

Abstract
Poly(ADP-ribose), or PAR, is a cellular polymer implicated in DNA/RNA metabolism, cell death, and cellular stress response via its role as a post-translational modification, signaling molecule, and scaffolding element. PAR is synthesized by a family of proteins known as poly(ADP-ribose) polymerases, or PARPs, which attach PAR polymers to various amino acids of substrate proteins. The nature of these polymers (large, charged, heterogeneous, base-labile) has made these attachment sites difficult to study by mass spectrometry. Here we propose a new pipeline that allows for the identification of mono(ADP-ribosyl)ation and poly(ADP-ribosyl)ation sites via the enzymatic product of phosphodiesterase-treated ADP-ribose, or phospho(ribose). The power of this method lies in the enrichment potential of phospho(ribose), which we show to be enriched by phosphoproteomic techniques when a neutral buffer, which allows for retention of the base-labile attachment site, is used for elution. Through the identification of PARP-1 in vitro automodification sites as well as endogenous ADP-ribosylation sites from whole cells, we have shown that ADP-ribose can exist on adjacent amino acid residues as well as both lysine and arginine in addition to known acidic modification sites. The universality of this technique has allowed us to show that enrichment of ADP-ribosylated proteins by macrodomain leads to a bias against ADP-ribose modifications conjugated to glutamic acids, suggesting that the macrodomain is either removing or selecting against these distinct protein attachments. Ultimately, the enrichment pipeline presented here offers a universal approach for characterizing the mono- and poly(ADP-ribosyl)ated proteome.
Keywords: poly(ADP-ribose), mono(ADP-ribose), pADPr, PAR, poly(ADP-ribose) polymerase, mass spectrometry, MS/MS, post-translational modifications, phosphodiesterase, phosphoproteomics, phosphoenrichment, IMAC, MOAC, macrodomain
Introduction
ADP-ribose (ADPr) is a post-translational modification that is synthesized by a family of ADP-ribosyltransferases,2 commonly known as poly(ADP-ribose) polymerases, or PARPs. These modifications are derived from the hydrolysis of NAD+ and exist in both the monomeric and polymeric forms, the latter of which is made up of 2–200 ADPr subunits. The canonical role for this polymer has been in the identification and repair of DNA nicks and double-stranded breaks via activation of the founding member of the PARP family, PARP-1.6 Indeed, this role has ushered in PARP-1 as a chemotherapeutic target, as the loss of PARP-1 sensitizes cells to genomic assault by established chemotherapeutic and radiation-based treatment.8 It is worth noting, however, that PAR’s cellular role has expanded beyond DNA repair into regulation of (among others) apoptosis,9 chromatin structure,10 synthesis of DNA/RNA,11 telomere maintenance,12 protein degradation,13 and microRNA activities.14 Not surprisingly, the increase in understanding of PAR’s biological roles has led to recognition of its therapeutic potential beyond modulation of DNA damage, including the treatment of necrosis and inflammation.15 PAR’s relative, mono(ADP-ribose), is far less studied but has received increasing attention due to a number of recent studies that have identified the enzymes which reverse mono(ADP-ribosylation) as well as novel roles for mono(ADP-ribose) in the cell.16 In an effort to aid in the understanding of the mono- and poly(ADP-ribosyl)ated proteome, we have looked to mass spectrometry to define the molecular basis of ADP-ribosylation and will begin by characterizing the poly(ADP-ribosylation) (or PARylation) activity of human PARP-1 (hPARP-1).
The hurdles that have kept mass spectrometry and proteomics from becoming universal tools for studying PARylation have to do with the physical properties of the modification itself: first, the modification can expand linearly or by branching and vary dramatically in length, resulting in a large, heterogeneous polymer without a defined mass. Second, many of the amino acid attachment sites are base-labile,17 preventing researchers from exposing the modified proteins or peptides to basic solutions, which are commonly used in proteomic sample preparations. Finally, the modification is dynamic, with basal levels existing below the level of detection of most molecular tools used in proteomics. One recently published approach to identify ADP-ribosylation sites by mass spectrometry paired boronate enrichment of ADP-ribosylated proteins with subsequent release of mono- and poly(ADP-ribose) from substrates by hydroxylamine.7 This elution strategy breaks ester bonds between the ADPr subunits and the carboxyl groups of aspartate and glutamate residues, leaving a characteristic 15.01 Da mass signature on the modified residue. Notably, this approach cannot identify nonacidic ADP-ribosylated residues and up to 33% of total ADP-ribosylated amino acid residues have been shown to be hydroxylamine-insensitive.18 In particular, lysine residues are important for the in vitro and in vivo activation of PARP-11,19 as well as substrate regulation by PARPs, for example, chromatin remodeling via PARylation of the lysine residues on histone tails.20
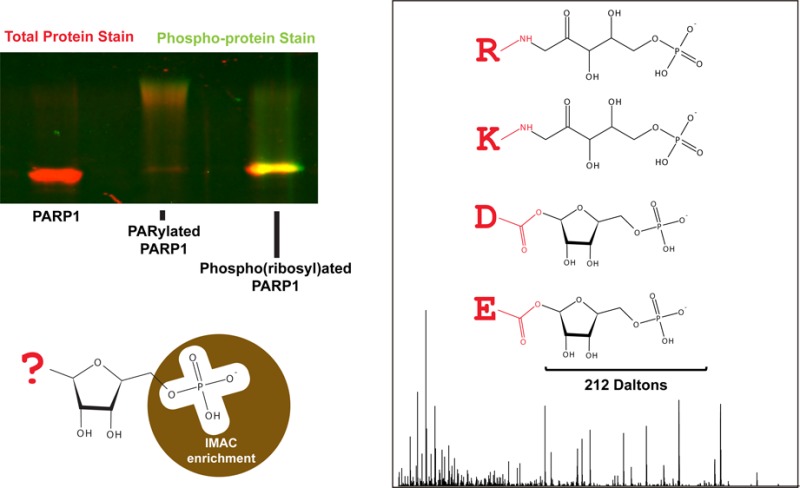
Because a global approach to identify all possible ADP-ribosylation sites is still needed, we have developed an enrichment protocol based on the digestion of ADPr by snake venom phosphodiesterase (SVP), a pyrophosphatase that cleaves ADPr subunits down to phospho(ribose) and 5′-AMP.21 This digestion produces a single phospho(ribose) group at the site of modification that can be identified by mass spectrometry as an adduct of 212.01 Da.22 Given the similarity of phospho(ribosyl) and phosphate groups, we reasoned that existing phosphoproteomic techniques may be used to enrich phospho(ribosyl)ated peptides. Indeed, a 2010 phosphoenrichment study that utilized immobilized metal affinity chromatography (IMAC) to enrich phosphopeptides was searched in 2012 for a coenrichment of ADPr or phosphoribose, both of which were found to have been enriched.23 More recently, Chapman et al. demonstrated the feasibility of this approach to identify PARylation sites on a purified, automodified human PARP-1.5 Here we have independently tested and validated this approach to identify ADP-ribosylation sites; we further compared three commercially available phosphoenrichment matrices and their use in enriching and characterizing phospho(ribosyl)ated peptides of hPARP-1 from a complex background of HeLa whole cell lysate. Finally, we have demonstrated the application of this method to identify endogenous mono- and poly(ADP-ribosyl)ation sites by mass spectrometry, yielding both known and novel acceptors of ADPr, including a number that identify ADPr on arginine residues.
Materials and Methods
Expression and Purification of HisPARP-1
The method was adapted from Langelier et al.24 In brief, 6 L of His-PARP-1 expressing DE3 cells was lysed in a cell homogenizer in the presence of 0.1% NP-40, 20 U/mL DNase I, 5 mM MgCl2, 1 μM bestatin, 1 μM pepstatin A, and 1× Roche cOmplete EDTA-free protease inhibitor. Lysate was cleared by centrifugation and loaded onto an ÄKTA FPLC (GE, 18-1900-26) with a pre-equilibrated 5 mL HisTrap FF Crude column (GE, 17-5286-01), where it was washed with 10 column volumes of loading buffer (20 mM sodium phosphate pH 7.4, 1 M NaCl, 0.5 mM TCEP, 40 mM imidazole pH 7.4, 1% glycerol, 1× Roche cOmplete EDTA-free protease inhibitor) before being eluted in 2 column volumes of elution buffer (20 mM sodium phosphate pH 7.4, 0.5 M NaCl, 0.5 mM TCEP, 0.5 M imidazole pH 7.4, 1% glycerol). Eluted samples were diluted 1:1 in heparin no-salt buffer (50 mM Tris pH 7.0, 0.1 mM tris(2-carboxyethyl)phosphine, 1% glycerol) and loaded onto a pre-equilibrated 5 mL heparin column (GE, 17-0407-01), washed with 5 volumes of low-salt buffer (50 mM Tris pH 7, 0.1 mM TCEP, 250 mM NaCl) and eluted over a gradient from 0 to 70% high-salt buffer (50 mM Tris pH 7, 0.1 mM TCEP, 1 M NaCl, 1% glycerol). Desired fractions were pooled and concentrated using a spin concentrator (30 000 MWCO, Amicon Z648035) before being loaded onto a pre-equilibrated size exclusion column (GE, Superdex 200/10/300 GL) in size purification buffer (25 mM HEPES pH 8, 0.1 mM TCEP, 150 mM NaCl); desired fractions were pooled and stored at −80 °C. All FPLC results were analyzed with UNICORN 5.01 (Build 318).
Purification of Snake Venom Phosphodiesterase I
The protocol was adapted from Oka et al.25 In brief, two 100 unit vials of Crotalus adamanteus phosphodiesterase I (Worthington, LS003926) were dissolved into 1 mL of loading buffer (10 mM Tris-Cl pH 7.5, 50 mM NaCl, 10% glycerol) and then loaded onto a pre-equilibrated 1 mL HiTrap blue sepharose column (GE, 17-0412-01), washed with 5 column volumes of loading buffer and then 5 column volumes of elution buffer (10 mM Tris-Cl pH 7.5, 50 mM NaCl, 10% glycerol, 150 mM potassium phosphate). Desired fractions were pooled, dialyzed against loading buffer, and stored at −80 °C. If enzyme preps were to be used to treat denatured proteins, an additional purification was needed to remove any contaminating proteases: samples were dialyzed into size exclusion chromatography buffer (10 mM Tris-Cl pH 7.3, 50 mM NaCl, 15 mM MgCl2, 1% glycerol) and resolved over a SuperDex 200/10/300 GL (GE Healthcare) using an ÄKTA FPLC (GE, 18-1900-26); desired fractions were pooled and stored at −80 °C. All FPLC results were analyzed with UNICORN 5.01 (Build 318).
Preparing Oligos for In Vitro PARP-1 Activation
Oligo sequences were from Langelier et al.24 Forward (GGGTGGCGGCCGCTTGGG) and reverse (CCCAAGCGGCCGCAACCC) oligos were mixed 1:1 in H2O, heated to 95 °C for 2 min, and then ramp-cooled to 25 °C over 45 min.
Automodification of HisPARP-1 In Vitro
HisPARP-1 was attached to Promega MagneHis beads (1 μg PARP-1/μL beads/5 μL attachment buffer) for 2 h at 4 °C in attachment buffer (50 mM Tris pH 7.4, 1% Tween, 0.2 mM DTT, 10% glycerol, 10 mM MgCl2). Beads were washed twice with 100 μL of wash buffer (50 mM sodium phosphate buffer pH 7.4, 200 mM NaCl, 5 mM imidazole pH 7.4) and then exposed to 30 μM (0.6% hot) 32P β-NAD+ in automodification buffer (20 mM Tris pH 7.5, 50 mM NaCl, 50 μM TCEP, 5 mM MgCl2, 1.2 μM annealed DNA) for 10 min, followed by a chase of 2 mM cold β-NAD+ for 60 min, all at 25 °C rotating at 500 rpm. For SDS-PAGE, beads were washed twice in 100 μL of wash buffer and eluted into 15 μL of 1× SDS-PAGE buffer, separated on an in-house 6–10% SDS-PAGE gel, fixed overnight (50% methanol, 10% acetic acid), washed for 30 min (H2O), stained with Pro-Q Diamond phosphoprotein stain (Invitrogen, MP 33300) for 1 h, destained for 3 × 30 min (20% acetonitrile, 50 mM sodium acetate pH 4), washed for 10 min (H2O), and imaged on a Fuji FLA7000 (filter: O580, wavelength: 532 nm). Pro-Q Diamond staining was validated based on comparison to Pro-Q Diamond PeppermintStick ladder (Life Technologies, P27167). Total protein was determined by Coomassie Blue staining (Invitrogen LC6060) and 32P-labeling was determined by overnight exposure against a phosphor-screen (GE, BAS-III 2040), followed by imaging on a Fuji FLA7000 (IP). Western blotting for poly(ADP-ribose) was performed by transferring proteins (Invitrogen XCell II Blot Module) from an in-house 6–10% SDS PAGE gel to a nitrocellulose membrane (Bio-Rad), and membranes were blocked in 5% milk in PBS before being incubated in primary antibody (anti-PAR, clone LP-9610 from BD Biosciences) for 1 h at room temperature, rinsed in PBS-T, and then incubated in secondary antibody (Anti-Rabbit 800 nm from LI-COR Biosciences) for 1 h before being imaged on an Odyssey CLx and analyzed in Image Studio (from LI-COR, version 2.0).
SVP Digestion of In Vitro PARylated HisPARP-1
One μg of PARylated HisPARP-1 was treated with 500 mUnits of purified SVP in SVP digestion buffer (50 mM Tris pH 7.5, 150 mM NaCl, 15 mM MgCl2, 20 mM 3-aminobenzamide) for 2 h at 25 °C, 500 rpm
Testing Loss of PARylation by Exposure to Phosphoelution Conditions
hPARP-1 was induced to automodify in vitro (as previously described) and mixed in a 1:2 ratio with BSA, and 1 μg hPARP–1/2 μg BSA was aliquoted and exposed to 5% ammonium hydroxide, 500 mM KH2PO4 pH 7, or automodification buffer (control) in a total volume of 10 μL for 5 min. Reactions were quenched by adding 1 mL of ice-cold precipitation buffer (0.02% deoxycholate, 4% Triton X-100, 10% TCA) and stored at −20 °C for 2 h before being pelleted by centrifugation at 4 °C and decanted. Pellets were washed with ice-cold acetone containing 20 μg/mL glycogen as a carrier, stored at −20 °C for 30 min, pelleted, decanted, dried by speedvac, and resuspended in SDS Running Buffer. For SDS-PAGE analysis, an equal volume of 2× SDS-PAGE buffer was added to samples for analysis on an in-house 6–10% tris-glycine gel.
Cell Culture
HeLa cells (Kyoto) were grown in arginine and lysine free DMEM (Pierce) containing 10% dialyzed FBS (Sigma), 0.4 mM arginine (13C615N4 from Cambridge, 12C614N4 from Sigma), and 0.8 mM lysine (13C615N2 from Isotec, 12C614N2 from Sigma). Trophoblast stem cells from PARG knockout mice (E3.5 from 129.SVJ mice, acquired from Dr. David Koh of Johns Hopkins University26) were grown in arginine- and lysine-free RPMI 1640 (Pierce) containing 16% dialyzed FBS (Sigma), 0.4 mM arginine (13C615N4 from Cambridge, 12C614N4 from Sigma), 0.8 mM lysine (13C615N2 from Isotec, 12C614N2 from Sigma), 1 mM sodium pyruvate (Life Technologies), 2 mM l-glutamine (Life Technologies), 25 units/mL penicillin (CellGro), 25 units/mL streptomycin (cellgro), 100 μM monothioglycerol (Sigma), 1 μg/mL heparin sulfate, 25 ng/mL FGF-4, and 0.5 mM benzamide (Sigma). PARG knockout cells were grown without benzamide for 48 h before harvesting. All cells were treated with 5 mM N-methyl-N′-nitro-N-nitrosoguanidine (MNNG, from AccuStandard) for 5 min before being washed three times with ice cold PBS (Gibco) and lysed in either 6 M guanidine-hydrochloride (Sigma), 8 M urea (Sigma) or lysis buffer (50 mM Tris pH 7.5, 0.4 M NaCl, 1 mM EDTA, 1× EDTA-free cOmplete protease-inhibitor from Roche, 1% NP-40, 1 μg/mL ADP-HPD, and 0.1% sodium deoxycholate). Cells lysed in either guanidine-hydrochloride or urea were subjected to sonication in an ice bath for 10 min with 30 s breaks between 30 s cycles (Bioruptor Standard). Cells lysed in lysis buffer were left on ice for 10 min. Following lysis, all cell debris was cleared by centrifugation.
PAR Enrichment by Macrodomain
Two mg of whole cell lysate in 1× lysis buffer (50 mM Tris pH 7.5, 0.4 M NaCl, 1 mM EDTA, 1× EDTA-free cOmplete protease-inhibitor from Roche, 1% NP-40, 1 μg/mL ADP-HPD, and 0.1% sodium deoxycholate) was incubated at 5 mg/mL with 40 μL of macrodomain-conjugated agarose beads (Tulip #2302) at 4 °C overnight before being washed three times with wash buffer (50 mM Tris pH 7.5, 0.4 M NaCl, and 0.1% sodium deoxycholate) and eluted by 8 M urea pH 7.
SVP Digestion of Endogenous Proteins with or without Protein Standard (hPARP-1)
All proteins were denatured in 8 M urea pH 7 for 10 min at 37 °C before being reduced in 1 mM Tris(2-carboxyethyl)phosphine (Life Technologies) for 10 min and then alkylated in 2 mM 2-chloroacetamide (Sigma) for 10 min in the dark. If automodified hPARP-1 is to be added as a standard, it is prepared the same way and added to the lysate background at this point. Samples were then diluted to a final concentration of 1 M urea, 50 mM NaCl, 15 mM MgCl2, 1 mM CaCl2, and 0.2 M Tris pH 7.3. Five μg of purified SVP were added for each mg of whole cell lysate and incubated for 2 h at 37 °C.
In-Solution Protein Digestion
Samples in 1 M urea, 0.2 M Tris-Cl pH 7.3, 1 mM CaCl2, 15 mM MgCl2, and 50 mM NaCl are treated with endoproteinase LysC (Wako) 1:50 enzyme/substrate. After 1 h, trypsin (Sigma) was added at a 1:50 enzyme/substrate ratio, and the entire reaction was incubated overnight. Reaction was stopped by adding an equal volume of desalting solvent A (5% acetonitrile, 0.1% TFA) and desalted on a C18 StageTip and eluted in desalting solvent B (80% acetonitrile, 0.1% TF) as in Rappsilber et al.27
Phosphoenriching Peptide Standards from HeLa Whole Cell Lysate Peptide Background
HeLa cells were scraped into 6 M Gnd-HCl, lysed by sonication, and cleared by centrifugation. 300 μg of protein was then reduced, alkylated, and in-solution digested by LysC and Trypsin, as described in “In-solution protein digestion”. To this mixture of peptides, 30 μg of peptides from automodified, SVP-treated hPARP-1 and 10 μg of peptides from bovine casein were added. This mixture was then sampled as input and split into 3 equal volumes that were enriched by either IMAC (Sigma PHOS-Select beads) or MOAC (GL Sciences or GlySci tips containing ZirChrom TiO2 beads). IMAC samples were enriched as in Villen et al. 2008;28 in brief, they were incubated for 1 h, shaking at 25 °C, on 50 μL of PHOS-Select beads in binding buffer (0.1% formic acid, 40% acetonitrile). These beads were then transferred to a pre-equilibrated StageTip,27 where they were washed with binding solvent three times, acidified with 1% FA, and eluted onto the StageTip with 0.5 M potassium phosphate pH 7, where they were acidified with 1% FA again and washed with desalting solvent A (5% acetonitrile, 0.1% TFA). They were then eluted with Desalting Solvent B (80% acetonitrile, 0.1% TFA). MOAC samples were enriched by either GL Sciences or GlySci TiO2 tips, both by their manufacturer’s protocols with the adaptation that they were eluted with 0.5 M potassium phosphate pH 7.
NanoLC–MS/MS Analysis
Peptides were separated on a Thermo-Dionex RSLCNano UPLC instrument with ∼10 cm × 75 μm ID fused silica capillary columns with ∼10 μm tip opening made in-house with a laser puller (Sutter) and packed with 3 μm reversed phase C18 beads (Reprosil-C18.aq, 120 Å, Dr. Maisch) with a 90 min gradient of 3–35% B at 200 nL/min. Liquid chromatography (LC) solvent A was 0.1% acetic acid and LC solvent B was 0.1% acetic acid, 99.9% acetonitrile. MS data were collected with a Thermo Orbitrap Elite. Data-dependent analysis was applied using Top5 selection, and fragmentation was induced by CID and HCD. Profile mode data were collected in all scans.
Database Search of MS/MS Spectra for Peptide and Protein Identification
Raw files were analyzed by MaxQuant version 1.4.0.8 using protein, peptide, and site FDRs of 0.01 and a score minimum of 40 for modified peptides and 0 for unmodified peptides and delta score minimum of 17 for modified peptides and 0 for unmodified peptides. Sequences were searched against the UniProt Human Database (definitions updated May 29, 2013). Endogenous phospho- and phosphoribose peptide lists were further restricted by a delta ppm of ±2σ from each respective data set (average and standard deviation were calculated from the complete tandem mass spectra (MS/MS) list of identified peptide precursors) and the expected heavy/light ratios (≥1 for heavy or light data sets, respectively). MaxQuant search parameters: Variable modifications included oxidation (M), acetylation (Protein N-term), phosphorylation (STY), and phosphoribosylation (DEKR). Phosphoribosylation (DEKR) allowed for neutral losses of H3PO4 (phosphoric acid, 97.98 Da) and C5H9PO7 (phosphoribose, 212.01 Da). Carbamidomethyl (C) was a fixed modification. Max-labeled amino acids were 3, max missed cleavages were 2, enzyme was Trypsin/P, max charge was 7, multiplicity was 2, and SILAC labels were Arg10 and Lys8.
Results
SVP Treatment of PARylated Substrates Generates Phospho(ribosyl)ated Proteins, Which Can Be Stained by a Phosphoprotein Dye, Pro-Q Diamond
As a model for protein PARylation we utilized hPARP-1, a poly(ADP-ribose) polymerase capable of autopoly(ADP-ribosyl)ation. Exposure of hPARP-1 to 32P-labeled β-NAD+ resulted in an increase in molecular weight of hPARP-1 above its unmodified mass of 113 kDa, which was correlated with the 32P signal observed in the autoradiograph, indicating incorporation of 32P-ADPr via hPARP-1 automodification (Figure 1a,b, lane 1 vs lane 2). Upon treatment with SVP (lane 3), the majority of the “smear” was lost by both Coomassie blue and 32P detection with an accompanied increase in the intensity of the Coomassie-stained band at the expected size of unmodified hPARP-1. This result demonstrates SVP’s ability to break down the polymer at pyrophosphate bonds, potentially reducing the polymer entirely to the phospho(ribosyl) group on the modified amino acid residue of PARylated proteins.
Figure 1.

Visualizing phospho(ribose) tags on hPARP-1. (a–c) hPARP-1 automodified in vitro upon exposure to 32P-labeled NAD+, PAR formation is evidenced by the 32P-labeled smear above unmodified hPARP-1 (arrowheads). Upon treatment with SVP, the smear diminishes while the native-sized hPARP-1 band reappears. Staining with the phosphostain Pro-Q Diamond indicates that this band is carrying phospho-groups, likely phosphoribose. (d,e) Pro-Q positive product of SVP-treated automodified hPARP-1 is susceptible to calf intestinal phosphatase (CIP) treatment.
Because of the similarity of phospho(ribose) and phosphate groups, we posited that the phospho(ribosyl)ated hPARP-1 might share properties with phosphoproteins. To test this hypothesis, we used the phosphoprotein gel stain, Pro-Q Diamond, to stain the polyacrylamide gel in Figure 1a (Figure 1c). While unmodified hPARP-1 and modified hPARP-1 were weakly stained with Pro-Q Diamond, the signal was significantly increased for SVP-treated hPARP-1 (Figure 1c, lanes 1–3). The phospho-specificity of the dye was confirmed with the two phosphoprotein controls, ovalbumin and β-casein, in the protein molecular weight ladder (Figure 1c, marker). To confirm that the staining associated with SVP-treated hPARP-1 is due to the presence of phosphate groups, we treated the samples with calf intestinal phosphatase (CIP). As expected, upon removal of the phosphate groups by CIP, the resultant ribosylated hPARP-1 was no longer stained by Pro-Q diamond (Figures 1d,e). These data suggest that SVP treatment of PARylated substrates produces phospho(ribose) groups and that the resultant phosphate groups may have similar physicochemical properties to phosphate groups in phosphoproteins. We then sought to examine our ability to enrich these phospho(ribose) groups using phosphopeptide enrichment strategies.
Neutral Phosphate Buffer Preserves Base-Labile ADP-Ribose Bonds and Serves as a Safe Alternative to Ammonia for Peptide Elution
Popular phosphoproteomic approaches use immobilized metal affinity chromatography (IMAC) or metal oxide affinity chromatography (MOAC) to enrich phosphopeptides, followed by elution with ammonium hydroxide. Unfortunately, ammonium hydroxide is highly basic and therefore releases ADPr from glutamic and aspartic acid residues.17 For this reason, we considered an alternative elution condition, neutral phosphate buffer, which has been used previously to competitively elute phosphopeptides.28 To assess ADPr stability in the presence of phosphate buffer 32P-labeled, automodified hPARP-1 was exposed to either 5% NH4OH, 0.5 M KH2PO4 pH 7 or control (automodification buffer containing 20 mM Tris pH 7.5). As can be seen in Figure 2, both the control and the neutral phosphate buffers maintained hPARP-1 in its PARylated form (smeared) while ammonia hydrolyzed PAR, returning much of the hPARP-1 to its native size by Coomassie (Figure 2a) and removing 32P-labeled PAR, as shown in the autoradiograph (Figure 2b). These results suggest that the standard alkaline conditions in phosphoproteomic elution protocols result in the loss of PARylation, while the neutral phosphate buffer preserves the ADPr-protein bond and should be a safe method to elute phospho(ribosyl)ated peptides from phospho-affinity matrices.
Figure 2.

Poly(ADP-ribose) is stable in the presence of neutral phosphate buffer. (a) Coomassie staining shows that PARylated hPARP-1 returns to its unmodified size upon treatment with ammonia for 5 min, while neutral phosphate retains the PARylation state as well as the control buffer (automodification buffer). (b) 32P-labeled PAR shows that the loss of PAR is correlated to the return of hPARP-1 to its native size. BSA (bovine serum albumin) was included as a carrier for sample cleanup by protein precipitation, which was the method applied to immediately quench the chemical exposure.
Quantitative Comparison of Phosphoproteomic Techniques in Coenriching Phospho(ribosyl)ated Peptides and Phosphopeptides
Next, we explored whether phospho(ribosyl)ated peptides can be enriched from cellular complex mixtures using phosphoenrichment matrices. SVP-treated hPARP-1 was mixed with HeLa cell lysate, which was SILAC29 labeled in “heavy” culture medium containing 13C6,15N2-lysine and 13C6,15N4-arginine (Supplementary Figure 1 in the Supporting Information). Because we expect the human hPARP-1 spectra to be derived from SVP-treated, unlabeled “light” hPARP-1 samples, we can verify the source of the peptide by SILAC state. As a positive phosphoenrichment control, peptides from known phosphoprotein standards, bovine caseins, were also added to the whole cell lysate background. The complex peptide mixture was subjected to three commercially available phosphoenrichment matrices: (1) Sigma PHOS-Select iron affinity gel (PS), (2) GL Science Titansphere Phos-TiO2 tips (GL), and (3) GlySci phosphopeptide NuTip using ZirChrom titanium dioxide beads (ZC). In each case, peptides were eluted with 0.5 M potassium phosphate buffer at pH 7.0 to preserve the labile bond between phospho(ribose) and acidic amino acids. Mass spectrometry data were collected on an Orbitrap, and fragmentation was induced by both collision-induced dissociation (CID) and higher-energy C-trap dissociation (HCD).
Overall, our complex background consisted of 44 655 peptides from 2148 proteins and included 3421 endogenous phosphopeptides. (See Supplementary Tables 2–4 in the Supporting Information.) Out of this background we identified 47 unique phosphopeptides from the spiked-in phosphoprotein standards (bovine caseins) using all enrichment techniques (Supplementary Table 1 in the Supporting Information, Figure 3a, and Supplementary Figure 2a,e in the Supporting Information). While PHOS-Select contributed the most unique peptide identifications (36%), both GL Sciences and Zirchrom found peptides that would not have otherwise been identified (2 and 13%, respectively). This stands in contrast with the 29 unique hPARP-1 phospho(ribosyl)ated peptides, of which nearly 60% were found exclusively through enrichment by PHOS-Select (Supplementary Table 1 in the Supporting Information, Figure 3b, and Supplementary Figure 2b,f in the Supporting Information), and only a single peptide (3%) was found solely by an alternative enrichment (ZirChrom). Further assessment of the PHOS-Select enrichment profile reveals that the 39 unique phosphopeptides and 28 unique phospho(ribose) peptides found in the PHOS-Select eluate entirely overlapped with the small number of peptides, which were found in the respective input and flowthrough analyses (Figure 3c,d and Supplementary Figure 2c,d,g,h in the Supporting Information).
Figure 3.

IMAC and MOAC enrichment of phospho- and phospho(ribose) peptides. IMAC (PHOS-Select, PS) was compared with MOAC (both ZirChrom, ZC and GL Sciences, GL) for enrichment of phosphopeptides (from bovine casein) and phospho(ribose) peptides (from hPARP-1) out of HeLa whole cell lysate background. (a,b) Unique phosphorylated (a) and phospho(ribosyl)ated (b) peptides identified in eluates from the three methods. (c,d) Unique phosphorylated (c) and phospho(ribosyl)ated (d) peptides identified in the unenriched (input), elution, and flowthrough from the IMAC method.
To determine whether the protocol proposed here is as robust for phospho(ribosyl)ated peptides as phosphopeptides, we performed a serial enrichment that included re-enriching the flowthrough sample multiple times to quantify the depletion of these two classes of target peptides (Figure 4). Automodified hPARP-1 was again used as the PAR standard; however, this time the PARylated hPARP-1 was denatured in 8 M urea, reduced, and alkylated prior to being added to the whole cell lysate background (Figure 4a). This denaturation step served to completely inactivate hPARP-1 (see Supplementary Figure 3 in the Supporting Information), thus allowing us to perform SVP digestion of the whole cell lysate and the PARylated standard in the same mixture. Furthermore, the His-tag on hPARP-1 allowed us to isolate a portion of the standard back out of the mixture both before and after SVP digestion; these samples served as a quality-control step as the loss of PAR and the formation of phospho(ribose) could be monitored by SDS-PAGE and Western blot. (See Figure 4b–d). As shown in Figure 4e, both classes of peptides are depleted from the background population at similar rates (as opposed to phosphopeptides being enriched preferentially prior to phospho(ribosyl)ated peptides), indicating that the IMAC method proposed is truly a dual enrichment of both phospho- and phospho(ribosyl)ated peptides. It should be noted that the peptides identified in this study include both those from the hPARP-1 standard as well as the endogenous phospho- and phospho(ribosyl)ation sites from the MNNG-treated murine PARG knockout cells used to generate the heavy-labeled complex background. For a complete list of endogenous phospho- and phospho(ribosyl)ated peptides, see Supplementary Tables 5 and 6 in the Supporting Information.
Figure 4.

Serial enrichment of phospho- and phospho(ribosyl)ated peptides out of a complex mixture. His-tagged, automodified PARP-1 was denatured in 8 M urea and spiked into heavy-labeled whole cell lysate from MNNG-treated murine PARG knockout cells before being treated by SVP and then digested to peptides and enriched three times in a row on IMAC beads (a). Samples were taken before and after SVP treatment, and the His-tagged PARP-1 was separated from the whole cell lysate by nickel-chelated agarose beads, allowing visualization of the total protein (b), PARylated His-PARP-1 (c), and phospho(ribosyl)ated His-PARP-1 (d). MS/MS analysis of the serial enrichments showed that the endogenous phospho-peptides and the phospho(ribosyl)ated PARP-1 peptides were depleted from the complex mixture at similar rates (e).
Characteristics of Phospho(ribosyl)ated peptides
Among the phospho(ribosyl)ated peptides identified from the hPARP-1 standard, 20 unique sites were modified. Many of these sites were outside of the automodification/BRCT domains that are known to be heavily PARylated30 (Table 1), and in fact, over one-third of the sites identified (7/20) are in the second zinc finger, which is not strictly required for PARP-1 activation.31 Of the 20 potential PARylation sites, 1 arginine, 3 lysine, 4 aspartate, and 12 glutamate residues were identified. While the basic sites may seem surprising we emphasize that the inherent NADase activity of PARP-132 has the potential to create free ADPr, a molecule that can spontaneously modify basic sites independent of PARP-1’s conjugation activity.33 Because this nonenzymatic mechanism of ADP-ribosylation is still under investigation, we believe the ability of this method to identify the presence of ADPr on both basic and acidic modifications will prove highly useful in elucidating methods of ADPr modification and automodification.
Table 1. PARP-1 Automodification Sites Identifieda.
| AA | no. | domain | novel | endogenous7 |
|---|---|---|---|---|
| E | 76 | ZF1 | N7 | Y |
| D | 112 | ZF2 | Y | |
| E | 116 | N5,7 | Y | |
| D | 145 | Y | ||
| E | 147 | N4,5 | ||
| E | 168 | N4,7 | Y | |
| E | 190 | N4,5,7 | Y | |
| D | 191 | Y | ||
| K | 239 | ZF3 | Y | |
| R | 452 | BRCT | Y | |
| E | 471 | N4,7 | Y | |
| E | 484 | N4,7 | Y | |
| K | 486 | Y | ||
| E | 488 | N3−5,7 | Y | |
| E | 491 | N3−5,7 | Y | |
| K | 498 | undefined | N1 | |
| E | 619 | WGR | Y | |
| E | 642 | N7 | Y | |
| D | 648 | N7 | Y | |
| E | 649 | Y |
A total of 20 automodification sites were identified on the PARP-1 standard used for assessing phosphoenrichment techniques. 12 of these sites were previously identified and are annotated as such. Those that were identified by Zhang et al. are known to be endogenous PARylation sites. ZF1 = Zinc Finger 1, ZF2 = Zinc Finger 2, BRCT = BRCA1 C-terminus, and WGR = tryptophan, glycine, arginine-rich.
Among the 20 hPARP-1 automodification sites identified, 10 were independently verified as endogenous sites in DNA damaged cells in a recent analysis.7 While most peptides presented with a single phospho(ribose), there were three examples of doubly phospho(ribosyl)ated peptides that demonstrated the ability of hPARP-1 to place these large, negatively charged polymers within close proximity of each other (Supplementary Spectra in the Supporting Information). A notable example of this is the dual modification of E488 and E491 PARP-1 automodification sites, which have been independently verified by a number of groups, including Zhang et al., who identified them as endogenous ADP-ribosylation sites (Table 1). Here we have shown the fragmentation patterns of the unmodified, singly modified and doubly modified forms of this peptide by both CID (Figure 5) and HCD (Supplementary Figure 4 in the Supporting Information), indicating the shift in molecular weight corresponding to a single (circle) or a double (square) phospho(ribose) group. The doubly modified peptide also demonstrates the potential for neutral loss of phosphoric acid (H3PO4 97.98 Da) and phospho(ribose) (C5H9PO7, 212.01 Da) from the parent ion upon fragmentation (Figure 5, Supplementary Figure 5 in the Supporting Information); these neutral losses were observed in 73% (16/22) of the spectra annotated for validation of the PARP-1 automodification sites (Supplementary Spectra in the Supporting Information), most often showing up in the presence of the modified form, indicating that neutral loss was not complete. Considering how common these neutral losses are, the authors advise including them in mass spectrometry search criteria.
Figure 5.

Proximal phospho(ribosyl)ation sites. E488 and E491 are previously characterized PARP-1 automodification sites, shown here in panels b and c, respectively, as compared with the unmodified form of the peptide shown in panel a. The doubly modified peptide (d) contains diagnostic fragments that carry one phosphoribose group (212.01 Da, circles) as well as those carrying two phosphoribose groups (424.02 Da, squares).
Our analysis identified three lysine modifications: two novel and one previously reported in a 2009 mutagenesis screen1 (Table 1). Two of these were found at the C-terminus of the peptide, suggesting that the phospho(ribosyl)ated residue did not prevent proteolytic cleavage at the modified lysine, in our case by a combination of LysC and trypsin. To confidently assign the novel PARylation site K486, its CID fragmentation pattern was compared with an unmodified version of the same peptide, revealing a b-ion series that was unmodified in both spectra and a y-ion series that contained the 212.01 Da shift indicative of a phospho(ribose) addition to every y-ion fragment (Figure 6). The extensive b- and y-ion series provide strong evidence of the phospho(ribose) modification on the peptide C-terminal lysine, demonstrating (1) the availability of phospho(ribosyl)ated lysines for protease cleavage and (2) the ability of PHOS-Select to enrich phospho(ribosyl)ated lysines.
Figure 6.

Phospho(ribosyl)ation on peptide terminal lysine. K486, a novel PARP-1 PARylation site identified in our analysis, is shown here at the peptide C-terminus (b). This fragmentation pattern is compared with that of the unmodified form (a) showing the characteristic 212.01 Da shift present in the entire y-series but absent from the b-series, validating the localization of phospho(ribose).
ADP-Ribosylation Sites Identified from Whole Cells
To establish a pipeline for identifying endogenous sites of mono- and poly(ADP-ribosyl)ation, HeLa cells were SILAC-labeled and then treated with the DNA damaging agent MNNG to induce PARylation before being subjected to an affinity pull-down by the mono- and poly(ADP-ribose) binding macrodomain from Af1521.34 ADP-ribosylated proteins were then denatured before being treated with SVP and then digested with a mixture of the proteases LysC and trypsin. These peptide mixtures were then split in half and either enriched over a charged or a stripped IMAC resin with the elution from the stripped resin serving as a nonspecific background control for the eluted peptides that had come off of the charged IMAC resin. (See Figure 7a.) Because both forward- and reverse-labeling patterns were used, the strongest hits from the database showed up in both populations, as demonstrated in Figure 7b,c. A representative spectrum for phosphoribosylated R4 from serine/arginine-rich splicing factor 2 (SRSF2) is shown with its parent ion in Figure 7b,d. Notably, the pipeline described in Figure 7a was performed in parallel on an MNNG-treated trophoblast stem cell line from a PARG knockout mouse model,26 producing 22 unique endogenous phospho(ribosyl)ated peptides, two of which (containing R4 from SRSF2 and R199 from heterogeneous nuclear ribonucleoprotein U, HNRNPU) overlapped with those found from the HeLa preparation. (See Supplementary Table 5 in the Supporting Information.) All of the phospho(ribosyl)ated peptides identified from these samples were found in the IMAC enriched fractions, indicating that macrodomain enrichment followed by SVP digestion was not sufficient for site identification.
Figure 7.

Endogenous ADP-ribosylation of arginine. To identify ADP-ribosylation sites from whole cells, we MNNG-treated HeLa cells that were either heavy (K8R10) or light (K0R0) labeled, affinity-enriched ADP-ribosylated proteins, treated these proteins with SVP to yield phosphoribose, and then digested these proteins to a peptide mixture that would then be enriched by either charged or stripped IMAC beads (a). Stripped IMAC beads from each population would serve as a background control for the reverse labeled peptides enriched over a charged matrix. This example shows the MS (b,c) spectra of both the heavy and light forms of R4 from serine/arginine-rich splicing factor 2 (SRSF2) as well as the annotated MS/MS of the light form (d). Serine (gray) carries a protein N-terminal acetylation, arginine (red) carries phospho(ribose).
To determine whether the macrodomain enrichment was necessary for site identification, we did the same analysis with the ADPr affinity purification omitted, again utilizing both the human wild-type and murine PARG knockout cell lines previously described. Twenty-two unique phospho(ribosyl)ated peptides were identified from these preparations, including the same HNRNPU peptide containing R199 found following macrodomain enrichment (it was again found in both cell lines), showing that the macrodomain enrichment is not only insufficient on its own for site identification but also that it is not necessary. Furthermore, a comparison of the macrodomain enriched versus unenriched data sets revealed a bias in the amino acids, which served as attachment sites for phospho(ribose); the macrodomain enrichment appears to have shifted the profile of ADP-ribosylated amino acids away from glutamic acid residues (Figure 8, source data can be found in the Supplementary Text and Supplementary Table 5 in the Supporting Information). This shift indicates that the macrodomain is either selecting against ADP-ribosylated glutamic acid in favor of other amino acid attachment sites or that it is actually removing the ADPr attachment from glutamic acids. The latter hypothesis lines up with recently published work showing that the macrodomain of Af1521 possesses ADP-ribosylhydrolase activity and suggests that this activity may be targeted toward glutamic acid sites of ADP-ribosylation.35
Figure 8.

Effect of macrodomain enrichment on the ADP-ribosylated amino acid profile. Unique phospho(ribosyl)ated peptides identified from whole cells, as detailed in Supplementary Table 5 in the Supporting Information, show a shift in the profile of amino acids carrying phospho(ribose) from both human wild-type (a) and murine PARG knockout cells (b).
Discussion
The expanding relevance of PARylation in cellular processes has led researchers to look beyond the canonical role of DNA repair when considering the consequences of altered PARylation levels.36 To this end, the most powerful tool for studying global changes in post-translational modifications continues to be systematic analyses of proteomes by mass spectrometry. Unfortunately the widespread use of proteomics and mass spectrometry has not yet been established in the field of PARylation due to challenges relating both to the modification itself, which may be labile, large, and highly charged, and to the low levels of PARylation that exist below the threshold of most analytical techniques. In response, enrichment techniques have been developed that have allowed researchers to study the PARylated proteome with the caveat that the identified proteins are either PAR acceptors or PAR binders; due to the lack of site identification in these studies, verification of which class these proteins belong to is both tedious and, in some cases, impossible.37 Recently, a study has demonstrated the feasibility of identifying mono- and poly(ADP-ribosyl)ation sites in a large proteomic screen that combines enrichment of ADP-ribosylated substrates by boronate chromatography with the removal of ADPr from substrates by hydroxylamine; this chemical treatment allows for subsequent identification of acidic ADP-ribosylation sites by the diagnostic 15.01 Da hydroxamic acid derivative left behind.7 The limitation in this study was that ADP-ribosylated lysine and arginine could not be detected as only acidic residues were left with the hydroxamic acid tag. In contrast, our pipeline can identify ADP-ribosylation attachment sites on both acidic and basic residues; it should also be noted that this universality allows for the discovery of novel amino acid attachment sites for ADPr beyond these acids and bases, the existence of which has not been ruled out. We believe our proposed method of enriching and identifying ADP-ribosylation sites addresses the need for a pipeline that is both global and definitive in identifying ADPr acceptors at the protein and amino acid levels.
The phosphoenrichment methods applied in this study have gained popularity in the phosphoproteomic field due to their high specificity and compatibility with both MALDI and ESI–LC–MS. MOAC has proven to enrich phosphopeptides more specifically than its predecessor, IMAC, likely due to the tighter binding of phosphate to the TiO2 microspheres (titanspheres) as compared with the chelated iron used by PHOS-Select IMAC resin.38 This tight binding, however, may explain the lack of phospho- and phospho(ribose) peptides found in the eluates from the TiO2 resins used here (GL Sciences and ZirChrom), which have an optimal elution pH between 9.2 and 9.4.39 IMAC elution is much more sensitive to competitive phosphate levels than it is to pH and does not have an optimal elution pH.40 We have demonstrated the stability of ADPr protein attachment sites in neutral phosphate buffer as compared with basic NH4OH and have restricted our elution conditions to ensure retention of phospho(ribose) on target peptides throughout the enrichment. This consideration may have favored the lower-affinity phosphoenrichment matrix, allowing for a single enrichment protocol capable of enriching both phospho- and phospho(ribose) peptides, perhaps at the expense of tightly bound phosphopeptides left on the TiO2 matrices. For thorough phosphopeptide analysis, it may be prudent to perform a parallel enrichment with optimal (i.e., basic) elution conditions from a TiO2 matrix.
While validating the presence of our phospho(ribosyl)ated protein sample, we discovered that the phosphoprotein SDS-PAGE gel stain, Pro-Q Diamond, can act as an indicator of phospho(ribose)-modified proteins. While we did not do any in-gel digests, the compatibility of Pro-Q Diamond with downstream LC–MS analysis41 suggests that isolation and identification of phopho(ribosyl)ated proteins as well as their PAR acceptor sites may be possible for researchers who wish to analyze changes in SDS-PAGE profiles. We believe this data-dependent approach would greatly complement the global analysis already offered by the phospho(ribose) ADP-ribosylation tag.
Optimization of our phosphoenrichment protocol presented us with a database of spectra identifying phospho(ribosyl)ated peptides from automodified hPARP-1, ultimately yielding 20 modified sites, eight of which are being reported for the first time. These spectra afforded us the opportunity to characterize phospho(ribosyl)ated peptides (and by extrapolation, ADP-ribosylation sites) with regard to their identification by CID- and HCD-assisted LC–MS/MS. First, we determined that multiple PARylation sites may exist within the same peptide, suggesting that hPARP-1 is capable of placing these large, highly charged polymers within an amino acid of each other (as in the hPARP-1 automodified peptide GFSLLATE*D*K; see Supplementary Spectra in the Supporting Information). The steric hindrance and charge-repulsion associated with neighboring PARylation sites may require a high level of flexibility from the protein, poly(ADP-ribose), or both. Second, it is worth noting that we identified two lysine hPARP-1 automodification sites at the C-terminal end of their respective peptides, indicating that these modified lysines were available for proteolytic digestion. (See Supplementary Table 2 in the Supporting Information.) Finally, fragmention by HCD and CID revealed the potential of phospho(ribose) to be partially or fully lost in the form of a phosphoric acid or phosphoribose, respectively. (See Supplementary Figure 5 in the Supporting Information.) This loss is not complete as the fragments portraying the neutral loss are often accompanied by otherwise-identical fragments that have maintained the full modification. In the future, these neutral loss fragments may serve as diagnostic indicators of peptide phospho(ribosyl)ation state. Recognition of these attributes will aid in the analysis of large, phospho(ribosyl)ated proteomes, which may present these characteristics that would allow them to be ignored by erroneous search parameters.
While demonstrating the application of this method to identify ADP-ribosylation sites from whole cells, we validated several known sites of ADP-ribosylation recently identified by a complementary mass spectrometry approach7 as well as a host of novel sites on both novel and known acceptors of mono- or poly(ADP-ribose). (See Supplementary Table 5 in the Supporting Information.) One of our most interesting hits lies on K350 of heterogeneous nuclear ribonucleoprotein A1 (HNRNPA1), a protein that was first shown to be poly(ADP-ribosyl)ated in whole cells in 1982 and 12 years later was shown to be one of the two major acceptors of ADPr in HeLa cells.42 More recently, PARylation of HNRNPA1 has been shown to affect splicing, stem-cell maintenance, and oocyte localization in drosophila, suggesting an interesting role for mammalian HNRNPA1 PARylation.43 While there have been several proteomic studies that have identified HNRNPA1 in poly or mono(ADP-ribosyl)ation purification schemes,37 this finding is the first indication of the site of PARylation on HNRNPA1 (spectrum annotated in Supplementary Figure 6 in the Supporting Information).
In summary, we have proposed and demonstrated the feasibility of a global, unbiased approach for characterizing the mono- and poly(ADP-ribosyl)ated proteome. Our technique, based on the digestion of ADPr down to its phospho(ribose) attachment site, allows for enrichment at the peptide level of both acidic and basic ADPr acceptor sites. Furthermore, we have shown that our method allows researchers to find sites of ADP-ribosylation without having to knock down ADPr hydrolases or perform an enrichment of the ADP-ribosylated proteome, steps that may otherwise introduce bias. Finally, this approach presents a unique opportunity to study the changes in the ADP-ribosylated proteome alongside the coenriched phosphoproteome. It is our hope that the accessibility of the techniques employed in this enrichment pipeline will allow researchers to characterize global ADP-ribosylation at the level of the amino acid, ultimately resulting in a greater understanding of both mono- and poly(ADP-ribose) function and regulation from the bottom up.
Acknowledgments
We thank Dr. John Pascal for providing the hPARP-1 expression plasmid used to synthesize all hPARP-1 protein used here24 as well as Dr. David Koh, who provided the PARG knockout cells used for endogenous ADP-ribose site identification.26 The research was funded by a Department of Defense Breast Cancer Research Program Idea Award #BC101881 (A.K.L.L.), a Journal of Cell Science Travelling Fellowship (C.M.D), an NCI training grant 5T32CA009110-35 (C.M.D), and NIDA P30DA028846-01 (S.-E.O).
Supporting Information Available
Supplementary Figure 1. Enriching phospho- and phospho(ribose) peptides from a complex background. Supplementary Figure 2. Co-enriching phospho(ribosyl)ated peptides by IMAC and MOAC. Supplementary Figure 3. PARP-1 is inactivated upon exposure to 8 M urea. Supplementary Figure 4. Multiple PAR sites can exist on the same peptide. Supplementary Figure 5. Neutral loss sequence from a doubly modified hPARP-1 peptide. Supplementary Figure 6. K350 of HNRNPA1 is phopho(ribosyl)ated. Supplementary Table 1. Phospho- and phospho(ribose) peptides identified from Casein and PARP1 standards, respectively. Supplementary Table 2. Phosphopeptides identified from HeLa whole cell lysate during phosphoenrichment comparison. Supplementary Table 3. All proteins identified during phosphoenrichment comparison (excluding reverse and contaminant hits). Supplementary Table 4. All peptides identified during phosphoenrichment comparison (excluding reverse and contaminant hits). Supplementary Table 5. Endogenous phospho(ribosyl)ated peptides identified from whole cells. Supplementary Table 6. Endogenous phosphorylated peptides identified from whole cells. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Altmeyer M.; Messner S.; Hassa P. O.; Fey M.; Hottiger M. O. Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res. 2009, 37113723–3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hottiger M. O.; Hassa P. O.; Luscher B.; Schuler H.; Koch-Nolte F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 354208–219. [DOI] [PubMed] [Google Scholar]
- Tao Z.; Gao P.; Liu H. W. Identification of the ADP-ribosylation sites in the PARP-1 automodification domain: analysis and implications. J. Am. Chem. Soc. 2009, 1314014258–14260. [DOI] [PubMed] [Google Scholar]
- Sharifi R.; Morra R.; Appel C. D.; Tallis M.; Chioza B.; Jankevicius G.; Simpson M. A.; Matic I.; Ozkan E.; Golia B.; Schellenberg M. J.; Weston R.; Williams J. G.; Rossi M. N.; Galehdari H.; Krahn J.; Wan A.; Trembath R. C.; Crosby A. H.; Ahel D.; Hay R.; Ladurner A. G.; Timinszky G.; Williams R. S.; Ahel I. Deficiency of terminal ADP-ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease. EMBO J. 2013, 3291225–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman J. D.; Gagne J. P.; Poirier G. G.; Goodlett D. R. Mapping PARP-1 auto-ADP-ribosylation sites by liquid chromatography-tandem mass spectrometry. J. Proteome Res. 2013, 1241868–1880. [DOI] [PubMed] [Google Scholar]
- De Lorenzo S. B.; Patel A. G.; Hurley R. M.; Kaufmann S. H. The Elephant and the Blind Men: Making Sense of PARP Inhibitors in Homologous Recombination Deficient Tumor Cells. Front. Oncol. 2013, 3, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Wang J.; Ding M.; Yu Y. Site-specific characterization of the Asp- and Glu-ADP-ribosylated proteome. Nat. Methods 2013, 1010981–984. [DOI] [PubMed] [Google Scholar]
- Underhill C.; Toulmonde M.; Bonnefoi H. A review of PARP inhibitors: from bench to bedside. Ann. Oncol. 2011, 222268–279. [DOI] [PubMed] [Google Scholar]
- David K. K.; Andrabi S. A.; Dawson T. M.; Dawson V. L. Parthanatos, a messenger of death. Front. Biosci. 2009, 14, 1116–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Murcia G.; Huletsky A.; Poirier G. G. Modulation of chromatin structure by poly(ADP-ribosyl)ation. Biochem. Cell Biol. 1988, 666626–635. [DOI] [PubMed] [Google Scholar]
- a Schreiber V.; Dantzer F.; Ame J. C.; de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nature reviews. Mol. Cell Biol. 2006, 77517–528. [DOI] [PubMed] [Google Scholar]; b Cesarone C. F.; Scarabelli L.; Giannoni P.; Gallo G.; Orunesu M. Relationship between poly(ADP-ribose) polymerase activity and DNA synthesis in cultured hepatocytes. Biochem. Biophys. Res. Commun. 1990, 17131037–1043. [DOI] [PubMed] [Google Scholar]
- Smith S.; Giriat I.; Schmitt A.; de Lange T. Tankyrase, a poly(ADP-ribose) polymerase at human telomeres. Science 1998, 28253931484–1487. [DOI] [PubMed] [Google Scholar]
- Arnold J.; Grune T. PARP-mediated proteasome activation: a co-ordination of DNA repair and protein degradation?. BioEssays 2002, 24111060–1065. [DOI] [PubMed] [Google Scholar]
- Leung A. K.; Vyas S.; Rood J. E.; Bhutkar A.; Sharp P. A.; Chang P. Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Mol. Cell 2011, 424489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagtap P.; Szabo C. Poly(ADP-ribose) polymerase and the therapeutic effects of its inhibitors. Nat. Rev. Drug Discovery 2005, 45421–440. [DOI] [PubMed] [Google Scholar]
- Feijs K. L.; Verheugd P.; Luscher B. Expanding functions of intracellular resident mono-ADP-ribosylation in cell physiology. FEBS J. 2013, 280153519–3529. [DOI] [PubMed] [Google Scholar]
- Cervantes-Laurean D.; Jacobson E. L.; Jacobson M. K. Preparation of low molecular weight model conjugates for ADP-ribose linkages to protein. Methods Enzymol. 1997, 280, 275–287. [DOI] [PubMed] [Google Scholar]
- Adamietz P.; Hilz H. Poly(adenosine diphosphate ribose) is covalently linked to nuclear proteins by two types of bonds. Z. Phys. Chem. 1976, 3574527–534. [DOI] [PubMed] [Google Scholar]
- Mao Z.; Hine C.; Tian X.; Van Meter M.; Au M.; Vaidya A.; Seluanov A.; Gorbunova V. SIRT6 promotes DNA repair under stress by activating PARP1. Science 2011, 33260361443–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messner S.; Altmeyer M.; Zhao H.; Pozivil A.; Roschitzki B.; Gehrig P.; Rutishauser D.; Huang D.; Caflisch A.; Hottiger M. O. PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic Acids Res. 2010, 38196350–6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara H.; Hasegawa S.; Fujimura S.; Shima T.; Sugimura T. Studies on poly (adenosine diphosphate ribose). V. Mechanism of hydrolysis of poly (adenosine diphosphate ribose) by snake venom phosphodiesterase. J. Biol. Chem. 1970, 245143606–3611. [PubMed] [Google Scholar]
- Hengel S. M.; Goodlett D. R. A Review of Tandem Mass Spectrometry Characterization of Adenosine Diphosphate-Ribosylated Peptides. Int. J. Mass Spectrom. 2012, 312, 114–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Matic I.; Ahel I.; Hay R. T. Reanalysis of phosphoproteomics data uncovers ADP-ribosylation sites. Nat. Methods 2012, 98771–772. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Huttlin E. L.; Jedrychowski M. P.; Elias J. E.; Goswami T.; Rad R.; Beausoleil S. A.; Villen J.; Haas W.; Sowa M. E.; Gygi S. P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010, 14371174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langelier M. F.; Planck J. L.; Servent K. M.; Pascal J. M. Purification of human PARP-1 and PARP-1 domains from Escherichia coli for structural and biochemical analysis. Methods Mol. Biol. 2011, 780, 209–226. [DOI] [PubMed] [Google Scholar]
- Oka J.; Ueda K.; Hayaishi O. Snake venom phosphodiesterase: simple purification with Blue Sepharose and its application to poly(ADP-ribose) study. Biochem. Biophys. Res. Commun. 1978, 804841–848. [DOI] [PubMed] [Google Scholar]
- Koh D. W.; Lawler A. M.; Poitras M. F.; Sasaki M.; Wattler S.; Nehls M. C.; Stoger T.; Poirier G. G.; Dawson V. L.; Dawson T. M. Failure to degrade poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc. Natl. Acad. Sci. U.S.A. 2004, 1015117699–17704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J.; Ishihama Y.; Mann M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 2003, 753663–670. [DOI] [PubMed] [Google Scholar]
- Villen J.; Gygi S. P. The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat. Protocols 2008, 3101630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong S. E.; Blagoev B.; Kratchmarova I.; Kristensen D. B.; Steen H.; Pandey A.; Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 2002, 15376–386. [DOI] [PubMed] [Google Scholar]
- Desmarais Y.; Menard L.; Lagueux J.; Poirier G. G. Enzymological properties of poly(ADP-ribose)polymerase: characterization of automodification sites and NADase activity. Biochim. Biophys. Acta 1991, 10782179–186. [DOI] [PubMed] [Google Scholar]
- Langelier M. F.; Planck J. L.; Roy S.; Pascal J. M. Crystal structures of poly(ADP-ribose) polymerase-1 (PARP-1) zinc fingers bound to DNA: structural and functional insights into DNA-dependent PARP-1 activity. J. Biol. Chem. 2011, 2861210690–10701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menard L.; Thibault L.; Poirier G. G. Reconstitution of an in vitro poly(ADP-ribose) turnover system. Biochim. Biophys. Acta 1990, 1049145–58. [DOI] [PubMed] [Google Scholar]
- Kharadia S. V.; Graves D. J. Relationship of phosphorylation and ADP-ribosylation using a synthetic peptide as a model substrate. J. Biol. Chem. 1987, 2623617379–17383. [PubMed] [Google Scholar]
- Karras G. I.; Kustatscher G.; Buhecha H. R.; Allen M. D.; Pugieux C.; Sait F.; Bycroft M.; Ladurner A. G. The macro domain is an ADP-ribose binding module. EMBO J. 2005, 24111911–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Rosenthal F.; Feijs K. L.; Frugier E.; Bonalli M.; Forst A. H.; Imhof R.; Winkler H. C.; Fischer D.; Caflisch A.; Hassa P. O.; Luscher B.; Hottiger M. O. Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases. Nat. Struct. Mol. Biol. 2013, 204502–507. [DOI] [PubMed] [Google Scholar]; b Jankevicius G.; Hassler M.; Golia B.; Rybin V.; Zacharias M.; Timinszky G.; Ladurner A. G. A family of macrodomain proteins reverses cellular mono-ADP-ribosylation. Nat. Struct. Mol. Biol. 2013, 204508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas S.; Chesarone-Cataldo M.; Todorova T.; Huang Y. H.; Chang P. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat. Commun. 2013, 4, 2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Gagne J. P.; Pic E.; Isabelle M.; Krietsch J.; Ethier C.; Paquet E.; Kelly I.; Boutin M.; Moon K. M.; Foster L. J.; Poirier G. G. Quantitative proteomics profiling of the poly(ADP-ribose)-related response to genotoxic stress. Nucleic acids Res. 2012, 40167788–7805. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Jungmichel S.; Rosenthal F.; Altmeyer M.; Lukas J.; Hottiger M. O.; Nielsen M. L. Proteome-wide Identification of Poly(ADP-Ribosyl)ation Targets in Different Genotoxic Stress Responses. Mol. Cell 2013, 5224272–285. [DOI] [PubMed] [Google Scholar]
- Aryal U. K.; Ross A. R. Enrichment and analysis of phosphopeptides under different experimental conditions using titanium dioxide affinity chromatography and mass spectrometry. Rapid Commun. Mass Spectrom. 2010, 242219–231. [DOI] [PubMed] [Google Scholar]
- Park S. S.; Maudsley S. Discontinuous pH gradient-mediated separation of TiO2-enriched phosphopeptides. Anal. Biochem. 2011, 409181–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart S. R.; Waterfield M. D.; Burlingame A. L.; Cramer R. Factors governing the solubilization of phosphopeptides retained on ferric NTA IMAC beads and their analysis by MALDI TOFMS. J. Am. Soc. Mass Spectrom. 2002, 1391042–1051. [DOI] [PubMed] [Google Scholar]
- Schulenberg B.; Goodman T. N.; Aggeler R.; Capaldi R. A.; Patton W. F. Characterization of dynamic and steady-state protein phosphorylation using a fluorescent phosphoprotein gel stain and mass spectrometry. Electrophoresis 2004, 25152526–2532. [DOI] [PubMed] [Google Scholar]
- a Kostka G.; Schweiger A. ADP-ribosylation of proteins associated with heterogeneous nuclear RNA in rat liver nuclei. Biochim. Biophys. Acta 1982, 6962139–144. [DOI] [PubMed] [Google Scholar]; b Prasad S.; Walent J.; Dritschilo A. ADP-ribosylation of heterogeneous ribonucleoproteins in HeLa cells. Biochem. Biophys. Res. Commun. 1994, 2042772–779. [DOI] [PubMed] [Google Scholar]
- a Ji Y.; Tulin A. V. Poly(ADP-ribosyl)ation of heterogeneous nuclear ribonucleoproteins modulates splicing. Nucleic Acids Res. 2009, 37113501–3513. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ji Y.; Tulin A. V. Poly(ADP-ribose) controls DE-cadherin-dependent stem cell maintenance and oocyte localization. Nat. Commun. 2012, 3, 760. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.