

Abstract
Hsp90 is a promising therapeutic target for the treatment of cancer. Novobiocin is the first Hsp90 C-terminal inhibitor ever identified and recent structure-activity relationship studies on the noviose sugar identified several commercially available amines as suitable surrogates. In an effort to further understand this region of the molecule, analogues containing various N′-amino substituents were prepared and evaluated against two breast cancer cell lines for determination of their efficacy. Compound 37j manifested the most potent anti-proliferative activity from these studies and induced Hsp90-dependent client protein degradation at mid nano-molar concentrations.
Keywords: Heat shock protein 90, Hsp90 inhibitors, Novobiocin analogues, Breast cancer
The 90 kDa heat shock protein is an ATP-dependent chaperone that belongs to the GHKL superfamily.1 It is one of the most abundant molecular chaperones in the cytosol and promotes the folding, activation and stabilization of more than 200 client proteins, approximately 50 of which are directly associated with cell growth and/or signaling pathways.2 Malignant or mutated oncogenic proteins, such as Her-2, Raf-1, Akt, CDK4, Src, Flt-3, hTert, c-Met, etc, are distributed amongst the six hallmarks of cancer and are highly dependent upon the Hsp90 protein folding machinery for their ability to promote cell survival, proliferation, and adaptation.3 In contrast to the homodimeric chaperone that is present in normal cells, Hsp90 exists as a heteroprotein super-chaperone complex in cancer cells and is bound to oncogenic proteins that are sensitive to physiological stress.4 In such scenarios, Hsp90 inhibition results in the simultaneous disruption of multiple oncogenic pathways and eventually leads to cancer cell death, while largely sparing normal cells. As a consequence, Hsp90 has emerged as a promising therapeutic target for the treatment of cancer.
Hsp90 contains three highly conserved domains: the 25 kDa N-terminus is responsible for ATPase activity, the 35 kDa middle domain is utilized for substrate recognition, and the 12 kDa C-terminus elicits dimerization and co-chaperone binding.5 The N-terminal domain has been extensively studied and 16 small molecules targeting this region have been evaluated in clinical trials.6,7 However, detriments such as heat shock induction and cytostatic activity associated with N-terminal inhibition has limited their potential use against cancer.8 However, some small molecules that bind to the C-terminal domain do not induce the pro-survival heat shock response, and in some cases, even cause Hsp70 and Hsp90 degradation.9–12 Because the Hsp90 C-terminus is responsible for mediating interactions with co-chaperones such as HOP (Hsp70-Hsp90 organization protein) and the immunophilins (eg. FK506 binding protein) to facilitate client protein maturation,13,14 small molecule modulation of this region exhibits activities not observed with N-terminal inhibitors.11,15
Novobiocin was first identified as an Hsp90 C-terminal inhibitor in 2000 by Neckers and coworkers.16 Subsequent modification to novobiocin has led to the elucidation of structure-activity relationships and analogues that exhibit superior inhibitory activity.17–21 Deletion of both the 4-hydroxy substituent on the coumarin ring and the 3′-carbamoyl group on noviose resulted in DHN2, which transformed novobiocin from a DNA gyrase inhibitor to a selective Hsp90 inhibitor (Figure 1).22 Subsequent replacement of the synthetically complex noviose sugar with readily available amines led to molecules represented by NA-1 (novobiocin analogue 1, Figure 1) and NA-2 (novobiocin analogue 2) that manifest increased anti-proliferative activity and solubility.21 During the course of studies aimed at the modification of these amines, Huang and coworkers developed a three-dimensional quantitative structure-activity relationship (3D-QSAR) model that suggested modifications to the amine may improve anti-proliferative activity.23 Consequently, second generation amino-analogues were designed, synthesized and evaluated against two breast cancer cell lines.
Figure 1.
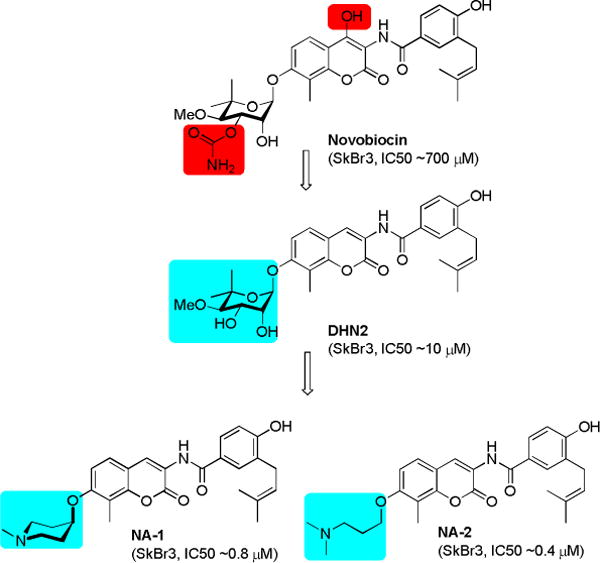
Evolution of novobiocin analogues.
SAR generated from novobiocin analogues suggested that a three-carbon linker between the 7-coumarin phenol and the amine was optimal.21 Although the anti-proliferative activity of NA-1 and NA-2 suggested flexibility of the amine moiety may be required, increasing the number of rotatable bonds is generally considered detrimental, due to entropic penalties. Therefore, novobiocin analogues containing rigid heterocyclic piperidine and pyrroridine derivatives were synthesized and evaluated. As illustrated in Figure 2, the piperidine or pyrrolidine ring system was linked to the 7-phenolic oxygen by 1–3 carbons.
Figure 2.
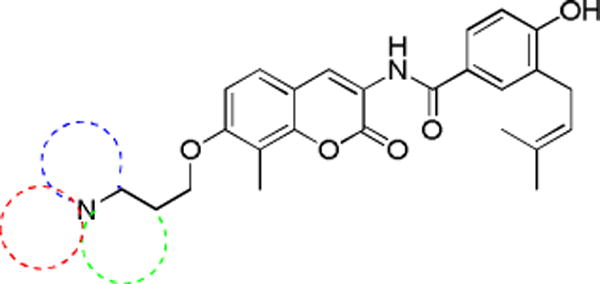
Proposed cyclic amino derivatives of novobiocin
These amines were assembled with the coumarin core in modular fashion utilizing Boc-protected secondary amines (1 and 3) or tertiary amines (2 and 4), which underwent Mitsunobu esterification with phenol 5 to afford 6–9.21 Subsequent hydrogenolysis to unmask the amine allowed for amide coupling with 4-(chlorocarbonyl)-2-(3-methylbut-2-en-1-yl)phenyl acetate (10) in the presence of pyridine afforded novobiocin C-linked heterocyclic analogues 11–14. Acid-catalyzed deprotection of 11 and 13 resulted in secondary amines 15 and 16. Finally, hydrolysis of ester 12 and 14–16 generated phenols 17–20 (Scheme 1).
Scheme 1.
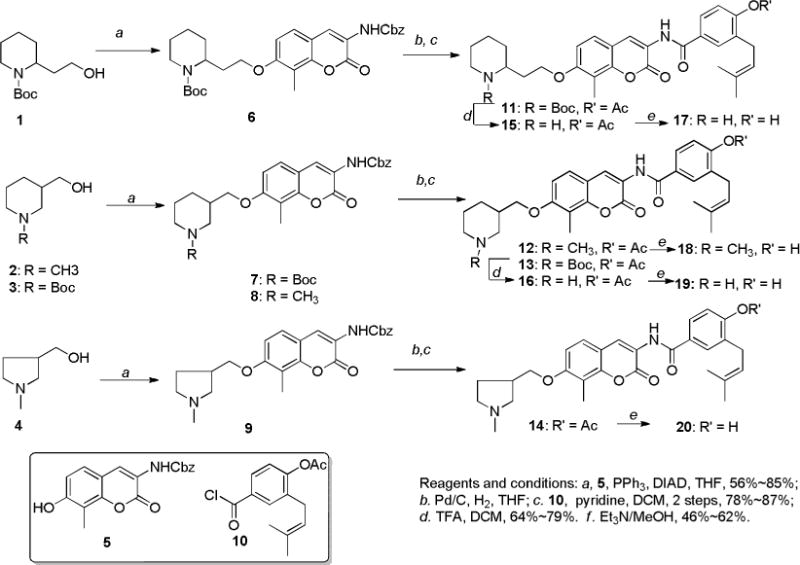
Synthesis of C-linked heterocyclic derivatives of novobiocin
Along with the C-linked heterocyclic derivatives (12, 14, 15–20), N-linked heterocycles (29–33) were also synthesized (Scheme 2). Compound 23 was regarded as a versatile intermediate and was prepared in 3 steps. Benzyl (7-hydroxy-8-methyl-2-oxo-2H-chromen-3-yl)carbamate (5) was treated with ten equivalents of 1,3-dibromopropane (21) in the presence of potassium carbonate to afford alkyl bromide 22. Subsequent hydrogenolysis followed by amide coupling with acid chloride 10 produced the corresponding amide 23. Displacement of the bromide in 23 with amines 24–28 in N,N-dimethylformamide and simultaneous hydrolysis of the phenolic ester gave amines 29–33.
Scheme 2.
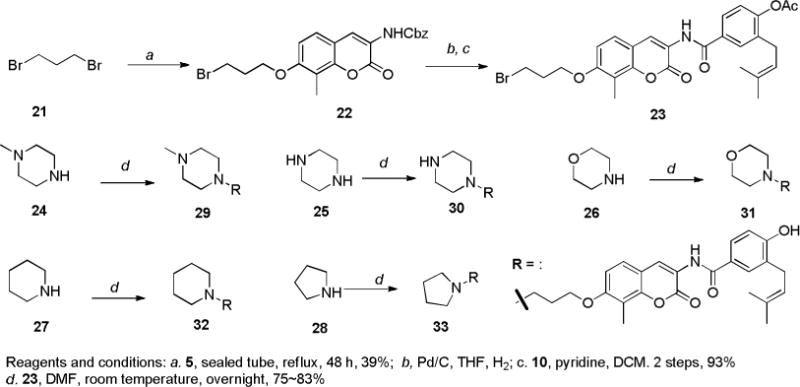
Synthesis of terminal cycloamine derivatives of novobiocin
Upon construction of the C- and N-linked heterocyclic novobiocin analogues, these molecules were evaluated for anti-proliferative activity against SKBr3 (estrogen receptor negative, Her2 over-expressing breast cancer cells) and MCF-7 (estrogen receptor positive breast cancer cells) cell lines. As shown in Table 1, the anti-proliferative activities manifested by C-linked heterocyclic analogues (12–20) were similar to NA-1, suggesting that various ring structures are accommodated within the binding site. Interestingly, the secondary amino analogues exhibit greater inhibitory activity than the corresponding tertiary amines (16 vs 12, 19 vs 18), which is further exacerbated against the MCF-7 cell line. Although piperizine analogues (29 and 30) maintained activity comparable to NA-1, compounds 31 and 32 were inactive at the highest concentration tested. However, shrinking the ring size to five atoms (33) restored activity, suggesting that hydrophobicity is limited in this region and that the inclusion of heteroatoms is necessary for further extension.
Table 1.
Anti-proliferative activities NA-1 analogues.
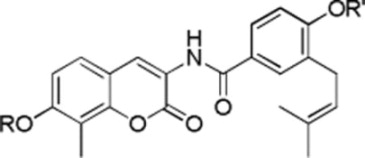
| ||||
|---|---|---|---|---|
| R | R′ | SKBr3 (μM) | MCF-7 (μM) | |
| KU-398 |
|
H | 0.76±0.17a | 1.09±0.10 |
| 12 |
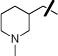
|
OAc | 0.74±0.01 | 1.65±0.11 |
| 14 |
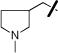
|
OAc | 0.88±0.09 | 1.88±0.45 |
| 15 |
|
OAc | 0.64±0.06 | 0.71±0.03 |
| 16 |
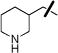
|
OAc | 0.62±0.04 | 0.82±0.07 |
| 17 |
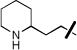
|
H | 0.64±0.06 | 0.71±0.03 |
| 18 |
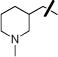
|
H | 1.30±0.63 | 2.64±0.45 |
| 19 |
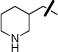
|
H | 0.60±0.01 | 0.79±0.10 |
| 20 |
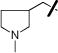
|
H | 0.92±0.56 | 1.55±0.04 |
| 29 |
|
H | 1.01±0.03 | 1.20±0.12 |
| 30 |
|
H | 0.82±0.02 | 0.93±0.21 |
| 31 |
|
H | >50 | >50 |
| 32 |
|
H | >50 | >50 |
| 33 |
|
H | 0.72±0.01 | 0.99±0.04 |
Values represent mean ± standard deviation for at least two separate experiments performed in triplicate
Data from the first set of compounds suggest there is limited hydrophobic space, however, further extension is possible, but requires an H-bond donor (e.g., 30), which cannot be replaced with a hydrophobic moiety (32) or an H-bond acceptor (31). In addition, Huang and co-workers suggested in their 3D-QSAR study that steric bulk N-substitution of NA-2 could lead to increased anti-proliferative activity.23 Therefore, analogues containing various linear and branched N-substitutions were pursued in an effort to maintain both the hydrophobicity and the H-bond donor properties. As shown in Scheme 3, compound 23 was treated with primary and secondary amines (34a–34o) to produce the corresponding secondary and tertiary amine derivatives containing various linear or branched chains (35a–35k). Similarly, treatment of 23 with amines 34l–34o afforded compounds 35j–35o that contain various H-bond properties to further verify whether H-bond donors or acceptors were favored.
Scheme 3.
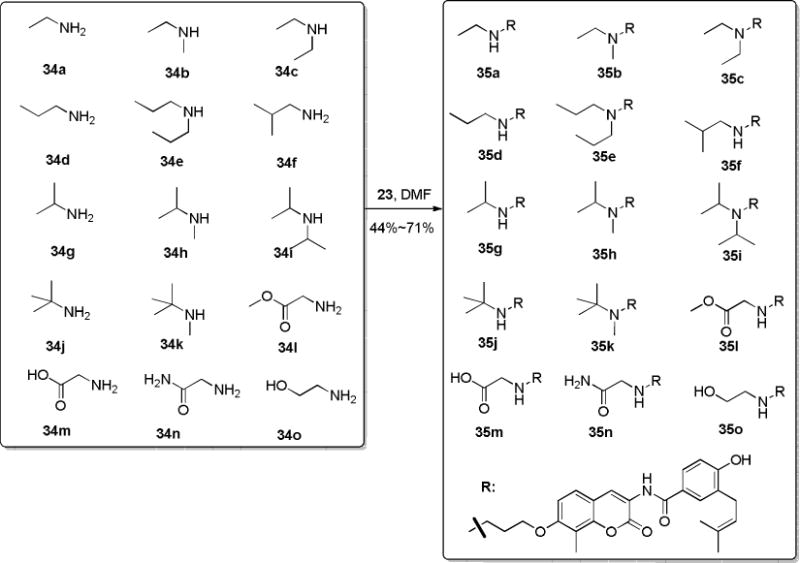
Synthesis of NA-2 analogues with linear and branched chains.
Anti-proliferative activity manifested by these analogues was evaluated against SKBr3 and MCF-7 cell lines as previously described. As shown in Table 2, a bulky group (up to 3 linear carbons, 35a, 35b, 35d, and 35f–35k except 35i) was generally well-tolerated in the binding site. It appears as though this hydrophobic chain can be linear (35a, 35b, and 35d) or branched (35f–35h, 35j and 35k). The diethylamine (35c) and diisopropylamine (35i) analogues exhibited decreased anti-proliferative activity against SKBr3 cells, while the diisopropylamine analogue manifested no activity (35e), indicating a limited amount of space is available. Glycine ester analogue 35l and amide analogue 35n exhibited decreased anti-proliferative activity against SKBr3 cells or MCF-7 cells while alcohol 35o retained activity against both cancer cell lines, supporting further extension into the binding site requires hydrogen bond donors. The significantly decreased activity of glycine acid analogue 35m may result from limited cell membrane penetration.
Table 2.
Anti-proliferative activities of NA-2 analogues
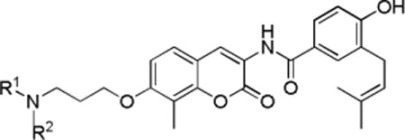
| ||||
|---|---|---|---|---|
| R1 | R2 | SKBr3 (μM) | MCF-7 (μM) | |
| KU-407 | CH3 | CH3 | 0.44±0.02a | 1.35±0.38 |
| 35a | CH2CH3 | H | 0.72±0.34 | 0.98±0.35 |
| 35b | CH2CH3 | CH3 | 0.73±0.06 | 2.19±0.50 |
| 35c | CH2CH3 | CH2CH3 | 4.90±0.42 | 6.39±0.04 |
| 35d | CH2CH2CH3 | H | 0.99±0.03 | 0.98±0.00 |
| 35e | CH2CH2CH3 | CH2CH2CH3 | >50 | >50 |
| 35f | CH2CH(CH3)2 | H | 1.03±0.02 | 1.32±0.06 |
| 35g | CH(CH3)2 | H | 0.75±0.26 | 0.74±0.16 |
| 35h | CH(CH3)2 | CH3 | 0.57±0.13 | 1.05±0.06 |
| 35i | CH(CH3)2 | CH(CH3)2 | 1.60±0.02 | 1.31±0.39 |
| 35j | C(CH3)3 | H | 0.66±0.06 | 0.95±0.10 |
| 35k | C(CH3)3 | CH3 | 0.88±0.10 | 1.14±0.11 |
| 35l | CH2COOMe | H | 1.06±0.11 | 2.82±0.27 |
| 35m | CH2COOH | H | 7.57±2.86 | 18.11±5.35 |
| 35n | CH2CONH2 | H | 2.29±0.47 | 1.46±0.35 |
| 35o | CH2CH2OH | H | 0.61±0.06 | 1.56±0.09 |
Values represent mean ± standard deviation for at least two separate experiments performed in triplicate
Although none of the first two sets of compounds showed significantly improved anti-proliferative activity, it is clear that steric bulk is tolerated in the binding site. Therefore, a set of NA-2 derivatives containing various rings (37a–37j) were prepared in a similar synthetic sequence, as described in Scheme 4.
Scheme 4.
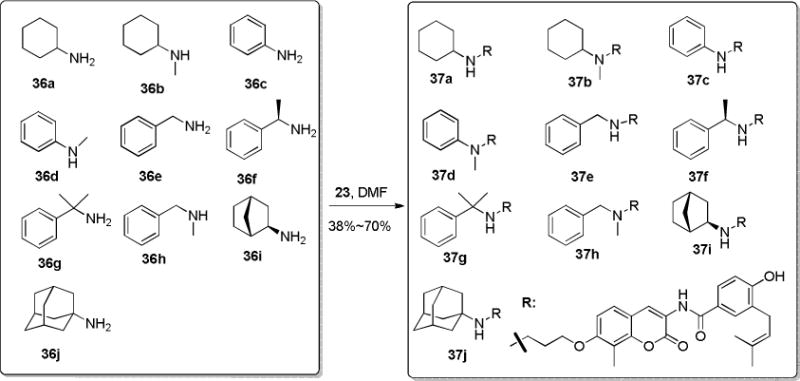
Synthesis of NA-2 derivatives with cyclic N-substitutions
The anti-proliferative data (Table 3) shows that cyclohexyl (37a and 37b), benzyl (37e), substituted benzyl (37f and 37g), and bicyclic alkyls (37i and 37j) are tolerated in the binding site. Aniline-containing analogues were inactive (37c and 37d), possibly due to the altered hydrogen bonding property manifested by amine due to the incorporation of the phenyl ring. Compound 37j, which contained an N-adamantyl substitution, manifested the most potent anti-proliferative activity against both SKBr3 and MCF-7 breast cancer cell lines, indicating that significant space is available, but the molecule must contain a free H-bond donor in the form of a secondary amine. In addition, 37j exhibited lower activity against normal cells (MRC5, 5.42 μM; HMLE, 15.05 μM), thus provide a considerable therapeutic window for the treatment of cancer.
Table 3.
Anti-proliferative activity of NA-2 derivatives with cyclic N-substitutions
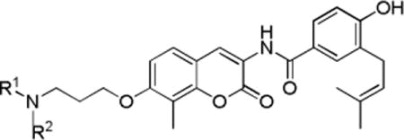
| ||||
|---|---|---|---|---|
| R1 | R2 | SKBr3 (μM) | MCF-7 (μM) | |
| 37a | Cyclohexyl | H | 0.66±0.12a | 1.07±0.35 |
| 37b | Cyclohexyl | CH3 | 0.97±0.03 | 1.08±0.07 |
| 37c | Phenyl | H | >50 | >50 |
| 37d | Phenyl | CH3 | >50 | >50 |
| 37e | Benzyl | H | 0.47±0.16 | 0.91±0.01 |
| 37f | (R)-1-phenylethyl | H | 1.00±0.31 | 1.45±0.20 |
| 37g | Cumenyl | H | 0.96±0.01 | 1.07±0.01 |
| 37h | Benzyl | CH3 | 10.73±0.49 | 11.52±4.16 |
| 37i |
|
H | 0.77±0.13 | 1.03±0.12 |
| 37j | Adamantyl | H | 0.31±0.04 | 0.32±0.03 |
Values represent mean ± standard deviation for at least two separate experiments performed in triplicate
To confirm these novobiocin analogues that contain amino groups in lieu of noviose manifest inhibitory activity through Hsp90 inhibition, western blot analyses of cell lysates following the administration of most active compound, 37j, were performed. As shown in Figure 3, the Hsp90-dependent client proteins, Raf and Akt, were degraded in MCF-7 cells in a concentration-dependent manner upon treatment with 37j. The non-Hsp90-dependent protein, actin, was not degraded upon the administration of 37j, indicating selective degradation of Hsp90-dependent proteins. In accordance with previous studies, Hsp90 levels remained constant at all concentrations tested, demonstrating that 37j does not induce the pro-survival heat shock response.
Figure 3.
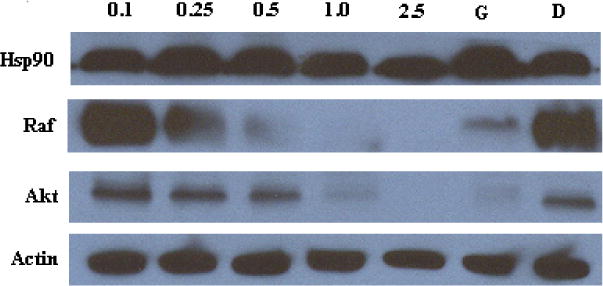
Western blot analyses of MCF-7 cell lysates for Hsp90 client protein degradation after 24 h incubation. Concentrations (in μM) of 37j are indicated above each lane. Geldanamycin (G, 500 nM) and DMSO (D) were employed respectively as positive and negative controls.
In conclusion, a library of novobiocin analogues containing a second generation of amino-appendages was designed, synthesized and evaluated. The results showed that a variety of bulky N-alkyl groups are well tolerated and can lead to compounds with efficacious activity against breast cancer cells. Compound 37j, a secondary amino derivative containing an N-adamatyl group, manifested the most potent anti-proliferative activity, which through western blot analyses confirmed this compound inhibits the Hsp90 protein folding machinery.
Acknowledgments
The authors gratefully acknowledge support of this project by the NIH/NCI (CA120458) and the DOD Prostate Cancer Research Program (QH815179).
References
- 1.Dutta RIM. Trends Biochem Sci. 2000;25:24. doi: 10.1016/s0968-0004(99)01503-0. [DOI] [PubMed] [Google Scholar]
- 2.Blagg BSJ, Kerr TA. Med Res Rev. 2006;26:310. doi: 10.1002/med.20052. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. Cell. 2011;144:646. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.Workman P. Trends Mol Med. 2004;10:47. doi: 10.1016/j.molmed.2003.12.005. [DOI] [PubMed] [Google Scholar]
- 5.Zhao H, Michaelis Mary L, Blagg Brian SJ. Adv Pharmacol. 2012;64:1. doi: 10.1016/B978-0-12-394816-8.00001-5. [DOI] [PubMed] [Google Scholar]
- 6.Jhaveri K, Taldone T, Modi S, Chiosis G. Biochim Biophys Acta-Mol Cell Res. 2012;1823:742. doi: 10.1016/j.bbamcr.2011.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim YS, Alarcon SV, Lee S, Lee MJ, Giaccone G, Neckers L, Trepel JB. Current Topics in Medicinal Chemistry. 2009;9:1479. doi: 10.2174/156802609789895728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vaughan CK, Neckers L, Piper PW. Nat Struct Mol Biol. 2010;17:1400. doi: 10.1038/nsmb1210-1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shelton SN, Shawgo ME, Matthews SB, Lu Y, Donnelly AC, Szabla K, Tanol M, Vielhauer GA, Rajewski RA, Matts RL, Blagg BSJ, Robertson JD. Mol Pharmacol. 2009;76:1314. doi: 10.1124/mol.109.058545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao HP, Brandt GE, Galam L, Matts RL, Blagg BSJ. Bioorg Med Chem Lett. 2011;21:2659. doi: 10.1016/j.bmcl.2010.12.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eskew JD, Sadikot T, Morales P, Duren A, Dunwiddie I, Swink M, Zhang XY, Hembruff S, Donnelly A, Rajewski RA, Blagg BSJ, Manjarrez JR, Matts RL, Holzbeierlein JM, Vielhauer GA. Bmc Cancer. 2011:11. doi: 10.1186/1471-2407-11-468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhao HP, Yan B, Peterson LB, Blagg BSJ. ACS Med Chem Lett. 2012;3:327. doi: 10.1021/ml300018e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cox MB, Johnson JL. Methods Mol Biol. 2011;787:45. doi: 10.1007/978-1-61779-295-3_4. [DOI] [PubMed] [Google Scholar]
- 14.Robson T, James IF. Drug Discovery Today. 2012;17:544. doi: 10.1016/j.drudis.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 15.Zhang L, Zhao HP, Blagg BSJ, Dobrowsky RT. J Proteome Res. 2012;11:2581. doi: 10.1021/pr300056m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LMJ. Biol Chem. 2000;275:37181. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- 17.Yu XM, Shen G, Neckers L, Blake H, Holzbeierlein J, Cronk B, Blagg BSJ. J Am Chem Soc. 2005;127:12778. doi: 10.1021/ja0535864. [DOI] [PubMed] [Google Scholar]
- 18.Burlison JA, Avila C, Vielhauer G, Lubbers DJ, Holzbeierlein J, Blagg BSJ. J Org Chem. 2008;73:2130. doi: 10.1021/jo702191a. [DOI] [PubMed] [Google Scholar]
- 19.Zhao HP, Kusuma BR, Blagg BSJ. ACS Med Chem Lett. 2010;1:311. doi: 10.1021/ml100070r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Donnelly AC, Zhao HP, Kusuma BR, Blagg BSJ. Medchemcomm. 2010;1:165. [Google Scholar]
- 21.Zhao H, Donnelly AC, Kusuma BR, Brandt GEL, Brown D, Rajewski RA, Vielhauer G, Holzbeierlein J, Cohen MS, Blagg BSJ. J Med Chem. 2011;54:3839. doi: 10.1021/jm200148p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Burlison JA, Neckers L, Smith AB, Maxwell A, Blagg BSJ. J Am Chem Soc. 2006;128:15529. doi: 10.1021/ja065793p. [DOI] [PubMed] [Google Scholar]
- 23.Huang XY, Shan ZJ, Zhai HL, Li LN, Zhang XY. J Chem Inform Mod. 2011;51:1999. doi: 10.1021/ci2002236. [DOI] [PubMed] [Google Scholar]