

Abstract
Most existing genome-wide association analyses are cross-sectional, utilizing only phenotypic data at a single time point, e.g. baseline. On the other hand, longitudinal studies, such as Alzheimer's Disease Neuroimaging Initiative (ADNI), collect phenotypic information at multiple time points. In this article, as a case study, we conducted both longitudinal and cross-sectional analyses of the ADNI data with several brain imaging (not clinical diagnosis) phenotypes, demonstrating the power gains of longitudinal analysis over cross-sectional analysis. Specifically, we scanned genome-wide single nucleotide polymorphisms (SNPs) with 56 brain-wide imaging phenotypes processed by FreeSurfer on 638 subjects. At the genome-wide significance level (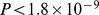 ) or a less stringent level (e.g.
) or a less stringent level (e.g. 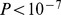 ), longitudinal analysis of the phenotypic data from the baseline to month 48 identified more SNP-phenotype associations than cross-sectional analysis of only the baseline data. In particular, at the genome-wide significance level, both SNP rs429358 in gene APOE and SNP rs2075650 in gene TOMM40 were confirmed to be associated with various imaging phenotypes in multiple regions of interests (ROIs) by both analyses, though longitudinal analysis detected more regional phenotypes associated with the two SNPs and indicated another significant SNP rs439401 in gene APOE. In light of the power advantage of longitudinal analysis, we advocate its use in current and future longitudinal neuroimaging studies.
), longitudinal analysis of the phenotypic data from the baseline to month 48 identified more SNP-phenotype associations than cross-sectional analysis of only the baseline data. In particular, at the genome-wide significance level, both SNP rs429358 in gene APOE and SNP rs2075650 in gene TOMM40 were confirmed to be associated with various imaging phenotypes in multiple regions of interests (ROIs) by both analyses, though longitudinal analysis detected more regional phenotypes associated with the two SNPs and indicated another significant SNP rs439401 in gene APOE. In light of the power advantage of longitudinal analysis, we advocate its use in current and future longitudinal neuroimaging studies.
Introduction
There has been increasing interest in genome-wide association studies (GWASs) with neuroimaging phenotypes. Alzheimer's Disease Neuroimaging Initiative (ADNI) provides a rich source of brain imaging, neuropsychological and genetic data, including genome-wide single nucleotide polymorphisms (SNPs) [1], [2]. In ADNI (or more specifically ADNI-1), while the subjects were followed up to 5 years, most of the previous GWAS analyses of brain-wide imaging phenotypes ignored the longitudinal data and mainly focused on only the baseline phenotypes [3]–[9]. In genome-wide association studies longitudinal analysis has been proposed and applied [10]–[14], and in particular its advantage over cross-sectional analysis has been established [15]. Hence, instead of using only the baseline structural MRI scans as phenotypes, we took advantage of the longitudinal imaging phenotypes measured at multiple time points from the baseline to 48 months, demonstrating the application of a linear mixed-effects model and its associated power gains. The advantage of longitudinal analysis is not surprising: assuming no SNP-age interactions, a cross-sectional study based on the baseline can only capture the mean differences of a phenotype across the (genetic) subgroups of subjects; in contrast, a longitudinal study offers the opportunity to estimate not only the mean values of the phenotype at the baseline, but also the rates of the changes of the phenotype in the genotypic groups. For example, as shown in Figure 1, the trajectories of the hippocampal volume appear to decline much faster for the subjects with the homozygotic minor alleles of SNP rs2075650 in gene TOMM40 than those from other two genotype groups. However, we also notice the variations in the rates (i.e. slopes) of the changes across the subjects, which call for a suitable statistical model to account for this source of variations. As to be shown, some alternative but popular and simpler models would fail for the longitudinal data here.
Figure 1. Trajectories of phenotype left hippocampus volume over time (in months) in three allele groups of SNP rs2075650.

Materials and Methods
Data
Data used in the preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). The ADNI was launched in 2003 by the National Institute on Aging (NIA), the National Institute of Biomedical Imaging and Bioengineering (NIBIB), the Food and Drug Administration (FDA), private pharmaceutical companies and non-profit organizations, as a $60 million, 5-year public-private partnership. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early Alzheimer's dementia (AD). Determination of sensitive and specific markers of very early AD progression is intended to aid researchers and clinicians to develop new treatments and monitor their effectiveness, as well as lessen the time and cost of clinical trials.
The Principal Investigator of this initiative is Michael W. Weiner, MD, VA Medical Center and University of California-San Francisco. ADNI is the result of efforts of many co-investigators from a broad range of academic institutions and private corporations, and subjects have been recruited from over 50 sites across the U.S. and Canada. The initial goal of ADNI was to recruit 800 subjects but ADNI has been followed by ADNI-GO and ADNI-2. To date these three protocols have recruited over 1500 adults, ages 55 to 90, to participate in the research, consisting of cognitively normal older individuals, people with early or late MCI, and people with early AD. The follow up duration of each group is specified in the protocols for ADNI-1, ADNI-2 and ADNI-GO. Subjects originally recruited for ADNI-1 and ADNI-GO had the option to be followed in ADNI-2. For up-to-date information, see www.adni-info.org.
Specifically, we started with the following data on 818 subjects in ADNI-1 [2]: FreeSurfer-processed brain imaging phenotypes, Illumina SNP genotypes and demographic information (including handedness, years of education, gender and age). For data quality control, following [4]. we adopted the following procedure: (1) including only non-Hispanic Caucasians; (2) checking each subject's identity and gender; (3) excluding the subjects with heavy missing values. At the end, we had 638 subjects remaining in the study.
The phenotypic data were processed with the FreeSurfer image analysis suite [16] by UCSF researchers [17]. Briefly, FreeSurfer version 4.3 was applied to T1 weighted structural MRI in the NiFTI format after being pre-processed by the Mayo Clinic [1]. Both longitudinal and cross-sectional registrations were used for the corresponding longitudinal and cross-sectional analyses. For cross-sectional processing, each scan was segmented according to an atlas defined by FreeSurfer, allowing for comparison between groups at a single time point. For longitudinal processing, for each subject with images at more than one time point, a within-subject template based on his/her average image was created using robust inverse consistent registration [18]. Then each subject's template was used to initialize the longitudinal image processing to increase the reliability and statistical power when measuring the brain changes over time [18].
For cross-sectional analysis, we used the phenotypic data only at the baseline, while for longitudinal analysis we used all the data for each subject, up to the measurements at the other five time points (months 6, 12, 24, 36 and 48) beyond the baseline. Since many subjects were not measured at some time points, we used only available data without imputation for either phenotypic or genotypic data.
Statistical models
For cross-sectional analysis of the baseline data, we use a (standard) linear regression model:
| (1) |
for subject  ,
, 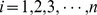 .
. 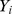 is the (regional imaging) phenotype of subject
is the (regional imaging) phenotype of subject  at the baseline;
at the baseline; 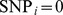 , 1 or 2 is the count of the minor allele for the SNP to be tested;
, 1 or 2 is the count of the minor allele for the SNP to be tested; 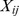 is one of the five covariates: left or right handedness, education in years, age at the baseline, gender and the baseline intracranial volume (ICV); and
is one of the five covariates: left or right handedness, education in years, age at the baseline, gender and the baseline intracranial volume (ICV); and 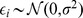 is an independent error term. The goal is to test the null hypothesis
is an independent error term. The goal is to test the null hypothesis 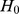 :
: 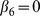 versus
versus 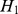 :
: 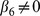 . We conduct single SNP-based analysis on each phenotype: each of the SNPs is tested one by one on each regional imaging phenotype sequentially.
. We conduct single SNP-based analysis on each phenotype: each of the SNPs is tested one by one on each regional imaging phenotype sequentially.
For longitudinal analysis, we use a linear mixed-effects model with a random slope and a random intercept [19]:
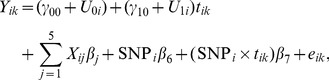 |
(2) |
for 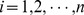 and
and 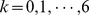 , where
, where 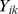 is the (regional imaging) phenotype value of subject
is the (regional imaging) phenotype value of subject  at time point
at time point 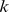 ,
, 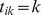 is the time point
is the time point 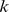 ,
, 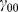 and
and 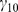 are the fixed intercept and fixed slope for time, and
are the fixed intercept and fixed slope for time, and 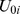 and
and 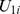 are random intercept and slope respectively:
are random intercept and slope respectively:
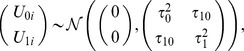 |
and 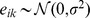 is an independent error term. Other terms are the same as in the standard linear regression model. The two random terms induce some complex and time-varying within-subject correlations and variances: for
is an independent error term. Other terms are the same as in the standard linear regression model. The two random terms induce some complex and time-varying within-subject correlations and variances: for 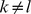 , we have
, we have
each of which is a function of time unless 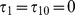 .
.
The goal in the longitudinal analysis is to test for no joint main- and interaction-effects of the SNP with 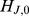 :
: 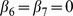 versus
versus 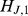 :
: 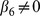 or
or 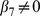 . This is the default test to be used in longitudinal analysis. Although the above test is preferred [19], sometimes we would like to see the separate contributions of the main effects of the SNP and the SNP-time interaction, thus we would conduct two separate tests. The first is to test for the zero main effects of the SNP with
. This is the default test to be used in longitudinal analysis. Although the above test is preferred [19], sometimes we would like to see the separate contributions of the main effects of the SNP and the SNP-time interaction, thus we would conduct two separate tests. The first is to test for the zero main effects of the SNP with 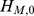 :
: 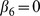 versus
versus 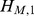 :
: 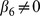 , and the second is for the zero SNP-time interaction effects with
, and the second is for the zero SNP-time interaction effects with 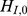 :
: 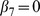 versus
versus 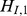 :
: 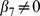 .
.
The purpose of introducing the random intercept and slope parameters is to take account of likely within-subject correlations among the multiple phenotypes for the same subject; in addition, the random slope parameter can account for heterogeneity of the slope parameters among the subjects. Figure 1 shows the trajectories of an imaging phenotype, volume of left hippocampus, for the three groups of the subjects with various genotypes of SNP rs2075650. It is clear that the longitudinal values of the imaging phenotype at the several time points for the same subject are more or less similar to each other as compared to the values from a different subject, suggesting the necessity of using the random subject-specific intercept term 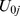 ; on the other hand, the rates of the change of the phenotype over time for different subjects may be different, implying the use of the random subject-specific slope parameter
; on the other hand, the rates of the change of the phenotype over time for different subjects may be different, implying the use of the random subject-specific slope parameter 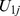 . We used the function lme ( ) in R package nlme to fit the linear mixed-effects model.
. We used the function lme ( ) in R package nlme to fit the linear mixed-effects model.
Alternative models for longitudinal analysis
A simpler linear mixed model contains only a random intercept term, which is perhaps more commonly used for longitudinal data:
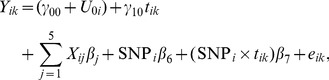 |
(3) |
for which the parameters are specified as in model (2). Compared to model (2), the random slope parameter 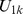 is missing in the new model (3). It is easy to verify that both models share the following mean function of the phenotype (conditional on the covariates):
is missing in the new model (3). It is easy to verify that both models share the following mean function of the phenotype (conditional on the covariates):
| (4) |
However, their variances are different: instead of having a time-varying within-subject covariance matrix for model (2), we have
| (5) |
for 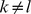 for model (3), suggesting a within-subject compound symmetry (CS) correlation matrix. Obviously model (3) is a special case of model (2).
for model (3), suggesting a within-subject compound symmetry (CS) correlation matrix. Obviously model (3) is a special case of model (2).
To compare model (3) against model (2), we can test the null hypothesis 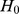 :
: 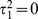 via a likelihood ratio test (LRT). The null distribution of the LRT statistic
via a likelihood ratio test (LRT). The null distribution of the LRT statistic 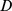 can be approximated by a mixture distribution,
can be approximated by a mixture distribution, 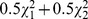 [20].
[20].
Alternatively, rather than using a linear mixed model, we can use generalized estimating equations (GEE) to draw inference for longitudinal data [21], [22]. GEE only requires to correctly specify a mean model and use a working within-subject correlation structure. Here we will use the mean model specified in (4), and use a working correlation matrix with a CS structure specified in (5). If a model-based covariance matrix is used, the validity of the GEE results depends on the correct specification of the working correlation structure; if a so-called sandwich or robust covariance matrix is used, then any working correlation can be used. We also note that, in the presence of missing phenotypes, the validity of GEE depends on the assumption of “missing completely at random” (MCAR), a stronger assumption than that of “missing at random” (MAR) required by linear mixed models; this may have implications in real data analysis in the presence of missing phenotypes, which is the case here. We used function geese ( ) from R package geepack to fit a GEE model.
Simulation set-ups
A longitudinal phenotype 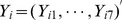 for each subject
for each subject  was simulated from the linear mixed-effects model with a random slope and a random intercept as specified in equation (2). Each simulated dataset consisted of
was simulated from the linear mixed-effects model with a random slope and a random intercept as specified in equation (2). Each simulated dataset consisted of 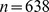 subjects. The phenotypes were generated either under
subjects. The phenotypes were generated either under 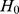 (i.e.
(i.e. 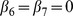 ) to investigate Type I errors or under
) to investigate Type I errors or under 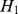 (i.e.
(i.e. 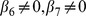 ) for power analysis; all parameters used were at at the maximum likelihood estimates of model (2) fitted to the original ADNI data. The same set of covariates (i.e. left or right handedness, education in years, age at the baseline, gender and ICV) were used.
) for power analysis; all parameters used were at at the maximum likelihood estimates of model (2) fitted to the original ADNI data. The same set of covariates (i.e. left or right handedness, education in years, age at the baseline, gender and ICV) were used.
To evaluate the robustness of model (2), we also simulated phenotypic data 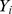 from model (3). The parameters used under
from model (3). The parameters used under 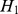 (i.e.
(i.e. 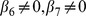 ) were half of the maximum likelihood estimates in model (3) fitted to the original ADNI data.
) were half of the maximum likelihood estimates in model (3) fitted to the original ADNI data.
Three most significant SNPs in Table 1, rs429358, rs2075650 and rs439401, were chosen to be used in simulations. Under each simulation set-up, 1000 sets of phenotypic data were independently generated and analyzed by five methods: a linear mixed-effects model with both a random slope and a random intercept (LMR-RSI); a linear mixed-effects model with only a random intercept term (LMR-RI); a marginal GEE model with a CS working correlation matrix and with the sandwich covariance estimator (GEE-Robust); a marginal GEE model with a CS working correlation matrix and with the model-based covariance estimator (GEE-Naive); a standard linear model for cross-sectional analysis at the baseline, testing for the main effects of an SNP (Baseline). We then estimated the empirical Type I error rate under 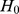 and empirical power under
and empirical power under 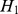 for each method.
for each method.
Table 1. Significant SNPs and each one's associated phenotype numbers at the significance level of 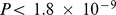 .
.
| Gene | # Phenotypes | |||
| SNP | (Chr) | Position | Longitudinal | Baseline |
| rs2075650 | TOMM40 | 45,395,619 | 3 | 1 |
| (19) | LHippVol: 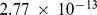
|
LHippVol: 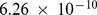
|
||
RCerebCtx: 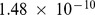
|
||||
LMeanTemp: 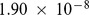
|
||||
| rs439401 | APOE | 45,414,451 | 1 | 0 |
| (19) | LMeanLatTemp: 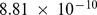
|
|||
| rs429358 | APOE | 45,411,941 | 42 | 4 |
| (19) | LHippVol: 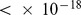
|
LHippVol: 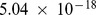
|
||
LEntCtx: 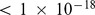
|
RHippVol: 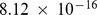
|
|||
LAmygVol: 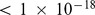
|
LAmygVol: 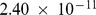
|
|||
| - | - | total | 46 | 5 |
Top 3 SNP-phenotype associations are listed with corresponding P-values.
Results
Data summary
After quality control, there were 638 subjects remaining in both cross-sectional and longitudinal data, though there were missing data for some phenotypes and in later follow-ups; see Table 2 for an example. Table 3 summarizes demographic information and intracranial volume (ICV) of the 638 subjects at the baseline. The P-values were calculated based on an F-test for a continuous variable or a Chi-squared test for a categorical variable for its mean or distributional differences among three diagnostic groups at the baseline: healthy normal subjects (HC), subjects with mild cognitive impairment (MCI) and patients with Alzheimer's dementia (AD). The table shows that the distributions of gender and mean years of education were significantly different among the three groups at the significance level 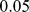 .
.
Table 2. The number (percentage) of non-missing observations at each time point in Figure 1.
| Month | 0 | 6 | 12 | 18 | 24 | 36 | 48 |
| #Obs | 635 (99.5%) | 616 (96.6%) | 574 (90%) | 246 (38.6%) | 462 (72.4%) | 263 (41.2%) | 56 (8.8%) |
Table 3. The baseline characteristics of 638 subjects, including gender, age, years of education, handedness (R/L) and intracranial volume (ICV).
| Name | HC | MCI | AD | P-value |
| number of subjects | 182 | 311 | 145 | - |
| Gender(M/F) | 103/79 | 204/107 | 80/65 | 0.0446 |
| Baseline age |
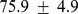
|
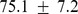
|
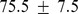
|
0.4153 |
| Education (years) |
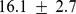
|
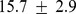
|
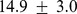
|
0.0005 |
| Hand(R/L) |
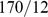
|
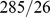
|
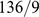
|
0.6392 |
| ICV |
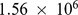
|
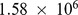
|
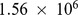
|
0.1463 |
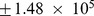
|
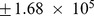
|
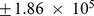
|
P-values were calculated to test for differences among the diagnostic groups, HC, MCI and AD.
We conducted cross-sectional and longitudinal analyses of each of about 500,000 SNPs with each of 56 neuroimaging phenotypes in some regions of interest (ROIs) as shown in Table 4 [4].
Table 4. 56 cortical thickness and volumetric phenotypes.
| Trait Name | Trait Description | Trait Name | Trait Description |
| AmygVol | Volume of amygdala | MidTemporal | Thickness of middle temporal gyrus |
| CerebCtx | Volume of cerebral cortex | Parahipp | Thickness of parahippocampal gyrus |
| CerebWM | Volume of cerebral white matter | PostCing | Thickness of posterior cingulate |
| HippVol | Volume of hippocampus | Postcentral | Thickness of postcentral gyrus |
| InfLatVent | Volume of inferior lateral ventricle | Precentral | Thickness of precentral gyrus |
| LatVent | Volume of lateral ventricle | Precuneus | Thickness of precuneus |
| EntCtx | Thickness of entorhinal cortex | SupFrontal | Thickness of superior frontal gyrus |
| Fusiform | Thickness of fusiform gyrus | SupParietal | Thickness of superior parietal gyrus |
| InfParietal | Thickness of inferior parietal gyrus | SupTemporal | Thickness of superior temporal gyrus |
| InfTemporal | Thickness of inferior temporal gyrus | Supramarg | Thickness of supramarginal gyrus |
| MeanCing | Mean thickness of caudal anterior | TemporalPole | Thickness of temporal pole |
| cingulate, isthmus cingulate, posterior | |||
| cingulate, and rostral anterior cingulate | |||
| MeanFront | Mean thickness of caudal midfrontal | MeanTemp | Mean thickness of inferior temporal, |
| rostral midfrontal, superior frontal, | middle temporal, superior temporal, | ||
| lateral orbitofrontal, and medial | fusiform, parahippocampal, ling- | ||
| orbitofrontal gyri and frontal pole | ual gyri temporal pole and transverse | ||
| temporal pole | |||
| MeanLatTemp | Mean thickness of inferior temporal, | MeanSensMotor | Mean thickness of precentral and |
| middle temporal, and superior | postcentral gyri | ||
| temporal gyri | |||
| MeanMedTemp | Mean thickness of fusiform, | MeanPar | Mean thickness of inferior and |
| parahippocampal, and lingual gyri, | superior parietal gyri, supramarginal | ||
| temporal pole and transverse | gyrus, and precuneus | ||
| temporal pole |
There are 2 phenotypes for each given phenotype name at the left and right sides of the brain respectively.
Figure 1 shows the longitudinal values of one phenotype, left hippocampus volume, for subjects in each of the three genotype groups for a (significant) SNP. We note the variation of the slope of the subject-specific phenotype trajectory, which calls for the use of a random slope parameter as specified in linear mixed model (2). We also note many missing values at some time points (Table 2); our analyses were all based on observed phenotypic (and genotypic) data without imputation.
Genome-wide association testing: a summary
Since a total number of 56 phenotypes were to be tested, taking the usual genome-wide significance level for a single phenotype at 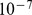 (or
(or 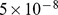 ), we used the Bonferroni method for multiple testing correction, yielding the significance cut-off at
), we used the Bonferroni method for multiple testing correction, yielding the significance cut-off at 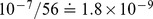 (or
(or 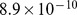 ).
).
Under the significance level 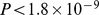 , a total number of 46 pairs of significant SNP-phenotype associations were detected in longitudinal analysis, including three SNPs, rs429358, rs2075650 and rs439401 that were associated with 3, 1 and 42 out of the total 56 phenotypes respectively (Table 1). In contrast, only 5 significant SNP-phenotype association pairs were detected in cross-sectional analysis at the baseline, involving only two SNPs, rs2075650 and rs429358, which were associated with only 1 and 4 phenotypes respectively. We reached the same conclusion with a more stringent significance level
, a total number of 46 pairs of significant SNP-phenotype associations were detected in longitudinal analysis, including three SNPs, rs429358, rs2075650 and rs439401 that were associated with 3, 1 and 42 out of the total 56 phenotypes respectively (Table 1). In contrast, only 5 significant SNP-phenotype association pairs were detected in cross-sectional analysis at the baseline, involving only two SNPs, rs2075650 and rs429358, which were associated with only 1 and 4 phenotypes respectively. We reached the same conclusion with a more stringent significance level 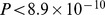 , as shown in Table 5.
, as shown in Table 5.
Table 5. Significant SNPs and each one's associated phenotype numbers at the level of 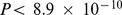 .
.
| Gene | # Phenotypes | |||
| SNP | (Chr) | Position | Longitudinal | Baseline |
| rs2075650 | TOMM40 | 45,395,619 | 2 | 1 |
| (19) | LHippVol: 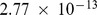
|
LHippVol: 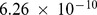
|
||
RCerebCtx: 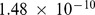
|
||||
| rs439401 | APOE | 45,414,451 | 1 | 0 |
| (19) | LMeanLatTemp: 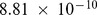
|
|||
| rs429358 | APOE | 45,411,941 | 40 | 4 |
| (19) | LHippVol: 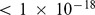
|
LHippVol: 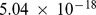
|
||
LEntCtx: 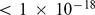
|
RHippVol: 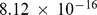
|
|||
LAmygVol: 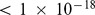
|
LAmygVol: 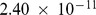
|
|||
| - | - | total | 43 | 5 |
Top 3 SNP-phenotype associations are listed with corresponding P-values.
If a less stringent significance level was employed, an even more substantial difference between the results of longitudinal analysis and that of cross-sectional analysis emerged. As shown in Table 6, at 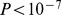 , seven SNPs, rs2075650, rs4902433, rs439401, rs11762610, rs1800627, rs11677350 and rs429358, were discovered to be associated with some phenotypes in longitudinal analysis; in contrast, only five SNPs, rs2075650, rs11677350, rs429358, rs2931352 and rs11875359 were detected from cross-sectional analysis. The same conclusion was reached with even a less stringent
, seven SNPs, rs2075650, rs4902433, rs439401, rs11762610, rs1800627, rs11677350 and rs429358, were discovered to be associated with some phenotypes in longitudinal analysis; in contrast, only five SNPs, rs2075650, rs11677350, rs429358, rs2931352 and rs11875359 were detected from cross-sectional analysis. The same conclusion was reached with even a less stringent 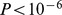 (not shown): one hundred and forty-seven significant SNP-phenotype association pairs were identified from longitudinal analysis, while only 47 pairs were found from cross-sectional analysis.
(not shown): one hundred and forty-seven significant SNP-phenotype association pairs were identified from longitudinal analysis, while only 47 pairs were found from cross-sectional analysis.
Table 6. Significant SNPs and each one's associated phenotype numbers at the level of 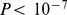 .
.
| SNP | L | I | M | B |
| rs2075650 | 25 | 12 | 3 | 1 |
LHippVol: 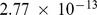
|
LHippVol: 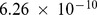
|
|||
RCerebCtx: 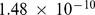
|
||||
LMeanTemp: 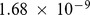
|
||||
| rs11677350 | 1 | 0 | 1 | 1 |
RCerebWM: 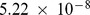
|
RCerebWM: 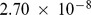
|
|||
| rs4902433 | 2 | 0 | 0 | 0 |
LMeanLatTemp: 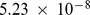
|
||||
LInfTemporal: 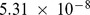
|
||||
| rs439401 | 6 | 10 | 0 | 0 |
RMeanLatTemp: 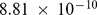
|
||||
RcerebCtx: 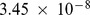
|
||||
RMeanTemp: 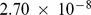
|
||||
| rs11762610 | 2 | 0 | 0 | 0 |
LFusiform: 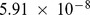
|
||||
LInfTemporal: 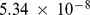
|
||||
| rs1800627 | 1 | 0 | 0 | 0 |
RAmygVol: 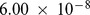
|
||||
| rs429358 | 46 | 40 | 5 | 5 |
LHippVol: 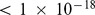
|
LHippVol: 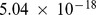
|
|||
LEntCtx: 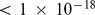
|
RHippVol: 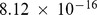
|
|||
LAmygVol: 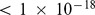
|
LAmygVol: 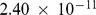
|
|||
| rs2931352 | 0 | 0 | 0 | 1 |
RParahipp: 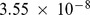
|
||||
| rs11875359 | 0 | 0 | 0 | 1 |
RInfLatVent: 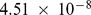
|
||||
| total | 83 | 62 | 9 | 9 |
In longitudinal analysis, column name “L”, “I” and “M” indicate the number of traits associated with the SNP from the longitudinal joint testing (i.e. with 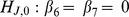 ), testing for interaction (i.e.
), testing for interaction (i.e. 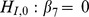 ) and testing for the main effects (i.e.
) and testing for the main effects (i.e. 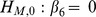 ); the column named “B” is for cross-sectional analysis of the baseline data. Top 3 SNP-phenotype association are listed with corresponding P-values.
); the column named “B” is for cross-sectional analysis of the baseline data. Top 3 SNP-phenotype association are listed with corresponding P-values.
In summary, both longitudinal and cross-sectional analyses indicated that both left and right hippocampus and amygdala were statistically significantly associated with SNP rs429358 in gene APOE at the genome-wide significance level 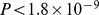 . In addition, SNP rs2075650 in gene TOMM40 was also identified to be associated with multiple phenotypes. These results are in agreement with some earlier studies. For example, both TOMM40 and APOE were known to be linked to Alzheimer's disease [23]–[26]. Finally, longitudinal analysis also detected another significant SNP rs439401, which also belongs to gene APOE and is very close to SNP rs4420638; SNP rs4420638 was shown to be related to late on-set Alzheimer's disease [27], [28], dyslipidemia [29], schizophrenia [30], myocardial infarction [31] and psychological stress [32].
. In addition, SNP rs2075650 in gene TOMM40 was also identified to be associated with multiple phenotypes. These results are in agreement with some earlier studies. For example, both TOMM40 and APOE were known to be linked to Alzheimer's disease [23]–[26]. Finally, longitudinal analysis also detected another significant SNP rs439401, which also belongs to gene APOE and is very close to SNP rs4420638; SNP rs4420638 was shown to be related to late on-set Alzheimer's disease [27], [28], dyslipidemia [29], schizophrenia [30], myocardial infarction [31] and psychological stress [32].
In addition, longitudinal analysis identified other four marginally significant SNPs that were missed by cross-sectional analysis at the significance level 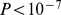 : rs1800627, rs11762610, rs4902433 and rs439401 on chromosomes 4, 7, 14 and 19 respectively. On the other hand, at the same significance level, cross-sectional analysis, but not longitudinal analysis, uncovered two marginally significant SNPs rs2931352 and rs11875359. Since these SNPs were identified under a less stringent significance level, they are only suggestive and need to be further replicated and validated.
: rs1800627, rs11762610, rs4902433 and rs439401 on chromosomes 4, 7, 14 and 19 respectively. On the other hand, at the same significance level, cross-sectional analysis, but not longitudinal analysis, uncovered two marginally significant SNPs rs2931352 and rs11875359. Since these SNPs were identified under a less stringent significance level, they are only suggestive and need to be further replicated and validated.
In addition to using the Bonferroni method to control the family-wise error rate, we also applied the false discovery rate (FDR) method [33] to adjust for multiple comparisons, as implemented in R function p. adjust (). The numbers of the significant SNP-phenotype associations at the various cut-offs of FDR are shown in Table 7. It is clear that at any given FDR level, longitudinal analysis identified a larger number of significant SNP-phenotype associations than that of cross sectional analysis. Note that, although an FDR (or q-value) is monotonic with the original unadjusted p-value, their relationship is not linear, which explains why at the FDR cut-offs of 0.035, 0.0011 and 0.0006, we reached the same numbers of the significant associations for longitudinal analysis, but not for cross sectional analysis, as those at the family-wise error rates of 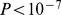 ,
, 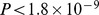 and
and 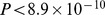 , respectively.
, respectively.
Table 7. The numbers of the significant SNP-phenotype associations at various levels of false discovery rate (FDR).
| FDR |
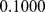
|
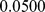
|
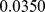
|
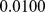
|
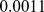
|
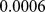
|
| Longitudinal | 112 | 90 | 83 | 64 | 46 | 43 |
| Baseline | 5 | 5 | 5 | 5 | 3 | 3 |
Genome-wide association testing: more details with some phenotypes
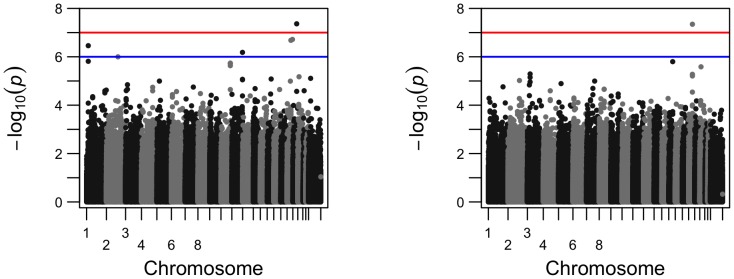
To visualize how the number of the significant SNPs increased from cross-sectional analysis to longitudinal analysis, we present their comparisons in Manhattan plots and Q-Q plots for two phenotypes, left hippocampus volume and right inferior lateral ventricle volume (Figures 2–5). For the volume of left hippocampus, 4 SNPs passed the significance level of 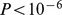 from longitudinal data analysis; in contrast, only 2 SNPs survived the threshold in cross sectional analysis. For volume of inferior lateral ventricle, there were respectively 6 and 2 SNPs passed the significance threshold of
from longitudinal data analysis; in contrast, only 2 SNPs survived the threshold in cross sectional analysis. For volume of inferior lateral ventricle, there were respectively 6 and 2 SNPs passed the significance threshold of 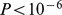 from longitudinal analysis and cross-sectional analysis.
from longitudinal analysis and cross-sectional analysis.
Figure 2. Comparison of the Manhattan plots for genome-wide p-values for phenotype left hippocampus volume from longitudinal analysis (left) and from cross-sectional analysis (right); SNP rs429358 is not included due to its small p-value.
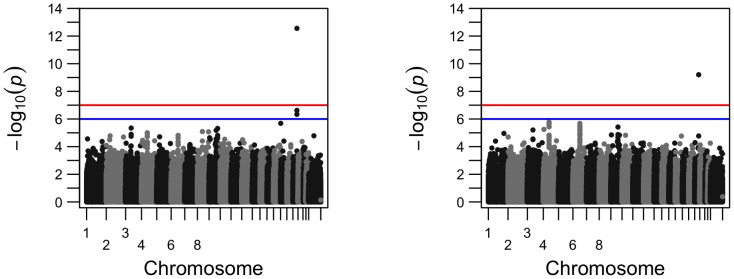
Figure 5. Comparison of the Q-Q plots for genome-wide p-values for phenotype volume of right inferior lateral ventricle from longitudinal analysis (left) and from cross-sectional analysis (right); SNP rs429358 is not included.
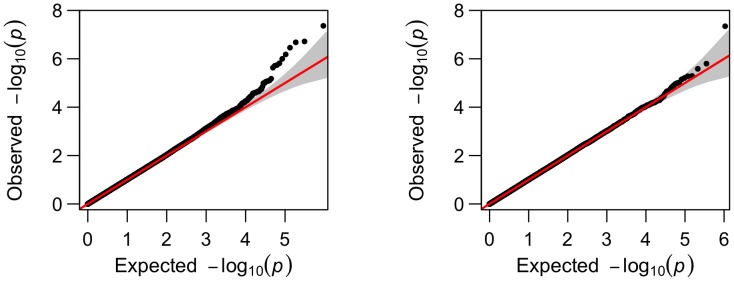
Figure 3. Comparison of the Q-Q plots for genome-wide p-values for phenotype left hippocampus volume from longitudinal analysis (left) and from cross-sectional analysis (right); SNP rs429358 is not included.
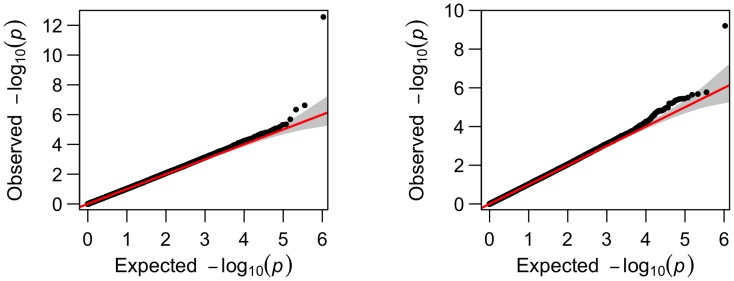
Figure 4. Comparison of the Manhattan plots for genome-wide p-values for phenotype volume of right inferior lateral ventricle from longitudinal analysis (left) and cross-sectional analysis (right); SNP rs429358 is not included.
Population stratification
To explore the possible existence of population stratification, we applied the principal component (PC) method [34]. Specifically, we first randomly selected 100,000 SNPs across the genome, then extracted the top ten PCs to be included as covariates in the linear mixed-effects model. For comparison, the Manhattan plots of the genome-wide p-values for phenotype volume of right amygdala are shown in Figure 6, while the Q-Q plots are in Figure 7. It is clear that whether or not to adjust for the top 10 PCs hardly made any difference to the overall results. Furthermore, we also calculated the genomic inflation factor  [35] in longitudinal data analysis. For phenotype volume of right amygdala, the inflation factor (
[35] in longitudinal data analysis. For phenotype volume of right amygdala, the inflation factor ( ) was estimated as 1.007 with the top 10 PCs, compared to 1.010 without PCs. A systematic examination of the inflation factors for all the 56 phenotypes without adjustment for PCs was also conducted: the estimated inflation factors ranged from 0.986 to 1.025 with mean 1.010 and standard error 0.014. Hence, in agreement with [4], there was no strong evidence for population stratification that would have questioned the validity of the genome-wide association results presented earlier.
) was estimated as 1.007 with the top 10 PCs, compared to 1.010 without PCs. A systematic examination of the inflation factors for all the 56 phenotypes without adjustment for PCs was also conducted: the estimated inflation factors ranged from 0.986 to 1.025 with mean 1.010 and standard error 0.014. Hence, in agreement with [4], there was no strong evidence for population stratification that would have questioned the validity of the genome-wide association results presented earlier.
Figure 6. Comparison of the Manhattan plots without (left) or with (right) top 10 PCs.
Figure 7. Comparison of the Q-Q plots without (left) or with (right) top 10 PCs.
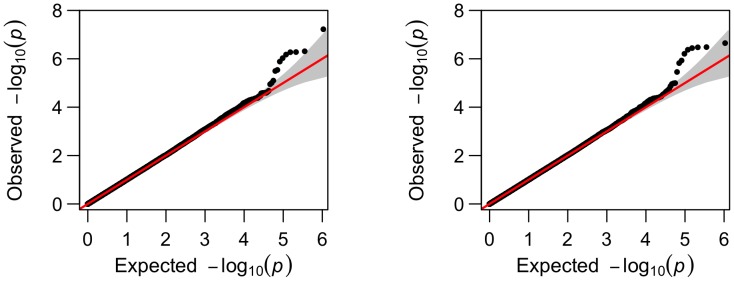
Alternative approaches to longitudinal analysis
We also applied a simpler linear mixed model (3) with only a random intercept term, and the corresponding GEE marginal model with a compound symmetry (CS) matrix as the working correlation structure. The results for phenotype left hippocampus volume are shown in Figure 8. Note that the results for the random-intercept model (3) and the GEE model with the model-based covariance matrix were very similar, as expected; both yielded severely inflated false positive rates with their estimated inflation factors much larger than 1. As analyzed in the Methods section, the problem was likely due to the mis-specification and thus under-estimation of the phenotype variances. In contrast, the GEE model with the sandwich estimator gave a much better controlled inflation factor, which however was still larger than that from the earlier linear mixed model (2); it could be due to the stronger “missing completely at random” assumption required by GEE, which might be violated here.
Figure 8. The Q-Q plots for genome-wide p-values for phenotype left hippocampus volume from longitudinal analysis based on (a) GEE with the sandwich covariance estimator (left, inflation factor = 1.070), (b) GEE with the model-based covariance estimator (middle, inflation factor = 2.077), and (c) linear mixed model with only a random intercept term (right, inflation factor = 1.976).

To establish the superiority of model (2) over model (3), we conducted the LRT on the null hypothesis 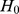 :
: 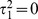 with phenotype left hippocampus volume and each of the 10196 SNPs on chromosome 19. The null hypothesis was rejected each time; the LRT statistics were large, ranging from 228.9 to 289.2 with a median of 279.9.
with phenotype left hippocampus volume and each of the 10196 SNPs on chromosome 19. The null hypothesis was rejected each time; the LRT statistics were large, ranging from 228.9 to 289.2 with a median of 279.9.
Simulation results
When simulated phenotypic data were generated from model (2), as shown in Table 8, it was confirmed that only LME-RSI, GEE-Robust and Baseline could satisfactorily control the Type I error, while the other two could not due to their use of mis-specified models. Note that a difference between the real data and simulated data was that there were no missing phenotypes in the latter. For power, as expected, the two methods for longitudinal analysis, LME-RSI and GEE-Robust, were almost equally powerful, both more powerful than cross-sectional analysis.
Table 8. Simulation results at significance level 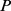 with different methods for phenotypic data generated from model (2).
with different methods for phenotypic data generated from model (2).
| Type I Error | ||||||
| Model | rs2075650 | rs439401 | ||||
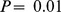
|
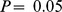
|
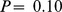
|
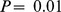
|
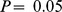
|
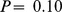
|
|
| LME-RSI | 0.007 | 0.044 | 0.087 | 0.007 | 0.039 | 0.097 |
| LME-RI | 0.071 | 0.177 | 0.258 | 0.090 | 0.190 | 0.276 |
| GEE-Robust | 0.008 | 0.045 | 0.089 | 0.008 | 0.045 | 0.106 |
| GEE-Naive | 0.082 | 0.189 | 0.257 | 0.102 | 0.191 | 0.286 |
| Baseline | 0.006 | 0.042 | 0.084 | 0.006 | 0.059 | 0.112 |
LME-RSI: a linear mixed-effects model with random slope and intercept; LME-RI: a linear mixed-effects model with only a random intercept term; GEE-Robust: GEE with the sandwich covariance estimator; GEE-Naive: GEE with the model-based covariance estimator; Baseline: a linear model at the baseline testing for the main effects of an SNP.
When simulated phenotypic data were generated from model (3), as shown in Table 9, the Type I error rates were generally controlled, though GEE-Naive might be too conservative. In terms of power, it is obvious that again longitudinal methods outperformed the cross sectional analysis of only the baseline data. Note that, since the CS working correlation structure used in GEE was correct, GEE-Naive also performed well. Most interestingly, though LME-RSI was fitted to a larger model (2) covering the true model (2) used in LME-RI, the power loss of LME-RSI was negligible when compared with LME-RI, showing the robustness of using model (2).
Table 9. Simulation results at significance level 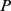 with different methods for phenotypic data generated from model (3).
with different methods for phenotypic data generated from model (3).
| Type I Error | ||||||||||||
| Model | rs2075650 | rs439401 | rs429358 | |||||||||
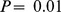
|
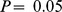
|
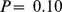
|
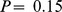
|
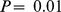
|
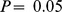
|
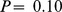
|
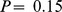
|
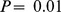
|
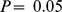
|
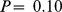
|
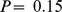
|
|
| LME-RSI | 0.006 | 0.038 | 0.079 | 0.136 | 0.007 | 0.043 | 0.096 | 0.143 | 0.007 | 0.043 | 0.097 | 0.140 |
| LME-RI | 0.006 | 0.039 | 0.079 | 0.135 | 0.008 | 0.043 | 0.096 | 0.147 | 0.007 | 0.044 | 0.096 | 0.141 |
| GEE-Robust | 0.004 | 0.040 | 0.088 | 0.135 | 0.007 | 0.052 | 0.104 | 0.148 | 0.009 | 0.045 | 0.100 | 0.145 |
| GEE-Naive | 0.000 | 0.012 | 0.031 | 0.043 | 0.001 | 0.010 | 0.033 | 0.059 | 0.001 | 0.010 | 0.023 | 0.044 |
| Baseline | 0.005 | 0.048 | 0.081 | 0.124 | 0.008 | 0.056 | 0.113 | 0.160 | 0.007 | 0.041 | 0.096 | 0.149 |
Discussion
We have conducted a genome-wide association scan on each SNP with each of 56 regional imaging phenotypes utilizing the ADNI-1 data. By taking advantage of the existing longitudinal imaging phenotypes, we have illustrated the power gains from longitudinal analysis of longitudinal phenotypes measured at multiple time points over cross-sectional analysis of only the baseline phenotypic data. In particular, application of a linear mixed-effects model to longitudinal phenotypic data identified a much larger number of SNP-phenotype associations, at both a genome-wide and other less stringent significance levels. The advantage of longitudinal analysis is expected due to its use of more data, compared to only the baseline data in cross-sectional analysis. Note that here our goal is, as usual, to identify genetic variants associated with a phenotype in whatever way. We also note that our longitudinal model is related to, but different from, modeling gene-age interactions [36]: in the mean model (4), there is no SNP-baseline age interaction. If desired, as an alternative approach one can model SNP-age interaction directly. Since almost all existing GWAS analyses of the ADNI-1 data are based on the baseline data with only few exceptions [37]–[39], we hope that our current study will help reinforce the message on the preference of longitudinal analysis over cross-sectional analysis. This issue will become even more compelling as more longitudinal phenotypic data, e.g. various neuroimaging phenotypes from ADNI-GO and ADNI-2, are being or will be collected.
We have only applied single SNP-based analyses, while there is increasing evidence of possible power gains with SNP-set analyses in cross-sectional studies [9], [40]. However, it remains to be done to extend some powerful SNP-set methods to mixed-effects models with longitudinal data, such as the variance-component or kernel methods, similarity-based tests and others [41]–[46]. Or, instead of controlling family-wise Type I error rate as approached here, one may apply some new methods to control false discovery rate (FDR) [47]. Furthermore, with the availability of DNA sequencing data, it will be useful to develop and apply new statistical tests to detect rare variant associations with longitudinal phenotypes, again based on some extensions of the methods for cross-sectional data [46], [48]–[53]. More generally, as pointed out by Lindquist [54], “Imaging genetics promises to be an important topic of future research, and to fully realize its promise, novel statistical techniques will be needed.” These are all interesting topics to be investigated.
Acknowledgments
The authors thank the reviewers for constructive and helpful comments. Data used in preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All the data are available from the ADNI website: adni.loni.usc.edu.
Funding Statement
This research was supported by NIH grants R01HL65462, R01HL105397, R01HL116720 and R01GM081535, and by the Minnesota Supercomputing Institute. Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12-2-0012). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The following institutions had no direct involvement in the funding of this present study, but are funders of ADNI: the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Alzheimer's Association; Alzheimer's Drug Discovery Foundation; BioClinica, Inc.; Biogen Idec Inc.; Bristol-Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; GE Healthcare; Innogenetics, N.V.; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated (Abraham et al. 2008) by the Laboratory for Neuro Imaging at the University of Southern California.
References
- 1. Jack CR, Bernstein MA, Fox NC, Thompson P, Alexander G, et al. (2008) The alzheimer's disease neuroimaging initiative (adni): Mri methods. Journal of Magnetic Resonance Imaging 27: 685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mueller SG, Weiner MW, Thal LJ, Petersen RC, Jack CR, et al. (2005) Ways toward an early diagnosis in alzheimers disease: The alzheimers disease neuroimaging initiative (adni). Alzheimer's & Dementia 1: 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Potkin SG, Guffanti G, Lakatos A, Turner JA, Kruggel F, et al. (2009) Hippocampal atrophy as a quantitative trait in a genome-wide association study identifying novel susceptibility genes for alzheimer's disease. PloS one 4: e6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shen L, Kim S, Risacher SL, Nho K, Swaminathan S, et al. (2010) Whole genome association study of brain-wide imaging phenotypes for identifying quantitative trait loci in mci and ad: A study of the adni cohort. Neuroimage 53: 1051–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stein JL, Hua X, Lee S, Ho AJ, Leow AD, et al. (2010) Voxelwise genome-wide association study (vgwas). Neuroimage 53: 1160–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stein JL, Hua X, Morra JH, Lee S, Hibar DP, et al. (2010) Genome-wide analysis reveals novel genes influencing temporal lobe structure with relevance to neurodegeneration in alzheimer's disease. Neuroimage 51: 542–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stein JL, Medland SE, Vasquez AA, Hibar DP, Senstad RE, et al. (2012) Identification of common variants associated with human hippocampal and intracranial volumes. Nature genetics 44: 552–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hibar DP, Stein JL, Kohannim O, Jahanshad N, Saykin AJ, et al. (2011) Voxelwise genewide association study (vgenewas): multivariate gene-based association testing in 731 elderly subjects. Neuroimage 56: 1875–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kohannim O, Hibar DP, Stein JL, Jahanshad N, Hua X, et al.. (2012) Discovery and replication of gene influences on brain structure using lasso regression. Frontiers in neuroscience 6. [DOI] [PMC free article] [PubMed]
- 10. Smith EN, Chen W, Kähönen M, Kettunen J, Lehtimäki T, et al. (2010) Longitudinal genome-wide association of cardiovascular disease risk factors in the bogalusa heart study. PLoS genetics 6: e1001094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fan R, Zhang Y, Albert PS, Liu A, Wang Y, et al. (2012) Longitudinal association analysis of quantitative traits. Genetic epidemiology 36: 856–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mei H, Chen W, Jiang F, He J, Srinivasan S, et al. (2012) Longitudinal replication studies of gwas risk snps influencing body mass index over the course of childhood and adulthood. PloS one 7: e31470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mukherjee B, Ko YA, VanderWeele T, Roy A, Park SK, et al. (2012) Principal interactions analysis for repeated measures data: application to gene–gene and gene–environment interactions. Statistics in medicine 31: 2531–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sikorska K, Rivadeneira F, Groenen PJ, Hofman A, Uitterlinden AG, et al. (2013) Fast linear mixed model computations for genome-wide association studies with longitudinal data. Statistics in medicine 32: 165–180. [DOI] [PubMed] [Google Scholar]
- 15. Costanza MC, Beer-Borst S, James RW, Gaspoz JM, Morabia A (2012) Consistency between cross-sectional and longitudinal snp: blood lipid associations. European journal of epidemiology 27: 131–138. [DOI] [PubMed] [Google Scholar]
- 16. Fischl B, Dale AM (2000) Measuring the thickness of the human cerebral cortex from magnetic resonance images. Proceedings of the National Academy of Sciences 97: 11050–11055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hartig M, Truran-Sacrey D, Raptentsetsang S, Schuff N, Weiner M (2012) Ucsf freesurfer overview and qc ratings. ADNI: Alzheimers Disease Neuroimaging Initiative, San Francisco.
- 18. Reuter M, Schmansky NJ, Rosas HD, Fischl B (2012) Within-subject template estimation for unbiased longitudinal image analysis. Neuroimage 61: 1402–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Benke K, Wu Y, Fallin D, Maher B, Palmer L (2013) Strategy to control type i error increases power to identify genetic variation using the full biological trajectory. Genetic epidemiology 37: 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stram DO, Lee JW (1994) Variance components testing in the longitudinal mixed effects model. Biometrics: 1171–1177. [PubMed]
- 21. Liang KY, Zeger SL (1986) Longitudinal data analysis using generalized linear models. Biometrika 73: 13–22. [Google Scholar]
- 22. Ziegler A, Kastner C, Blettner M (1998) The generalised estimating equations: an annotated bibliography. Biometrical Journal 40: 115–139. [Google Scholar]
- 23. Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, et al. (1997) Effects of age, sex, and ethnicity on the association between apolipoprotein e genotype and alzheimer disease: a meta-analysis. Jama 278: 1349–1356. [PubMed] [Google Scholar]
- 24. Holland D, Brewer JB, Hagler DJ, Fennema-Notestine C, Dale AM, et al. (2009) Subregional neuroanatomical change as a biomarker for alzheimer's disease. Proceedings of the National Academy of Sciences 106: 20954–20959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sanan D, Weisgraber K, Russell S, Mahley R, Huang D, et al. (1994) Apolipoprotein e associates with beta amyloid peptide of alzheimer's disease to form novel monofibrils. Isoform apoe4 associates more efficiently than apoe3. Journal of Clinical Investigation 94: 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, et al. (1993) Binding of human apolipoprotein e to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset alzheimer disease. Proceedings of the National Academy of Sciences 90: 8098–8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Abraham R, Moskvina V, Sims R, Hollingworth P, Morgan A, et al. (2008) A genome-wide association study for late-onset alzheimer's disease using dna pooling. BMC medical genomics 1: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Beecham GW, Martin ER, Li YJ, Slifer MA, Gilbert JR, et al. (2009) Genome-wide association study implicates a chromosome 12 risk locus for late-onset alzheimer disease. The American Journal of Human Genetics 84: 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y, Zhou D, Zhang Z, Song Y, Zhang D, et al.. (2010) Effects of genetic variants on lipid parameters and dyslipidemia in chinese population. Journal of lipid research: jlr–P007476. [DOI] [PMC free article] [PubMed]
- 30. Verbrugghe P, Bouwer S, Wiltshire S, Carter K, Chandler D, et al. (2012) Impact of the reelin signaling cascade (ligands–receptors–adaptor complex) on cognition in schizophrenia. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics 159: 392–404. [DOI] [PubMed] [Google Scholar]
- 31. Shiffman D, Kane JP, Louie JZ, Arellano AR, Ross DA, et al. (2008) Analysis of 17,576 potentially functional snps in three case–control studies of myocardial infarction. PLoS One 3: e2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kring SII, Barefoot J, Brummett BH, Boyle SH, Siegler IC, et al. (2011) Associations between apoe variants and metabolic traits and the impact of psychological stress. PloS one 6: e15745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B (Methodological): 289–300.
- 34. Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, et al. (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nature genetics 38: 904–909. [DOI] [PubMed] [Google Scholar]
- 35. Devlin B, Roeder K (1999) Genomic control for association studies. Biometrics 55: 997–1004. [DOI] [PubMed] [Google Scholar]
- 36. Shete S, Chen J, Zhou X, Amos C (2000) Modeling age x major gene interaction by a variance component approach. Genetic epidemiology 21: S849–53. [DOI] [PubMed] [Google Scholar]
- 37. Melville SA, Buros J, Parrado AR, Vardarajan B, Logue MW, et al. (2012) Multiple loci influencing hippocampal degeneration identified by genome scan. Annals of neurology 72: 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Silver M, Janousova E, Hua X, Thompson PM, Montana G (2012) Identification of gene pathways implicated in alzheimer's disease using longitudinal imaging phenotypes with sparse regression. NeuroImage 63: 1681–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang H, Nie F, Huang H, Yan J, Kim S, et al. (2012) From phenotype to genotype: an association study of longitudinal phenotypic markers to alzheimer's disease relevant snps. Bioinformatics 28: i619–i625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ge T, Feng J, Hibar DP, Thompson PM, Nichols TE (2012) Increasing power for voxel-wise genome-wide association studies: the random field theory, least square kernel machines and fast permutation procedures. NeuroImage 63: 858–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wessel J, Schork NJ (2006) Generalized genomic distance–based regression methodology for multilocus association analysis. The American Journal of Human Genetics 79: 792–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wei Z, Li M, Rebbeck T, Li H (2008) U-statistics-based tests for multiple genes in genetic association studies. Annals of human genetics 72: 821–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Pan W (2009) Asymptotic tests of association with multiple snps in linkage disequilibrium. Genetic epidemiology 33: 497–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tzeng JY, Zhang D (2007) Haplotype-based association analysis via variance-components score test. The American Journal of Human Genetics 81: 927–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wu MC, Lee S, Cai T, Li Y, Boehnke M, et al. (2011) Rare-variant association testing for sequencing data with the sequence kernel association test. The American Journal of Human Genetics 89: 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fan R, Wang Y, Mills JL, Wilson AF, Bailey-Wilson JE, et al. (2013) Functional linear models for association analysis of quantitative traits. Genetic epidemiology 37: 726–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xie J, Cai TT, Li H (2011) Sample size and power analysis for sparse signal recovery in genome-wide association studies. Biometrika 98: 273–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Basu S, Pan W (2011) Comparison of statistical tests for disease association with rare variants. Genetic epidemiology 35: 606–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dering C, Hemmelmann C, Pugh E, Ziegler A (2011) Statistical analysis of rare sequence variants: an overview of collapsing methods. Genetic epidemiology 35: S12–S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Luo L, Boerwinkle E, Xiong M (2011) Association studies for next-generation sequencing. Genome research 21: 1099–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen LS, Hsu L, Gamazon ER, Cox NJ, Nicolae DL (2012) An exponential combination procedure for set-based association tests in sequencing studies. The American Journal of Human Genetics 91: 977–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee S, Emond MJ, Bamshad MJ, Barnes KC, Rieder MJ, et al. (2012) Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. The American Journal of Human Genetics 91: 224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhao Z, Wang W, Wei Z (2013) An empirical bayes testing procedure for detecting variants in analysis of next generation sequencing data. The Annals of Applied Statistics 7: 2229–2248. [Google Scholar]
- 54. Lindquist MA (2008) The statistical analysis of fmri data. Statistical Science 23: 439–464. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All the data are available from the ADNI website: adni.loni.usc.edu.