.1074/jbc.M311981200. [DOI] [PubMed] [Google Scholar]
- 36.Fidler IJ. The pathogenesis of cancer metastasis: the “seed and soil” hypothesis revisited. Nat Rev Cancer. 2003;3:453–58. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 37.Witz IP. Yin-yang activities and vicious cycles in the tumor microenvironment. Cancer Res. 2008;68:9–13. doi: 10.1158/0008-5472.CAN-07-2917. [DOI] [PubMed] [Google Scholar]
- 38.Wiseman BS, Werb Z. Stromal effects on mammary gland development and breast cancer. Science. 2002;296:1046–49. doi: 10.1126/science.1067431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glasow A, Kiess W, Anderegg U, Berthold A, Bottner A, Kratzsch J. Expression of Leptin (Ob) and Leptin Receptor (Ob-R) in Human Fibroblasts: Regulation of Leptin Secretion by Insulin. J Clin Endocrinol Metab. 2001;86:4472–79. doi: 10.1210/jcem.86.9.7792. [DOI] [PubMed] [Google Scholar]
- 40.Torday JS, Sun H, Wang L, Torres E. Leptin mediates the parathyroid hormone-related protein paracrine stimulation of fetal lung maturation. Am J Physiol Lung Cell Mol Physiol. 2002;282:L405–10. doi: 10.1152/ajplung.2002.282.3.L405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin TC, Lee TC, Hsu SL, Yang CS. The Molecular Mechanism of Leptin Secretion and Expression Induced by Aristolochic Acid in Kidney Fibroblast. Plos one. 2011;6:e16654. doi: 10.1371/journal.pone.0016654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ishikawa M, Kitayama J, Nagawa H. Enhanced expression of leptin and leptin receptor (OB-R) in human breast cancer. Clin Cancer Res. 2004;10:4325–31. doi: 10.1158/1078-0432.CCR-03-0749. [DOI] [PubMed] [Google Scholar]
- 43.Garofalo C, Koda M, Cascio S, Sulkowska M, Kanczuga-Koda L, Golaszewska J, et al. Increased expression of leptin and the leptin receptor as a marker of breast cancer progression: possible role of obesity-related stimuli. Clin Cancer Res. 2006;12:1447–53. doi: 10.1158/1078-0432.CCR-05-1913. [DOI] [PubMed] [Google Scholar]
- 44.Catalano S, Marsico S, Giordano C, Mauro L, Rizza P, Panno ML, et al. Leptin enhances, via AP-1, expression of aromatase in the MCF-7 cell line. J Biol Chem. 2003;278:28668–76. doi: 10.1074/jbc.M301695200. [DOI] [PubMed] [Google Scholar]
- 45.Bennett PA, Lindell K, Karlsson C, Robinson IC, Carlsson LM, Carlsson B. Differential expression and regulation of leptin receptor isoforms in the rat brain: effects of fasting and oestrogen. Neuroendocrinology. 1998;67:29–36. doi: 10.1159/000054295. [DOI] [PubMed] [Google Scholar]
- 46.Lindell K, Bennett PA, Itoh Y, Robinson IC, Carlsson LM, Carlsson B. Leptin receptor 5’untranslated regions in the rat: relative abundance, genomic organization and relation to putative response elements. Mol Cell Endocrinol. 2001;172:37–45. doi: 10.1016/s0303-7207(00)00382-8. [DOI] [PubMed] [Google Scholar]
- 47.Li L, Dragulev B, Zigrino P, Mauch C, Fox JW. The invasive potential of human melanoma cell lines correlates with their ability to alter fibroblast gene expression in vitro and the stromal microenvironment in vivo. Int J Cancer. 2009;125:1796–804. doi: 10.1002/ijc.24463. [DOI] [PubMed] [Google Scholar]
- 48.Tsujino T, Seshimo I, Yamamoto H, Ngan CY, Ezumi K, Takemasa I, et al. Stromal myofibroblasts predict disease recurrence for colorectal cancer. Clin Cancer Res. 2007;13:2082–90. doi: 10.1158/1078-0432.CCR-06-2191. [DOI] [PubMed] [Google Scholar]
- 49.Infante JR, Matsubayashi H, Sato N, Tonascia J, Klein AP, Riall TA, et al. Peritumoral fibroblast SPARC expression and patient outcome with resectable pancreatic adenocarcinoma. J Clin Oncol. 2007;25:319–25. doi: 10.1200/JCO.2006.07.8824. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.