

Abstract
Many degenerative retinal diseases illustrate retinal inflammatory changes that include infiltration of microglia and macrophages into the subretinal space. In the current study, we examined the role of chemokines in the Abca4-/-Rdh8-/- mouse model of Stargardt disease and the Mertk-/- mouse model of retinitis pigmentosa. PCR array analysis of 84 chemokines and related molecules revealed 84.6-fold elevated expression of Ccl3 (MIP-1a) 24 h after light exposure in Abca4-/-Rdh8-/- mice. Only MIP-1 chemokines, including Ccl3 and Ccl4, displayed peak expression 24 h after light exposure, and peaked earlier than the other chemokines. Secretion of Ccl3 was documented only in microglia whereas both microglia and RPE cells produced Ccl2. Exposure of Cx3Cr1gfp/ΔAbca4-/-Rdh8-/- mice to intense light resulted in the appearance of Cx3Cr1GFP+ monocytes in the subretinal space. To address the in vivo role of CCL3 in retinal degeneration, Ccl3-/-Abca4-/-Rdh8-/- mice and Ccl3-/-Mertk-/- mice were generated. Following intense light exposure, Ccl3-/-Abca4-/-Rdh8-/- mice displayed persistent retinal inflammation with appearance of Iba-1-positive cells in the subretinal space, severe photoreceptor cell death and increased Ccl4 expression compared with Abca4-/-Rdh8-/- mice. In contrast, Ccl3-/-Abca4-/-Rdh8-/- mice exhibited a milder retinal inflammation and degeneration than Abca4-/-Rdh8-/- mice in age-related chronic retinal degeneration under room light conditions. The deficiency of Ccl3 also attenuated the severity of retinal degeneration in Mertk-/- mice. Taken together, our results indicate that Ccl3 has an essential role in regulating the severity of retinal inflammation and degeneration in these mouse models.
INTRODUCTION
Clinical and experimental evidence suggests an important role for inflammation in retinal degeneration (1-4). Microglial cells are resident macrophages in the central nervous system and play a key role in this pathological inflammatory process (2). Therefore, elucidating the activities and functions of these inflammatory cells is essential in expanding our knowledge of the pathology of retinal degeneration. Understanding the role of chemokines in retinal degeneration is particularly important as they not only dictate the migration of inflammatory cells, but are also potential drivers of retinal degenerative conditions in humans and mice (5, 6).
The visual process is initiated in photoreceptors by activation of rhodopsin through the photo-conversion of visual chromophore 11-cis-retinal to all-trans-retinal. This isomeric conversion initiates a signaling cascade which ultimately propagates the visual stimulus to the brain. To maintain vision, all-trans-retinal is recycled for regeneration of 11-cis-retinal via the visual cycle which is a series of biochemical reactions in photoreceptors and in retinal pigment epithelial (RPE) cells (7, 8). Even though all-trans-retinal is an essential source for regeneration of rhodopsin, delayed clearance of all-trans-retinal is closely associated with retinal disorders (9-11). Clearance of all-trans-retinal in photoreceptors occurs in two steps: translocation of all-trans-retinal from the inside to the outside of photoreceptor outer segment (POS) discs by the ATP-binding cassette transporter 4 (ABCA4) (12) and reduction of all-trans-retinal to all-trans-retinol in the cytosolic lumen of photoreceptor outer segments by retinol dehydrogenase 8 (RDH8) (13, 14). We previously developed a model of retinal degeneration in mice mediated by all-trans-retinal in which Abca4 and Rdh8 are deleted (15, 16). This model reproduces many features of human Stargardt- and AMD-like retinal phenotype characterized by lipofuscin accumulation, drusen formation, complement activation, photoreceptor/RPE atrophy and choroidal neovascularization (16). The uniqueness of this model is that the Abca4-/-Rdh8-/- mouse not only displays age-related chronic degeneration under room light conditions but also displays acute retinal degeneration when exposed to intense light (15). Our previous study showed that Abca4-/-Rdh8-/- mice exposed to intense light exhibited increases in several pro-inflammatory molecules (2). However, these increases were transient, and all inflammatory molecules returned to the basal level 7 days after light exposure. Pro-inflammatory molecules increased in age-related chronic degenerated retinas of Abca4-/-Rdh8-/- mice, but many anti-inflammatory molecules were also shown to increase in these mice. These anti-inflammatory molecules included: complement factor H (CFH), a regulator of the complement cascade, Arginase, liver (ARG1), a marker of M2 macrophages which possesses anti-inflammatory properties (17), and Transforming growth factor, beta 1 (TGFB), a regulator of the immune cascade. These findings point to the paradoxical nature of the inflammatory response in Abca4-/-Rdh8-/- mice.
In this study, we present data indicating that CCL3 is a critical regulator of retinal inflammation which is associated with severity of retinal degeneration.
MATERIALS AND METHODS
Animals
Abca4-/-Rdh8-/- mice were generated as described previously, and all mice were genotyped by an established method (16). Only mice with the leucine variation at amino acid 450 of RPE65 were used. Rd8 mutation on Crb1 gene was also assessed (18) and only mice without this mutation were employed in this study. Ccl3-/-, Ccl2-/-, Mertk-/- and Cx3Cr1gfp/Δ mice were obtained from Jackson Lab (Bar Harbor, Maine). Genotyping for Ccl3 was performed with primers; for wild type forward; 5’-ATGAAGGTCTCCACCACTGC-3’, reverse; 5’-AGTCAACGATGAATTGGCG-3’, for mutant forward; 5’-TAAAGCATGCTCCAGACT-3’ and reverse, 5’-CAAAGGCTGCTGGTTTCAAA-3’ (19). Genotyping for Ccl2 was performed with primers; for wild type forward; 5’- TGACAGTCCCCAGAGTCACA-3’, common reverse; 5’-TCATTGGGATCATCTTGCTG -3’, for mutant forward; 5’-GCCAGAGGCCACTTGTGTAG-3’, for Mertk was performed with primers; for wild type forward; 5’-GCTTTAGCCTCCCCAGTAGC-3’, reverse; 5’-GGTCACATGCAAAGCAAATG-3’, for mutant forward; 5’-CGTGGAGAAGGTAGTCGTACATCT-3’ and reverse; 5’-TTTGCCAAGTTCTAATTCCATC-3’, and for Cx3Cr1 was performed with primers; for wild type, 5’–TCCACGTTCGGTCTGGTGGG-3’ and 5’–GGTTCCTAGTGGAGCTAGGG–3’; and Cx3cr1–GFP mutant, 5’–GATCACTCTCGGCATGGACG–3’ and 5’–GGTTCCTAGTGGAGCTAGGG–3’ according to the protocol from Jackson Lab. C57BL/6 or littermate control mice were used as WT mice. Equal numbers of males and females were used. All mice were housed in the animal facility at the School of Medicine, Case Western Reserve University, where they were maintained either under complete darkness or in a 12 h light (~10 lux)/12 h dark cycle environment. Experimental manipulations in the dark were done under dim red light transmitted through a Kodak No. 1 safelight filter (transmittance >560 nm). All animal procedures and experiments were approved by the Case Western Reserve University Animal Care Committees and conformed to both the recommendations of the American Veterinary Medical Association Panel on Euthanasia and the Association of Research for Vision and Ophthalmology.
Induction of light damage
Mice were dark-adapted for 48 h before exposure to light. Light damage was induced by exposing mice to 10,000 lux of diffuse white fluorescent light (150 W spiral lamp; Commercial Electric, Cleveland, OH) for 30 min or 15 min. Before such light exposure, pupils of mice were dilated with a mixture of 0.5% tropicamide and 0.5% phenylrphrine hydrochloride (Midorin-P®, Santen Pharmaceutical Co., Ltd, Osaka, Japan) and after exposure animals were kept in the dark until evaluation.
Histological analysis
All procedures to make sections for immunohistochemistry (IHC) and light microscopy were performed using a previously described method (14). The following antibodies were used for IHC; rabbit anti-Iba1 Ab (1:400, Wako, Chuo-ku, Osaka, Japan), rabbit anti-Glial Fibrillary Acidic Protein Ab (GFAP; 1:400, Dako, Carpinteria, CA), mouse anti–rhodopsin 1D4 Ab (1:100, gift from Dr. Robert Molday, University of British Columbia, Vancouver, Canada), anti-T cell Ab (anti-CD3; 1: 200, Dako), anti-CD45 Ab (1: 200, Abcam, Cambridge, MA), monoclonal anti-mouse neutrophil Ab (NIMP-R14; 1: 200, Abcam) and Alexa 488–conjugated peanut agglutinin (PNA; 1:200, Invitrogen). Images of IHC were captured by a confocal microscope (LSM, Carl Zeiss, Thornwood, NY).
Flat-mount RPE preparation for immunostaining
All procedures to make flat-mount RPE were described previously (2). Rabbit anti-ZO-1 Ab (1:200, Invitrogen) was used. Size of RPE cells was measured by LSM image browser (Carl Zeiss, Thornwood, NY).
Scanning laser ophthalmoscopy (SLO) imaging and ultra-high resolution spectral domain optical coherence tomography (SD-OCT)
HRAII (Heidelberg Engineering, Heidelberg, Germany) for SLO and ultra-high resolution SD-OCT (Bioptigen, Research Triangle Park, NC) were employed for in vivo imaging of mouse retinas. Mice were anesthetized by intraperitoneal injection of a mixture (20 μl/g body weight) containing ketamine (6 mg/ml) and xylazine (0.44 mg/ml) in 10 mM sodium phosphate, pH 7.2, with 100 mM NaCl. Pupils were dilated with mixture of 0.5% tropicamide and 0.5% phenylephrine hydrochloride (Midorin-P®, Santen Pharmaceutical Co., Ltd). Five pictures acquired in the B-scan mode were used to construct each final averaged SD-OCT image. SD-OCT images were scored using our previously established scoring system (20).
Isolation of primary RPE and retinal microglial cells
Primary mouse RPE cells and retinal microglial cells were prepared from 2-week-old mice. Enucleated eyes were incubated with 2% dispase (Invitrogen) in Dulbecco’s Modified Eagle Medium (DMEM) (Invitrogen) for 1 h at 37 °C, and neural retinas and eyecups were separated under a surgical microscope (ILLUMIN-i, Endure Medical, Cumming, GA). The RPE layer was peeled from eyecups, passed through 70-μm and 40-μm nylon mesh filters (Falcon Plastics, Brookings, SD), and cultured in DMEM containing MEM non-essential amino acids (Invitrogen), penicillin/streptomycin (Invitrogen), 20 mM HEPES, pH 7.0, and 10% fetal bovine serum. To enrich microglial cells, neural retinas were homogenized and cultured in DMEM containing MEM non-essential amino acids (Invitrogen), penicillin/streptomycin (Invitrogen), 20 mM HEPES, pH 7.0, and 10% fetal bovine serum for 7 days at 37 °C. Adherent cells to the plastic surface were treated with 0.05% trypsin (Invitrogen), and less adhesive cells were collected as microglial cells.
Enzyme-linked immunosorbent assay (ELISA)
Production of Ccl3 and Ccl4 from retinal primary cells was quantified by ELISA kits (Ccl2; MJE00 and Ccl3; MMA00) purchased from R&D systems (Minneapolis, MN) with 50 μl of cell culture supernatants of primary RPE or microglial cells. Concentrated cell lysates were prepared with NP-40 lysis buffer containing 20 mM Tris, pH 8.0, 137 mM NaCl, 10% glycerol and 1% NP-40. Then protein concentration was measured with a BCA protein assay kit (Pierce, Rockford, IL). Production of Ccl3, Ccl4 and Il1 β was also quantified by ELISA kits from R&D systems (Ccl4; MMB00 and IL1β; MLB00C) with mouse eyes. Two eyes from one mouse were homogenized in 500 μl of PBS with protease inhibitor cocktails (Roche) by a glass/glass homogenizer. The homogenates (50 μl) were used for the quantification. A single data point was obtained from each mouse (2 eyes).
Fundus fluorescein angiography
Fluorescein Sodium (ANGIOFLUOR®, Alliance Pharmaceuticals, Inc, Richmond, TX) was diluted in PBS to 25 mg/ml and injected 2.5 mg/100μl/mouse via intraperitoneal injection 10 min prior to taking images. Fundus fluorescein angiography was performed by HRAII (Heidelberg Engineering).
Quantitative RT- PCR (qRT-PCR)
All procedures for qRT-PCR were described previously (2). Following primers were used for analyses: Ccl3 (231 bp), forward 5’-CTGCCCTTGCTGTTCTTCTC-3’, reverse 5’-CTTGGACCCAGGTCTCTTTG-3’; Ccl4 (196 bp), forward 5’-GCCCTCTCTCTCCTCTTGCT-3’, reverse 5’-GTCTGCCTCTTTTGGTCAGG-3’; Ccl2 (187 bp), forward 5’-GCTGACCCCAAGAAGGAATG-3’, reverse 5’-GTGCTTGAGGTGGTTGTGGA-3’; Ccr1 (206 bp), forward 5’-TTCCTCCTCTGGACCCCCTA-3’, reverse 5’-TTGAAACAGCTGCCGAAGGT-3’; Ccr5 (172 bp), forward 5’-GCTGCCTAAACCCTGTCATC-3’, reverse 5’-TCATGTTCTCCTGTGGATCG-3’; Ccr2 (227 bp), forward 5’-ATTCTCCACACCCTGTTTCG-3’, reverse 5’-ATGCAGCAGTGTGTCATTCC-3’; Ccl12 (188 bp), forward 5’-CAGTCCTCAGGTATTGGCTGGA-3’, reverse5’-TCCTTGGGGTCAGCACAGAT-3’; Cxcl10 (154 bp), forward 5’-CCTCATCCTGCTGGGTCTG-3’, reverse 5’-CTCAACACGTGGGCAGGA-3’; Il1β (167 bp), forward 5’-CCTGCAGCTGGAGAGTGTGG-3’, reverse 5’-CCAGGAAGACAGGCTTGTGC-3’; Nox2 (207 bp), forward 5’-TCGAAAACTCCTTGGGTCAG-3’, reverse 5’-TGCAGTGCTATCATCCAAGC-3’; Arg1 (181 bp) forward 5’-CGCCTTTCTCAAAAGGACAG-3’, reverse 5’- ACAGACCGTGGGTTCTTCAC-3’; Transforming growth factor, beta 1 (Tgfb, 185 bp) forward 5’- TGAGTGGCTGTCTTTTGACG-3’, reverse 5’-GGTTCATGTCATGGATGGTG-3’;Gapdh (150 bp), forward 5’-GTGTTCCTACCCCCAATGTG-3’, reverse 5’- AGGAGACAACCTGGTCCTCA-3’. Relative expression of genes was normalized by housekeeping gene Gapdh.
qRT-PCR based RNA expression analysis
RNA expression analysis was performed by RT2 Profiler PCR Array System provided by SABiosciences (Frederic, MD). Fold changes were calculated after the data normalized to 5 housekeeping genes. Total RNA was purified from 16 retinas of 4-week-old Abca4-/-Rdh8-/- and WT mice at each time point by RiboPure kit (Ambion, Austin, TX).
Data analysis
Data representing the means ± S.D. for the results of at least three independent experiments were compared by the one-way analysis of variance test.
RESULTS
Ccl3 expression shows the greatest increase in the retina of light exposed Abca4-/-Rdh8-/- mice
To investigate the involvement of chemokines in light induced acute retinal degeneration of Abca4-/-Rdh8-/- mice, PCR array analysis for 84 different chemokines and related molecules was performed (Table I). Retinas were harvested from Abca4-/-Rdh8-/- and WT mice 24 h and 7 days after light exposure at 10,000 lux for 30 min. Expression levels of dark adapted Abca4-/-Rdh8-/- and WT mice were used as a basal control for light exposed Abca4-/-Rdh8-/- and WT mice, respectively. Ccl2, Ccl3, Ccl4, Ccl12 and Cxcl10 expression increased 10-fold or higher in light exposed Abca4-/-Rdh8-/- mice when compared to dark adapted mice. Ccl3 expression at 24 h showed 84.6-fold increase as the greatest change of all tested genes. Only Cxcl10 increased 10-fold or higher in light exposed WT mice.
Table I.
Expression of chemokines and their related molecules in mouse retinas after light exposure
| genes | Abca4-/- Rdh8-/- mice1, 2 | WT | |||
|---|---|---|---|---|---|
| (24 h) | (7 days) | (24 h) | (7 days) | ||
| CC chemokine | Ccl2 | 30.52 | 1.13 | -1.40 | -2.93 |
| Ccl3 | 84.59 | 27.14 | 1.83 | -2.09 | |
| Ccl4 | 34.29 | 6.52 | 1.41 | -4.26 | |
| Ccl5 | 4.45 | 3.61 | 4.68 | 1.80 | |
| Ccl7 | 9.56 | -2.94 | -2.04 | -3.12 | |
| Ccl8 | -2.60 | -3.39 | -3.00 | -10.26 | |
| Ccl11 | -4.13 | -9.28 | -3.21 | -6.49 | |
| Ccl12 | 13.57 | 1.92 | 1.73 | -3.28 | |
| CC chemokine receptor | Ccr2 | 5.95 | 7.84 | -9.32 | -25.05 |
| Ccr3 | 29.04 | 31.76 | -1.21 | -1.19 | |
| Ccr6 | -3.01 | -3.39 | -1.23 | -2.76 | |
| Ccr7 | 1.62 | 3.88 | -1.51 | -1.44 | |
| Ccr10 | 2.46 | 3.51 | 7.62 | 3.43 | |
| CXC chemokine | Cxcl1 | 4.68 | -1.36 | 1.62 | -2.93 |
| Pf4 | -5.16 | -1.68 | -3.55 | -3.57 | |
| Cxcl5 | 7.26 | -2.01 | 8.96 | 8.53 | |
| Cxcl10 | 21.10 | 1.73 | 13.15 | -2.00 | |
| CXC chemokine receptor | Cxcr5 | 3.32 | 5.09 | 1.48 | 1.10 |
| Interleukin | Il1b | 6.44 | 1.47 | -1.25 | 1.22 |
| Il1f6 | -3.45 | -1.82 | -4.06 | -2.77 | |
| Il1f8 | -15.87 | -32.42 | 2.97 | 1.22 | |
| Il4 | 8.88 | 3.07 | 4.61 | 2.97 | |
| Il16 | 3.01 | 1.16 | -1.74 | -2.93 | |
| Il20 | -3.76 | -3.05 | -1.45 | -3.79 | |
| Interleukin receptor | Il1r1 | 1.69 | 1.46 | 1.64 | 1.61 |
| Il1r2 | -5.14 | -6.36 | -2.67 | -1.77 | |
| Il5ra | 4.35 | 6.16 | 2.91 | 2.52 | |
| Il6ra | 6.47 | 1.26 | -2.01 | -2.21 | |
| Il8rb | 4.65 | -4.75 | -3.21 | -2.93 | |
| Il10ra | -1.42 | 4.06 | -1.36 | -1.01 | |
| Others | Ifng | 3.58 | 2.59 | -3.21 | -2.93 |
| Tgfb1 | 1.74 | 2.63 | 3.62 | 3.03 | |
| Itgb2 | 1.88 | 4.58 | 1.00 | -1.10 | |
| Spp1 | 1.56 | 3.34 | -2.51 | -2.76 | |
| Tnfrsf1a | 5.47 | 3.14 | 2.74 | 1.84 | |
| Bcl6 | 3.39 | 1.84 | 3.28 | 2.78 | |
Fold changes more than 3 compared to data obtained from dark adapted mice are presented. The data was normalized to the housekeeping genes (Gusb, Gapdh, Actb, Hprt1 and Hsp90ab1).
Fold changes more than 10 are presented in bold text.
Minus signs (-) indicate reduced expression.
Mice were exposed to 10,000 lux white light for 30 min after 48 h of dark adaptation. Before such light exposure, pupils of mice were dilated. Mice were kept in the dark until evaluations.
RNA was purified from 16 retinas of 4-week-old mice.
Expression of MIP-1 genes peaks earlier than others after light treatment in Abca4-/-Rdh8-/- mice
To understand the dynamic nature of the expression of these inflammatory markers that showed 10-fold or higher changes, a time course measurement was conducted. The time course analyses using quantitative PCR (qPCR) for chemokines and chemokine receptors (Fig. 1) revealed that only MIP-1 transcripts, including Ccl3 and Ccl4, peaked 24 h after light exposure in Abca4-/-Rdh8-/- mice. In contrast, Ccl2, Cxcl10 and Ccl12 had a different temporal profile, peaked 3 days after light treatment. Light exposed WT mice did not show a significant increase in these chemokines at either 24 h or 3 days after light exposure. Chemokine receptors were also investigated to determine any changes in expression levels. Ccr1, which is a receptor for Ccl3, Ccr2, which is a receptor of Ccl2 and Cx3cr1 peaked 3 days after light exposure in Abca4-/-Rdh8-/- mice. Ccr5, a receptor for Ccl3 and Ccl4 peaked 12 h and continuously increased until 3 days after light exposure in Abca4-/-Rdh8-/- mice.
Figure 1. Distinct profiles in expression of Ccl3 and Ccl2 in the retina of Abca4-/-Rdh8-/- mice after light exposure.
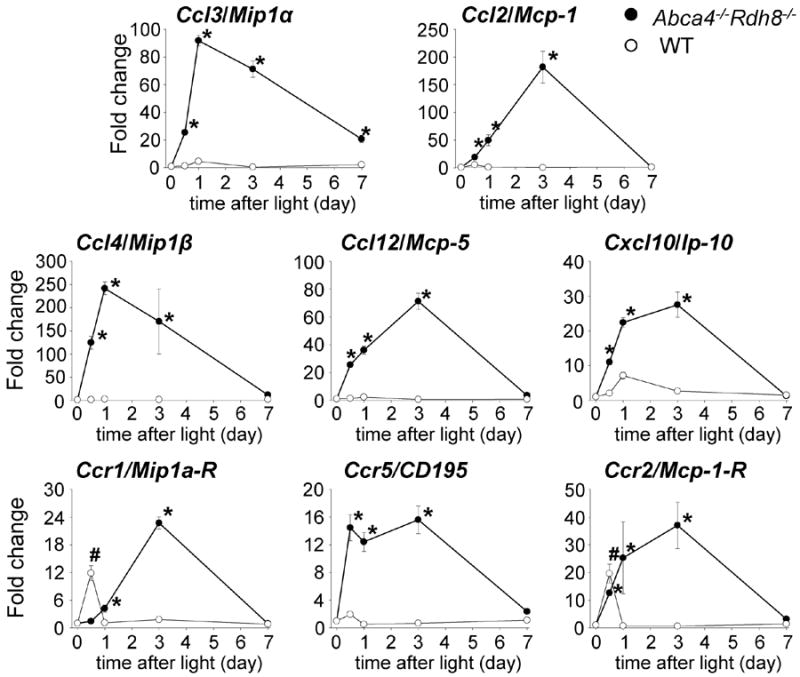
qRT-PCR was performed with RNA purified from 16 retinas of 4-week-old Abca4-/-Rdh8-/- and WT mice at each time point. Fold changes in expression to no light exposed Abca4-/-Rdh8-/- or WT mice are presented. The expression of each gene was normalized to the housekeeping gene Gapdh. Error bars indicate S.D. of the means (n = 3). * indicates P < 0.05 vs no light exposed Abca4-/-Rdh8-/-mice. # indicates P < 0.05 vs no light exposed WT mice.
To determine the likely source of Ccl3 and Ccl2 in the retina, primary cultured RPE cells and microglia were isolated from 2-week-old Abca4-/-Rdh8-/- mice (2) and co-incubated with photoreceptor outer segments (POS), which activate microglia/macrophages via TLR4 (2), lipopolysaccharide (LPS), a ligand of TLR4, and Pam3CSK4, a ligand of TLR1/2. Protein amounts of Ccl3 and Ccl2 from these cells were measured by ELISA. Only primary microglia revealed an increase in Ccl3 secretion (supplemental Fig. S1), whereas both primary RPE and microglial cells increased their Ccl2 secretion, indicating that microglia and monocytes are the dominant cells producing Ccl3 rather than RPE.
Low grade chronic inflammation in retinas of Abca4-/-Rdh8-/- mice
Given the different gene expression patterns of these factors in the retina, we sought to understand if they have distinct roles in acute versus chronic retinal degeneration in Abca4-/-Rdh8-/- mice (16). Expression levels of the chemokine and chemokine receptor transcripts were examined in 4-week-, 6-month- and 12-month-old Abca4-/-Rdh8-/- and WT mice to assess the changes associated with chronic degeneration of the retina under room light conditions (Fig. 2). Compared to 4-week-old mice, expression of MIP-1 related genes, including Ccl3, Ccl4 and Ccr1, was increased at the mRNA level in 6-month-old and decreased in 12-month-old Abca4-/-Rdh8-/- mice, although Ccr5 which is also a receptor of Ccl3 and Ccl4 was increased till 12-month-old. In contrast, monocyte chemotactic protein (MCP) related genes, including Ccl2 and Ccl12, were increased at 12 months of age. Ccr2, a receptor of the Ccl2 ligand, was also increased at age of 6 and 12 months. Cxcl10 displayed a similar expression pattern as MIP-1 genes. These data indicate the presence of low grade chronic inflammation in the retina of aged Abca4-/-Rdh8-/- mice as observed in human degenerative retinal diseases (1).
Figure 2. Low grade chronic inflammation in Abca4-/-Rdh8-/- mice.

qRT-PCR was performed with RNA purified from 16 retinas of 4-week-old, 6-month-old and 12-month-old Abca4-/-Rdh8-/- and WT mice. The expression of genes in Abca4-/-Rdh8-/- mice was compared with WT mice after normalization to the housekeeping gene Gapdh, and presented by fold changes. Error bars indicate S.D. of the means (n = 3). * indicates P < 0.05 vs 4-week-old mice.
Delayed clearance of subretinal microglia in Ccl3-/-Abca4-/-Rdh8-/- mice compared with Abca4-/-Rdh8-/- mice
Because of the distinct expression pattern of MIP-1 genes, Ccl3 and Ccl4, and other chemokines, we hypothesized that MIP-1 gene products have a distinct role in all-trans-retinal mediated retinal degeneration. To directly assess this hypothesis, Ccl3-/-Abca4-/-Rdh8-/- mice were generated by crossing Abca4-/-Rdh8-/- with Ccl3-/- mice. Littermate controls were also generated from this mouse cross. To examine the role of CCL3 in acute retinal degeneration, Ccl3-/-Abca4-/-Rdh8-/-, Abca4-/-Rdh8-/- and WT mice were exposed to 10,000 lux light for 30 min. This light exposure caused an increased number of autofluorescent (AF) spots which was detected by in vivo SLO which provides a high-quality images by acquiring florescent signals of the retina with a horizontal/confocal view (21) (Fig. 3A). We found significantly more AF spots in Ccl3-/-Abca4-/-Rdh8-/- mice when compared to Abca4-/-Rdh8-/- mice 14 and 21 days after light exposure. In comparison Ccl3-/- mice did not develop retinal degeneration by the same light exposure condition as Abca4-/-Rdh8-/- mice, and displayed similar resistance to light induced damage as WT mice (supplemental Fig. S2).
Figure 3. Extended appearance of microglia/macriphage in the subretinal space in Ccl3-/-Abca4-/-Rdh8-/- mice after light exposure.
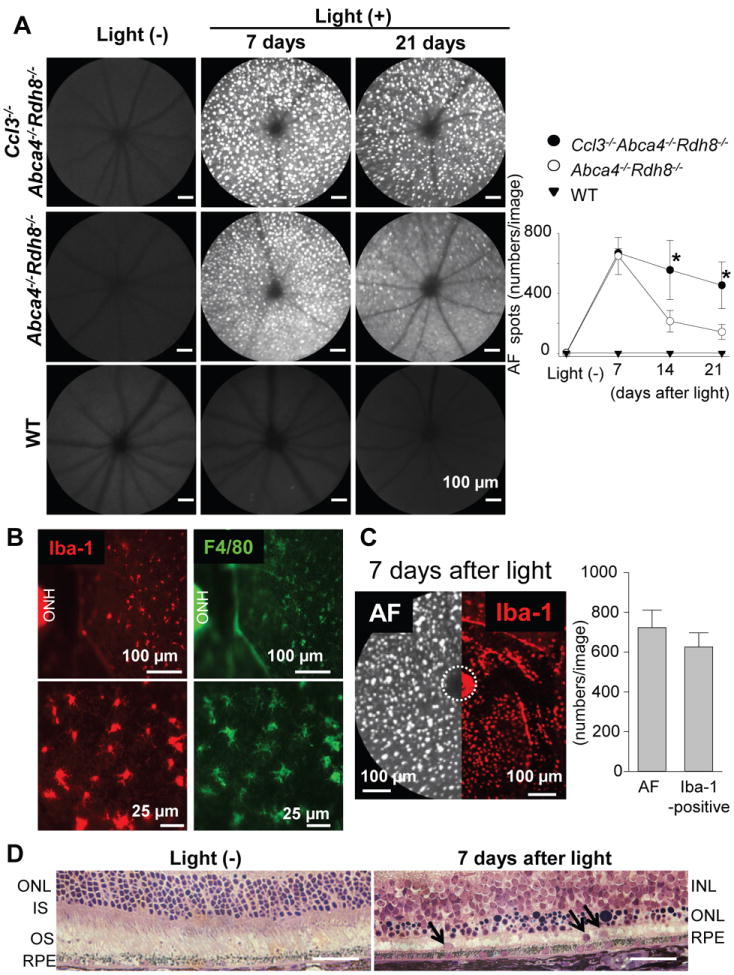
Ccl3-/-Abca4-/-Rdh8-/-, Abca4-/-Rdh8-/- and WT mice at 4-6-week-old age were exposed to 10,000 lux light for 30 min. A. Retinal images were captured by in vivo SLO. Images were taken at 7, 14, and 21 days after light exposure (left). Bars indicate 100 μm. Numbers of autofluorescent (AF) spots of each image were counted (right). Error bars indicate S.D. of the means (n > 6).* indicates P < 0.05 vs light exposed Abca4-/-Rdh8-/- mice. B. RPE flat-mounts of Abca4-/-Rdh8-/- mice were prepared 7 days after light exposure and stained with anti-Iba-1 (left) and anti-F4/80 (right) Abs. Lower panels show magnified images. ONH, optic nerve head. C. SLO image (left) and flat-mount IHC with anti-Iba-1 Ab (right) in the same magnification is presented. Abca4-/-Rdh8-/- mice were exposed to 10,000 lux for 30 min and kept in the dark for 7 days. Numbers of AF spots in SLO images (per image) and Iba-1-positive cells in IHC in the same size area were counted (right graph). ONH was circled by broken line. D. Retinal cross section of Abca4-/-Rdh8-/- mice without light exposure and 7 days after light exposure at 10,000 lux for 30 min was prepared by Epon-embedment. Arrows indicate cells in the subretinal space. Bars indicate 50 μm. ONL, outer nuclear layer; IS, inner segment; OS, outer segment; INL, inner nuclear layer.
Flat-mount retinas from these mice 7 days after light exposure displayed many infiltrated cells into the subretinal space. These infiltrated cells stained positive for both Iba-1 (a marker for microglia/macrophage) and F4/80 (a marker for macrophage) (Fig. 3B). Immunohistochemical analysis with anti-Iba-1, anti-NIMP-R14 (for neutrophil) and anti-CD3 (for T cell) Abs revealed that most of the subretinal cells were Iba-1-positve cells (Table. II, supplemental Fig. S3). A comparison of a SLO image and a flat-mount image with Iba-1 Ab staining is presented in Fig. 3C. The number of AF spots obtained using SLO and counting Iba-1-positive cells in flat-mount retinas gave similar results. Retinal histology revealed loss of photoreceptor layers and nuclei of what appear to be infiltrating cells in the subretinal space in Abca4-/-Rdh8-/- mice 7days after light exposure (Fig. 3D).
Table II.
Populations of subretinal cells 7 and 21 days after light exposure at 10,000 lux for 30 min
| mouse models | days after light | Iba-1-positive (%) | NIMP-R14-positive3 (%) | CD3-positive (%) | counted cells1/8 slides |
|---|---|---|---|---|---|
| Abca4-/-Rdh8-/- | 7 days | 97.9 ± 0.9 | 2.1 ± 0.9 | 0 | 191.5 ± 13.5 |
| Abca4-/-Rdh8-/- | 21 days | 90.3 ± 4.2 | 6.9 ± 1.0 | 2.4 ± 2.2 | 59.7 ± 18.6 |
| Ccl3-/-Abca4-/-Rdh8-/- | 7 days | 98.6± 2.8 | 1.3 ± 0.5 | 0 | 198.2± 21.7 |
| Ccl3-/-Abca4-/-Rdh8-/- | 21 days | 91.4 ± 6.2 | 5.4 ± 0.7 | 2.8± 1.5 | 147.7 ± 24.1 |
|
|
|||||
| GFP-positive (%) | counted cells2 | ||||
|
|
|||||
| Cx3Cr1gfp/ΔAbca4-/-Rdh8-/- | 7 days | 100 | 1034 | ||
Cryosections were prepared from every 200 μm distance from the edge to edge (8 slides/eye), and IHC was performed with anti-Iba-1 Ab for microglia/macrophage, anti-Nimp-R14 Ab for neutrophil and anti-CD3 Ab for T cell. Numbers of subretinal cells were counted from these sections, and the ratio of these cells was calculated.
Flat-mount eyes were prepared and subretinal cells with GFP and autofluorescent signals were counted under fluorescent microscope.
NIMP-R14 recognizes Ly6C in addition to Ly6G, and can therefore react with activated microglia/macrophages. As shown in supplemental Fig S3, subretinal cells with stronger signals over background or weakly stained cells were counted as positive cells.
As a second approach, we generated Abca4-/-Rdh8-/- mice crossed with Cx3Cr1gfp/Δ mice in which their monocytes express GFP (22). Flat-mount eyes after peeled off the neural retina were examined. Light-exposed Abca4-/-Rdh8-/- mice did not show GFP-positive cells on the apical side of RPE layer, whereas Cx3Cr1gfp/ΔAbca4-/-Rdh8-/- mice after induction of retinal light damage demonstrated GFP-positive cells above the RPE layers (Fig. 4A). GFP-positive cells were not detected before light exposure in either Cx3Cr1gfp/ΔAbca4-/-Rdh8-/- or in Abca4-/-Rdh8-/- mice, indicating translocation of microglial/macrophage cells from the inner retina to the subretinal space. Ramified shaped GFP-positive cells were observed only in the inner retina prior to light exposure, and no GFP+ cells were detected in the subretinal space (Fig. 4B, upper). In contrast, eyes of Cx3Cr1gfp/ΔAbca4-/-Rdh8-/- mice 7 days after light exposeure displayed increased numbers of more rounded GFP-positive cells in the deeper retina close to the RPE layer (Fig. 4B, lower). Multiple GFP-positive cells were observed in the ONL layers as well (Fig. 4C) and GFP positive cells were not detected in the subretinal layers (Table II). SLO images of Cx3Cr1gfp/ΔAbca4-/-Rdh8-/- mice with and without light exposure showed GFP signals from the inner plexiform layer where resting microglial cells normally reside (Fig. 4D, upper). Although there were no GFP signals from the level of the subretinal space (OS~RPE) before light exposure, GFP and AF signals were detected in the subretinal space of Cx3Cr1gfp/ΔAbca4-/-Rdh8-/- mice following light exposure (Fig. 4D, lower). As the number of AF spots in light exposed Abca4-/-Rdh8-/- mice is similar to that of GFP+ cells in Cx3Cr1gfp/ΔAbca4-/-Rdh8-/- mice, it is likely that the AF spots detected with SLO are consistent with microglial cells. RPE flat-mounts from light exposed Ccl3-/-Abca4-/-Rdh8-/- mice displayed enlarged RPE cells and reduced expression of tight junction protein, Zo-1, compared to light exposed and no light exposed Abca4-/-Rdh8-/- mice, thus indicating a loss of RPE cell integrity and viability (supplemental Fig. S4). Taken together, these data indicate delayed clearance of microglia and loss of RPE cell integrity in the absence of Ccl3.
Figure 4. Translocation of monocytes during light-induced retinal degeneration.
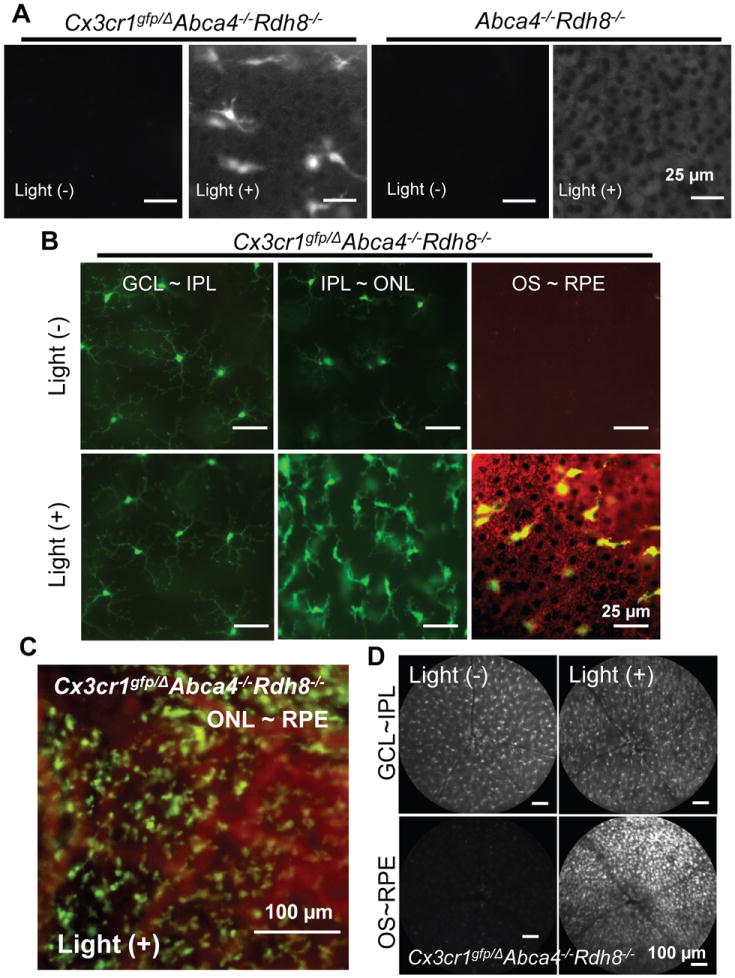
A. Flat-mount RPE was examined 7 days after light exposure at 10,000 lux for 30 min under a fluorescent microscope with 4-week-old Cx3Cr1gfp/Δ Abca4-/-Rdh8-/- and Abca4-/-Rdh8-/- mice. Bars indicate 25 μm. B. Shapes of GFP-positive cells were examined in 4-week-old Cx3Cr1gfp/ΔAbca4-/-Rdh8-/- mice by flat-mount eyes 7 days after exposure to light at 10,000 lux for 30 min at different depth of the retina. GFP signals are shown in green in the left and right panels and autofluorescent signals are shown in red. Bars indicate 25 μm. C. GFP-positive cells at the ONL~RPE level is presented. Cx3Cr1gfp/ΔAbca4-/-Rdh8-/- mice were exposure to light at 10,000 lux for 30 min and flat-mount retina was prepared 7 days after light. Bar indicates 100 μm. D. SLO images were captured from 4-week-old Cx3Cr1gfp/ΔAbca4-/-Rdh8-/- mice before and 7 days after light exposure at 10,000 lux for 30 min Bars indicate 100 μm.
Increased expression of chemokines and the other inflammatory molecules in Ccl3-/-Abca4-/-Rdh8-/- mice after light exposure
Light exposed Ccl3-/-Abca4-/-Rdh8-/- and Abca4-/-Rdh8-/- mice displayed severe retinal degeneration, although there was a different clearance rate of subretinal infiltrating cells between the two above mentioned mouse lines (Fig. 5A). To elucidate the molecular changes causing the delay in clearance of subretinal microglia/macrophages in Ccl3-/-Abca4-/-Rdh8-/- mice, mRNA levels of chemokines and other inflammatory molecules in the retina were examined. Although Ccl3 was completely diminished, Ccl4, which has a sequence homology of ~ 60% with the murine Ccl3 gene, was increased in Ccl3-/-Abca4-/-Rdh8-/- mice compared to Abca4-/-Rdh8-/- mice at 3 days and 7 days after light exposure (Fig. 5B). Absolute expression levels of Ccl3, Ccl4, Ccl2 and Il1b against the house keeping Gapdh gene at 7 days after light are also shown (Fig. 5C). Inflammatory molecules, excluding Ccl3, were increased in Ccl3-/-Abca4-/-Rdh8-/- mice compared to Abca4-/-Rdh8-/- mice. Expression of M1 and M2 microglia/macrophage related molecules were also evaluated (Fig. 5D), because M1 macrophages contribute to inflammation whereas M2 macrophages have an anti-inflammatory role (23). Nox2, a prototypic M1 microglia/macrophage marker (24) was increased in both Ccl3-/-Abca4-/-Rdh8-/- and Abca4-/-Rdh8-/- mice at 3 days after light exposure. However, expression of Arg1, a marker of M2 microglia/macrophage (17), was increased only in Abca4-/-Rdh8-/- mice at 3 days after light. Tgfb, an immunosuppressive factor and component of the immune-privilege in the eye (25), was elevated in Abca4-/-Rdh8-/- mice at 3 days but returned to basal levels at 7 days after light exposure.
Figure 5. Elevated mRNA expressions of inflammatory molecules in the retinas of light exposed Ccl3-/-Abca4-/-Rdh8-/- mice.
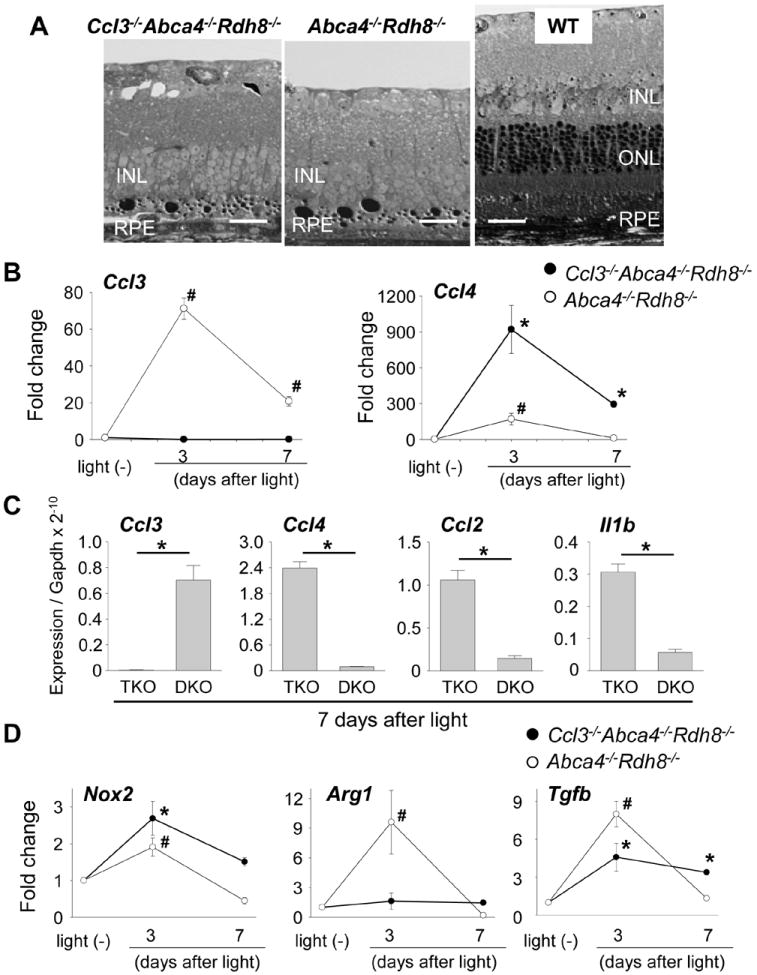
A. Severity of retinal degeneration in 4-week-old Ccl3-/-Abca4-/-Rdh8-/-, Abca4-/-Rdh8-/- and WT mice after light exposure at 10,000 lux for 30 min was examined by epon-embedment sections 7 days after light exposure. B. RNA samples were collected from 16 retinas of 4-week-old Ccl3-/-Abca4-/-Rdh8-/- (TKO) and Abca4-/-Rdh8-/- (DKO) at each point. Ccl3 and Ccl4 mRNA expression levels were examined by qRT-PCR. Data are normalized by Gapdh expression and shown by fold change. Error bars indicate S.D. of the means (n = 3). # indicates P < 0.05 vs no light exposed Abca4-/-Rdh8-/- mice. C. Absolute expression levels of Ccl3, Ccl4, Ccl2 and Il1b mRNA at 7 days after light were shown when compared to Gapdh expression. Error bars indicate S.D. of the means (n = 3). * indicates P < 0.05 between each group. D. RNA samples were collected from 16 retinas of mice at each point. Expression levels of Nox2, a marker of M1 microglia/macrophage, Arg1, a marker of M2 microglia/macrophage, and Tgfb are normalized by Gapdh expression and shown by fold change. Error bars indicate S.D. of the means (n = 3). * indicates P < 0.05 vs no light exposed Ccl3-/-Abca4-/-Rdh8-/- mice. # indicates P < 0.05 vs no light exposed Abca4-/-Rdh8-/- mice.
Severe retinal degeneration develops in Ccl3-/-Abca4-/-Rdh8-/- mice after brief light exposure
Because Ccl3-/-Abca4-/-Rdh8-/- and Abca4-/-Rdh8-/- mice both exhibited similar levels of retinal degeneration after 30 min of light exposure at 10,000 lux, it is unclear what impact Ccl3 has on light-induced photoreceptor cell death. However, since Ccl3-/-Abca4-/-Rdh8-/- mice showed prolonged retinal inflammation after light exposure, we examined the effect of Ccl3 on mice exposed to 10,000 lux light for 15 min, which is only half the duration of our usual light exposure period. Ccl3-/-Abca4-/-Rdh8-/- mice exhibited severe retinal degeneration compared with Abca4-/-Rdh8-/- mice 7 days after light exposure (Fig. 6A). Furthermore, Ccl3-/-Abca4-/-Rdh8-/- mice exposed to 15 min of light showed higher Iba-1-positive cell accumulation in the subretinal space and increased numbers of AF spots compared with Abca4-/-Rdh8-/- mice (Fig. 6B, C). Additional production of Ccl3 after light exposure at 10,000 lux for 15 min was observed in Abca4-/-Rdh8-/- mice, whereas Ccl3 production was not detected in Ccl3-/-Abca4-/-Rdh8-/- mice by ELISA (Fig. 6D). Further, elevated production of Ccl4 was documented in Ccl3-/-Abca4-/-Rdh8-/- mice compared with Abca4-/-Rdh8-/- mice 1 and 3 days after light exposure. Increased production of Il1β (130.8±53.5 pmol/eye in Ccl3-/-Abca4-/-Rdh8-/- mice vs 46.2±8.8 pmol/eye in Abca4-/-Rdh8-/- mice) was also detected 1 day after light exposure in Ccl3-/-Abca4-/-Rdh8-/- mice. Taken together, these data indicate that Ccl3 deficiency exacerbates acute light-induced retinal degeneration with more production of Ccl4 in Abca4-/-Rdh8-/- mice.
Figure 6. Severer retinal degeneration in Ccl3-/-Abca4-/-Rdh8-/- mice than in Abca4-/-Rdh8-/- mice after 15 min of light exposure.
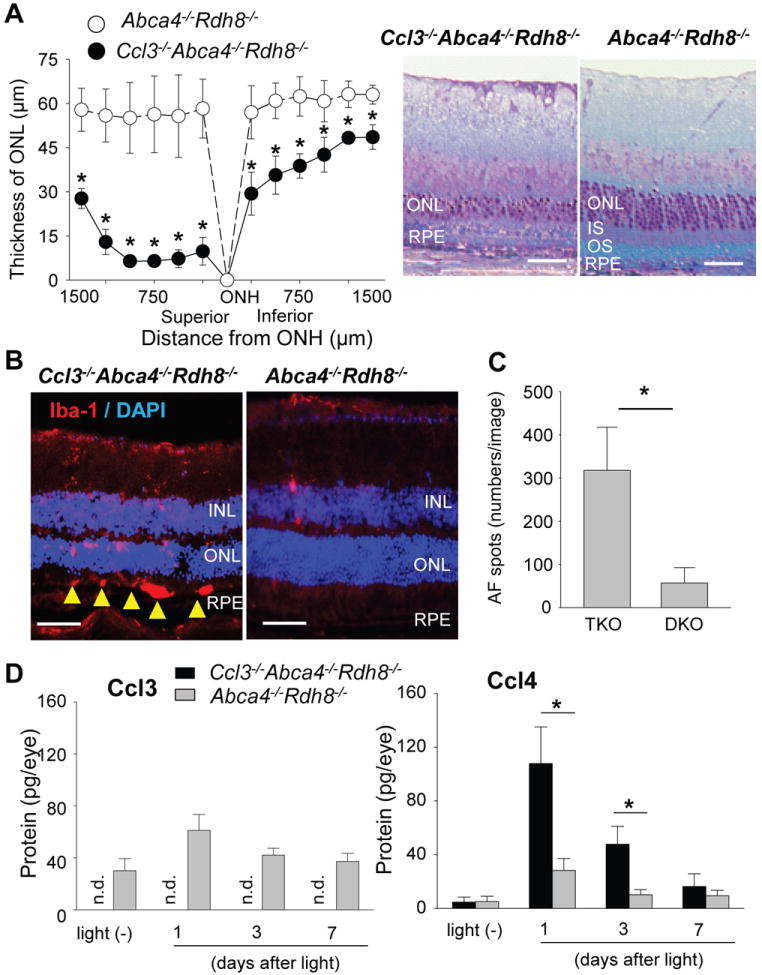
Four-6-week-old Ccl3-/-Abca4-/-Rdh8-/- (TKO) and Abca4-/-Rdh8-/- (DKO) mice were exposed to 10,000 lux light for 15 min. A. Thickness of ONL was measured by SD-OCT (left) and retinal sections (right) were prepared at 7 days after light exposure. Error bars indicate S.D. of the means (n > 6).* indicates P < 0.05 vs light exposed Abca4-/-Rdh8-/- mice. Bars indicate 50 μm. ONL, outer nuclear layer; IS, inner segments; OS, outer segments; RPE, retinal pigment epithelium. B. IHC by using anti-Iba-1, a marker of microglia/macrophage, 7 days after light exposure for 15 min are presented. Yellow arrowheads indicate infiltrated microglia/macrophages in the subretinal space. Bars indicate 30 μm. INL, inner nuclear layer; ONL, outer nuclear layer; RPE, retinal pigment epithelium. C. Numbers of AF spots were counted by using SLO. Error bars indicate S.D. of the means (n > 6). * indicates P < 0.05. D. Production of Ccl3 and Ccl4 was quantified by ELISA with eyes before and 1, 3 and 7 days after light exposure at 10,000 lux for 15 min. Two eyes from one mouse were homogenized in 500 μl of PBS with proteinase inhibitors, and the homogenates (50 μl) were used for the quantification. Error bars indicate S.D. of the means (n > 3 mice) * indicates P < 0.05. n.d. indicates not detectable.
Ccl3 deficiency moderates age-related retinal degeneration in Abca4-/-Rdh8-/- mice
Our previous studies showed that age-related retinal degeneration in Abca4-/-Rdh8-/- mice is characterized by chronic and sustained low grade retinal inflammation (2, 16). To elucidate the role of CCL3 in chronic degeneration compared with light induced acute degeneration, the retinal phenotype of 6-month-old Ccl3-/-Abca4-/-Rdh8-/- and Abca4-/-Rdh8-/- mice were examined. We found severe retinal degeneration in Abca4-/-Rdh8-/- mice compared with WT mice by SD-OCT which displays a tangential view of the retina with ultrahigh-resolution in vivo (26). Retinal degeneration in 6-month-old Ccl3-/-Abca4-/-Rdh8-/- mice resembled WT mice (Fig. 7A and 7B), indicating reversal of the Abca4-/-Rdh8-/- phenotype in the absence of Ccl3. GFAP expression was also weaker in 6-month-old Ccl3-/-Abca4-/-Rdh8-/- mice than in Abca4-/-Rdh8-/- mice (Fig. 7A lower), indicating milder reactive gliosis against retinal inflammation. Further, the number of AF spots, which represent subretinal microglial/macrophage cells (2), was also decreased in 6-month-old Ccl3-/-Abca4-/-Rdh8-/- mice (Fig. 7C). Production of Ccl3, Ccl4 and Il1β was quantified with eyes of 6-month-old mice by ELISA (Fig. 7D). Ccl3-/-Abca4-/-Rdh8-/- mice showed no production of Ccl3, but 139.1 ± 41.1 pg/eye of Ccl3 was detected in Abca4-/-Rdh8-/- mice. Basal Ccl3 production was observed in WT mice between mice at 6 months of age (36.8 ± 0.93 pg/eye) and 6 weeks of age (37.3 ± 6.3 pg/eye). Decreased production of Ccl4 and Il1β was noted in Ccl3-/-Abca4-/-Rdh8-/- retina compared to Abca4-/-Rdh8-/- retina. Collectively, these findings indicate that Ccl3 enhances age-related retinal degeneration and inflammation in Abca4-/-Rdh8-/- mice, which is the opposite effect that Ccl3 has on acute retinal degeneration caused by light.
Figure 7. Ccl3 deficiency attenuated age-related retinal degeneration in Abca4-/-Rdh8-/- mice.
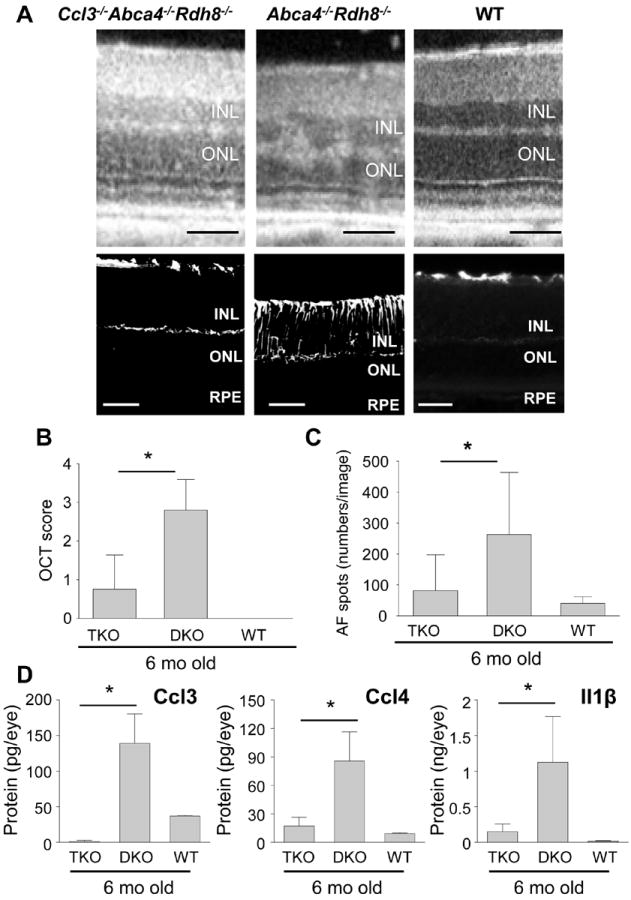
A. Retinal phenotype of Ccl3-/-Abca4-/-Rdh8-/- (TKO) and Abca4-/-Rdh8-/- (DKO) mice at 6-month old age were examined by in vivo SD-OCT imaging (upper panels), and IHC staining by GFAP, a marker for Müller cell gliosis (lower panels). Bars indicate 50 μm. INL, inner nuclear layer; ONL, outer nuclear layer; RPE, retinal pigment epithelium. B. Severity of retinal degeneration was evaluated by established scoring system (20). * indicates P < 0.05. C. Numbers of AF spots at 6-month of age were counted. Error bars indicate S.D. of the means (n > 5) * indicates P < 0.05. D. Production of Ccl3, Ccl4 and Il1β was quantified by ELISA. Two eyes of 6-month-old mice were homogenized in 500 μl of PBS with proteinase inhibitors, and the homogenates (50 μl) were used for the quantification. Error bars indicate S.D. of the means (n > 3 mice) * indicates P < 0.05.
Ccl3 deficiency results in increased photoreceptor survival in a murine model of retinitis pigmentosa
The role of CCL3 in retinal degeneration was further examined in the Mertk-/- mouse model of retinitis pigmentosa. Mutations in the MERTK gene cause retinal dystrophies in humans and in animal models (27). MERTK belongs to a family of receptor tyrosine kinases that includes AXL and TYRO3, and plays an indispensable role in the clearance of photoreceptor debris by RPE phagocytosis (28). Accumulation of photoreceptor debris in the subretinal space due to RPE phagocytosis deficiency is closely associated with the photoreceptor cell death seen in the Royal College of Surgeons (RCS) rat with disabled Mertk and in Mertk-/- mice (29). To determine if the retinal degenerative phenotype of Mertk-/- mice was altered in the absence of Ccl3, Ccl3-/-Mertk-/- mice were generated, and the thickness of ONL in Ccl3-/-Mertk-/-, Mertk-/- and WT mice was assessed at 5 weeks and 8 weeks of age by in vivo SD-OCT imaging. Mertk-/- mice showed degraded ONL compared with WT mice; however, Ccl3-/-Mertk-/- mice had increased ONL thickness when compared with Mertk-/- mice, but was still less than WT mice (Fig. 8A). Representative retinal histology images of 5-week-old Ccl3-/-Mertk-/-, Mertk-/- and WT mice are shown (Fig. 8B). Fewer numbers of Iba-1-positive cells were noted in Ccl3-/-Mertk-/- mice than in Mertk-/- mice at 5 and 8 weeks of age (Fig. 8C). Since inflammation in damaged retinas affects the integrity of the inner blood-retinal barrier (2), the integrity of this barrier in 8-week-old Ccl3-/-Mertk-/-, Mertk-/- and WT mice was examined by fluorescent angiography. Whereas Ccl3-/-Mertk-/- mice showed only weak fluorescent dye leakage from the optic nerve head (ONH), Mertk-/- mice showed increased leakage not only from ONH but also from retinal vessels (Fig. 8D). The incidence of fluorescent dye leakage in Ccl3-/-Mertk-/-, Mertk-/- and WT mice from the ONH were 33.3, 83.3 and 0%, respectively. These findings indicate that the deficiency of Ccl3 contributed to milder retinal degeneration in the Mertk-/- mouse model of retinitis pigmentosa.
Figure 8. Ccl3 deficiency protects the retina from degeneration in the mouse model for retinitis pigmentosa.
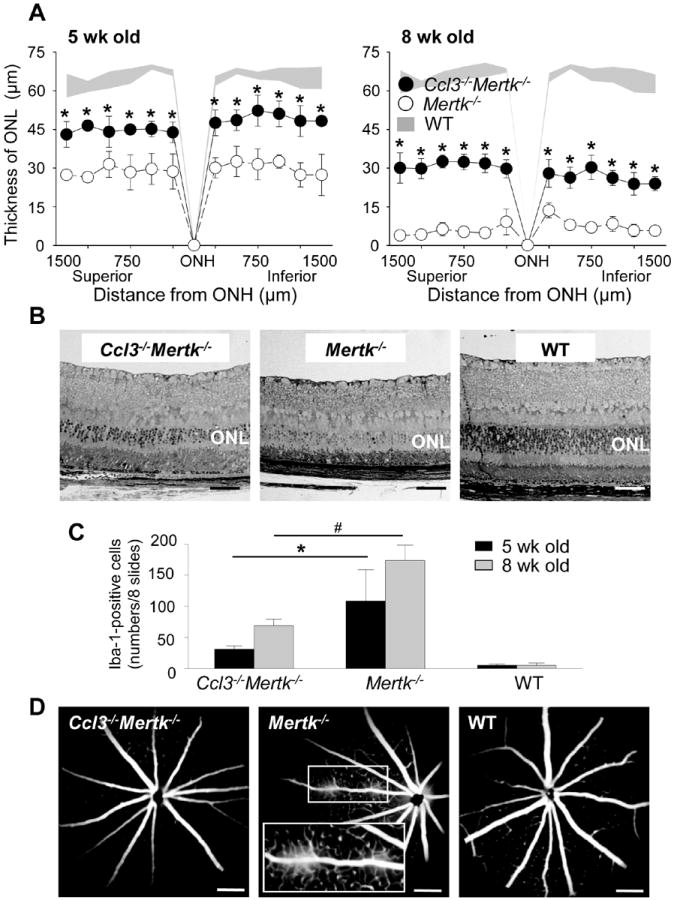
Ccl3-/-Mertk-/- mice were established by crossing between Mertk-/- and Ccl3-/- mice. Littermate Mertk-/- mice were used as control. A. Thickness of ONL from Ccl3-/-Mertk-/-, Mertk-/- and WT mice at 5-week and 8-week of age were measured by SD-OCT. Error bars indicate S.D. of the means (n > 6). * indicates P < 0.05 vs Mertk-/- mice. B. Images of retinal histology from 5-week-old Ccl3-/-Mertk-/-, Mertk-/- and WT mice are shown. Bars indicate 50 μm. ONL, outer nuclear layer. C. Iba-1-positive cells in the subretinal space were counted using cryosections from Ccl3-/-Mertk-/-, Mertk-/- and WT mice at 5-week and 8-week of age. These cryosections were prepared every 200 μm distance from the edge to edge (8 slides/eye), and IHC was performed with anti-Iba-1 Ab. Error bars indicate S.D. of the means (n > 6) *, # indicates P < 0.05. D. Fluorescent angiography from 8-week-old Ccl3-/-Mertk-/-, Mertk-/- and WT mice are presented. Magnified images of solid line inset shown in the broken rectangle.
Ccl2 deficiency protects the retina from degeneration in mouse models
To further elucidate the role of CCL3 and CCL2 in the pathophysiology of retinal degeneration, Ccl2-/-Abca4-/-Rdh8-/- mice were generated by crossing Abca4-/-Rdh8-/- with Ccl2-/- mice. Ccl2-/-Mertk-/- mice were also generated. After light exposure at 10,000 lux for 30 min, Ccl2-/-Abca4-/-Rdh8-/- mice showed better preservation of the ONL (Fig. 9A, left) and fewer AF spots/Iba-1-positive cells in the subretinal space (Fig. 9A, right) compared to Abca4-/-Rdh8-/- mice. Less severe age-related retinal degeneration with fewer AF spots was also observed in Ccl2-/-Abca4-/-Rdh8-/- mice compared with Abca4-/-Rdh8-/- mice at 6 months of age (Fig. 9B). In addition, Ccl2-/-Mertk-/- mice revealed a more intact ONL when compared to Mertk-/- mice at 5 weeks and 8 weeks of age (Fig. 9C). Therefore, loss of Ccl2 resulted in milder retinal degeneration in all three models of retinal degeneration.
Figure 9. Ccl2 deficiency protects the retina from degeneration in Abca4-/-Rdh8-/- mice and Mertk-/- mice.
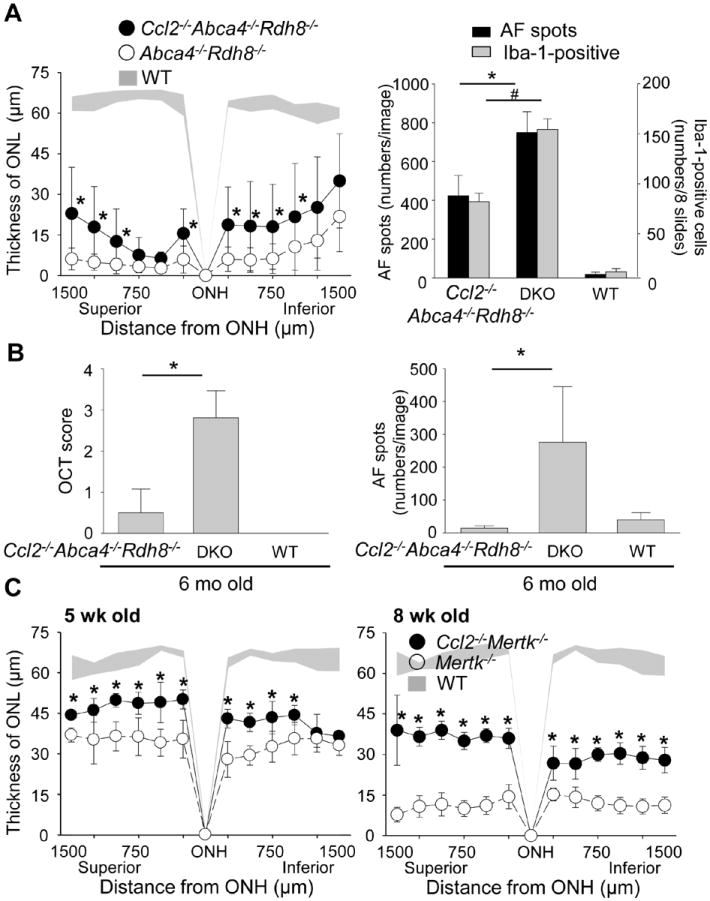
A. Ccl2-/-Abca4-/-Rdh8-/-, Abca4-/-Rdh8-/- (DKO) and WT mice at 4-6-week-old age were exposed to 10,000 lux light for 30 min. Thickness of ONL was measured by SD-OCT at 7 days after light exposure (left). Error bars indicate S.D. of the means (n > 6).* indicates P < 0.05 vs light exposed Abca4-/-Rdh8-/- mice. Numbers of AF spots of each image of SLO and Iba-1-postitive cells in the subretinal space were counted 7 days after light exposure (right). Cryosections were prepared every 200 μm distance from the edge to edge (8 slides/eye), and IHC was performed with anti-Iba-1 Ab. Error bars indicate S.D. of the means (n > 6).*, # indicates P < 0.05. B. Ccl2-/-Abca4-/-Rdh8-/-, Abca4-/-Rdh8-/- (DKO) and WT were kept under regular light conditions (12h ~10 lux/12 h dark), and retinal phenotype of these mice were characterized at the age of 6 month. Severity of retinal degeneration was evaluated by established scoring system (20) with in vivo OCT imaging (left). * indicates P < 0.05. Numbers of AF spots was counted by in vivo SLO imaging (right). * indicates P < 0.05. C. Thickness of ONL from Ccl2-/-Mertk-/-, Mertk-/- and WT mice at 5-week and 8-week old age were measured by SD-OCT. Error bars indicate S.D. of the means (n > 6). * indicates P < 0.05 vs Mertk-/- mice.
DISCUSSION
Our previous studies with the retinal degeneration model of Abca4-/-Rdh8-/- mice implicated a role for RPE-derived chemokines and cytokines in recruitment of tissue microglia from the inner retina to the subretinal space (2). Although the increased number of Iba-1 cells is likely due to infiltration, we cannot exclude the possibility that an increase in Iba-1+ cells is not due to cell proliferation. Retinal vascular endothelial cells are also a potential source of these chemokines in vivo, especially in recruitment of monocytes into the inner retina. In the current study, transcript level analysis of chemokines in the retina of Abca4-/-Rdh8-/- mice revealed selective elevation of Ccl3 and Ccl4 24 h after light exposure among tested chemokines. WT mice did not develop light-induced retinal degeneration and increased production of Ccl3 and Ccl4 was not documented. Further, although most chemokines have overlapping targets in terms of receptor binding, by generating Ccl3-/-Abca4-/-Rdh8-/- mice, we demonstrated a non-redundant role for CCL3 in retinal degeneration.
CCL3 is likely produced by subretinally translocated tissue microglia from the inner retina where they normally reside, and this can be a trigger for additional monocyte infiltration from the circulation via inner retinal blood vessels. Prolonged activation of resident microglia is also observed in experimental Herpes Encephalitis, experimental autoimmune encephalomyelitis and myocardial infarction (30-32).
The ingestion of POS by RPE cells is essential for the maintenance of retinal health (33). However, RPE cells can also contribute to retinal inflammation when POS phagocytosis is disrupted or abnormal (1). Our previous study implicated RPE cells as a source of chemokines that contribute to migration of tissue microglia from the inner retina to the subretinal space (2). These microglial cells also ingest photoreceptor debris, and thereby display increased autofluoresence signals and increased production of pro-inflammatory and chemotactic cytokines. This results in further infiltration of monocytes from the circulation and promotes their migration from capillaries in the inner retina to the subretinal space. Previous studies using GFP-chimeras of myeloid cells had increased accumulation of monocytes in the subretinal space after light induced retinal damage (34). In addition to RPE cells and microglia, other cells produce Ccl3 including astrocytes and Müller cells {Jensen, 2013 #173;Shamsuddin, 2011 #174}. Of particular consideration is that light exposed Abca4-/-Rdh8-/- mice showed reactive gliosis in these cells (2). Their interaction with the vascular endothelium may also be associated with chemokine production as observed in neural inflammation in the CNS (35, 36).
When Ccl3-/-Abca4-/-Rdh8-/- and littermate Abca4-/-Rdh8-/- mice were exposed to 10,000 lux of light for 30 min, light exposed Ccl3-/-Abca4-/-Rdh8-/- mice showed delayed clearance of infiltrated microglia from the subretinal space compared to Abca4-/-Rdh8-/- mice. Although light exposed Ccl3-/-Abca4-/-Rdh8-/- mice did not secrete any Ccl3, a substantial increase of Ccl4 was observed in these mice compared to Abca4-/-Rdh8-/- mice after light exposure. Furthermore, Ccl3-/-Abca4-/-Rdh8-/- mice also displayed an increase of Ccl2 and Il1b, which are hallmarks of retinal inflammation after light (1). These observations indicate persistent retinal inflammation, which contributes to the severity of retinal degeneration in light exposed Ccl3-/-Abca4-/-Rdh8-/- mice.
Results of the current study also show unchanged expression of Arg1, a marker of M2 macrophages in Ccl3-/-Abca4-/-Rdh8-/- mice before and after light exposure, whereas changes in expression of the Nox2 gene, which is a prototypic M1 marker (24) were observed in Ccl3-/-Abca4-/-Rdh8-/- and Abca4-/-Rdh8-/- mice. CCR2-associated M1 monocytes contribute to the digestion of damaged tissue, whereas Cx3Cr1-associated M2 monocytes promote healing in cases of myocardial infarction (30-32). Accumulating evidence in other chronic diseases implies that the lack of Arg1 elevation in light exposed Ccl3-/-Abca4-/-Rdh8-/- mice balances the local environment to inflammatory state and thus contributes to persistent retinal inflammation. We additionally showed data that supports a non-redundant role for CCL3 in a second model of retinal degeneration the Mertk-/- mouse. Ccl3-/-Mertk-/- mice displayed a less severe and less frequent impairment of the blood-retinal-barrier compared to Mertk-/- mice.
Macrophage/microglia has M1 and M2 sub-populations, which may regulate severity of AMD pathology (37, 38). M1 and M2 cells are also reported to play important roles in other inflammation models for many degenerative diseases (39). Persistent inflammation after traumatic brain injury is known to promote progression to Alzheimer disease (40). The pro-inflammatory M1 type microglia-based inflammatory mechanism has been shown to be involved in progression of post-trauma brain injury to Alzheimer disease for over decades.
Studies of the Alzheimer’s disease model also implicated a role for CCL2-CCR2 interaction in activation of tissue microglial cells (39), and in the pathogenesis of age-related retinal degeneration in Ccl2-/- and Ccr2-/- mice (6, 41, 42). Conversely, CCL2 and CCR2 were found to have a harmful role in chronic oxidative stress induced or inherited retinal degeneration as Cc2 and Ccr2 gene knockout mice had less severe retinal degeneration (43, 44). In the current study, increased expression of Ccl2 in light-exposed Abca4-/-Rdh8-/- mice was observed, in addition to decreased retinal degeneration in Ccl2-/-Abca4-/-Rdh8-/- mice and in the Ccl2-/-Mertk-/- mouse were demonstrated. These data indicate that in our models, CCL2-CCR2 activation accelerates inflammation, possibly by recruiting M1 rather than M2 cells, and that preceded CCL3 production could affect CCL2-CCR2 interaction in degenerative conditions. Differential chemokine networks modulate the severity of disease phenotype and improved understanding in each disease and disease state could largely contribute to future care of retinal diseases.
CCL3 and its receptors, CCR1 and CCR5, are therapeutic targets for treatment of human immunodeficiency virus infection, multiple sclerosis, rheumatoid arthritis, diabetes, endometriosis, organ transplant rejection and multiple myeloma (45). Given the current findings, CCL3, CCR1 and CCR5 are also potential targets for therapeutic intervention in retinal degeneration; however, further studies are required to determine the role of this chemokine at each disease stage. Taking these results into consideration, a direct inhibition of microglial cells using drugs for anti-microglial activation, such as minocycline (46), might be beneficial to treat inflammation in degenerative retinal diseases.
In conclusion, this study revealed that production of chemokines was closely associated with degenerative retinal changes and microglia/macrophage translocation into the subretinal space. Regulatory mechanism of chemokine networks were differed in models of retinal degeneration. CCL3 showed a distinct role in pathogenesis of retinal degeneration under acute and chronic conditions in mouse models. Although a preceding increase in CCL3 from retinal microglial cells suggests the role of CCL3 as a potential master regulator of retinal inflammation, paradoxical effects of this chemokine in relationship to retinal degeneration could also explain the complex of pathology observed in retinal degeneration and other neurodegenerative diseases.
Supplementary Material
Acknowledgments
We thank Drs. K. Palczewski, D. Mustafi, B. Kevany, Z. Dong, S. Howell (Case Western Reserve Univ.), Y. Hirohashi (Sapporo Medical Univ.) and T. Sakai (Jikei University School of Medicine) for their comments and technical supports.
This work was supported by funding from the NIH (EY022658, EY019031, EY019880, EY021126, and EY11373), Research to Prevent Blindness Foundation, Foundation Fighting Blindness, and Ohio Lions Eye Research Foundation.
ABBREVIATIONS
- ABCA4
ATP-binding cassette transporter 4
- AF
autofluorescence
- ARG1
Arginase liver
- GFAP
Glial Fibrillary Acidic Protein
- IHC
immunohistochemistry
- MERTK
c-mer proto-oncogene tyrosine kinase
- MIP-1
Macrophage Inflammatory Proteins 1
- ONL
outer nuclear layer
- RDH8
retinol dehydrogenase 8
- RPE
retinal pigment epithelium
- SD-OCT
spectral-domain optic coherence tomography
- SLO
scanning laser ophthalmoscopy
References
- 1.Xu H, Chen M, Forrester JV. Para-inflammation in the aging retina. Pro Retin Eye Res. 2009;28:348–368. doi: 10.1016/j.preteyeres.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 2.Kohno H, Chen Y, Kevany BM, Pearlman E, Miyagi M, Maeda T, Palczewski K, Maeda A. Photoreceptor proteins initiate microglial activation via Toll-like receptor 4 in retinal degeneration mediated by all-trans-retinal. J Biol Chem. 2013;288:15325–15341. doi: 10.1074/jbc.M112.448712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mustafi D, Maeda T, Kohno H, Nadeau JH, Palczewski K. Inflammatory priming predisposes mice to age-related retinal degeneration. J Clin Invest. 2012;122:2989–3001. doi: 10.1172/JCI64427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shiose S, Chen Y, Okano K, Roy S, Kohno H, Tang J, Pearlman E, Maeda T, Palczewski K, Maeda A. Toll-like receptor 3 is required for development of retinopathy caused by impaired all-trans-retinal clearance in mice. J Biol Chem. 2011;286:15543–15555. doi: 10.1074/jbc.M111.228551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Combadiere C, Feumi C, Raoul W, Keller N, Rodero M, Pezard A, Lavalette S, Houssier M, Jonet L, Picard E, Debre P, Sirinyan M, Deterre P, Ferroukhi T, Cohen SY, Chauvaud D, Jeanny JC, Chemtob S, Behar-Cohen F, Sennlaub F. CX3CR1-dependent subretinal microglia cell accumulation is associated with cardinal features of age-related macular degeneration. J Clin Invest. 2007;117:2920–2928. doi: 10.1172/JCI31692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ambati J, Anand A, Fernandez S, Sakurai E, Lynn BC, Kuziel WA, Rollins BJ, Ambati BK. An animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nat Med. 2003;9:1390–1397. doi: 10.1038/nm950. [DOI] [PubMed] [Google Scholar]
- 7.von Lintig J, Kiser PD, Golczak M, Palczewski K. The biochemical and structural basis for trans-to-cis isomerization of retinoids in the chemistry of vision. Trends Biochem Sci. 2010;35:400–410. doi: 10.1016/j.tibs.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kiser PD, Golczak M, Maeda A, Palczewski K. Key enzymes of the retinoid (visual) cycle in vertebrate retina. Biochim Biophys Acta. 2012;1821:137–151. doi: 10.1016/j.bbalip.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Allikmets R, Shroyer NF, Singh N, Seddon JM, Lewis RA, Bernstein PS, Peiffer A, Zabriskie NA, Li Y, Hutchinson A, Dean M, Lupski JR, Leppert M. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science. 1997;277:1805–1807. doi: 10.1126/science.277.5333.1805. [DOI] [PubMed] [Google Scholar]
- 10.Allikmets R, Singh N, Sun H, Shroyer NF, Hutchinson A, Chidambaram A, Gerrard B, Baird L, Stauffer D, Peiffer A, Rattner A, Smallwood P, Li Y, Anderson KL, Lewis RA, Nathans J, Leppert M, Dean M, Lupski JR. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet. 1997;15:236–246. doi: 10.1038/ng0397-236. [DOI] [PubMed] [Google Scholar]
- 11.Janecke AR, Thompson DA, Utermann G, Becker C, Hubner CA, Schmid E, McHenry CL, Nair AR, Ruschendorf F, Heckenlively J, Wissinger B, Nurnberg P, Gal A. Mutations in RDH12 encoding a photoreceptor cell retinol dehydrogenase cause childhood-onset severe retinal dystrophy. Nat Genet. 2004;36:850–854. doi: 10.1038/ng1394. [DOI] [PubMed] [Google Scholar]
- 12.Quazi F, Lenevich S, Molday RS. ABCA4 is an N-retinylidene-phosphatidylethanolamine and phosphatidylethanolamine importer. Nat Commun. 2012;3:925. doi: 10.1038/ncomms1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rattner A, Smallwood PM, Nathans J. Identification and characterization of all-trans-retinol dehydrogenase from photoreceptor outer segments, the visual cycle enzyme that reduces all-trans-retinal to all-trans-retinol. J Biol Chem. 2000;275:11034–11043. doi: 10.1074/jbc.275.15.11034. [DOI] [PubMed] [Google Scholar]
- 14.Maeda A, Maeda T, Imanishi Y, Kuksa V, Alekseev A, Bronson JD, Zhang H, Zhu L, Sun W, Saperstein DA, Rieke F, Baehr W, Palczewski K. Role of photoreceptor-specific retinol dehydrogenase in the retinoid cycle in vivo. J Biol Chem. 2005;280:18822–18832. doi: 10.1074/jbc.M501757200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maeda A, Maeda T, Golczak M, Chou S, Desai A, Hoppel CL, Matsuyama S, Palczewski K. Involvement of all-trans-retinal in acute light-induced retinopathy of mice. J Biol Chem. 2009;284:15173–15183. doi: 10.1074/jbc.M900322200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maeda A, Maeda T, Golczak M, Palczewski K. Retinopathy in mice induced by disrupted all-trans-retinal clearance. J Biol Chem. 2008;283:26684–26693. doi: 10.1074/jbc.M804505200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 18.Mattapallil MJ, Wawrousek EF, Chan CC, Zhao H, Roychoudhury J, Ferguson TA, Caspi RR. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci. 2012;53:2921–2927. doi: 10.1167/iovs.12-9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu YP, Proia RL. Deletion of macrophage-inflammatory protein 1 alpha retards neurodegeneration in Sandhoff disease mice. Proc Natl Acad Sci U S A. 2004;101:8425–8430. doi: 10.1073/pnas.0400625101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maeda A, Golczak M, Chen Y, Okano K, Kohno H, Shiose S, Ishikawa K, Harte W, Palczewska G, Maeda T, Palczewski K. Primary amines protect against retinal degeneration in mouse models of retinopathies. Nat Chem Biol. 2012;8:170–178. doi: 10.1038/nchembio.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mainster MA, Timberlake GT, Webb RH, Hughes GW. Scanning laser ophthalmoscopy. Clinical applications. Ophthalmology. 1982;89:852–857. doi: 10.1016/s0161-6420(82)34714-4. [DOI] [PubMed] [Google Scholar]
- 22.Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Novak ML, Koh TJ. Macrophage phenotypes during tissue repair. J Leukoc Biol. 2013;93:875–881. doi: 10.1189/jlb.1012512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liao B, Zhao W, Beers DR, Henkel JS, Appel SH. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp Neurol. 2012;237:147–152. doi: 10.1016/j.expneurol.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niederkorn JY. See no evil, hear no evil, do no evil: the lessons of immune privilege. Nat Immunol. 2006;7:354–359. doi: 10.1038/ni1328. [DOI] [PubMed] [Google Scholar]
- 26.Cense B, Nassif N, Chen T, Pierce M, Yun SH, Park B, Bouma B, Tearney G, de Boer J. Ultrahigh-resolution high-speed retinal imaging using spectral-domain optical coherence tomography. Opt Express. 2004;12:2435–2447. doi: 10.1364/opex.12.002435. [DOI] [PubMed] [Google Scholar]
- 27.Charbel Issa P, Bolz HJ, Ebermann I, Domeier E, Holz FG, Scholl HP. Characterisation of severe rod-cone dystrophy in a consanguineous family with a splice site mutation in the MERTK gene. Br J Ophthalmol. 2009;93:920–925. doi: 10.1136/bjo.2008.147397. [DOI] [PubMed] [Google Scholar]
- 28.Seitz HM, Camenisch TD, Lemke G, Earp HS, Matsushima GK. Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J Immunol. 2007;178:5635–5642. doi: 10.4049/jimmunol.178.9.5635. [DOI] [PubMed] [Google Scholar]
- 29.Nandrot EF, Dufour EM. Mertk in daily retinal phagocytosis: a history in the making. Adv Exp Med Biol. 2010;664:133–140. doi: 10.1007/978-1-4419-1399-9_16. [DOI] [PubMed] [Google Scholar]
- 30.Getts DR, Terry RL, Getts MT, Muller M, Rana S, Shrestha B, Radford J, Van Rooijen N, Campbell IL, King NJ. Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. J Exp Med. 2008;205:2319–2337. doi: 10.1084/jem.20080421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marques CP, Cheeran MC, Palmquist JM, Hu S, Urban SL, Lokensgard JR. Prolonged microglial cell activation and lymphocyte infiltration following experimental herpes encephalitis. J Immunol. 2008;181:6417–6426. doi: 10.4049/jimmunol.181.9.6417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204:3037–3047. doi: 10.1084/jem.20070885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kevany BM, Palczewski K. Phagocytosis of retinal rod and cone photoreceptors. Physiology (Bethesda) 2010;25:8–15. doi: 10.1152/physiol.00038.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joly S, Francke M, Ulbricht E, Beck S, Seeliger M, Hirrlinger P, Hirrlinger J, Lang KS, Zinkernagel M, Odermatt B, Samardzija M, Reichenbach A, Grimm C, Reme CE. Cooperative phagocytes: resident microglia and bone marrow immigrants remove dead photoreceptors in retinal lesions. Am J Pathol. 2009;174:2310–2323. doi: 10.2353/ajpath.2009.090023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Engelhardt B. Immune cell entry into the central nervous system: involvement of adhesion molecules and chemokines. J Neurol Sci. 2008;274:23–26. doi: 10.1016/j.jns.2008.05.019. [DOI] [PubMed] [Google Scholar]
- 36.Minagar A, Carpenter A, Alexander JS. The destructive alliance: interactions of leukocytes, cerebral endothelial cells, and the immune cascade in pathogenesis of multiple sclerosis. Int Rev Neurobiol. 2007;79:1–11. doi: 10.1016/S0074-7742(07)79001-3. [DOI] [PubMed] [Google Scholar]
- 37.Cao X, Shen D, Patel MM, Tuo J, Johnson TM, Olsen TW, Chan CC. Macrophage polarization in the maculae of age-related macular degeneration: a pilot study. Pathol Int. 2011;61:528–535. doi: 10.1111/j.1440-1827.2011.02695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nussenblatt RB, Ferris F., 3rd Age-related macular degeneration and the immune response: implications for therapy. Am J Ophthalmol. 2007;144:618–626. doi: 10.1016/j.ajo.2007.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.El Khoury J, Luster AD. Mechanisms of microglia accumulation in Alzheimer’s disease: therapeutic implications. Trends Pharmacol Sci. 2008;29:626–632. doi: 10.1016/j.tips.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 40.Giunta B, Obregon D, Velisetty R, Sanberg PR, Borlongan CV, Tan J. The immunology of traumatic brain injury: a prime target for Alzheimer’s disease prevention. J Neuroinflammation. 2012;9:185. doi: 10.1186/1742-2094-9-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen M, Forrester JV, Xu H. Dysregulation in retinal para-inflammation and age-related retinal degeneration in CCL2 or CCR2 deficient mice. PLoS One. 2011;6:e22818. doi: 10.1371/journal.pone.0022818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chan CC, Ross RJ, Shen D, Ding X, Majumdar Z, Bojanowski CM, Zhou M, Salem N, Jr, Bonner R, Tuo J. Ccl2/Cx3cr1-deficient mice: an animal model for age-related macular degeneration. Ophthalmic Res. 2008;40:124–128. doi: 10.1159/000119862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suzuki M, Tsujikawa M, Itabe H, Du ZJ, Xie P, Matsumura N, Fu X, Zhang R, Sonoda KH, Egashira K, Hazen SL, Kamei M. Chronic photo-oxidative stress and subsequent MCP-1 activation as causative factors for age-related macular degeneration. J Cell Sci. 2012;125:2407–2415. doi: 10.1242/jcs.097683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo C, Otani A, Oishi A, Kojima H, Makiyama Y, Nakagawa S, Yoshimura N. Knockout of ccr2 alleviates photoreceptor cell death in a model of retinitis pigmentosa. Exp Eye Res. 2012;104:39–47. doi: 10.1016/j.exer.2012.08.013. [DOI] [PubMed] [Google Scholar]
- 45.Ribeiro S, Horuk R. The clinical potential of chemokine receptor antagonists. Pharmacol Ther. 2005;107:44–58. doi: 10.1016/j.pharmthera.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 46.Cukras CA, Petrou P, Chew EY, Meyerle CB, Wong WT. Oral minocycline for the treatment of diabetic macular edema (DME): results of a phase I/II clinical study. Invest Ophthalmol Vis Sci. 2012;53:3865–3874. doi: 10.1167/iovs.11-9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.