

Abstract
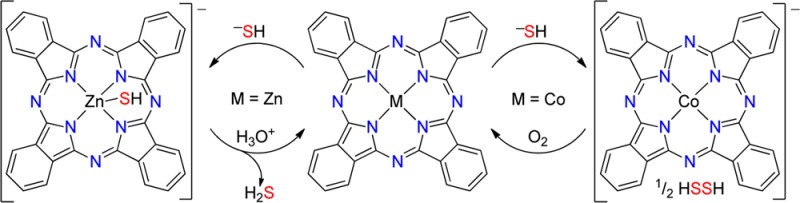
Hydrogen sulfide (H2S) is an important signaling molecule that exerts action on various bioinorganic targets. Despite this importance, few studies have investigated the differential reactivity of the physiologically relevant H2S and HS– protonation states with metal complexes. Here we report the distinct reactivity of H2S and HS– with zinc(II) and cobalt(II) phthalocyanine (Pc) complexes and highlight the chemical reversibility and cyclability of each metal. ZnPc reacts with HS–, but not H2S, to generate [ZnPc-SH]−, which can be converted back to ZnPc by protonation. CoPc reacts with HS–, but not H2S, to form [CoIPc]−, which can be reoxidized to CoPc by air. Taken together, these results demonstrate the chemically reversible reaction of HS– with metal phthalocyanine complexes and highlight the importance of H2S protonation state in understanding the reactivity profile of H2S with biologically relevant metal scaffolds.
Short abstract
The protonation state of H2S influences its reactivity with different metal phthalocyanine (Pc) complexes. Both ZnPc and CoPc react with H2S in a chemically reversible manner, with redox-inactive ZnPc binding HS− and redox-active CoPc undergoing reduction. The [ZnPc-SH]− product can be reverted to ZnPc by protonation, and [CoIPc]− can be redoxidized to CoPc with air.
Hydrogen sulfide (H2S) is an endogenously produced molecule that plays important and diverse roles in both vasoregulation and neurotransmission, as well as other physiological processes.1−10 As a gaseous small-molecule signaling agent, endogenous H2S joins NO and CO as a gasotransmitter, and all three mediate important functions through action on bioinorganic targets.7−10 Unlike NO and CO, however, H2S exists in different protonation states at physiological pH, which can facilitate lipid and water solubility in the diprotic (H2S) and monoanionic (HS–) forms, respectively. Furthermore, the redox potential, nucleophilicity, and tendency to form insoluble metal salts also vary with the H2S protonation state, thus complicating reactivity with transition-metal centers.3 Despite its widespread importance, the coordination chemistry of H2S with bioinspired transition-metal scaffolds remains underexplored by comparison to CO and NO.11 Although H2S binding to ruthenium- and iron-based complexes have been reported,11−16 investigations of isolated porphyrinoid scaffolds remain limited.17−20 Motivated by the growing interest in the biochemical functions of H2S and the lack of information on the differential reactivity of H2S and HS– in bioinorganic contexts, we report here the differential reactivity of H2S and HS– toward metal phthalocyanine (Pc) complexes and highlight the chemically reversible reactions of HS– with these platforms.
Phthalocyanines are planar, aromatic porphyrin derivatives that have been used previously as models of bioinorganic reactivity including the reversible binding of NO, CO, and O2 to heme mimics21 and the reduction of CO.22 Metal phthalocyanine complexes have characteristic UV–vis spectroscopic signatures23 including the Q band (600–700 nm), which provides information on the oxidation state and binding modes of the central metal ion, as well as the B band (300–400 nm) and window region (400–550 nm), which provide information about bound ligands and the metal oxidation state.24 On the basis of these characteristics, as well as the solubility23 and redox properties,24 we viewed ZnPc and CoPc as promising initial platforms on which to investigate the differential reactivity of H2S and HS– with redox-inactive and -active metal complexes.
Because of its redox inactivity, we reasoned that the treatment of ZnPc with H2S or HS– would result in metal ligation rather than metal-based redox chemistry. To probe such reactivity, we titrated ZnPc in tetrahydrofuran (THF) with H2S gas (up to 100 equiv or by bubbling for 15 min) but failed to observe any reaction by UV–vis spectroscopy. By contrast, titration of ZnPc in THF with NaSH dissolved in dimethyl sulfoxide (DMSO) resulted in clean conversion to a new species, as evidenced by a 5 nm bathochromic shift of the Q band, the appearance of a broad absorbance centered at 410 nm, and well-anchored isosbestic points at 329, 381, and 667 nm (Figure 1a). Control experiments titrating ZnPc in THF with DMSO, H2O, KOH in DMSO, H2S in DMSO, or S8 failed to change the ZnPc UV–vis spectrum. The addition of aqueous NaSH to ZnPc in THF resulted in reactivity identical with that of the DMSO experiments, suggesting that the availability of weakly acidic protons does not influence the reactivity. Similarly, the addition of [NBu4][BH4], a stronger reductant than H2S or HS–,25 failed to change the UV–vis spectrum of ZnPc, suggesting that HS–-mediated reduction of the metal or ligand was not occurring. To probe the binding stoichiometry, we constructed a Job plot by monitoring changes in absorbance as a function of the ZnPc and NaSH molar ratios, which resulted in data consistent with 1:1 binding (Figure S2 in the Supporting Information, SI). Taken together with the above experiments, these studies suggest the formation of a [ZnPc-SH]− adduct upon treatment of ZnPc with HS–.
Figure 1.
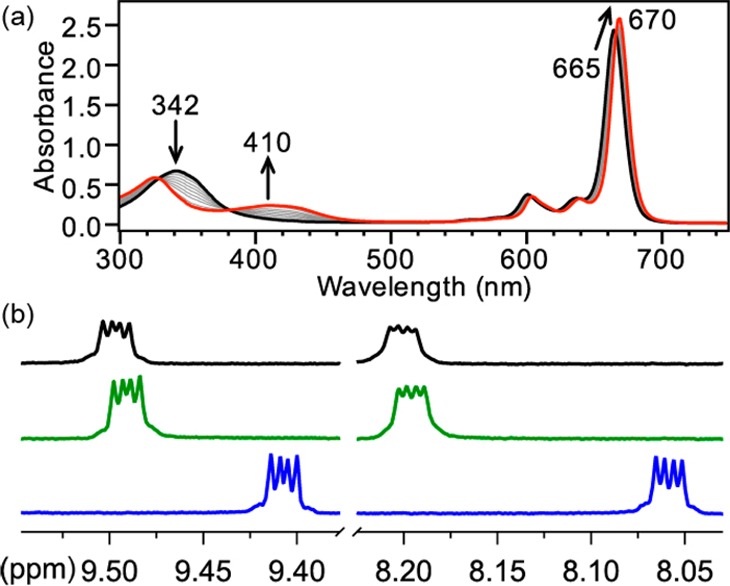
(a) UV–vis titration of ZnPc (6.3 μM in THF, black) with NaSH (0.25 equiv increments of 8 mM NaSH in DMSO up to 5 equiv). (b) 1H NMR (600 MHz, THF-d8) spectra of 600 μM ZnPc (top, black), 600 μM ZnPc with 2 equiv of KOH in DMSO-d6 (middle, green), and 600 μM ZnPc with 2 equiv of NaSH in DMSO-d6 (bottom, blue).
To confirm that HS– was binding to the zinc(II) center and not reacting with the Pc ring directly, we used 1H NMR spectroscopy to investigate changes in the Pc resonances upon reaction with NaSH. Treatment of ZnPc in THF-d8 with 2 equiv of NaSH in DMSO-d6 resulted in an upfield shift in the Pc 1H NMR resonances from 9.59 and 8.20 ppm to 9.41 and 8.06 ppm, respectively (Figure 1b). Furthermore, the dd splitting pattern of the Pc ring is maintained upon treatment with NaSH, indicating that C4 rotational symmetry is preserved. This symmetry preservation precludes the possibility of HS– nucleophilic addition or HS• radical addition into the Pc ring because such an addition would lower the overall symmetry of the complex and subsequently increase the complexity of the coupling. Treatment of ZnPc in THF-d8 with 2 equiv of KOH in DMSO-d6 failed to change the 1H NMR spectrum of ZnPc significantly, indicating that the changes in the chemical shift upon treatment of ZnPc with HS– were not simply derived from acid–base chemistry (Figure 1b).
Because ZnPc binds HS– but not H2S, we reasoned that bound HS– should be acid-labile, thus allowing for chemically reversible coordination of HS– by the addition of a suitable proton source (Scheme 1). To test this hypothesis and to demonstrate the chemically reversible binding of HS– to ZnPc, we first generated [ZnPc-SH]− in situ by treating ZnPc in THF with 10 equiv of NaSH in DMSO and then added an equimolar amount of AcOH. As predicted, the characteristic spectral features of [ZnPc-SH]− at 410 and 670 nm reverted to the 342 and 665 nm absorbances corresponding to the parent ZnPc (Figure 2). A further addition of NaSH in DMSO regenerated the 410 and 670 nm [ZnPc-SH]− spectral features.26
Scheme 1.
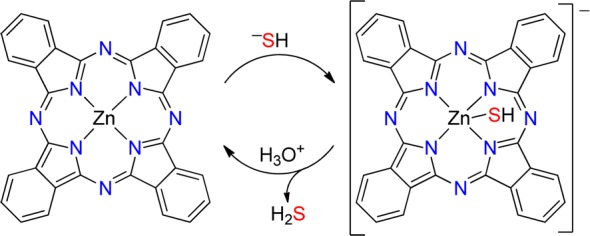
Figure 2.
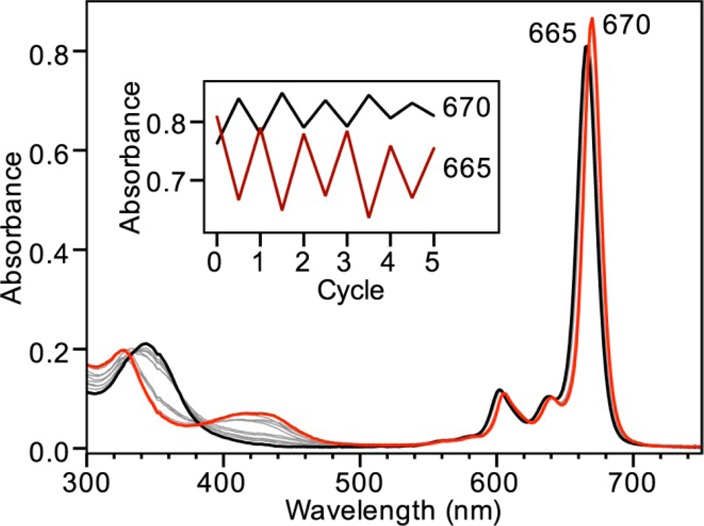
UV–vis spectra of ZnPc (2 μM in THF, black) treated with 10 equiv of NaSH in DMSO (red). Treatment with 10 equiv of AcOH regenerates the original ZnPc spectrum. This system can be cycled numerous times (inset).26
Having established that redox-inactive ZnPc binds HS– but not H2S, we next investigated the reactions of HS– and H2S with redox-active CoPc. We chose CoPc because of its well-defined and readily monitored redox states of blue CoIIPc and green [CoIPc]−.27,28 Paralleling the chemistry observed for ZnPc, CoPc does not react with H2S gas (up to 100 equiv or by bubbling for 15 min). Titration of CoPc in THF with NaSH in DMSO, however, resulted in a significant bathochromic shift of the Q band from 656 to 697 nm, the emergence of a broad absorbance at 467 nm centered in the window region, and well-anchored isosbestic points at 316, 370, 555, and 676 nm (Figure 3). These new absorbances match the reported spectrum of [CoIPc]−27 and also match the spectrum of [CoIPc]− generated from CoPc and [NBu4][BH4] (Figure S1 in the SI). A Job plot constructed by monitoring the absorbance at 467 nm as a function of the CoPc and HS– molar ratio is consistent with a 1:1 reaction of CoPc with HS– (Figure S3 in the SI). This reaction stoichiometry, as well as previous work using CoPc to oxidize thiolates to disulfides, is consistent with the initial oxidation of HS– to HSSH with potential conversion to further oxidation products (Scheme 2).29−33
Figure 3.
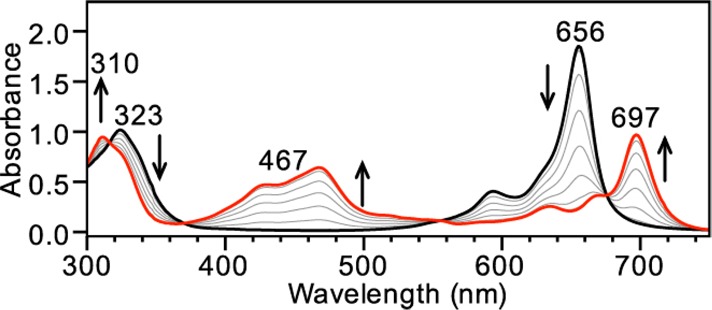
UV–vis titration showing the reduction of CoPc (7 μM in THF, black) to [CoIPc]− (red) by NaSH (1 equiv increments of 21.7 mM NaSH in DMSO up to 10 equiv).
Scheme 2.
On the basis of the observed HS–-mediated reduction of CoPc, we reasoned that the observed reactivity could be reversed by oxidation with atmospheric O2 to generate a chemically reversible and cycleable system. To demonstrate this redox cycling, we first treated a THF solution of CoPc with 10 equiv of NaSH under N2 to generate [CoIPc]− and then exposed the solution to air, which resulted in rapid oxidation back to the parent CoPc (Figure 4). The subsequent addition of NaSH regenerates [CoIPc]−. If protected from O2 under a N2 atmosphere, the [CoIPc]− product is stable and does not spontaneously revert to CoPc. Unlike ZnPc, this chemically reversible reaction with HS– results in a color change that can be easily detected by the naked eye (Figure 4, inset), highlighting the potential for future use in chemically reversible colorimetric HS– detection.
Figure 4.
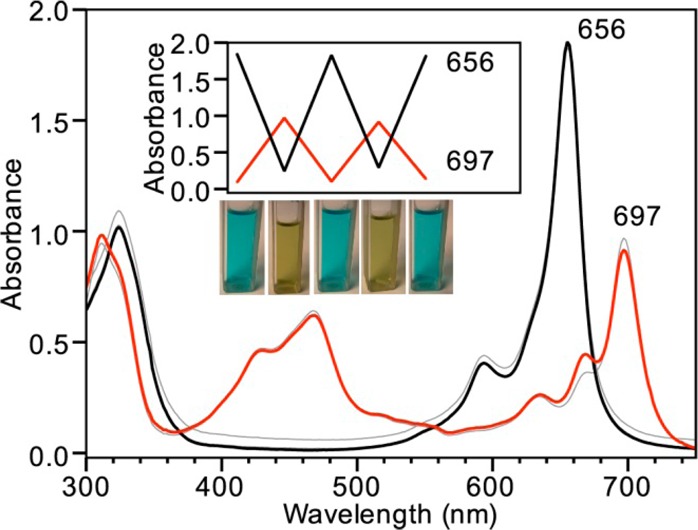
UV–vis spectra of CoPc (7 μM in THF, black trace, blue cuvette) after treatment with 10 equiv of NaSH in DMSO (red trace, green cuvette). Subsequent exposure to atmospheric O2 regenerates CoPc. The inset shows changes in the Q band, corresponding to three cycles of treatment with HS– followed by exposure to air.
Taken together, these studies with ZnPc and CoPc demonstrate the differential reactivity of HS– and H2S toward metal centers and highlight how these changes in a protonation state can be used to generate chemically reversible HS– ligation, in the case of ZnPc. Additionally, these examples of chemical reversibility clarify the fundamental reaction chemistry of porphyrin-derived scaffolds with H2S and expand the fundamental understanding of how H2S interacts with biologically relevant metal scaffolds. To further expand on this chemistry, we are currently pursuing water-soluble derivatives for chemically reversible anaerobic H2S detection, which will be reported in due course.
Acknowledgments
This work was supported by the Oregon Medical Research Foundation and the National Institute of General Medical Sciences (Grant R00GM092970). The NMR facilities at the University of Oregon are supported by NSF/ARRA (Grant CHE-0923589).
Supporting Information Available
Experimental procedures, UV–vis data, Job plots, and 1H NMR data for ZnPc and CoPc after reaction with NaSH. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Blackstone E.; Morrison M.; Roth M. B. Science 2005, 308, 518. [DOI] [PubMed] [Google Scholar]
- Czyzewski B. K.; Wang D. N. Nature 2012, 483, 494–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabil O.; Banerjee R. J. Biol. Chem. 2010, 285, 21903–21907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu K.; Lee S. W.; Bian J. S.; Low C. M.; Wong P. T. Neurochem. Int. 2008, 52, 155–165. [DOI] [PubMed] [Google Scholar]
- Shatalin K.; Shatalina E.; Mironov A.; Nudler E. Science 2011, 334, 986–990. [DOI] [PubMed] [Google Scholar]
- Yang G.; Wu L.; Jiang B.; Yang W.; Qi J.; Cao K.; Meng Q.; Mustafa A. K.; Mu W.; Zhang S.; Snyder S. H.; Wang R. Science 2008, 322, 587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe K.; Kimura H. J. Neurosci. 1996, 16, 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. Q.; Xin H.; Zhu Y. Z. Acta Pharmacol. Sin. 2007, 28, 1709–1716. [DOI] [PubMed] [Google Scholar]
- Mustafa A. K.; Gadalla M. M.; Snyder S. H. Sci. Signaling 2009, 2, re2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. Physiol. Rev. 2012, 92, 791–896. [DOI] [PubMed] [Google Scholar]
- James B. R. Pure Appl. Chem. 1997, 69, 2213–2220. [Google Scholar]
- English D. R.; Hendrickson D. N.; Suslick K. S.; Eigenbrot C. W.; Scheidt W. R. J. Am. Chem. Soc. 1984, 106, 7258–7259. [Google Scholar]
- Galardon E.; Roger T.; Deschamps P.; Roussel P.; Tomas A.; Artaud I. Inorg. Chem. 2012, 51, 10068–10070. [DOI] [PubMed] [Google Scholar]
- Ma E. S.; Rettig S. J.; Patrick B. O.; James B. R. Inorg. Chem. 2012, 51, 5427–5434. [DOI] [PubMed] [Google Scholar]
- Ma E. S. F.; Rettig S. J.; James B. R. Chem. Commun. 1999, 2463–2464. [Google Scholar]
- Reboucas J. S.; James B. R. Inorg. Chem. 2013, 52, 1084–1098. [DOI] [PubMed] [Google Scholar]
- Pavlik J. W.; Noll B. C.; Oliver A. G.; Schulz C. E.; Scheidt W. R. Inorg. Chem. 2010, 49, 1017–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reboucas J. S.; Patrick B. O.; James B. R. J. Am. Chem. Soc. 2012, 134, 3555–3570. [DOI] [PubMed] [Google Scholar]
- Meininger D. J.; Caranto J. D.; Arman H. D.; Tonzetich Z. J. Inorg. Chem. 2013, 52, 12468–12476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collman J. P.; Ghosh S.; Dey A.; Decreau R. A. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 22090–22095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collamati I.; Ercolani C.; Rossi G. Inorg. Nucl. Chem. Lett. 1976, 12, 799–802. [Google Scholar]
- Lieber C. M.; Lewis N. S. J. Am. Chem. Soc. 1984, 106, 5033–5034. [Google Scholar]
- Ghani F.; Kristen J.; Riegler H. J. Chem. Eng. Data 2012, 57, 439–449. [Google Scholar]
- Leznoff C. C.; Lever A. B. P.. Phthalocyanines: Properties and Applications; Wiley-VCH: New York, 1996; Vols. 1–4. [Google Scholar]
- Harris D. C.Quantitative Chemical Analysis, 8th ed.; W. H. Freeman and Company: New York, 2010. [Google Scholar]
- The solution becomes naturally buffered, so each addition of NaSH or AcOH required more equivalents.
- Day P.; Hill H. A. O.; Price M. G. J. Chem. Soc. A 1968, 90–91. [Google Scholar]
- Clack D. W.; Yandle J. R. Inorg. Chem. 1972, 11, 1738–1742. [Google Scholar]
- Fischer H.; Schulz-Ekloff G.; Wohrle D. Chem. Eng. Technol. 1997, 20, 624–632. [Google Scholar]
- Pereira-Rodrigues N.; Cofre R.; Zagal J. H.; Bedioui F. Bioelectrochemistry 2007, 70, 147–154. [DOI] [PubMed] [Google Scholar]
- Qi X. H.; Baldwin R. P. J. Electrochem. Soc. 1996, 143, 1283–1287. [Google Scholar]
- Rao T. V.; Rao K. N.; Jain S. L.; Sain B. Synth. Commun. 2002, 32, 1151–1157. [Google Scholar]
- Faddeenkova G. A.; Kundo N. N. Russ. J. Appl. Chem. 2003, 76, 1946–1950. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.