

Abstract
The interaction that occurs between molecules is a dynamic process that impacts both structural and conformational properties of the ligand and the ligand binding site. Herein, we investigate the dynamic cross-talk between a protein and the ligand as a source for new opportunities in ligand design. Analysis of the formation/disappearance of protein pockets produced in response to a first-generation inhibitor assisted in the identification of functional groups that could be introduced onto scaffolds to facilitate optimal binding, which allowed for increased binding with previously uncharacterized regions. MD simulations were used to elucidate primary changes that occur in the Hsp90 C-terminal binding pocket in the presence of first-generation ligands. This data was then used to design ligands that adapt to these receptor conformations, which provides access to an energy landscape that is not visible in a static model. The newly synthesized compounds demonstrated anti-proliferative activity at ~150 nanomolar concentration. The method identified herein may be used to design chemical probes that provide additional information on structural variations of Hsp90 C-terminal binding site.
Keywords: Drug-Design, Flexibility, Allostery, MD simulations, Dynamics-Based Design, Hsp90
Introduction
Proteins carry out functions and participate in interaction networks by dynamically switching amongst diverse conformational sub-states. This process favors binding to various partners and allows biomolecules to tune their function for various conditions. In this context, perturbations induced by ligand-binding, covalent modification, or protein-protein interaction can lead to the activation of specific conformational states.1,2
Allosteric events represent one mechanism by which conformational regulation can be achieved: Modifications at one site are propagated throughout the protein and the net result can shift the structural population towards a dynamic state that elicits specific functions.3 Understanding the molecular determinants of allosteric tuning can then facilitate the discovery and characterization of novel binding sites and provide new opportunities for the design of molecules that selectively interfere with functional sub-states. Identification of such determinants could translate into the modulation of specific cell functions by perturbing protein conformations.4–6 In this model, the basic requirement is identification of protein residues that can be targeted for interference with specific conformations, which can be elucidated through understanding the internal dynamics of allosteric coordination between different parts of the protein.6,7 Such insights could then be used to analyze the receptor’s conformation in the presence of a ligand. This interaction can result in the identification of protein flexibility that allows ligand binding while simultaneously dictating the conformation of the binding site.8,9
Characterization of this dynamic process could then be exploited for the development of compounds that allow for the extension of binding interactions into previously uncharacterized regions. The formation/disappearance of such pockets in response to first-generation ligands could then define the structural and physico-chemical basis for the introduction of functionalities that facilitate binding to the new shape.
This strategy requires the evaluation of protein dynamics as well as the consideration of multiple receptor structures during the design process. It is worth noting that the inclusion of protein flexibility in drug development is increasingly accepted to overcome the limitations of single-structure-based approaches. Several articles and reviews have demonstrated the nature of the receptor model can affect the performance of a docking-based screen. Moreover, ligands may bind to receptor conformations that occur infrequently and may not be captured in crystal structure studies. This is true even for enzymatic active sites wherein the presence of a ligand can dramatically influence the position and orientation of catalytic residues. In addition, the location of water molecules can also represent a problem, as dynamic diffusion in the binding site can establish interactions between the protein and the ligand.10–17 In this context, several experimental and computational studies have assessed the predictive power of using multiple receptor conformations for docking and screening, resulting in improved results over the use of single crystal structures.12,14 From a computational point of view, using Monte Carlo (MC) as well as Molecular Dynamics (MD) based methods can account for protein flexibility, as the former focuses mainly on alternative rotameric conformations in the binding site and the latter, generates ensembles of receptor structures in solution, to screen for sites of preferential interactions with small molecule probes.
In this article, we initiate these concepts and investigate the feasibility of exploiting the structural distortions induced by first generation ligands on a binding site that is not accessible in the unbound form and guide the rational development of novel small molecules that exhibit improved properties. In this article, we focus on allosteric inhibitors that target the C-terminal binding site of the Heat Shock Protein 90 (Hsp90) molecular chaperone.
Hsp90 represents a paradigmatic example of a protein whose activities are strictly dependent on conformational dynamics and on ligand-mediated transitions amongst conformational sub-states.18,19 Crystal structures of the chaperone from different organisms highlighted a common modular organization of the N- (NTD), middle (M) and C-terminal (CTD) domains.20–22 The chaperone activity depends upon ATP binding and hydrolysis at the N-terminus, which is coupled to a conformational cycle that involves opening and closing of a molecular clamp formed by association of the NTDs.23 Although the exact mechanism of coupling between ATP-hydrolysis and client protein folding is still a matter of debate, structural and biochemical data support a model in which nucleotide binding at the NTD propagates a conformational signal to the CTD, which induces a conformational rearrangement that brings the two NTDs into close association in the ATP-bound state, but not in the ADP-bound or apo states.20–22,24,25 The CTDs act as a hinge for this “molecular clamp” mechanism.
From a biological point of view, Hsp90 is a nodal protein at the crossroad of multiple cell functions where it orchestrates integrated cellular pathways that are required for cell development and maintenance.26,27 The chaperone plays a key role in cancer cells, where it is overexpressed compared to normal cells. As a consequence, Hsp90 inhibition has been intensely pursued as an anti-cancer target. In fact, 17 inhibitors that target the N-terminal ATP-binding site have entered clinical trials.26 However, clinical trials have shown these drugs to be limited by various ailments, including poor solubility and toxicity unrelated to Hsp90 inhibition.28
A potential strategy to overcome these limitations is to regulate Hsp90 function by targeting allosteric sites on the chaperone that are distinct from the N-terminal ATP binding site. Coumarin antibiotics, in particular novobiocin, were identified as the first generation of Hsp90 C-terminal inhibitors in 2000.29–31 However the low inhibitory activity manifested by novobiocin (~700 μM in SKBr3 cells) is not sufficient for clinical application. Yet, it provided a starting point for the development of more efficacious C-terminal inhibitors.
We have set out to design new C-terminal targeted inhibitors by exploiting a computational approach that relies on the identification of druggable sites distal from the active site 6 via Molecular Dynamics (MD) simulations.32,33 In our model, allosteric pathways are defined by the identification of distal residues that move coherently within the active site of a protein. Towards this objective, we defined the coordination propensity (CP) between any two residues within a protein as a function of their distance fluctuation. Analysis of the long-range CP patterns of residue-pairs identifies the amino acids contributing to the activation/inhibition of relevant motions. These residues, often belonging to different domains, may change their long-distance CP in a cooperative manner in the presence/absence of a ligand.32,33 Controlling these residues with small molecules provide a mechanism to influence their motion through an allosteric mechanism. Therefore, this approach can be used to identify new druggable sites, to design potential allosteric ligands, and to explore the allosteric effect of these molecules on protein function, which adds an additional dimension to structure-based drug design.
In the case of Hsp90, we have been able to identify an allosteric site, distant from the ATP-binding pocket and located in the C-terminal domain, against which several ligands were successfully screened. These ligands showed good anticancer activity along with the ability to perturb physical interactions between Hsp90 and client proteins.6 These efforts demonstrated that the detailed analysis of the microscopic determinants of functionally-oriented internal dynamics can be translated into the discovery of novel functional modulators.
In this article, analysis of the dynamic response of Hsp90 to the presence of novobiocin in the allosteric pocket was used to characterize the formation/disappearance of pockets in the ligand-protein interaction region. The resulting structures were then probed by docking a range of known, CTD-targeted novobiocin analogues (Figure 1, 2). The aim of this step was to determine whether productive binding interactions could be established by these derivatives and whether such poses could support structure-activity relationships. Next, the generated docking poses and binding-pocket conformational changes were used to guide the design of new inhibitors that contain functional groups selected to exhibit an optimal fit within the ligand-induced conformations of the binding site. Such molecules provide information on the structural variations of the binding site in response to the protein’s function. Finally, the newly designed ligands were evaluated for their antiproliferative activity against cancer cells, and demonstrated improved activity compared to the original leads.
Figure 1. Strategy for the identification of new allosteric inhibitors using protein dynamics.

Identification of the putative C-terminal allosteric binding site from the analysis of the dynamics of the activated state of Hsp90 bound to ATP. Docking of Novobiocin, the first known lead to bind Hsp90-CTD, in this binding site and long-timescale all-atom MD simulations of the complex. The ensemble-based approach ensures the incorporation of receptor flexibility in the characterization of the ligand-induced conformational changes of the binding site. The new structures of the binding site have been probed with a series of analogues of known activity; this analysis allowed the identification of structure-activity relationships. This information has been exploited to design new derivatives with chemical modifications and functional groups that can reach into the cavities. Finally, selected hits have been tested experimentally
Figure 2. Two different orientations of Novobiocin.

(van der Waal representation, each atom of the ligand is represented by a sphere scaled to its van der Waals radius) in the binding site (white-chain A, grey-chain B). The Cα carbons of the representative structures of the second, third and fourth clusters have been superimposed to one of the first cluster (only residues 596-611 (chain A) of the loop L are shown: blue - first cluster, cyan - second cluster, orange – third cluster and red - fourth cluster). Drifting of loop L allows the helical coil (in yellow - residues 411-435, chain B) connecting the M-large and M-small domains of chain B to come closer to the binding site, together with an amphipathic loop in the M-large domain of chain B (in yellow - residues 329-339).
Overall, the results demonstrate the dynamics-based approach could represent an effective strategy to guide the discovery and evolution of second-generation inhibitors by exploiting the salient aspects of conformational protein-ligand cross-talk. Although we have developed and tested it in the context of allosteric inhibitors, this methodology may be applicable to other cases in which an initial protein-ligand complex has been identified.
Materials and methods
Docking calculations
The initial setup for the receptor preparation before docking runs was performed using the Schrödinger’s ‘Protein Preparation Wizard’ program (see www.Schrodinger.com), starting from the most representative protein conformation of previous MD simulations of Hsp90 in the ATP bound state.6 Bond orders and atomic charges were assigned and hydrogen atoms were added. The assignments of protonation states for basic and acidic residues were based on the optimization of hydrogen bonding patterns. The final minimization of the protein was performed with the Preparation Wizard default.
The shape and properties of the resulting binding site were mapped onto a grid with dimensions of 36 Å (enclosing box) and 14 Å (ligand diameter midpoint box), centered on residues 474-487, 502-503, 591-599, 602-603, 652-657 from chain A and residues 502-504, 591-595, and 656-662 from chain B (Hsp90 residues numbering as in the PDB entry 2CG9).20
Rigid receptor and flexible ligand docking calculations were performed using the program Glide (version 5.8 Schrödinger, LLC, New York, NY, 2012).34,35 Docking calculations were performed in Standard Precision mode (SP) with standard OPLS-AA (2001) force field,36 non-planar conformations of amide bonds were penalized, Van der Waals radii were scaled by 0.80 and the partial charge cut off was fixed to 0.15. No further modifications were applied to the default settings.
Molecular dynamics simulations
The best pose of the docking run of the ATP-bound state of Hsp90 and Novobiocin has been employed as a starting point for all-atom MD simulations in explicit water. The simulation and the analysis of the trajectory were performed using the GROMACS software package 37 using the GROMOS96 53A6 force field.38 This force field was chosen to ensure consistency with previous studies we carried out on the allosteric properties of Hsp90.32,33 In order to remove any bad contacts, the complex was initially minimized in vacuo by multiple minimizations (200 steps steepest descent plus 200 steps conjugate gradient). Subsequently, the system was solvated in a tetrahedral solvation box containing around 573000 particles. The SPC water model was used for solvation 39 and Na+ counterions were added to ensure electroneutrality. In order to allow the solvent molecules to relax around the solute, the system was minimized with position restraints on the protein and ligand and just minimizing the positions of water and ions (500 steps steepest descent plus 200 steps conjugate gradient). After this minimization stage, all the system was equilibrated by a 100 ps of MD run in the NVT ensemble to allow the temperature reaching a plateau at 300 K. The first equilibration run was followed by a second 100 ps run in the NPT ensemble to adjust the pressure and the density of the system, by weak coupling to a bath of constant pressure (P0 = 1 bar, coupling time τP = 0.5 ps).40 The first 20 ns of the trajectory were not used in the subsequent analysis in order to minimize convergence artifacts. Equilibration of the trajectory was checked by monitoring the equilibration of quantities, such as the root-mean-square deviation (rmsd) with respect to the initial structure, the internal protein energy, and fluctuations were calculated on different time intervals. The electrostatic term was described by using the particle mesh Ewald algorithm. The LINCS 41 was used to constrain all bond lengths. For the water molecules, the SETTLE algorithm 42 was used. A dielectric permittivity, ε = 1, and a time step of 2 fs were used. All atoms were given an initial velocity obtained from a Maxwellian distribution at the desired initial temperature of 300 K. In all simulations, the temperature was maintained close to the intended values by weak coupling to an external temperature bath 40 with a coupling constant of 0.1 ps. The proteins and the rest of the system were coupled separately to the temperature bath. This protocol resulted in 100 ns MD run. The structural cluster analysis was carried out using the method described by Daura and co-workers 43 with a cutoff of 0.3 nm on the entire protein.
Chemistry
3′-chloro-6-methoxy-[1,1′-biphenyl]-3-carboxylic acid (14a): General procedure for the synthesis of 14a and 14b
[1,1′-Bis(diphenylphosphino)ferrocene]dichloropalladium(II) (58 mg, 0.08 mmol) and 2 mol/L aqueous potassium carbonate (2 mL) were added to a solution of methyl 3-iodo-4-methoxybenzoate (12, 584 mg, 2.0 mmol) and 2-chlorophenylboroni acid (13, 624 mg, 4.0 mmol). The resulting mixture was stirred at 110 °C for 12 hours before cooling down to room temperature. Solvents were evaporated under vacuum and the brown residue was purified by column chromatography using pure dichloromethane as eluent to give biarylester as a thick oil, which was directly subject to hydrolysis using sodium hydroxide (400 mg, 10 mmol) in methanol (10 mL) and water (10 mL) to afford acid 14a as a white amorphous solid (451 mg, 2 steps, 86%). 1H NMR (500 MHz, DMSO-d6) δ 8.31 (s, 1H), 8.22 (dd, J = 8.2, 2.4 Hz, 1H), 8.02 (dd, J = 8.7, 2.2 Hz, 1H), 7.97 (d, J = 0.7 Hz, 1H), 7.92 (d, J = 2.2 Hz, 1H), 7.74 (t, J = 8.0 Hz, 1H), 7.28 (d, J = 8.7 Hz, 1H), 3.88 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 166.94, 159.39, 147.63, 138.69, 135.85, 131.64, 131.55, 129.74, 127.06, 123.78, 123.68, 122.13, 111.72, 56.09. HRMS (ESI+) m/z: [M + Na+] calcd for C14H11ClNaO3 285.0294; found 285.0296.
6-methoxy-3′-nitro-[1,1′-biphenyl]-3-carboxylic acid (14b)
Acid 14b was obtained as a light brown amorphous solid (414 mg, 2 steps, 76%). 1H NMR (500 MHz, DMSO-d6) δ 7.93 (d, J =8.3 Hz, 1H), 7.86 (s, 1H), 7.53 – 7.48 (s, 1H), 7.45 – 7.34 (m, 3H), 7.09 (d, J = 8.6 Hz, 1H), 3.81 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 169.48, 157.77, 140.24, 132.62, 131.66, 130.86, 129.94, 129.28, 128.87, 127.92, 127.05, 126.79, 110.78, 55.78. HRMS (ESI+) m/z: [M + Na+] calcd for C14H11NNaO5 296.0535; found 296.0532.
1-(3′-chloro-6-methoxy-[1,1′-biphenyl]-3-yl)-3-(8-methyl-7-((1-methylpiperidin-4-yl)oxy )-2-oxo-2H-chromen-3-yl)urea (18a): General procedure for the synthesis of 18a and 18b
Triethlyamine (264 mg, 2.62 mmol) was added to a solution of acid 14a (180 mg, 0.65 mmol) and diphenylphosphoryl azide (115 mg, 0.65 mmol) in acetonitrile (15 mL). The resulting mixture was stirred at room temperature for 1 hour before being concentrated to dryness. The colorless residue was purified by flash column chromatography on silica using dichloromethane as eluent to give benzoic azide 15a, which was then dissolved in benzene and converted to isocyanate 16a after refluxing for 8 hours and removal of solvent. 16a was dried under vacuum and dissolved in dry dichloromethane without further purification. To it was added aniline 17 (41 mg, 0.14 mmol) and the resulting mixture was stirred at room temperature for 24 hours before concentrated to dryness. The brown residue was purified by column chromatography using dichloromethane and methanol (10:1) as eluent to afford 18a as light brown amorphous solid (65 mg, 83%). 1H NMR (500 MHz, Chloroform-d) δ 8.80 (s, 1H), 8.45 (t, J = 2.0 Hz, 1H), 8.24 (dd, J = 8.3, 2.3 Hz, 1H), 7.97 (t, J = 8.6 Hz, 1H), 7.93 (d, J = 2.3 Hz, 1H), 7.87 (dd, J = 7.7, 1.7 Hz, 1H), 7.63 (dd, J = 8.3, 7.7 Hz, 1H), 7.36 (d, J = 8.4 Hz, 1H), 7.12 (d, J = 8.7 Hz, 1H), 6.89 (d, J = 8.7 Hz, 1H), 4.55 (m, 1H), 3.94 (s, 3H), 2.77 (m, 2H), 2.60 (m, 2H), 2.44 (s, 3H), 2.36 (s, 3H), 2.26 – 2.12 (m, 2H), 2.08 – 1.92 (m, 2H). 13C NMR (126 MHz, CDCl3) δ 165.33, 159.78, 159.71, 156.92, 149.67, 148.31, 139.06, 135.77, 130.28, 129.35, 129.19, 128.87, 126.64, 125.91, 124.85, 124.73, 122.61, 121.77, 115.40, 113.64, 111.38, 110.62, 71.44, 56.24, 52.03, 45.98, 30.18, 8.63. HRMS (ESI+) m/z: [M + H+] calcd for C30H31ClN3O5 548.1952; found 548.1955.
1-(6-methoxy-3′-nitro-[1,1′-biphenyl]-3-yl)-3-(8-methyl-7-((1-methylpiperidin-4-yl)oxy)-2-oxo-2H-chromen-3-yl)urea (18b)
Urea 18b was obtained as a light brown amorphous solid (162 mg, 84%). 1H NMR (400 MHz, Chloroform-d) δ 8.38 (s, 1H), 8.37 (t, J = 2.0 Hz, 1H), 8.12 (dd, J = 8.2, 2.3 Hz, 1H), 7.84 (dt, J = 7.8, 1.3 Hz, 1H), 7.53 (t, J = 8.0 Hz, 1H), 7.47 (d, J = 2.7 Hz, 1H), 7.39 (dd, J = 8.8, 2.7 Hz, 1H), 7.23 (d, J = 8.6 Hz, 1H), 6.94 (d, J = 8.9 Hz, 1H), 6.81 (d, J = 8.7 Hz, 1H), 4.49 (m, 1H), 3.77 (s, 3H), 2.73 (m, 2H), 2.57 (m, 2H), 2.39 (s, 3H), 2.24 (s, 3H), 2.05 (m, 2H), 1.93 (m, 2H). 13C NMR (126 MHz, DMSO) δ 158.45, 154.84, 152.54, 151.22, 147.82, 147.10, 139.18, 135.10, 128.44, 128.34, 127.15, 124.22, 123.39, 122.43, 122.08, 120.95, 120.44, 119.79, 119.47, 113.70, 111.37, 109.84, 70.43, 55.13, 50.60, 44.20, 28.72, 7.44. HRMS (ESI+) m/z: [M + H+] calcd for C30H31N4O7 559.2193; found 559.2196.
1-(3′-amino-6-methoxy-[1,1′-biphenyl]-3-yl)-3-(8-methyl-7-((1-methylpiperidin-4-yl)oxy) -2-oxo-2H-chromen-3-yl)urea (18c)
Nitrourea 18b (56 mg, 0.1 mmol) was dissolved in methanol (10 mL) and stirred under hydrogen atmosphere for 4 hours. The mixture was filtered and filtrate was concentrated to dryness to give anime 18c as a light brown amorphous solid (45 mg, 85%). 1H NMR (500 MHz, Chloroform-d) δ 8.30 (s, 1H), 7.32 (dd, J = 8.8, 2.8 Hz, 1H), 7.21 (d, J = 2.7 Hz, 1H), 7.18 (d, J = 8.4 Hz, 1H), 7.07 (d, J = 7.9 Hz, 1H), 6.85 – 6.81 (m, 2H), 6.79 (t, J = 2.0 Hz, 1H), 6.75 (d, J = 8.7 Hz, 1H), 6.59 (dd, J = 8.0, 2.4 Hz, 1H), 4.46 (m, 1H), 3.66 (s, 3H), 2.74 (m, 2H), 2.65 (m, 2H), 2.39 (s, 3H), 2.20 (s, 3H), 2.03 (m, 2H), 1.89 (m, 4H). 13C NMR (126 MHz, CDCl3+CH3OH) δ 159.65, 155.52, 153.46, 152.69, 148.69, 145.76, 139.12, 131.58, 131.31, 129.24, 128.77, 125.08, 123.69, 122.68, 121.67, 120.30, 120.05, 116.74, 114.41, 114.15, 112.01, 110.37, 70.22, 55.92, 51.19, 44.95, 29.06, 8.12. HRMS (ESI+) m/z: [M + H+] calcd for C30H33N4O5 529.2451; found 529.2452.
Results
The methodology proposed for the dynamics-based design of allosteric inhibitors of the Hsp90 C-terminus is as follows: 1) Identification of the (allosteric) binding site; 2) generation of the protein-novobiocin complex; 3) performing long-timescale all-atom MD simulations of the complex; 4) characterization of the ligand-induced conformational changes of the binding site to identify new cavities and pockets; 5) probing new structures of the pocket with a series of analogues with known activity; and 6) exploitation of this information to design derivatives containing functional groups that can reach into identified cavities.
Moreover, once generated, the new pocket structures can be used to guide the assembly of an inhibitor through fragment based strategies or as docking targets for high-throughput in silico screening (Figure 1).
Identification of the C-terminal binding site
The allosteric binding site in the Hsp90 C-terminal domain was identified by analyzing the extent in which distinct subdomains of the full-length chaperone are mechanically coupled, and hence capable of propagating signals that upon nucleotide binding to NTD causes a conformational response in distal regions. We developed a general theoretical model to identify the “hot spots” associated with the allosteric communication between a binding site and a distal region that may be involved with function. Cross-talk between the N- and C-domains was investigated by defining the coordination propensity (CP) between any two residues as a function of fluctuation of their distance components. Low CP values are related to efficiently communicating residues.6,32,33 In the ATP-bound, active state, long-range coordination from the binding site was directed to residues at the CTD interface. Such residues span the dimerization core of the C-terminal domain and the C-terminal loops near the boundary of the M-domain.6,32,33
Importantly, a consistent dynamic response of the C-terminal hot spots to the presence of ATP was observed for three structurally different representatives of the Hsp90 family, namely Hsp90, Grp94 and HtpG.33 These data, which correlate well with experimental H-D exchange and mutational analyses, support the long-range nucleotide-dependent modulation of structural dynamics in the C-terminal region.
The hot spot C-terminal region was subjected to structural investigation and one pocket (named Pocket A) was consistently detected in all representative MD conformations. The properties of the pocket were shown to be suitable to accommodate small compounds that interact directly with the hot spot allosteric residues. Analysis of their chemical properties provided pharmacophore models that recapitulated complementary interactions necessary for productive binding to the C-terminal pocket. Such models were then used to screen the NCI repository which identified two compounds that were demonstrated experimentally to inhibit several important protein-protein interactions and to interrupt biological pathways fundamental to cancer cell proliferation.6
To date, no experimental co-crystal structures of Hsp90-CTD inhibitors bound to the protein are available, most likely due to the high conformational flexibility of the complex. The poor pharmacological potency of first generation analogues of novobiocin, together with the inability to obtain crystal structures with Hsp90-CTD inhibitors have hampered the discovery of more potent analogues. This approach attempts to overcome these limitations by defining a putative binding site through molecular modeling. The identification of a binding site allows visualization of the putative binding mode of the ligand as a basis for structure-based drug design.
Generating the structure of the Hsp90/Novobiocin complex
The starting point for this design strategy was generation of a model of Hsp90 in complex with novobiocin. Novobiocin was docked with Glide (v 5.8, Schrödinger, LLC, New York, NY, 2012) 34,35 into the newly discovered CTD allosteric pocket (see the “Matherials and Methods” section), using the GlideScore SP scoring function. The putative binding site was held rigid while the ligand was free to move. Several poses of novobiocin in the binding site were analyzed to identify interactions between the ligand and the protein.
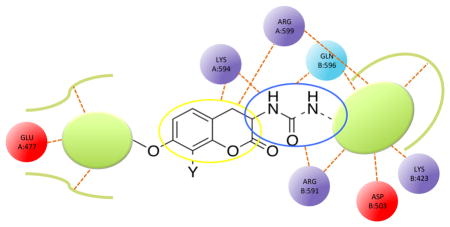
The putative binding site in the C-terminal domain is mainly constituted by secondary structural elements; part of the three-helix coil forming the dimerization interface and a helix strand segment, residues 587-597 (Hsp90 residues numbering as in the pdb entry 2CG9), that is associated with dimeric interactions with its equivalent in the other protomer. Some of the residues in the extended loop connecting the C-terminal three-stranded beta-sheet (residues 596-611 of chain A, named loop L) and the helix strand segment in chain A participate in interactions with the ligand. In particular: the 4-hydroxy substituent of the coumarin core can form hydrogen bonds with Ser657 B, Arg591 B and Glu477 B, while the carbonyl group can make one or two hydrogen bonds with the side chain of Arg599 A. The amide nitrogen can bind the side chain of Glu477 B and the hydroxyl group in the benzamide side chain can establish a hydrogen bond interaction with Asp503 B. The noviose sugar can form several hydrogen bonds with the backbone of Phe656 A, Asp659 B, Glu477 A, Gly476 A and with the side chain of Arg591 A. The coumarin core permits interaction between the carbonyl group and Arg591 B. Finally, the prenyl group accommodates in a hydrophobic cavity formed by the side chains of residues Ile505 B, Ile592 B, Leu598 B (Figure 2, 3).
Figure 3. Structures of Novobiocin bound to Hsp90 in the first most populated cluster obtained from MD simulation.

Upper panel: 2D diagram of the interactions between Novobiocin and the representative structure of the first cluster. The ligand is displayed as a 2D structure while residues are represented as colored spheres according to their properties, labeled with the residue name and residue number. Ligand interactions with the residues are drawn as lines, colored by interaction type. Solvent exposure is indicated by the break in the line drawn around the pocket. Lower panel: 3D representation of the molecular surface formed by protein residues which have atoms closer than 5 Angstroms to the ligand. The molecular surface is colored according to the residue properties (blue, positively charged residues; red, negatively charged residues, cyan, polar residues; green, hydrophobic residues; gray, glycine).
Dynamic reorganization of the Lead-CTD allosteric pocket
The best pose of novobiocin in the putative CTD binding site was selected by ranking docking solutions using the “Emodel” scoring function, which has been optimized for the comparison of conformers.34,35 The best pose of novobiocin in complex with Hsp90 was used as a starting point for extensive MD simulations in explicit solvent. Cluster analysis of the trajectories was used to identify the most representative conformations from the protein’s conformational ensemble. Trajectory snapshots were grouped into 22 clusters, with the first four accounting for 75% of the structural diversity. Table 1 provides the details of cluster population and structural properties in terms of RMSD distributions for the whole structure and for the pocket. Table 2 provides a summary of the residues contacted by the ligand during conformational evolution.
Table 1.
Percentage of the total number of conformations captured by the four most representative clusters and RMSD mean and distributions within the clusters.
| Cluster | Size/Total Population | Mean RMSD (Å) | Global RMSD (Å) (Backbone) Reference:Cluster1 | Pocket RMSD (Å) (Backbone) Reference:Cluster1 |
|---|---|---|---|---|
| 1 | 25 % | 0.290 | - | - |
| 2 | 19 % | 0.291 | 3.821 | 1.955 |
| 3 | 17 % | 0.290 | 5.884 | 2.545 |
| 4 | 15 % | 0.295 | 6.390 | 2.563 |
Table 2.
Summary of contacts between novobiocin and different ligand-induced Hsp90 conformations.
| Arg591 B | Lys594 A | Asp503B | Lys423 B | Gulu477 A | Hydrophobic channel | Gln596 B | Arg599B | Hydrophobic pocket | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||
| H-bonds | Hydroph int | ||||||||||
|
|
|||||||||||
| Aromatic side chain | 1 | ✓ | ✓ | ✓ | |||||||
| 2 | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||
| 3 | ✓ | ✓ | ✓ | ✓ | |||||||
| 4 | ✓ | ✓ | |||||||||
| 5 | ✓ | ✓ | |||||||||
| 6 | ✓ | ✓ | |||||||||
| 7 | ✓ | ✓ | |||||||||
| 8 | ✓ | ✓ | ✓ | ✓ | ✓ | ||||||
| 9 | ✓ | ✓ | |||||||||
| 10 | ✓ | ✓ | |||||||||
| 11 | ✓ | ✓ | ✓ | ✓ | |||||||
|
| |||||||||||
| Amide/Urea | 1 | ✓ | ✓ | ✓ | |||||||
| 2 | ✓ | ✓ | ✓ | ||||||||
| 3 | ✓ | ✓ | ✓ | ||||||||
| 4 | ✓ | ||||||||||
| 5 | ✓ | ||||||||||
| 6 | ✓ | ||||||||||
| 7 | ✓ | ||||||||||
| 8 | ✓ | ✓ | |||||||||
| 9 | ✓ | ✓ | |||||||||
| 10 | ✓ | ✓ | |||||||||
| 11 | ✓ | ✓ | |||||||||
|
| |||||||||||
| sugar mimic molety | 1 | ✓ | |||||||||
| 2 | ✓ | ||||||||||
| 3 | ✓ | ||||||||||
| 4 | ✓ | ||||||||||
| 5 | ✓ | ✓ | |||||||||
| 6 | ✓ | ✓ | |||||||||
| 7 | ✓ | ✓ | |||||||||
| 8 | ✓ | ✓ | |||||||||
| 9 | ✓ | ✓ | |||||||||
| 10 | ✓ | ✓ | |||||||||
| 11 | ✓ | ✓ | |||||||||
|
| |||||||||||
| Coumarin core | 1 | ✓ | |||||||||
| 2 | ✓ | ✓ | |||||||||
| 3 | ✓ | ✓ | |||||||||
| 4 | ✓ | ✓ | |||||||||
| 5 | ✓ | ||||||||||
| 6 | ✓ | ||||||||||
| 7 | ✓ | ||||||||||
Representative structures of the first four clusters were visually inspected (Figures 2 to 6) to identify important conformational changes that occur in response to ligand binding, which was used to modify activity of the ligand.
Figure 6. Structures of Novobiocin bound to Hsp90 in the fourth most populated cluster obtained from MD simulation.

Upper panel: 2D diagram of the interactions between Novobiocin and the representative structure of the fourth cluster. The ligand is displayed as a 2D structure while residues are represented as colored spheres according to their properties, labeled with the residue name and residue number. Ligand interactions with the residues are drawn as lines, colored by interaction type. Solvent exposure is indicated by the break in the line drawn around the pocket. Lower panel: 3D representation of the molecular surface formed by protein residues which have atoms closer than 5 Angstroms to the ligand. The molecular surface is colored according to the residue properties (blue, positively charged residues; red, negatively charged residues, cyan, polar residues; green, hydrophobic residues; gray, glycine).
As mentioned above, the putative binding site in the C-terminal domain consists of mainly secondary structural elements that do not undergo large conformational changes and therefore maintain their position during dynamics. In contrast, the extended loop L drifts away from the binding pocket and fluctuates back and forth (Figure 2). The opening of loop L appears to be influenced by electrostatic repulsion between the side chains of Asp600 A, belonging to loop L, and Asp330 B, belonging to an amphipathic loop (residues 329-339) that projects from the M-large domain. However, opening of loop L is also restricted by the electrostatic attraction between residue Arg599 A and residue Asp330 B, resulting in a motion that influences the binding site.
In the most populated cluster, the cavity that accommodates the noviose sugar on novobiocin remains mostly hydrophobic in character (Figure 3), but unlike the starting structure used for MD simulations, the side chain of Glu477 A points towards the interior, suggesting the potential to establish ionic interactions with the ligand.
In this structure, loop L is most distal from the binding site. Consequently, the side chain of Arg599 A is not available for binding. However, movement allows the helical coil (residues 411-435, chain B) connecting the M-large and M-small domains of chain B (residues 273-409 and 435-525 respectively) to come closer to the binding site, along with an amphipathic loop in the M-large domain of chain B (residues 329-339). In this manner, the side chains of Leu331 B and Phe329 B join the small hydrophobic cavity that accommodates the prenyl side chain of novobiocin and together, form a larger hydrophobic channel. Moreover, during these dynamics, residues Met589 B and Met593 B (belonging to the helix stretch of the binding site) orient their side chains toward the interior of the binding pocket adding to the formation of this hydrophobic channel (Figure 3). Exclusion of the Arg599 A side chain and reorientation of these residues allow the protein to acquire a hydrophobic character, which could be further stabilized via the inclusion of a hydrophobic group. The side chain of Lys423 B is pushed away from this cavity and instead points towards the binding site, providing a potential H-bond or an ionic interaction with the ligand (Figure 3).
In the representative structure of the second most populated cluster, residues of the putative binding site present similarly to that of the first cluster. Movement of loop L (Figure 4) results in a conformational change, wherein residues Gln596 B and Lys594 A expose their side chains in the binding pocket and favor stabilization of the hydroxyl and/or carbonyl groups on the coumarin core (docking runs showed that productive interactions with the binding site can be established by the coumarin core in two positions, one rotated by 180 degrees with respect to the other) as well as the oxygen and nitrogen of the amide bond. These residues substitute for the original interaction observed with Arg599 A and strengthen binding interactions. Moreover, Lys594 A establishes a cation-pi interaction with the benzene ring of the coumarin core. Lys423 B projects its side chain towards the binding site similar to the first cluster.
Figure 4. Structures of Novobiocin bound to Hsp90 in the second most populated cluster obtained from MD simulation.

Upper panel: 2D diagram of the interactions between Novobiocin and the representative structure of the second cluster. The ligand is displayed as a 2D structure while residues are represented as colored spheres according to their properties, labeled with the residue name and residue number. Ligand interactions with the residues are drawn as lines, colored by interaction type. Solvent exposure is indicated by the break in the line drawn around the pocket. Lower panel: 3D representation of the molecular surface formed by protein residues which have atoms closer than 5 Angstroms to the ligand. The molecular surface is colored according to the residue properties (blue, positively charged residues; red, negatively charged residues, cyan, polar residues; green, hydrophobic residues; gray, glycine).
In the representative structure of the third most populated cluster, loop L is in an intermediate position in which the side chains of Arg599 A and Asp330 B are attracted to each other (Figure 5). In this cluster, the steric bulk of Arg599 blocks formation of the hydrophobic channel observed in the most populated clusters, although the helical coil (residues 411-435, chain B) connecting the M-large and M-small domains of chain B and the amphipathic loop in the M-large domain of chain B (residues 329-339) has begun to approach the binding site, which results in a large hydrophobic pocket that accommodates the prenyl appendage on novobiocin (residues Ile592 B, Ile505 B, Leu598 B, Met 589 B and Met583 B join Leu427 B of the helical coil and Leu331-Phe332 B of the amphipathic loop). This structural arrangement allows Lys594 A to establish a cation-pi interaction with the benzene ring of the coumarin core and Arg599 B with the benzamide side chain of novobiocin.
Figure 5. Structures of Novobiocin bound to Hsp90 in the third most populated cluster obtained from MD simulation.

Upper panel: 2D diagram of the interactions between Novobiocin and the representative structure of the third cluster. The ligand is displayed as a 2D structure while residues are represented as colored spheres according to their properties, labeled with the residue name and residue number. Ligand interactions with the residues are drawn as lines, colored by interaction type. Solvent exposure is indicated by the break in the line drawn around the pocket. Lower panel: 3D representation of the molecular surface formed by protein residues which have atoms closer than 5 Angstroms to the ligand. The molecular surface is colored according to the residue properties (blue, positively charged residues; red, negatively charged residues, cyan, polar residues; green, hydrophobic residues; gray, glycine).
In the representative structure of the fourth most populated cluster, loop L still remains in the closed conformation (Figure 6). Three charged aminoacids, Lys423 B, Arg599A and Asp600 B project their side chain towards the binding site, interrupting the sequence of amino acids that form the small hydrophobic pocket that accommodates the prenyl group on novobiocin. The presence of these amino acids suggests the potential to introduce hydrogen bond donor/acceptor groups onto the benzamide side chain. Electron-withdrawing groups could exploit the close proximity of Lys423 B, which is observed in 3 out of 4 analyzed protein conformations.
In all representative structures, residues in the binding site that belong to the three-helix coil of chain A, which forms the dimerization interface (residues 587-597, chain A) maintain a similar position in the starting structure, indicating the stability of this region and the availability of charged/polar amino acids (as Ser657 B, Arg591 B, Glu477 B and Asp503 B) to establish H-bond or ionic interactions with the ligand.
Docking of compounds with known activity. Using chemical probes to probe the structural plasticity of the allosteric site
Next, a range of molecules with known activities derived from novobiocin were selected in order to probe the four most populated clusters through docking runs (Figure 7, 8). Analysis of the interactions established by each ligand with the protein serves to verify whether the conformations of the proposed binding sites can provide a model that accounts for the observed activities. This analysis can also be useful for the identification of important interactions that should be maintained, while suggesting potential modifications that can improve efficacy.
Figure 7.

Structures of Novobiocin analogues used to probe the structural plasticity of the allosteric site and their experimental IC50 against SKBr3 cells.
Figure 8. Structural representation of compound 10 (see Figure 7) bound to Hsp90.

Top: the protein is shown in ribbon representation and it is colored according to the secondary structure elements. Bottom: molecular surface formed by protein residues which have atoms closer than 5 Angstroms to the ligand. The molecular surface is colored according to the residue properties (blue, positively charged residues; red, negatively charged residues, cyan, polar residues; green, hydrophobic residues).
All of the analyzed derivatives represent a potent coumarin scaffold identified from prior investigation 44 that demonstrated the 4-hydroxy substituent is detrimental to Hsp90 inhibitory activity. Analogous studies aimed at elucidating structure-activity relationships have also determined that while the sugar moiety is important for interactions with Hsp90, not all functionalities, such as the 3′-carbamoyl group, are necessary. Moreover they demonstrated that simplified sugar mimics such as alkyl amines can be suitable replacements of the noviose moiety. Selected molecules have been chosen with two different sugar surrogates to determine whether the proposed binding sites can explain the differences in observed activities. Amide side chains containing an aromatic ring have also been shown to be important for improved anti-proliferative activity.45–48 Therefore, molecules containing different aromatic side chains that include heteroatoms and various appendages were investigated.
As described above, the most populated cluster is characterized by formation of a hydrophobic channel due to separation of loop L from the binding site. All compounds analyzed in this pose present their aromatic side chains into this channel, suggesting that this interaction can play an important role in binding affinity.
Compounds 1, 2 and 3, which differ only in composition the aromatic amide side chains, exhibit similar binding modes: The amide oxygen and nitrogen are involved in two mutually exclusive H-bound interactions with the side chain of Arg591 B and the backbone carbonyl group of Lys594 A, respectively.
The 3-prenylated 4-hydroxybenzoic acid moiety of 1 and the biphenyl side chains of 2 and 3 are accommodated in the binding site similarly, even though the biphenyl side chain projects farther into the hydrophobic channel as compared to the prenylated derivative. The phenol on the benzamide side chain of compound 1 can produce hydrogen bond interactions with Asp503 B, while 2 presents the side chain towards Lys423 B via the methoxy group on the first phenyl ring. However, in agreement with the experimental activity data, compound 3 appears favored because it can establish the same interaction as compound 2 and it can bind the backbone carbonyl group of several amino acids within the hydrophobic channel. The sugar moiety establishes similar interactions in all three compounds. The coumarin methoxy group of 4 binds complementary with the side chain of Lys594 A, and pulls the biphenyl side chain away from the hydrophobic channel.
Similar interactions are established with compounds 5–7, which exhibit better anti-proliferative activities. The structural model of the binding site suggests that this effect is due to the piperidine moiety, which provides a strong ionic interaction with the negatively charged amino acid Glu477 A (Figure 8).
Compounds 8–11 can occupy the hydrophobic channel similar to the other compounds because the longer urea moiety (compared to the amide bond) compensates for the shorter aromatic amide side chain. Several poses of these compounds suggest that Arg591 B can establish a cation-pi interaction with the benzene ring of the aromatic side chain. The NH groups of the urea moiety can make one or two hydrogen bonding interactions with the carbonyl group of Lys594 A. Poses of the most potent compounds, 8 and 11, show the chloride atom to be stabilized by interactions with the side chain of Lys423 B.
Similar binding modes and interactions were observed for all compounds in the representative structure of the second most populated cluster, which is characterized by the presence of side chains belonging to Gln596 B, Lys594 A and Lys423 B. In particular, compounds 2 and 3 are favored with respect to 1, because the oxygen atom of the methoxyl/hydroxyl groups on the second phenyl ring in the benzamide side chain can bind the side chain of Gln596 B, and Lys423 B can establish a cation-pi interaction with the same phenyl ring. At variance with the case of compound 2, these interactions were consistently observed in all the binding poses of compound 3, because the hydroxyl group, which binds Gln596 B, favors the right positioning of the phenyl ring compared to the methoxy group. Furthermore, the anti-proliferative activities of ligands bearing the piperidine moiety can be explained by the potential to establish strong ionic interactions with Glu477 A (Figure 8). The urea moiety presented in 8–11 appears to compensate for the smaller aromatic side chain contained in these compounds, although they do not completely fill the hydrophobic channel, suggesting that a longer side chain could lead to enhanced activity. The chloride atom on the benzene side chains of 8 and 11 can interact favorably with the side chain of Lys423 B.
As mentioned previously, the third most populated cluster contains a sterically bulky Arg599 that partially blocks formation of the hydrophobic channel that accommodates the aromatic side chain. Consequently, the binding poses of the benzamide side chain show greater variability. Arg599 B and Gln596 B, which bind the carbonyl group of the coumarin core and the NH group of the amide bond or urea moiety respectively, appear important for the binding of all ligands. The hydrophobic interaction is determined by the first benzene ring with a methoxy group in 4-position as shown with 2–4 and 6–7. The latter is accommodated in a small pocket formed by Leu427 B, Leu331 B, Ile 505 B, Phe332 B and Pro504 B. Compounds 2 and 3 can be stabilized by a cation-pi interaction between Lys594 B and the benzene ring of the coumarin core. Moreover, the phenol on the second benzene ring of compound 2 can bind the backbone carbonyl group of Leu331 B in the M-large domain, favoring cation-pi interactions with Arg399 B. Unlike 2 and 3, the steric bulk represented by the methoxy group in the coumarin core of compound 4 prevents Lys594 B from establishing additional interactions. Also in this protein conformation, Glu477 projects its side chain into the binding site, and stabilizes the piperidine ring. The urea moiety allows the aromatic side chain to reach the small hydrophobic pocket formed by Leu427 B, Leu331 B, Ile 505 B, Phe332 B and Pro504 B. However, this model cannot provide rationale to explain the anti-proliferative activities manifested by 8 and 11, which contain a chloride atom in lieu of the aromatic side chain.
In the fourth most populated cluster, residues Lys423 B, Arg599A and Asp600 B project their side chain towards the binding site. These amino acids, together with the small hydrophobic cavity formed by residues Ile505 B, Ile592 B and Leu598 B, guide the aromatic side chain of all compounds. In particular, they establish interactions through H-bonds donor/acceptor groups with the side chains of 1–8. The piperidine ring can be stabilized by Glu447 B, as observed in the other protein sub-states. The urea moiety of 8–11 allows these compounds to fit into the binding site similar to others, whereby the closed-state conformation of the binding site (similar to the starting conformation used for MD) permits the single phenyl ring in the aromatic side chain to reach into the small hydrophobic cavity. The chloride atoms in 8 and 11 can interact with the protein in different positions, including Arg599 B or the side chain of Lys423 B.
Design of new derivatives
Analysis of the conformations elicited by the putative C-terminal binding site during dynamics studies with novobiocin and docking runs with known inhibitors provide valuable information for the design of new derivatives that manifest improved activities.
The new molecules were designed by modifying the coumarin core of novobiocin according to aforementioned SAR analyses and substituting noviose with N-methyl-piperidine, which is known to increase anti-proliferative activity.
As mentioned above, all representative structures of the most populated clusters exhibit a hydrophobic pocket surrounding the noviose sugar. Moreover, docking runs on derivatives containing the N-methyl-piperidine suggest that the side chain of Glu477 A could be responsible for the increased activity manifested by these compounds (Figure 8).
In all analyzed structures, the hydrophobic pocket that accommodates the aromatic side chain increases its volume during MD simulations. In the most populated cluster, it becomes a large channel. Formation of this large hydrophobic area and inspection of docking poses suggest that a hydrophobic, bulky, and extended substituent at the right distance from the piperidine ring can improve interactions. The shape and volume of this hydrophobic channel suggest that this substituent can be appended to compounds containing the urea moiety, which will optimize fitting to the binding site. We tested this hypothesis by appending biphenyl substituents to compounds containing the urea linker (18a–c in Scheme 1). As mentioned previously, polar/charged amino acids (in particular Asp503 B, Lys423 B) project their side chains alongside this hydrophobic area suggesting the potential to introduce additional hydrogen bond donor/acceptors or electron-withdrawing groups onto the benzamide side chain.
Scheme 1.

Synthesis of new novobiocin analogues.
Synthesis and Activities
The synthesis of these compounds is straightforward. As shown in Scheme 1, the key intermediate 16 was synthesized in four steps: Suzuki coupling between methyl 3-iodo-4-methoxyl benzoate 12 and phenyl boronic acid 13 gave the biaryl ester in good yield. Upon hydrolysis, the esters were converted to corresponding benzoic acids 14, which were then converted to the benzoic azides 15 enlisting diphenylphosphoryl azide in the presence of triethylamine. Subsequent Curtius rearrangement afforded the isocyanates 16, which upon reaction with amine 17 produced ureas 18a and 18b in good yield. The nitro containing compound 18b was reduced to aniline 18c with Pd/C under hydrogen atmosphere.
Biological evaluation of these compounds for establishment of anti-proliferative activity was performed against SKBr3 (estrogen receptor negative, HER2 over-expressing breast cancer cells) and MCF-7 (estrogen receptor positive breast cancer cells) cell lines. As shown in Table 3, these three analogues manifested sub micro-molar anti-proliferative activity against both cancel cell lines, an improvement over previously published compounds. It appears that installation of an electron-withdrawing group on the second phenyl ring is more beneficial than an electron-donating group.
Table 3.
Anti-proliferative activity for new novobiocin analogues.
| SKBr3 (μM) | MCF-7 (μM) | |
|---|---|---|
| 18a | 0.13±0.01 | 0.47±0.05 |
| 18b | 0.16±0.01 | 0.36±0.06 |
| 18c | 0.36±0.01 | 0.67±0.09 |
Discussion
The ability to optimize lead compounds is an essential goal for drug development programs to increase the rate of success. The design and discovery of new drugs largely depends on a thorough understanding of the dynamic and complex behavior of biological targets. 8,12,49–53 In this context, methods based on computational biophysics and chemistry can give significant, yet partially unexplored, contributions.8,10,54–56 In particular, it is now widely accepted that conformational flexibility plays a key role in all biomolecular recognition processes, as well as protein function.2,3 Since proteins are flexible systems that exist in different conformational states with similar energies, often times only small barriers exist between these states.57 A ligand can selectively bind one conformer or it can bind different closely related conformation, conformational plasticity and dynamic adaptation can also be used to explain the ability of a binding site to accommodate different ligands or to force related ligands to populate different binding orientations.58 The selection of specific conformational states combined with the modulation of protein dynamics upon ligand binding serves as the basis of allosteric regulation.58 For example, binding distant from the active site can shift the protein population towards an active/inactive conformation by inducing specific fluctuation patterns that extend throughout the entire three-dimensional architecture.
Given the importance of dynamics in protein-ligand recognition, binding and allostery, it is important to account for protein flexibility in the process of drug discovery and to design lead compounds that exhibit specific biological activities.
MD simulations have proven to be a reliable method for exploring protein flexibility at the atomic level required for molecular design.12,59,60 Moreover, methods aimed at characterizing the ligand-dependent conformational dynamics of proteins have led to the discovery of novel druggable sites in addition to further understanding of previously identified sites. Therefore, MD simulations can generate conformational ensembles that elucidate the principal modes of motion in solution and can pinpoint structural rearrangements that reveal binding sites that are not readily available in a “one-structure representation”.
In this paper, we have built upon previous studies that have focused on the identification of an allosteric binding site in the Hsp90 C-terminal domain in an effort to develop a new strategy for the development of allosteric regulators. In particular, the dynamic changes within the allosteric site was determined by the use of a first-generation ligand which led to the development of a new generation of Hsp90 inhibitors that exhibited improved inhibitory activities.
Our hypothesis is that ligand binding induces a conformational rearrangement of the receptor-binding pocket that is essential to the recognition process and accessible on the energy landscape of the protein. Therefore this cannot be sampled on the MD timescale in the absence of ligand. In order to facilitate the exploration of these protein motions for the design of new inhibitors, we docked novobiocin, the first known inhibitor to bind Hsp90-CTD, into the putative C-terminal binding site of activated Hsp90 and used this complex structure as a starting point for MD simulations. Protein conformations observed during the dynamic process of the protein-novobiocin complex was then used to design new derivatives. Specifically, information obtained in the inhibitor-based modulation of Hsp90 structural dynamics were exploited to identify positions wherein the addition of specific chemical functionalities could allow for extended binding interactions into previously uncharacterized regions. The formation/disappearance of pockets due to the conformational response of the protein to the first-generation inhibitor defined the stereochemical properties of functional groups that were needed for inclusion into existing scaffolds, and to facilitate optimal fitting.
The representative structures of the most populated clusters obtained from the MD trajectory were then selected to represent the most accessible protein conformations in complex with the ligand. These clusters represented the basis for identification of complementary interactions that could establish productive binding with similar ligands.
We then probed the proposed site by docking a set of novobiocin derivatives with known activities into the new, ligand-induced conformation of the C-terminal binding site. The objective was to verify whether these molecules could be accommodated in the site and to determine wether a rational correlation existed between the experimental activities and the interactions they established. The representative structures of the four most populated clusters of novobiocin/Hsp90 complex were used identify the conformational variability and plasticity of Hsp90. Conformational rearrangements of the protein that caused the appearance/disappearance of different functionalities in the binding pocket were taken into, account for the observed activities of these derivatives. A good correlation between the calculated and experimental activities 61 validated the model and provided potential properties for the allosteric site.
Analysis of the docking poses identified the most important protein-ligand interactions (e.g those interactions that are present in the majority of poses) that should be maintained during the design of new inhibitors.
In a previous work, we determined quantitative structure-activity relationships using the same set of novobiocin analogues,61 which together with the identification of the most important protein-ligand interactions, analysis of the site dynamics, and evaluation of positional persistence during simulation were then used to design new novobiocin analogues.
The designed inhibitors were examined trough the same docking protocol as used for the first generation of compounds and the most promising molecules were selected for synthesis. Importantly, some of the synthesized compounds manifested improved anti-proliferative activity compared to the original lead, which led to ~150 nanomolar inhibitory activity. The improved activities manifested by the designed molecules represent an encouraging experimental validation of this approach. It is worth noting that while validation via a series of experimental mutation and design-based data, 6,61–63 the model presented here may only offer a partial view of the mechanism by which novobiocin and novobiocin-based derivatives bind the Hsp90 C-terminal domain. The dynamic structures cannot quantitatively explain the structure-activity relationships of dimeric novobiocin derivatives or coumermycin A analogues,48,64 nor can they fully account for the results of photoaffinity labeling studies using novobiocin photoaffinity probes.65
It is important to also note that this model represents a limited view of the vast conformational space accessible to this flexible molecular chaperone. This implies that other Hsp90 structures (such as the open structure) can be populated in solution and accommodate ligands in slightly different poses, such as that derived from the photoaffinity studies.65 In fact, the ligands used for these studies do not contain the charged azido group. Presence of this charged group may also influence the orientation of ligands in the binding site. The intrinsic limitation of modeling strategies and of MD methods in sampling large protein conformational changes restrain our ability to access the large conformational changes required for transitions from the closed to the open structures.
Our structural and local dynamic model for binding and design and the fact that large conformational changes should be considered to fully explain Hsp90 activity can be reconciled in the context of a conformational selection model for binding. The presence of a first-generation ligand favors protein motions that shift the conformational population towards states that are not accessible in the unbiased simulations of the unbound form. In this scenario, new ligands could select different conformations on the protein energy landscape on which to bind. The relative weight of such conformations depends upon the conditions in which the protein is studied (solution, temperature…). The fact that we have obtained good structure-activity relationships and validated predictions for multiple examples using the closed structure suggests that this approach can capture the essential dynamic and structural requirements necessary for a small molecule binding.
In this context, it must be highlighted that different simulative approaches, based on enhanced sampling methods, could reveal deformations useful for lead optimization. Replica Exchange MD (REMD) 66,67 simulations, for instance, could sample significant structural changes of the allosteric site. However, efficient exchange between neighboring replicas requires overlap of the potential energies sampled at nearby simulation temperatures, resulting in the use of high numbers of replicas when studying protein systems in explicit solvent. This would clearly hamper the application of the method to our (large) system, as it results in huge demands in terms of computing power. A viable and computationally cheaper alternative could by the use of accelerated Molecular Dynamics (aMD),13 an extended biased potential MD approach that has been shown to efficiently study systems at time-scales several orders of magnitude greater than those accessible by the use of standard classical MD methods.
Overall, we suggest that the best solution to explore the mechanisms of coupling between binding and protein fluctuations for the purpose of drug design is represented by a combination of enhanced sampling methods with the analysis of ligand induced deformations that was presented in this article. It is important to note, that different conformational search strategies based on Monte Carlo (MC) approaches could also be used.14 However, in our experience as well as others’,14 MC techniques optimally report on the changes in the rotameric states of residues in contact with the ligand and in the immediate neighborhood of the binding site, but they are less suitable for capturing the cooperative and large-scale modifications that were observed in our MD simulations.
A second important caveat is that the method presented herein, is not amenable for use in large-scale screening efforts. The increase in computer power and the availability of new high-performance computing solutions at relatively low prices, represented e.g. by GPU based computing, could provide the opportunity to include atomic level description of molecular motions in drug design and to support additional medicinal chemistry efforts.
Despite the limitations of time-span and conformational space accessible in simulations, this methodology appears general and can be adapted to the study of the dynamic ligand-protein cross-talk for other systems as well, such as the study of mechanisms of endogenous ligand regulation and evolution of protein dynamics. For example, a recent paper by Bhabha et al.68 demonstrated that bacterial and vertebrate dihydrofolate reductases (DHFR) use different dynamic mechanisms to perform their function. Such mechanisms depend upon the differential flexibility of important loops for ligand/cofactor binding and release, and on the differences in the mobility of enzyme subdomains, which stems from variations in specific regions of the primary sequences. Our methodology could be applied to investigate, at the atomic level, the species-dependent correlations between the presence of the cofactor and/or substrate and the microscopic conformational fluctuations of the binding site that facilitate global and slow functional motions of the enzyme.
In summary, our goal was to use information obtained from protein dynamic studies to guide the modification of existing hits to identify molecules with improved activity. Relevant functionalities were identified through MD simulations, docking simulations and QSAR analysis. Some of the compounds synthesized exhibit good antiproliferative activities against two breast cancer cell lines, which validated this approach and provides an attractive starting point for the rational development of new Hsp90 inhibitors. The results presented here I provide the potential to expand the chemical diversity space of Hsp90 antagonists on a rational level, and into account both, takes structural and dynamic properties simultaneously.
Acknowledgments
GC acknowledges funding from AIRC (Associazione Italiana Ricerca sul Cancro) through the grant: IG 11775; and from Fondazione Cariplo through grant 2011.1800 for the RST call–“Premio fondazione cariplo per la ricerca di frontiera”. The CNR-MIUR Flagship Project “Interomics” is also acknowledged.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
References
- 1.Swain JF, Gierasch LM. Curr Opin Struct Biol. 2006;16:102–108. doi: 10.1016/j.sbi.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Smock RG, Gierasch LM. Science. 2009;324:198–203. doi: 10.1126/science.1169377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tsai CJ, del Sol A, Nussinov R. Mol BioSyst. 2008;5:207–216. doi: 10.1039/b819720b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hardy JA, Wells JA. Curr Op Struct Biol. 2004;14:706–715. doi: 10.1016/j.sbi.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 5.Zorn JA, Wells JA. Nat Chem Biol. 2010;6:179–188. doi: 10.1038/nchembio.318. [DOI] [PubMed] [Google Scholar]
- 6.Morra G, Neves MAC, Plescia CJ, Tsutsumi S, Neckers L, Verkhivker G, Altieri DC, Colombo G. J Chem Theory and Computation. 2010;6:2978–2989. doi: 10.1021/ct100334n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.del Sol A, Tsai CJ, Ma B, Nussinov R. Structure. 2009;17:1042–1050. doi: 10.1016/j.str.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lerner MG, Bowman AL, Carlson HA. Journal of Chemical Information and Modeling. 2007;47:2358–2365. doi: 10.1021/ci700167n. [DOI] [PubMed] [Google Scholar]
- 9.Bowman AL, Lerner MG, Carlson HA. Journal of the American Chemical Society. 2007;129:3634–3640. doi: 10.1021/ja068256d. [DOI] [PubMed] [Google Scholar]
- 10.Carlson HA, McCammon JA. Molecular Pharmacology. 2000;57:213–218. [PubMed] [Google Scholar]
- 11.Wong CF, Kua J, Zhang Y, Straatsma TP, McCammon JA. Proteins. 2005;61:850–858. doi: 10.1002/prot.20688. [DOI] [PubMed] [Google Scholar]
- 12.Baron R, McCammon JA. Annu Rev Phys Chem. 2013;64:151–175. doi: 10.1146/annurev-physchem-040412-110047. [DOI] [PubMed] [Google Scholar]
- 13.Markwick PRL, McCammon JA. Phys Chem Chem Phys. 2011;13:20053–20065. doi: 10.1039/c1cp22100k. [DOI] [PubMed] [Google Scholar]
- 14.Lexa KW, Carlson HA. Q Rev Biophys. 2012;45:301–343. doi: 10.1017/S0033583512000066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bottegoni G, Kufareva I, Totrov M, Abagyan R. Journal of Computer-Aided Molecular Design. 2008;22:311–325. doi: 10.1007/s10822-008-9188-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cavasotto CN, Kovacs JA, Abagyan RA. Journal of the American Chemical Society. 2005;127:9632–9640. doi: 10.1021/ja042260c. [DOI] [PubMed] [Google Scholar]
- 17.Abagyan RA, Totrov MM, Kuznetsov DA. J Comp Chem. 1994;15:488–506. [Google Scholar]
- 18.Krukenberg KA, Street TO, Lavery LA, Agard DA. Q Rev Biophys. 2011;44:229–255. doi: 10.1017/S0033583510000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Richter K, Buchner J. Cell. 2006;127:251–253. doi: 10.1016/j.cell.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 20.Ali MMU, Roe SM, Vaughan CK, Meyer P, Panaretou B, Piper PW, Prodromou C, Pearl LH. Nature. 2006;440:1013–1017. doi: 10.1038/nature04716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shiau AK, Harris SF, Southworth DR, Agard DA. Cell. 2006;127:329–340. doi: 10.1016/j.cell.2006.09.027. [DOI] [PubMed] [Google Scholar]
- 22.Dollins DE, Warren JJ, Immormino RM, Gewirth DT. Mol Cell. 2007;28:41–56. doi: 10.1016/j.molcel.2007.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mickler M, Hessling M, Ratzke C, Buchner J, Hugel T. Nat Struct Mol Biol. 2009;16:281–286. doi: 10.1038/nsmb.1557. [DOI] [PubMed] [Google Scholar]
- 24.Krukenberg KA, Forster F, Rice LM, Sali A, Agard DA. Structure. 2008;16:755–765. doi: 10.1016/j.str.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Southworth DR, Agard DA. Mol Cell. 2008;32:631–640. doi: 10.1016/j.molcel.2008.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trepel JB, Mollapour M, Giaccone G, Neckers L. Nat Rev Cancer. 2010;10:537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Workman P, Burrows FJ, Neckers L, Rosen N. Ann NY Acad Sci. 2007;1113:202–216. doi: 10.1196/annals.1391.012. [DOI] [PubMed] [Google Scholar]
- 28.Messaoudi S, Peyrat JF, Brion JD, Alami M. Anticancer Agents Med Chem. 2008;8:761–782. doi: 10.2174/187152008785914824. [DOI] [PubMed] [Google Scholar]
- 29.Marcu MG, Chadli A, Bouhouche I, Catelli M, Neckers LM. J Biol Chem. 2000;275:37181–37186. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- 30.Marcu MG, Schulte TW, Neckers L. JNCI. 2000;92:242–248. doi: 10.1093/jnci/92.3.242. [DOI] [PubMed] [Google Scholar]
- 31.Donnelly A, Blagg BSJ. Curr Med Chem. 2008;15:2702–2717. doi: 10.2174/092986708786242895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morra G, Verkhivker GM, Colombo G. PLOS Comp Biol. 2009;5:e1000323. doi: 10.1371/journal.pcbi.1000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morra G, Potestio R, Micheletti C, Colombo G. Plos Comput Biol. 2012;8:e1002433. doi: 10.1371/journal.pcbi.1002433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Journal of Medicinal Chemistry. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 35.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Journal of Medicinal Chemistry. 2006;49:6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 36.Jorgensen WL, Maxwell DS, Tirado-Rives J. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 37.Hess B, Kutzner C, van der Spoel D, Lindahl E. J Chem Theory and Computation. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 38.Scott WRP, Hunenberger PH, Tironi IG, Mark AE, Billeter SR, Fennen J, Torda AE, Huber T, Kruger P, Gunsteren WFV. J Phys Chem A. 1999;103:3596–3607. [Google Scholar]
- 39.Berendsen HJC, Grigera JR, Straatsma PR. J Phys Chem. 1987;91:6269–6271. [Google Scholar]
- 40.Berendsen HJC, Postma JPM, Gunsteren WFv, Nola AD, Haak JR. J Chem Phys. 1984;81:3684–3690. [Google Scholar]
- 41.Hess B, Bekker H, Fraaije JGEM, Berendsen HJC. J Comp Chem. 1997;18:1463–1472. [Google Scholar]
- 42.Miyamoto S, Kollman PA. J Comp Chem. 1992;13:952–962. [Google Scholar]
- 43.Daura X, Gademann K, Jaun B, Seebach D, Gunsteren WFv, Mark AE. Angew Chemie Intl Ed. 1999;38:236–240. [Google Scholar]
- 44.Donnelly AC, Mays JR, Burlison JA, Nelson JT, Vielhauer G, Holzbeierlein J, Blagg BS. J Org Chem. 2008;73:8901–8920. doi: 10.1021/jo801312r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu XM, Shen G, Neckers L, Blake H, Holzbeierlein J, Cronk B, Blagg BSJ. J Am Chem Soc. 2005;127:12778–12779. doi: 10.1021/ja0535864. [DOI] [PubMed] [Google Scholar]
- 46.Burlison JA, Neckers L, Smith AB, Maxwell A, Blagg BSJ. J Am Chem Soc. 2006;128:15529–15536. doi: 10.1021/ja065793p. [DOI] [PubMed] [Google Scholar]
- 47.Donnelly A, Zhao H, Kusuma BR, Blagg BSJ. MedChemComm. 2010;1:165–170. [Google Scholar]
- 48.Zhao HP, Donnelly AC, Kusuma BR, Brandt GEL, Brown D, Rajewski RA, Vielhauer G, Holzbeierlein J, Cohen MS, Blagg BSJ. J Med Chem. 2011;54:3839–3853. doi: 10.1021/jm200148p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arkin MR, Wells JA. Nat Rev Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 50.Workman P. Curr Cancer Drug Targets. 2003;3:297–300. doi: 10.2174/1568009033481868. [DOI] [PubMed] [Google Scholar]
- 51.Gestwicki JE, Crabtree GR, Graef IA. Science. 2004;306:865–9. doi: 10.1126/science.1101262. [DOI] [PubMed] [Google Scholar]
- 52.Muzzi A, Masignani V, Rappuoli R. Drug Discovery Today. 2007;12:429–439. doi: 10.1016/j.drudis.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 53.Lerner MG, Meagher KL, Carlson HA. J Comp Aided Mol Des. 2008;22:727–736. doi: 10.1007/s10822-008-9231-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Carlson HA, Smith RD, Khazanov NA, Kirchhoff PD, Dunbar JB, Benson ML. Journal of Medicinal Chemistry. 2008;51:6432–6441. doi: 10.1021/jm8006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carlson HA, Masukawa KM, Rubins K, Bushman FD, Jorgensen WL, Lins RD, Briggs JM, McCammon JA. Journal of Medicinal Chemistry. 2000;43:2100–2114. doi: 10.1021/jm990322h. [DOI] [PubMed] [Google Scholar]
- 56.Nichols SE, Baron R, McCammon JA. Methods Mol Biol. 2012;819:93–103. doi: 10.1007/978-1-61779-465-0_7. [DOI] [PubMed] [Google Scholar]
- 57.Chennubhotla C, Yang Z, Bahar I. Mol BioSyst. 2008;4:287–292. doi: 10.1039/b717819k. [DOI] [PubMed] [Google Scholar]
- 58.Boehr DD, Nussinov R, Wright PE. Nat Chem Biol. 2009;5:789–796. doi: 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kua J, Zhang YK, McCammon JA. Journal of the American Chemical Society. 2002;124:8260–8267. doi: 10.1021/ja020429l. [DOI] [PubMed] [Google Scholar]
- 60.Adcock SA, McCammon JA. Chem Rev. 2006;106:1589–1615. doi: 10.1021/cr040426m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhao H, Moroni E, Yan B, Colombo G, Blagg BSJ. ACS Medicinal Chemistry Letters. 2013;4:57–62. doi: 10.1021/ml300275g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Retzlaff M, Stahl M, Eberl HC, Lagleder S, Beck J, Kessler H, Buchner J. EMBO Rep. 2009;10:1147–1153. doi: 10.1038/embor.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Allan RK, Mok D, Ward BK, Ratajczak T. J Biol Chem. 2006;281:7161–7171. doi: 10.1074/jbc.M512406200. [DOI] [PubMed] [Google Scholar]
- 64.Kusuma BR, Peterson LB, Zhao HP, Vielhauer G, Holzbeierlein J, Blagg BSJ. J Med Chem. 2011;54:6234–6253. doi: 10.1021/jm200553w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Matts RL, Dixit A, Peterson LB, Sun L, Voruganti S, Kalyanaraman P, Hartson SD, Verkhivker GM, Blagg BSJ. ACS Chem Biol. 2011;6:800–807. doi: 10.1021/cb200052x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nymeyer H, Garcia AE. Proc Natl Acad Sci USA. 2003;100:13934–13939. doi: 10.1073/pnas.2232868100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yoshida K, Yamaguchi T, Okamoto Y. Chem Phys Lett. 2005;41:2280–2284. [Google Scholar]
- 68.Bhabha G, Ekiert DC, Jennewein M, Zmasek CM, Tuttle LM, Kroon G, Dyson HJ, Godzik A, Wilson IA, Wright PE. Nat Struct Mol Biol. 2013;20:1243–1249. doi: 10.1038/nsmb.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]