

Abstract
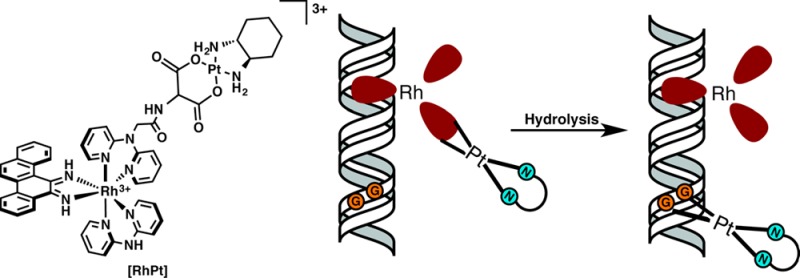
We report the synthesis and characterization of a bimetallic conjugate (RhPt) in which an oxaliplatin derivative is tethered to a rhodium metalloinsertor through an aminomalonate leaving group ligand. The complex interacts with DNA through metalloinsertion at a base pair mismatch followed by formation of a covalent Pt–DNA adduct. Characterization of RhPt in mismatch repair-deficient HCT116O cells reveals increased cytotoxicity compared to cisplatin and oxaliplatin as well as relative to the unconjugated rhodium and platinum counterparts. Caspase and poly-ADP ribose polymerase inhibition assays indicate that RhPt induces apoptotic cell death. Inductively coupled plasma mass spectrometry (ICP-MS) experiments reveal that RhPt exhibits enhanced cellular uptake properties that contribute to its increased efficacy.
Short abstract
We report the synthesis and characterization of a bifunctional DNA metalloinsertor conjugate (“RhPt”). The bimetallic complex contains a rhodium mismatch recognition component tethered to a platinum anticancer agent and exhibits dual binding to DNA involving both metalloinsertion at a base pair mismatch and covalent cross-linking of platinum. In mismatch repair-deficient cells, RhPt exhibits enhanced cellular uptake and cytotoxicity over traditional platinum therapeutics.
Platinum anticancer compounds are among the most successful and most widely used chemotherapeutics to date.1 However, cancers that exhibit deficiencies in the DNA mismatch repair (MMR) pathway, including 15% of sporadic colorectal cancer cases and 18% of all solid tumors, have encountered limited success in treatment with classical platinum therapeutics.2,3 Such cancers are largely resistant to cisplatin and only marginally responsive to oxaliplatin.4,5
Rhodium metalloinsertors may offer a promising strategy in the development of new therapies for such cancers. These bulky, octahedral complexes bind specifically to DNA base pair mismatches,6 which are amplified in cells with defective MMR machinery.2,3 Metalloinsertors exhibit cytotoxicity preferentially in MMR-deficient cells, and the extent of this selectivity correlates with mismatch binding affinity and localization to the nucleus, where they target mismatches in genomic DNA.7,8
The design of bifunctional drug conjugates is a burgeoning field in chemotherapy, especially as a strategy to circumvent resistance.9 Here, we present a bimetallic oxaliplatin–metalloinsertor conjugate (RhPt) that displays dual DNA binding behavior. Additionally, RhPt exhibits enhanced cytotoxicity and cellular uptake in MMR-deficient HCT116O cancer cells compared to first-line platinum therapeutics as well as its unconjugated subunits. The cytotoxicity of RhPt appears to be triggered by an apoptotic cell death pathway, and its potency is attributed to the improved cellular uptake of the complex.
The RhPt conjugate, shown in Figure 1, consists of a trisheteroleptic Rh(III) scaffold tethered to an oxaliplatin derivative by an aminomalonate leaving group ligand. RhPt was constructed via a linear synthesis in which the rhodium scaffold was first functionalized with the aminomalonate, followed by complexation to platinum (see the Supporting Information).10 The platinum unit employs the same (1R,2R)-1,2-diaminocyclohexane nonleaving group ligand as oxaliplatin and, therefore, is expected to form the same DNA adducts.11 The rhodium unit contains a sterically expansive 5,6-chrysene diimine ligand (chrysi), which is responsible for the recognition of DNA mismatches.12 Too wide to intercalate into well-matched DNA, the chrysi complexes instead target thermodynamically destabilized mismatches from the minor groove, ejecting the bases from the duplex in a binding mode known as metalloinsertion.13 The aminomalonate linker is tethered to one of the noninserting ancillary ligands, which allows the conjugate to remain intact temporarily but ultimately enables the release of platinum, via hydrolysis, for DNA binding. The biological activity of RhPt was compared to several complexes, including cisplatin and oxaliplatin (Figure 1). The rhodium hydrolysis product, Rh(Amal), was included as a control to test whether the biological activity of RhPt may be attributed to the intact conjugate and not premature hydrolysis of the subunits. The unconjugated platinum complex, Pt(Amal) was included to explore the effects of the aminomalonate ligand on activity.
Figure 1.

Chemical structures of complexes studied.
DNA binding studies were performed with RhPt and radiolabeled duplex DNA containing a CC mismatch (Figure 2). As RhPt does not cleave DNA upon irradiation, a competition titration was carried out using rac-[Rh(bpy)2chrysi]3+, which does photocleave DNA at the site of a mismatch.6 RhPt inhibits photocleavage by rac-[Rh(bpy)2chrysi]3+ at the mismatched site; this indicates that RhPt binds specifically to the mismatch via metalloinsertion. The binding affinity of RhPt for a CC mismatch was determined to be 1.1 × 107 M–1 (Figure S1, Supporting Information), comparable to that of monomeric metalloinsertors.8 DNA binding by the conjugate also involves the formation of covalent adducts. Platination of the DNA is indicated by the appearance of bands with reduced electrophoretic mobility, located above the unmodified parent bands (Figures 2 and S2, Supporting Information).14 Platinum binding to DNA was further characterized by dimethyl sulfate (DMS) footprinting. DMS methylates the N7 position of guanine, resulting in cleavage at those sites upon treatment with piperidine.15 At a 1:1 molar ratio of DNA and RhPt (Figure 2), the guanine residues on the labeled strand are protected from cleavage, signifying the formation of platinum 1,2-intrastrand adducts at the N7 positions of the d(GpG) site, as is the case for oxaliplatin.16,17 That RhPt performs noncovalent metalloinsertion and covalent platinum binding establishes the bifunctionality of the conjugate. The lack of interplay between the rhodium and platinum binding modes suggests that each subunit functions independently and without inhibition of the other; that is, platinum binding does not alter the apparent equilibrium of metalloinsertion in the minor groove, nor does rhodium binding impede DNA platination.
Figure 2.
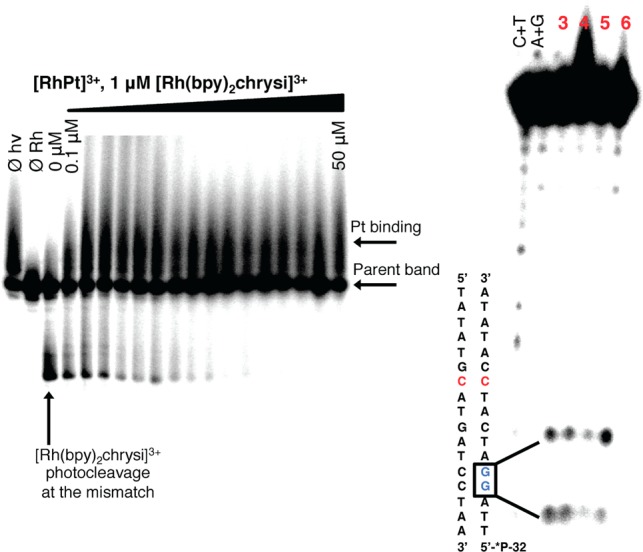
DNA binding of RhPt. Left: Competition titration of increasing concentrations of RhPt (0–50 μM) with 1 μM rac-[Rh(bpy)2chrysi]3+ on 1 μM 5′-[32P] labeled 17mer duplex DNA with a CC mismatch. Controls without irradiation (Øhν), and without Rh (ØRh) were included. RhPt inhibits photocleavage by [Rh(bpy)2chrysi]3+ at the mismatched site. Right: DMS footprinting of duplex DNA containing a CC mismatch. Lanes (left to right): Maxam–Gilbert sequencing (C+T; A+G); 3, DMS alone; 4, oxaliplatin (1 μM); 5, RhPt (1 μM); 6, RhPt (50 μM). Bands of high electrophoretic mobility indicate cleavage at guanine residues; covalent binding of platinum to guanine inhibits cleavage.
The antiproliferative effects of RhPt were explored in the isogenic human colorectal carcinoma cell lines HCT116N (MMR-proficient) and HCT116O (MMR-deficient) using an antibody assay for DNA synthesis.18 RhPt exhibits antiproliferative activity similar to that of oxaliplatin and considerably outperforms cisplatin, which preferentially targets HCT116N cells (Figure S3, Supporting Information). RhPt does not preferentially target either cell line. However, it is active at submicromolar concentrations and, in fact, is more potent than either of its unconjugated subunits (Figure S4, Supporting Information). Furthermore, RhPt exhibits enhanced cytotoxicity (LC50 = 9 μM) over cisplatin (30 μM) and oxaliplatin (28 μM) in MMR-deficient cells and is also substantially more potent than Rh(Amal) (43 μM) and Pt(Amal) (57 μM) (Table S1 and Figure S5, Supporting Information).
Cellular uptake was examined via inductively coupled plasma mass spectrometry (ICP-MS) (Figure 3). The cellular uptake of both rhodium and platinum for RhPt generally exceeds that of the monomeric complexes, with RhPt displaying high initial uptake that decreases over time, possibly due to an efflux mechanism.19 Furthermore, the differences in uptake between RhPt and hydrolysis product Rh(Amal) suggest that the conjugate does not hydrolyze prior to entry into the cell and is taken up in its intact form. Overall, it would appear that RhPt possesses enhanced cellular uptake properties not inherent to either subunit alone. The localization of each complex was also examined (Table S1, Supporting Information). Little differentiation is observed in the subcellular distribution of the complexes, with the notable exception of a substantial increase in the nuclear rhodium concentration of RhPt compared to Rh(Amal). Curiously, this enhanced nuclear targeting of rhodium does not result in cell-selective activity.
Figure 3.
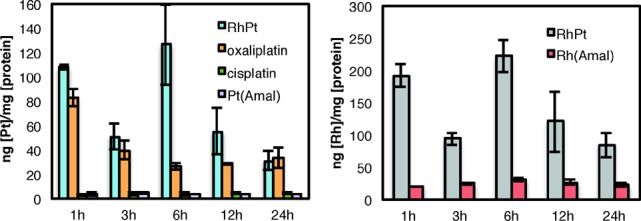
Cellular accumulation of metal complexes in HCT116O cells. Rh and Pt counts were normalized to cellular protein: (left) whole-cell uptake of Pt complexes as a function of time; (right) whole-cell uptake of Rh for RhPt and hydrolysis product Rh(Amal).
To further understand the biological activity of RhPt, we examined the mechanism of cell death. It has been previously established that rhodium metalloinsertors trigger necrosis dependent upon DNA repair protein poly-ADP ribose polymerase (PARP).7 Cytotoxicity studies revealed that the viability of cells treated with RhPt does not increase in the presence of PARP inhibitor DPQ,20 suggesting PARP-independent cell death (Figure 4).21 The assay was also performed with the caspase inhibitor Z-VAD-FMK.22 The viability of RhPt-treated cells increases in the presence of Z-VAD-FMK, signifying that RhPt triggers caspase-dependent death (Figure 4). This is consistent with studies of platinum cytotoxicity generally; it is well established that cisplatin and oxaliplatin typically trigger apoptosis.23 This result may, in part, explain the lack of cell-selectivity observed for RhPt. By initiating apoptosis, rather than necrosis, it is possible that the highly selective biological response to mismatch recognition by rhodium is overridden by the effects of high concentrations of platinum in the cell.
Figure 4.
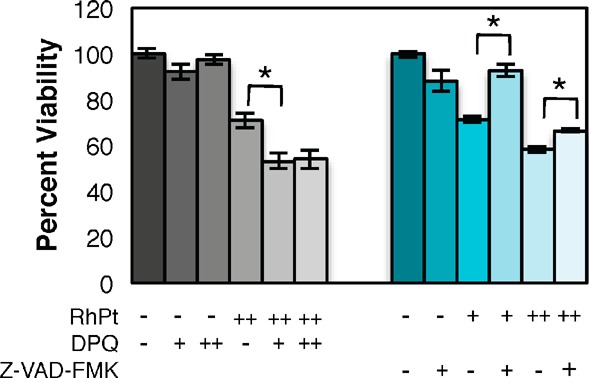
Cell viability in HCT116O cells after 72 h with PARP and caspase inhibitors. Viability is normalized to untreated controls. Left (gray): PARP inhibition assay. Cells were treated with 0 or 20 μM RhPt and 0, 25, or 50 μM DPQ. DPQ does not increase the viability of cells treated with RhPt. Right (blue): Caspase inhibition assay. Cells were treated with 0, 10, or 20 μM RhPt and 0 or 20 μM Z-VAD-FMK. Z-VAD-FMK increases viability in RhPt-treated cells. Addition of either inhibitor alone does not affect viability. *p < 0.0001(unpaired two-tailed t test).
In this work, we examined the biological effects of conjugation of a DNA metalloinsertor with a platinum drug. In vitro, the complex successfully exhibits bifunctionality via dual DNA binding. In MMR-deficient cells, this strategy affords enhanced cellular uptake and potency over the individual subunits as well as versus traditional chemotherapeutics. However, RhPt is not without its limitations. The platinum subunit appears to dominate the cellular response, resulting in a loss of cell selectivity. Nevertheless, the biological analysis of RhPt provides insight into the behavior of bifunctional DNA targeting agents as well as a foundation for the design of future conjugates that are both potent and selective in their cellular targeting.
Acknowledgments
We are grateful for instrumentation made available by the Caltech Environmental Analysis Center.
Supporting Information Available
Experimental procedures and supporting figures. This material is available free of charge via the Internet at http://pubs.acs.org.
Financial support from the NIH (GM033309) for this work is gratefully acknowledged. We also thank the NIH for a training grant (NIH/NRSA 5T32GM7616-33) to A.G.W.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Wang D.; Lippard S. J. Nature Rev. Drug Discovery 2005, 4, 307. [DOI] [PubMed] [Google Scholar]
- Arzimanoglou I. I.; Gilbers F.; Barber H. R. K. Cancer 1998, 82, 1808–1820. [DOI] [PubMed] [Google Scholar]
- Lawes D. A.; SenGupta S.; Boulos P. B. Eur. J. Surg. Oncol. 2003, 29, 201–212. [DOI] [PubMed] [Google Scholar]
- Aebi S.; Fink D.; Gordon R.; Kim H. K.; Zheng H.; Fink J. L.; Howell S. B. Clin. Cancer Res. 1997, 3, 1763–1767. [PubMed] [Google Scholar]
- Ibrahim A.; Hirschfeld S.; Cohen M. H.; Griebel D. J.; Williams G. A.; Pazdur R. Oncologist 2004, 9, 8–12. [DOI] [PubMed] [Google Scholar]
- Jackson B. A.; Barton J. K. J. Am. Chem. Soc. 1997, 119, 12986. [Google Scholar]
- a Ernst R. J.; Song H.; Barton J. K. J. Am. Chem. Soc. 2009, 131, 2359. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ernst R. J.; Komor A. C.; Barton J. K. Biochemistry 2011, 50, 10919–10928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor A. C.; Schneider C. J.; Weidmann A. G.; Barton J. K. J. Am. Chem. Soc. 2012, 134, 19223–19233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisnovsky S. P.; Wilson J. J.; Radford R. J.; Pereira M. P.; Chan M. R.; Laposa R. R.; Lippard S. J.; Kelley S. O. Chem. Biol. 2013, 20, 1323–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RhPt and Rh(Amal) each showed only one peak by HPLC; thus, all studies were carried out on the racemic complexes.
- Woynarowski J. M.; Faivre S.; Herzig M. C.; Arnett B.; Chapman W. G.; Trevino A. V.; Raymond E.; Chaney S. G.; Vaisman A.; Varchenko M.; Juniewicz P. E. Mol. Pharmacol. 2000, 58, 920–927. [DOI] [PubMed] [Google Scholar]
- Jackson B. A.; Barton J. K. Biochemistry 2000, 39, 6176–6182. [DOI] [PubMed] [Google Scholar]
- a Pierre V. C.; Kaiser J. T.; Barton J. K. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zeglis B. M.; Pierre V. C.; Barton J. K. Biochemistry 2009, 48, 4247–4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- No difference in platination levels was observed in DNA binding experiments with well-matched DNA.
- Brabec V.; Leng M. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adenine methylation is also observed with DMS treatment, but there is no evidence of platinum binding at these sites.
- At 50 μM RhPt, only one guanine is protected, possibly due to distortions to the DNA that impede the formation of 1,2-d(GpG) adducts.
- Koi M.; Umar A.; Chauhan D. P.; Cherian S. P.; Carethers J. M.; Kunkel T. A.; Boland C. R. Cancer Res. 1994, 54, 4308. [PubMed] [Google Scholar]
- Eckford P. D. W.; Sharom F. J. Chem. Rev. 2009, 109, 2989. [DOI] [PubMed] [Google Scholar]
- DPQ = 3,4-dihydro-5[4-(1-piperindinyl)butoxy]-1(2H)-isoquinoline. See:Costantino G.; Macchiarulo A.; Camaioni E.; Pellicciari R. J. Med. Chem. 2001, 44, 3786–3794. [DOI] [PubMed] [Google Scholar]
- In fact, a significant decrease in viability is observed during combination treatment of RhPt and DPQ, although the reasons for this cooperative effect are unclear at present.
- By irreversibly binding to the active site of caspases, Z-VAD-FMK inhibits apoptosis. See:Vandenabeele P.; Vanden Berghe T.; Festjens N. Sci. STKE 2006, 358, pe44. [DOI] [PubMed] [Google Scholar]
- a Siddik Z. H. Oncogene 2003, 22, 7265. [DOI] [PubMed] [Google Scholar]; b Arango D.; Wilson A. J.; Shi Q.; Corner G. A.; Arañes M. J.; Nicholas C.; Lesser M.; Mariadason J. M.; Augenlicht L. H. Br. J. Cancer 2004, 91, 1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.