

Abstract
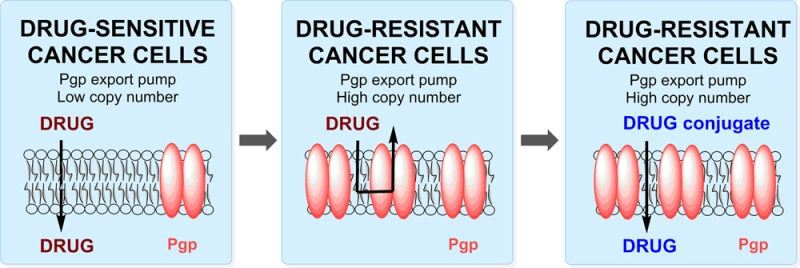
Multidrug resistance (MDR) is a major cause of chemotherapy failure in the clinic. Drugs that were once effective against naïve disease subsequently prove ineffective against recurrent disease, which often exhibits an MDR phenotype. MDR can be attributed to many factors; often dominating among these is the ability of a cell to suppress or block drug entry through upregulation of membrane-bound drug efflux pumps. Efflux pumps exhibit polyspecificity, recognizing and exporting many different types of drugs, especially those whose lipophilic nature contributes to residence in the membrane. We have developed a general strategy to overcome efflux-based resistance. This strategy involves conjugating a known drug that succumbs to efflux-mediated resistance to a cell-penetrating molecular transporter, specifically, the cell-penetrating peptide (CPP), d-octaarginine. The resultant conjugates are discrete single entities (not particle mixtures) and highly water-soluble. They rapidly enter cells, are not substrates for efflux pumps, and release the free drug only after cellular entry at a rate controlled by linker design and favored by target cell chemistry. This general strategy can be applied to many classes of drugs and allows for an exceptionally rapid advance to clinical testing, especially of drugs that succumb to resistance. The efficacy of this strategy has been successfully demonstrated with Taxol in cellular and animal models of resistant cancer and with ex vivo samples from patients with ovarian cancer. Next generation efforts in this area will involve the extension of this strategy to other chemotherapeutics and other MDR-susceptible diseases.
Keywords: molecular transporters, cell-penetrating peptide (CPP), guanidinium-rich molecular transporters, octaarginine, multidrug resistance, cancer, drug delivery, P-glycoprotein
Introduction
The development of treatments for multidrug resistant (MDR) disease is one of the greatest challenges in medicine. MDR is a ubiquitous problem, being associated with diseases such as cancer, bacterial, parasitic, and viral infections. The World Health Organization estimates, for example, that there were over 450,000 new MDR tuberculosis cases worldwide in 2012 alone.1,2 For cancer, chemotherapy fails in over 90% of patients with metastatic cancer, an outcome driven in large measure by MDR.3
MDR is a multifaceted problem caused by a number of different mechanisms, including drug efflux, target mutation,4 cell cycle effects, drug metabolism,5 apoptosis evasion,6 and altered membrane permeation.5,7 Drug efflux is often a dominating mechanism in many MDR cases across disease types, including cancer,5 malaria,8 and tuberculosis.9 Drug efflux arises from an energy-dependent transport of a drug out of the cell or its membrane mediated by membrane-associated protein pumps, such as P-glycoprotein (Pgp, also known as ABCB1 or MDR1) (Figure 1).10 While MDR and strategies to overcome it are subjects of considerable breadth associated with many diseases, in this review, emphasis will be placed on MDR cancer, the role of Pgp efflux pumps in disrupting the effective treatment of cancer, and strategies for overcoming this efflux-based resistance.
Figure 1.
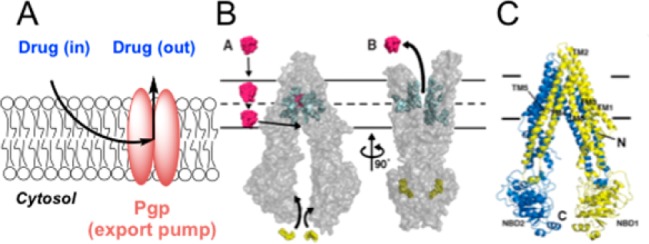
(A) Schematic of drug efflux by Pgp export. As a lipophilic drug enters the membrane, it encounters the intramembrane access point of Pgp. (B) Aller’s model of substrate transport by Pgp.15 The drug first enters the membrane, where it encounters and then enters Pgp (B,A), and then the conformational change of Pgp expels the drug out of the cell (B,B).15 (C) Crystal structure of Pgp. Panels B and C are reprinted with permission from ref (15). Copyright 2009 American Association for the Advancement of Science.
Several efflux pumps are known to be associated with MDR,5 the most prevalent of which is Pgp.11 As a member of the adenosine triphosphate-binding cassette (ABC) superfamily, Pgp is a promiscuous transmembrane protein that exhibits the remarkable ability to recognize and export numerous xenobiotics and toxins from the cell. In striking contrast to the impressive structural specificity of many proteins for their substrates, Pgp exhibits polyspecificity, recognizing and exporting numerous structurally diverse molecules with molecular weights from 330 to 4000 Da.12,13 This uncanny ability to recognize structurally different agents is attributed to a selectivity filter based upon physical properties and a well-placed location in the plasma membrane. Its substrate molecules, while differing in structure, are generally hydrophobic in nature and thus partition into the nonpolar membrane, where Pgp resides.14 These hydrophobic substrates include widely used classes of chemotherapeutic agents, such as taxanes, vinca alkaloids, anthracyclins, kinase inhibitors, and camptothecins that often are rendered ineffective due to Pgp export.5 In 2009, a ground-breaking study by Aller and colleagues reported the crystal structure of mouse Pgp, which shares 87% sequence homology with human Pgp, to a resolution of 3.8 Å.15 This unique study revealed that Pgp has portals open to both the cytoplasm and the inner leaflet of the cell membrane, allowing for effective collection and expulsion of lipophilic substrates that are preferentially solubilized in the membrane (Figure 1B,C). An unfortunate outcome of drugs designed for passive diffusion across the nonpolar membrane of a cell is that they are often substrates for Pgp export due to their residence time in the membrane.
Many studies link Pgp expression to resistant and recurrent cancers. However, one of the challenges associated with quantifying the extent of this link is the difficulty of measuring Pgp expression levels.16 Despite these challenges, taxane resistance caused by Pgp efflux has been well characterized in vitro and has been shown to play an important role in recurrent ovarian17 and breast18 cancers. Increased expression of Pgp has also been found to track with a poor response to taxane-based therapy in nonsmall-cell lung cancer.19,20 It has additionally been shown that recurrent ovarian cancers have higher levels of Pgp expression on a population basis.17
Of great significance in efforts to understand how some cells evade chemotherapy, efflux pumps have been more broadly implicated in the proposed stem cell-like behavior of certain cancer cell populations,21 which is of importance in emerging theories on cancer resistance. While the cancer stem cell hypothesis is for some still a subject of debate, there have been many recent, high-profile studies in several different cancer types that further support its role in cancer.22−24 The not uncommon view that cancer cells are largely homogeneous and that recurrence occurs when debulking chemotherapy causes or selects upregulation of resistance factors (Figure 2A) is giving way to emerging evidence for stem cell-like behavior of many cancer cells. The cancer stem cell hypothesis proposes that there is a heterogeneous mix of cancer cells in a tumor, and some cells can regenerate the entire tumor, like embryonic stem cells can generate an entire organism.21 The stem-like cancer cells already have high expression levels of efflux-pumps and other resistance factors and are thus not cleared by initial rounds of chemotherapy. As a result, the cancer stem-like cells seed disease recurrence, and the recurrent disease is thus chemoresistant (Figure 2B). Efflux pump expression, including Pgp, has long been considered a hallmark of stem cells.25−27 Moreover, high levels of pump expression have also recently been found for a variety of stem cell-like tumor cells, such as leukemia28 and osteosarcomas.29 The cancer stem cell hypothesis implies that without utilizing or developing therapies that can avoid or overcome efflux-mediated resistance, including Pgp, chemotherapy would only reduce tumor burden and slow disease progression but not eradicate the pool of progenitor cancer cells.
Figure 2.

Two theories on the origin of chemotherapy-resistant cancers. (A) Conventional and (B) cancer stem cell hypothesis21 of cancer resistance.
Several distinct strategies have been pursued to abrogate Pgp-mediated resistance in multidrug resistant cancer. These strategies include:
-
1.
Development of new agents or modification of existing therapeutic agents such that they are no longer Pgp substrates;
-
2.
Pgp pump inhibition with coadministration of existing chemotherapeutics;
-
3.
Inhibition of Pgp expression through the use of RNA interference;
-
4.
Attachment or complexation of an existing chemotherapeutic to a drug delivery agent, including molecular transporters such as the cell-penetrating peptide, d-octaarginine, to overcome Pgp-mediated efflux.
Each strategy is described in more detail below, with a focus on the significant and broad-spectrum opportunity that cell-penetrating agents such as peptides and other molecular transporters provide as a means to overcome pump-mediated efflux in MDR disease with a potentially rapid path to the clinic.
1. Development of New Agents to Avoid Efflux-Mediated MDR
Of the strategies for overcoming MDR noted above, the search for new agents that are not Pgp substrates has justifiably attracted attention. This search has taken two forms: the identification of new leads or the modification of existing drugs. The former, based largely on screening natural product or synthetic libraries, is attractive but has a long path to the clinic. Paclitaxel, for example, took approximately 30 years to advance from first “hit” to clinical use.30 An example of a new drug derived by modification of an existing drug is the newly approved taxane, cabazitaxel (trade name Jevtana), a Sanofi-Aventis therapy that was approved for hormone-refractory prostate cancer in 2010 (Figure 3).31 This new semisynthetic taxane was found to have similar potency and side effect profiles to other taxanes, but it displays a decreased susceptibility to efflux by Pgp.32 Because of this decreased susceptibility to Pgp export, it showed clinical activity in women with taxane-resistant metastatic breast cancer.33 However, it is not yet approved for this indication.
Figure 3.
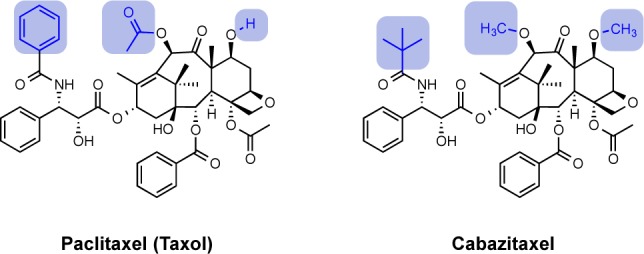
Structures of paclitaxel (Taxol) and its analogue, cabazitaxel, a substrate less susceptible to Pgp export, with differing functional groups highlighted.32
In addition to the development of small molecule candidates that avoid Pgp efflux, considerable effort has gone into the development of nanomaterials that do not serve as Pgp substrates, such as Tat-functionalized nanocrystalline silver agents.34
2. Pgp Pump Inhibition to Overcome Drug Efflux
In addition to tuning existing drugs or finding new ones, another strategy to overcome efflux-mediated resistance, under investigation for over 30 years, is based on inhibiting Pgp with small molecules.35 Toward this end, many modulators/inhibitors of ABC transporters have been developed, but cytotoxicity and the complex pharmacokinetics of interdependent drugs (codosed inhibitor and anticancer drugs) have slowed the clinical implementation of this approach. An important and challenging aspect of inhibiting Pgp export is that export pumps are required for normal cell function.5 Several generations of pump inhibitors have been developed and tested, and they are extensively covered in recommended reviews.5,6,35−38 As brief context for this review, the first compound identified as a Pgp inhibitor was verapamil, a calcium channel blocker, whose advancement was hampered by cardiovascular side effects.39,40 Other first-generation inhibitors included the immunosuppressant, cyclosporine A,41 and the antimalarial, quinine.42 These Pgp inhibitors generally displayed low affinity and low specificity for Pgp, thus requiring high doses that often elicited undesired side effects. Second generation Pgp inhibitors were developed through structural modifications of the first generation agents. While they displayed improved Pgp modulation, they also showed adverse pharmacokinetic interactions with cytochrome P450 (CYP450), leading to both decreased efficacy and increased toxicity.6,43 On the basis of extensive structure–activity relationships (SAR) studies on previous inhibitors, the third generation Pgp inhibitors were designed to optimize the pharmacophoric elements necessary for Pgp specificity and to avoid pharmacokinetic interactions with known chemotherapeutics.36 Additionally, CPP–chlorambucil conjugates have also been reported to function as Pgp inhibitors.44
Although there have been several Pgp inhibitors tested in clinical trials, significant clinical benefit has yet to be achieved.45 Pgp pump inhibition remains, however, an actively pursued strategy for overcoming MDR.
3. Inhibition of Pgp Expression Through the Use of RNA Interference
There are other experimental strategies under investigation for the inhibition of Pgp. One recent strategy is to achieve formal Pgp inhibition by blocking Pgp synthesis through the use of RNA interference (RNAi).46,47 RNAi is an endogenous mechanism by which post-transcriptional gene silencing is achieved in a sequence-specific manner using double-stranded RNA molecules, including short interfering RNA (siRNA).48 Relative to small molecule Pgp inhibitors, RNAi technology circumvents many adverse pharmacokinetic effects that could result from drug interactions. The specificity of RNAi could also reduce off-target effects observed with small molecule inhibitors. Although there are several potential advantages to RNAi-mediated inhibition of Pgp expression, there are numerous complex barriers to achieving successful clinical use of siRNA, the first and foremost being delivery of siRNA into cells. Most reported studies on silencing export pumps with siRNA employ commercially available transfection reagents, largely limiting their findings to in vitro studies.49−51 The first reported animal model for Pgp downregulation utilized chemically-modified Stealth RNAi against Pgp in nude mice bearing lung carcinoma tumors.52 Transfection was achieved using electroporation, resulting in ∼60% reduction in tumor size after 2 weeks of cotreatment with vinorelbine. A 2010 study by Patil and co-workers reported a dual agent poly(d,l-lactide-co-glycolide) nanoparticle, encapsulating both Taxol and a Pgp-targeted siRNA.53 A 50% reduction in tumor size was observed after 16 days of treatment, demonstrating the feasibility of systemic administration. It is important to note, however, that Pgp is critical for normal physiological function,5 and thus nonselective inhibition can cause an on-target toxicity common to all Pgp inhibitors.
4. Molecular Transporters for Overcoming Efflux-Mediated MDR
A distinct strategy for overcoming Pgp-mediated efflux and associated MDR is based on the use of drug delivery agents that modify the mechanism of cellular entry of a drug and thus avoid Pgp export. This strategy avoids the need for Pgp inhibition and the attendant challenges with the “double drugging” of interdependent drugs as described previously. Not long after the discovery of the importance of Pgp in MDR, researchers hypothesized that changing the mechanism of uptake of a drug might overcome Pgp-mediated resistance.54 Št’astný and co-workers conjugated the targeting moieties anti-CD71, antithymocyte globulin, anti-CD4, and transferrin to N-(2-hydroxypropyl)methacrylamide (HPMA) copolymers containing doxorubicin.55 The results of this study indicated that by changing the mechanism of cellular uptake of the drug, conjugation to targeting agents could partially overcome Pgp-mediated MDR. Doxorubicin has also been conjugated directly to the targeting agent, transferrin, and subsequently shown to have activity in multidrug-resistant KB cell lines.56 However, this cytotoxicity appears to be via a different mechanism than the parent doxorubicin.57 Transferrin has also been used in conjunction with doxorubicin-encapsulated liposomes to overcome a Pgp-based MDR phenotype.54,58 Though these studies support the potential of using targeting moieties to alter the uptake mechanisms of a chemotherapeutic, thereby avoiding MDR, there has been limited work to investigate these agents in vivo.
Conjugation of a chemotherapeutic to a molecular transporter changes both its physical properties and its mechanism of cellular uptake,59,60 thus serving as a general strategy to evade Pgp efflux. Currently, there are only a few examples of this strategy: the use of Taxol–oligoarginine conjugates to overcome resistance in vitro, in vivo, and ex vivo in malignant human ascites;61,62 the use of conjugates of doxorubicin with several different cell-penetrating peptides such to overcome MDR,63,64 and phage-discovered CPP conjugates of methotrexate.65 These studies on this approach will be expanded upon and discussed in more detail in the following sections. In brief, drug–transporter conjugation represents a powerful and general strategy to not only improve drug formulation and control drug release but also target drug delivery and most importantly to overcome resistant disease. By fixing the shortcomings of a known drug, this drug-rescue strategy could have a fast track to clinical trials. It can also be used to overcome formulation, distribution, and targeting problems associated with drug leads and screening strategies.
Molecular transporters are a class of agents that enable or enhance the passage of drugs or probes across biological barriers.59,66−68 While often referred to, using a structurally limiting terminology, as cell-penetrating peptides, the term “molecular transporters”, introduced by us in 2000, serve to more generally cover the many structural classes of molecules that exhibit cell-penetrating behavior. This terminology framed the more general expectation and now widely demonstrated finding that many nonpeptidic systems could function in a fashion similar or superior to cell-penetrating peptides. Indeed, Wender, Rothbard, and co-workers first reported that the uptake of the CPP, Tat49–57 (RKKRRQRRR), is a function of its arginine content and not its peptide backbone and more generally proposed that transport into the cell is due to the number and spatial array of its guanidinium groups.69 Multiple mechanisms have been advanced to explain the paradoxical behavior of an oligocation and thus highly water-soluble and polar agent crossing the nonpolar cell membrane to enter cells.59 Both adaptive translocation and endocytotic mechanisms have been proposed and evidence indicates that some proposed mechanisms work simultaneously.70 Robust uptake is generally observed for a variety of cell types, although there are differences in rates and mechanisms of uptake depending on cell line, cargo, and transporter variations.59 Mechanistically, the guanidinium groups are proposed to form bifurcated hydrogen bonds with cell surface anions (e.g., carboxylates, sulfates, and phosphates), and the resultant, charge-neutralized complexes are driven inward by the polarization of the membrane or by encapsulation in an endosome.60 Guanidinium-rich molecular transporters thus function as “polarity chameleons”, being highly water-soluble in the extracellular milieu but becoming nonpolar upon complexation with membrane components, allowing for rapid cellular uptake.
The finding by Wender, Rothbard, and co-workers that the number and spatial array of guanidinium group controls cellular uptake provided a blueprint for the design of the first cell-penetrating guanidinium-rich peptoid transporters,69 followed by oligocarbamates,71 dendrimers,72,73 oligocarbonates,74 carbohydrates,75 and other transporters.66 Cell-penetrating guanidinium-rich molecular transporters have been shown to enable or enhance cellular uptake of small molecules, probes, imaging agents, metals, peptides, proteins, siRNA, DNA plasmids, quantum dots, and vesicles.59,66,67 In addition to passage across cell membranes, guanidinium-rich molecular transporters have also been shown to cross skin, ocular, buccal, and blood–brain barriers, and have been advanced into clinical trials.66
The use of molecular transporters to improve or change the cellular uptake profile of drugs and probes has been exploited for many different applications from basic research to clinical evaluation.66 It is noteworthy that guanidinium-rich conjugates are both highly water-soluble and rapidly enter cells, characteristics of great importance in avoiding Pgp export associated with MDR (Table 1). Their properties and performance thus provide a powerful and general strategy for avoiding Pgp-based efflux. Our working hypothesis was that the properties and thus cellular uptake of a drug, which, because of its lipophilicity, would spend time in a membrane and thus be a substrate for Pgp export, could be altered by conjugation to a transporter to avoid Pgp export and promote cellular entry. Because of conjugation to the oligo-cationic transporter, the drug conjugate would be rendered highly water-soluble, spend little time in the nonpolar membrane, and rapidly enter cells, thus evading Pgp export (Figure 4).
Table 1. Advantages of Molecular Transporter-Taxol Conjugates over Free Taxol;61,62 These Advantages May Be Extended to Many Approved Small Molecule Therapeutics.

Conjugates are discrete chemical entities released only inside cells.
Release rate is controlled by linker design.
Enhanced release rate in high reducing environment of cancer cells.
Figure 4.

Lipophilic drugs that are substrates for Pgp export, upon conjugation to molecular transporters, afford drug–transporter conjugates that are highly water-soluble, are not recognized by Pgp pumps, and rapidly enter cells.61
Early work in this area was largely limited to nonreleasable conjugates of doxorubicin and its derivatives. Temsamani and co-workers tested doxorubicin coupled to two different cell-penetrating peptidic transporters, d-penetratin and pegelin, 16 and 18 amino acids in length, respectively, to form nonreleasable conjugates that displayed improved accumulation in resistant K562/ADR cells.76 Liang and Yang also synthesized a nonreleasable doxorubicin conjugate,77 utilizing the cell-penetrating Tat peptide.78−80 They found improved cell kill of the Tat conjugate relative to free doxorubicin in drug-resistant MCF-7 cells and suggested that this improvement was likely due to a change in the mechanism of uptake relative to the free drug alone.77 Aroui and co-workers also reported a comparative study on doxorubicin conjugated to either Tat or penetratin, which showed different cell kill profiles depending upon both CPP identity and cell line.64 Additionally, doxorubicin was conjugated to a small series of proprietary peptides, named Vectocell peptides, that were developed to transport molecules across cell membranes via endocytosis.81 The authors found that a peptide–doxorubicin conjugate coupled through an ester linkage showed increased antitumoral activity against both doxorubicin-sensitive and -resistant cancer models.63 This Vectocell conjugate also displayed better efficacy than doxorubicin alone in a partially drug-resistant in vivo tumor model. Lindgren and colleagues also reported the nonreleasable conjugation of methotrexate to a CPP identified by phage display, which killed resistant breast cancer cells more efficiently than unconjugated drug.65
Though this early work had focused largely on nonreleasable transporter conjugates, we sought to design and test a system that would allow release of a cargo, preferably only inside of the cell. Such a strategy would allow an inactive drug conjugate to be loaded into a cell, after which the free, active drug would be released at a rate controlled by linker design. To explore this idea, in 2006, we introduced an analytical method that allows one to measure in real time, in vitro and in vivo, cellular uptake, cargo release, and cargo turnover using an optical probe (luciferin) as the cargo and luciferase as its intracellular target (Figure 5A).82 The first generation strategy was based on a disulfide-containing linker that would allow for controllable release of cargo only inside the cell, mediated by the higher intracellular concentrations of glutathione (GSH).82−84 In this approach, a disulfide bond between the transporter and cargo (luciferin) is cleaved by GSH, producing a free thiol that cyclizes into a pendant ester or carbonate, releasing free cargo (luciferin), which is then turned over by firefly luciferase with emission of one photon/turnover event. Only free luciferin is a substrate for luciferase, and every turnover event is marked by release of a photon that is counted by an extracellular charge-coupled device (CCD) camera. Thus, the process emulates and measures the equivalent of drug delivery, release, and turnover with a surrogate drug probe (luciferin). This real time quantification method was shown to be effective in both cells82 and animals.85
Figure 5.

Analytical method to measure the real-time uptake, release, and turnover of transporter–drug surrogate (A, luciferin;82 B, coelenterazine H61) conjugates with a GSH-cleavable linker in both cell and animals.
In 2008, we sought to explore the generality and effectiveness of this transporter conjugation strategy in overcoming Pgp-mediated efflux with an octaarginine conjugate of coelenterazine H (Figure 5B).61 Coelenterazine H is a bioluminescent substrate for intracellular Renilla luciferase and emits a photon with each turnover event. As a lipophilic heterocycle, it is also a substrate for Pgp-mediated efflux.86 Ovarian cancer cells (OVCA-429) with a low expression level of Pgp exports pumps, when treated with either coelenterazine H or a releasable coelenterazine–transporter conjugate, exhibit a bioluminescent signal indicating that both the free probe and probe conjugate enter cells.61 However, when the same experiment was repeated with an MDR ovarian cancer cell line, OVCA-429T, bioluminescence was observed for the coelenterazine conjugate but not for coelenterazine H alone. Significantly, the octaarginine–coelenterazine H conjugate produced similar bioluminescent signals in both resistant and nonresistant cell lines, indicating that it is not affected by Pgp export. As further evidence that a Pgp-evading mechanism was operative in overcoming the resistant phenotype, cotreatment with a known Pgp inhibitor, cyclosporine A, restored the bioluminescent signal for coelenterazine H but did not significantly affect the signal output from the octaarginine–coelenterazine conjugate. In addition to providing evidence of the ability of transporters to avoid Pgp-mediated efflux, these studies suggest that this approach might have generality for many probes and drugs that are Pgp substrates.
On the basis of these results, we set out to examine whether the efficacy of a known drug that succumbs to Pgp-based resistance could be restored simply through releasable conjugation to a molecular transporter. Taxol was selected because of its widespread use against many different types of cancers and its known tendency to develop tumor resistance. Taxol is widely used for the treatment of lung, breast, and ovarian carcinomas,30 as well as for Kaposi’s sarcoma and others.87 While collected patient data is difficult to assess, the market size for taxanes, a measure of its clinical utility, is estimated to be $3 billion annually.88 However, despite this clinical and commercial success, Taxol is ineffective against various types of resistant cancer. As a nonpolar drug, Taxol is soluble in nonpolar membranes, thus having a higher residency time there and a higher probability of Pgp-mediated efflux from the cell.89 Because less drug gets into the cell and thus to its target, Taxol shows reduced activity against MDR tumors.17,18,20 Increasing the dose of Taxol to offset efflux loss and force more intracellular accumulation is not an option, as that would cause greater off-target toxicities. However, a releasable Taxol–transporter conjugate that is not a substrate for Pgp export would evade export and release free Taxol in a cell at a concentration and rate determined by linker design (Figure 4).61,82 Of additional significance, octaarginine conjugates of Taxol, in contrast to the free drug, are highly water-soluble,90 allowing for dramatically reduced formulation volume, and thus administration times, and eliminating the need for toxic excipients, such as Cremophor EL.91
The release of free Taxol from these conjugates is induced by the cleavage of a disulfide bond by intracellular glutathione, which has a higher concentration in ovarian tumor-derived ascites than in the surrounding lymphocytes, macrophages, and mesothelial cells.92 Further, by releasing Taxol only inside a cell at a rate controlled by linker design, these conjugates allow for sustained release, thereby avoiding bolus effects and minimizing repeated or prolonged administration procedures that place burdens on both patients and clinicians.
In our efforts to examine whether the efficacy of Taxol, which succumbs to Pgp-based resistance, could be restored through conjugation to a molecular transporter, we focused our studies on models of epithelial ovarian cancer. Ovarian cancer is the leading cause of death for gynecologic malignancies, with 140,000 deaths and 220,000 new cases diagnosed each year worldwide.93 The current standard front-line therapy for advanced ovarian cancer is surgical elimination of all visible diseases, if technically feasible, followed by a combination platinum- and taxane-based regimen.94 While the majority of patients respond initially to this therapy, most patients subsequently develop recurrent disease. This recurrent disease is often chemotherapy resistant, with poor survival outcomes. Neither extending the time of treatment with Taxol, nor adding a third chemotherapeutic agent to the platinum/taxane front-line therapy improve overall survival.95,96 Recently, the National Cancer Institute recommended the administration of Taxol via intraperitoneal (IP) injection due to promising clinical results for ovarian cancer treatment.97 IP administration has several advantages including localized delivery directly to the targeted tissue, minimizing systemic exposure, which reduces off-target effects. In addition, IP-administered Taxol has a much longer dwell time, thus providing a “depot” effect or a metronomic chemotherapy. Interestingly, Futaki and co-workers recently showed that intravenous (IV) administration of d-octaarginine to tumor-xenografted mice resulted in high accumulation of the transporter in tumor xenografts,98 a preliminary indication that IV-injectable formulations of transporter–drug conjugates could also be used for treating certain resistant tumors.
In 2008, our lab demonstrated that these Taxol–octaarginine conjugates outperform Taxol alone in a panel of both Taxol-sensitive and Taxol-resistant ovarian cancer cell lines and enhance survival in tumor-bearing animals.61 Taxol_octaarginine conjugates, along with their hydrolytic stability (measured as a half-life under assay conditions) and release rates (measured as a half-life under reducing conditions) are shown in Table 2.61,62 Taxol was attached to the octaarginine transporter at either the C2′ or the C7 position using a series of bioactivatable disulfide linkers (Table 2). The two positions of attachment were selected due to their expected difference in activity upon modification. Modification of the free alcohol at the C2′ position of Taxol is known to significantly diminish efficacy;99 thus, drug release would be necessary for activity. Previous studies have shown Taxol can undergo modification at C7 without a significant loss of activity;30 thus, C7–transporter conjugates were also prepared. The linkers used in these conjugates varied in substitution at the α position of the ester linkage attached to Taxol, from unhindered to geminal dimethyl substituents. Further variations included connectivity through a carbonate linkage and varying the length of the linker. The conjugates used for these studies displayed a wide range of stabilities under physiological conditions, with half-lives ranging from hours to weeks, though they all rapidly release free drug in a reducing environment (minutes to hours). In short, they can be readily tuned for shelf stability and for intracellular release.
Table 2. Taxol–Transporter Conjugates, Linkers, and Relative Stabilities;62 Hydrolytic Stability Is Measured As Half-Life under Assay Conditions, and Release Rates Are Measured As a Half-Life under Reducing Conditions.
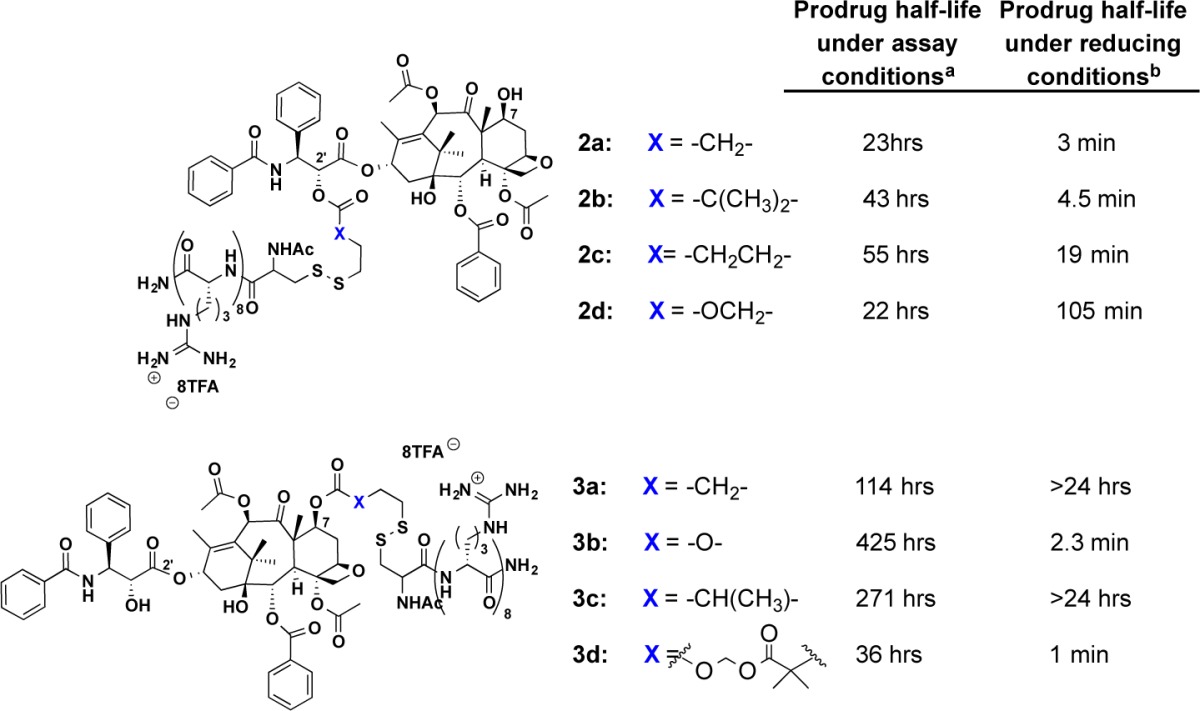
Assay conditions for prodrug stability: HBS, pH 7.4, 37 °C.
Assay conditions for prodrug stability: 10 mM dithiothreitol (DTT), HBS, pH 7.4, 37 °C.
A variety of human ovarian cancer cell lines were tested, including Taxol-sensitive UCI-101, SCOV-3, OVCA429, and OVCA433 cells, along with Taxol-resistant variants, such as OVCA429T, OVCA433T, and MCF-7-Pgp.61 In all cell lines, the releasable Taxol–octaarginine conjugates outperformed Taxol alone, with major differences (4–100-fold) in resistant cell lines. A Tubulin polymerization assay and a cell cycle assay of the conjugates were used to determine whether Taxol and the Taxol conjugates shared a common mechanism of action (Figure 6). As expected, the greatest loss in cell viability was found for cells in G2/M interphase for both conjugates and Taxol alone. In vivo, mice inoculated with either UCI-101 ovarian tumor cells or with Taxol-resistant OVCA429T cells showed improved survival when dosed IP with the releasable Taxol–octaarginine conjugates as compared to Taxol alone (Figure 7). The choice of conjugates to evaluate in each study was made to minimize the number of mice used while maximizing results obtained. Compound 3a was chosen for evaluation in the resistant OVCA429 tumor model because it produced the highest percentage of cells arrested in the G2/M phase in the resistant 429T line (Figure 6B). Because of multiple differences between 2a and 3a (e.g., location of transporter attachment, hydrolytic stability, release rate, etc.), 2a and 3a were not directly compared in these studies.
Figure 6.

Mechanisms of action of Taxol–octaarginine conjugates. (A) Tubulin polymerization assay. Conjugates were compared to free Taxol for the ability to polymerize free tubulin, as assessed by measuring increase in turbidity (absorbance at 350 nm).61 (B) Cell cycle assay. Conjugates were compared to free Taxol for the ability to kill cells through the same cell-cycle arrest mechanism.61 *Conditions under which octaarginine conjugates produce a significantly higher percentage of cells in G2/M phase than Taxol alone. Reproduced with permission from ref (61). Copyright 2008 National Academy of Sciences.
Figure 7.

Life extension graphs (Kaplan–Meier survival curves) for tumor-bearing mice treated with either Taxol or its octaarginine derivatives.61 (A) One × 107 UCI-101 tumor cells expressing luciferase were implanted into the peritoneal cavity of athymic nu/nu mice 7 days before treatment. Mice were treated with IP injections of 5 (left) or 10 mg/kg (right) of Taxol or equimolar amounts of its derivatives [octaarginine conjugated to C2′, 2a or C7, 3a, positions] on days 0, 5, and 10. Tumor burden was monitored by bioluminescence imaging (n = 8 per group). C2′ conjugate, 2a, produces significantly greater survival than Taxol at both 5 mg/kg (P = 0.0039) and 10 mg/kg (P = 0.047). (B) Taxol-sensitive (OVCA-429) and Taxol-resistant (OVCA-429T) cancers, when treated with free Taxol and releasable Taxol–transporter conjugates. Mice were implanted with 1 × 107 OVCA-429 or OVCA-429T cells expressing luciferase and subsequently treated (7 days later) with 5 mg/kg of Taxol or equimolar amounts of an octaarginine C7-conjugated derivative 3a on days 0, 5, and 10. Tumor burden was measured by bioluminescence imaging (n = 8 per group). The octaarginine conjugate 3a produces significantly better survival rates than Taxol in OVCA-429T cells (P = 0.0002). Reproduced with permission from ref (61). Copyright 2008 National Academy of Sciences.
An issue of further clinical significance that can be beneficially impacted with transporter technology is related to the controllable biodistribution of conjugates. By design, one can generate conjugates that stay where administered or others that distribute because uptake is made slower. Illustrative of this point, the biodistribution and pharmacokinetics of octaarginine–luciferin conjugates were evaluated in real time in living luciferase-expressing animals.61 Luciferin and releasable luciferin–octaarginine conjugates were administered via IP injection into mice constitutively expressing firefly luciferase, and their respective real time biodistributions were compared (Figure 8). Because of the rapid cellular uptake of the octaarginine conjugate, it remained localized near the site of administration within the IP cavity (Figure 8, center panel). Luciferin conjugates with control peptide, tetralysine, which does not display significant cellular uptake, stayed localized in the IP cavity but provided significantly poorer uptake into cells, as shown by the greatly reduced signal from released luciferin (Figure 8, left panel). In dramatic contrast, free luciferin, when injected IP, distributed over the entire animal (Figure 8, right panel). For solid tumors, the localization and uptake of the octaarginine conjugates could allow for greater tumor accumulation and correspondingly reduced systemic toxicity. Discussions of this study can be read in the works of Fonesca, Hampton, and others.100−102
Figure 8.
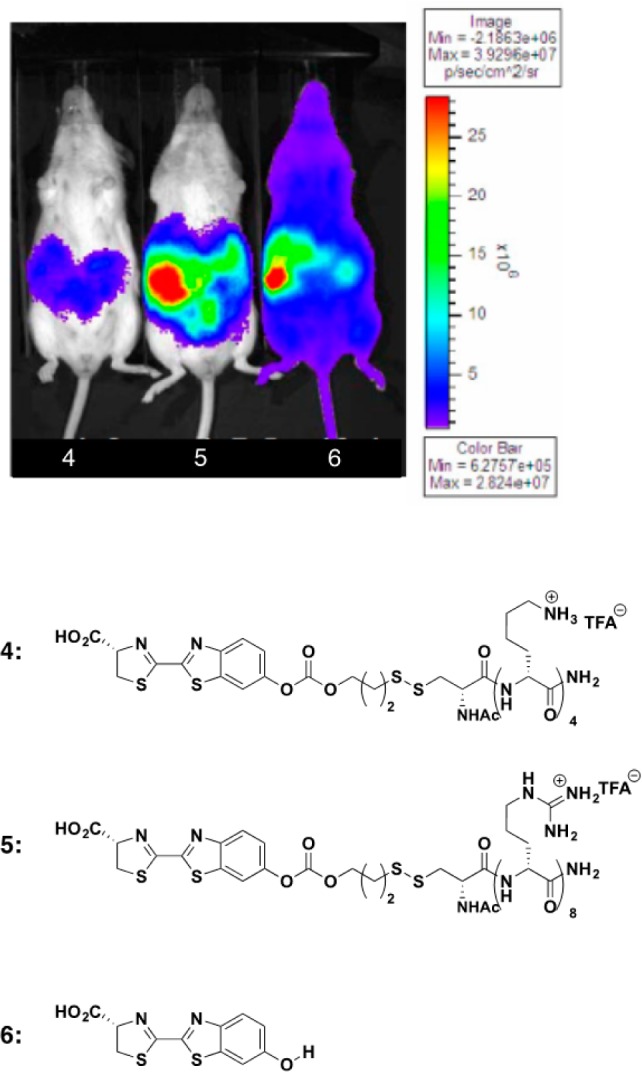
Real-time uptake, release, and biodistribution of IP-administered transporter–luciferin conjugates.61 Control transporter conjugate, luciferin−tetralysine (4, left); active luciferin−octaarginine conjugate (5, center); and free luciferin (6, right). Reproduced with permission from ref (61). Copyright 2008 National Academy of Sciences.
In a significant step toward a clinical approach to treating resistant ovarian cancer, we recently demonstrated that the Taxol–octaarginine conjugates showed effective cell kill in ex vivo ascites from drug-resistant ovarian cancer patients in multiple subtypes of ovarian carcinoma, including clear cell carcinoma and serous/papillary carcinoma (Figure 9).62 All Taxol–octaarginine conjugates tested in ex vivo ascite cells displayed similar levels of cell kill, outperforming Taxol alone, which was inactive against these resistant cell samples from ovarian cancer patients. Of note, preliminary toxicity assays against normal human neutrophils with these conjugates indicate that the octaarginine conjugates were not significantly more toxic than Taxol alone.62 As with all new agents, additional studies will address toxicity questions, including hematologic toxicity, with respect to both IV and IP administration, though recent successful studies of IV administration indicate that, at least for certain concentrations and formulations,99 an appropriate therapeutic window would be accessible.
Figure 9.
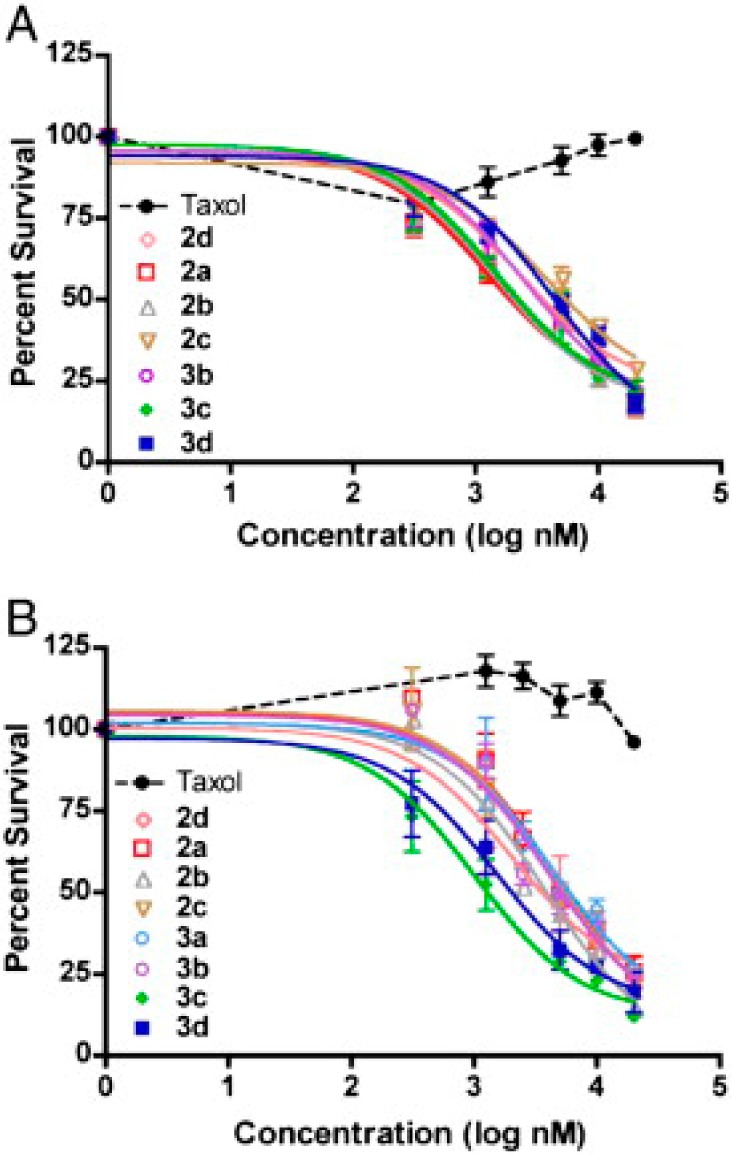
Dose–response curves of Taxol and Taxol–transporter conjugates against two ex vivo subtypes of ovarian carcinoma.62 The Taxol conjugates outperformed Taxol in 9 of 9 patient samples: (A) clear cell carcinoma and (B) serous papillary carcinoma. Reproduced with permission from ref (62). Copyright 2012 Elsevier.
Though all conjugates in these studies utilized a cleavable disulfide linker, other bioactivatable mechanisms are readily amenable to incorporation for transporter–drug cleavage or targeting, such as protease-, esterase-, or phosphatase-cleavable moieties.103−105 As demonstrated with the disulfide linkers, the rate of release of free drug can be controlled and tuned for specific applications. Another advantage of designing releasable transporter–drug conjugates is the ability to limit release to strictly intracellular release, which would serve to minimize side effects.
Outlook
Multidrug resistance remains a major challenge in the treatment of a number of the world’s most devastating diseases, including cancer, tuberculosis, and malaria. Strategies to address this resistance could have a profoundly beneficial impact on treating disease. In this review, we have focused on the use of molecular transporter–drug conjugates as a powerful and potentially general strategy to overcoming multidrug resistance mediated by Pgp-export pumps.
As discussed herein, transporter–drug conjugates often offer huge advantages over free drug alone and, in the case of known drugs that succumb to resistance, a remarkably fast path to clinical trials. Guanidinium-rich transporter–drug conjugates are water-soluble and thus offer many advantages in formulation and administration, as evident with Taxol, which can be formulated in small volumes of water and thus more rapidly administered, a benefit to both patients and clinicians. For many drugs, as exemplified by Taxol, the solubilizing effect allows one to avoid using toxic excipients (e.g., Cremophor EL). While Taxol complexed with proteins (e.g., Abraxane)106 or with nanoparticles53,107 can be used for formulation and delivery, it is unclear whether they overcome Pgp-based resistance. Perhaps most importantly, in contrast to Taxol–protein complexes or nanoparticle complexes, drug–transporter conjugates are discrete, single molecule entities and thus provide greater batch-to-batch consistency and fewer regulatory problems. An often under-appreciated aspect of releasable drug conjugates is that they can be tuned to control drug payout from minutes to days, allowing for the significant clinical benefit of sustained release and avoiding peak–trough issues associated with free drug dosing and clearance. Finally and most importantly, drug–transporter conjugates dramatically change the properties of lipophilic drugs thereby evading Pgp-based resistance. Though not yet experimentally explored, these transporter–drug conjugates should have application to other cancer types rendered difficult to treat due to export pump-mediated resistance, such as breast18 and nonsmall-cell lung cancer.19,20
Though the work performed by our lab and others has been primarily focused on anthracyclines and taxanes, there are many other approved small molecule chemotherapeutics that suffer from efflux-mediated resistance, both in cancer and in other diseases. Because it has been shown that transporter–drug conjugates largely assume the physical properties of the transporter, this strategy should be broadly applicable to these other therapeutic classes in addressing efflux-related resistance.
While the current transporter–drug conjugates outperform the drug alone and are thus poised for clinical evaluation, it is expected for many cancers that transporter–drug conjugates could also be targeted to diseased tissue. The work described above showed passive targeting due to higher GSH levels in some cancer cells. Similarly, a recent study by Futaki and co-workers showed that octaarginine–drug conjugates show higher accumulation in tumor tissue.98 To achieve, where necessary, even higher levels of intratumoral drug concentration and selectivity, more direct targeting efforts are possible. Foundational work by both the Tsien and Wender groups has shown that “kinetic targeting” with transporters provides a complementary strategy to thermodynamic targeting of monoclonal antibodies. Cationic guanidinium-rich transporters conjugated to an oligoanion sequence through a cleavable linking peptide do not enter cells due to charge neutralization that inactivates uptake. However, when the linker is cleaved by an overexpressed cell-surface protease expressed on tumor cells, the transporter–drug conjugate freely enters proximate cells.103−105 The Tsien lab explored the use of a matrix metallo-protease-cleavable linker sequence,104,105 while our lab utilized a prostate-specific antigen sequence.103 MMP-cleavable sequences have also been used with doxorubicin for cancer.108 However, no one has explored this strategy in resistant cancers to date. Ligand conjugates such as folate or transferrin have also been explored by others, and nanoparticles have also been recognized for their potential as drug delivery agents to target and deliver chemotherapeutics to resistant cancers.107
The opportunity to improve the formulation, administration, release, targeting, and efficacy of a chemotherapeutic drug by conjugation to a molecular transporter is significant, especially in connection with treating resistant disease. This transporter-enabled strategy can greatly improve the prognosis of patients bearing resistant tumors. In the case of known drugs that succumb to resistance, this concept provides the basis for an exceptionally fast path to the clinic as conjugate synthesis is rapid and tunable, and the strategy does not require exploring a new mode of action but rather improving upon an existing drug. This strategy can be applied to a large number of existing chemotherapeutic agents, as well as drug leads, and its use extends to other important diseases where efflux-mediated resistance is manifest.
Acknowledgments
Support of this work to P.A.W. through Grants NIH-CA031841 and NIH-CA031845 from the National Institutes of Health and to E.G.S. and J.R.V. through fellowships from the National Science Foundation (E.G.S. and J.R.V.) and the Stanford Graduate Fellowship (E.G.S.) are acknowledged.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- World Health Organization. Towards Universal Access to Diagnosis and Treatment of Multidrug-Resistant and Extensively Drug-Resistant Tuberculosis by 2015. In WHO Progress Report 2011 WHO/HTM/TB/2011.3; World Health Organization: Geneva, Switzerland, 2011. [Google Scholar]
- Silva P. E. A. D.; Palomino J. C. Molecular Basis and Mechanisms of Drug Resistance in Mycobacterium Tuberculosis: Classical and New Drugs. J. Antimicrob. Chemother. 2011, 66, 1417–1430. [DOI] [PubMed] [Google Scholar]
- Longley D. B.; Johnston P. G. Molecular Mechanisms of Drug Resistance. J. Pathol. 2005, 205, 275–292. [DOI] [PubMed] [Google Scholar]
- Abouzeed Y. M.; Baucheron S.; Cloeckaert A. ramR Mutations Involved in Efflux-Mediated Multidrug Resistance in Salmonella Enterica Serovar Typhimurium. Antimicrob. Agents Chemother. 2008, 52, 2428–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szakács G.; Paterson J. K.; Ludwig J. A.; Booth-Genthe C.; Gottesman M. M. Targeting Multidrug Resistance in Cancer. Nat. Rev. Drug Discovery 2006, 5, 219–234. [DOI] [PubMed] [Google Scholar]
- Colabufo N. A.; Berardi F.; Cantore M.; Contino M.; Inglese C.; Niso M.; Perrone R. Perspectives of P-Glycoprotein Modulating Agents in Oncology and Neurodegenerative Diseases: Pharmaceutical, Biological, and Diagnostic Potentials. J. Med. Chem. 2010, 53, 1883–1897. [DOI] [PubMed] [Google Scholar]
- Mohandas N.; Winardi R.; Knowles D.; Leung A.; Parra M.; George E.; Conboy J.; Chasis J. Molecular Basis for Membrane Rigidity of Hereditary Ovalocytosis. A Novel Mechanism Involving the Cytoplasmic Domain of Band 3. J. Clin. Invest. 1992, 89, 686–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowman A. F.; Karcz S.; Galatis D.; Culvenor J. G. A P-Glycoprotein Homologue of Plasmodium Falciparum Is Localized on the Digestive Vacuole. J. Cell Biol. 1991, 113, 1033–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues L.; Machado D.; Couto I.; Amaral L.; Viveiros M. Contribution of Efflux Activity to Isoniazid Resistance in the Mycobacterium Tuberculosis Complex. Infect. Genet. Evol. 2012, 12, 695–700. [DOI] [PubMed] [Google Scholar]
- Kartner N.; Riordan J. R.; Ling V. Cell Surface P-Glycoprotein Associated with Multidrug Resistance in Mammalian Cell Lines. Science 1983, 221, 1285–1288. [DOI] [PubMed] [Google Scholar]
- Sharom F. J. ABC Multidrug Transporters: Structure, Function and Role in Chemoresistance. Pharmacogenomics 2008, 9, 105–127. [DOI] [PubMed] [Google Scholar]
- Lam F. C.; Liu R.; Lu P.; Shapiro A. B.; Renoir J.-M.; Sharom F. J.; Reiner P. B. B-Amyloid Efflux Mediated by P-Glycoprotein. J. Neurochem. 2001, 76, 1121–1128. [DOI] [PubMed] [Google Scholar]
- Schuetz E. G.; Beck W. T.; Schuetz J. D. Modulators and Substrates of P-Glycoprotein and Cytochrome P4503A Coordinately up-Regulate These Proteins in Human Colon Carcinoma Cells. Mol. Pharmacol. 1996, 49, 311–318. [PubMed] [Google Scholar]
- Gatlik-Landwojtowicz E.; Äänismaa P.; Seelig A. Quantification and Characterization of P-Glycoprotein–Substrate Interactions. Biochemistry 2006, 45, 3020–3032. [DOI] [PubMed] [Google Scholar]
- Aller S. G.; Yu J.; Ward A.; Weng Y.; Chittaboina S.; Zhuo R.; Harrell P. M.; Trinh Y. T.; Zhang Q.; Urbatsch I. L.; Chang G. Structure of P-Glycoprotein Reveals a Molecular Basis for Poly-Specific Drug Binding. Science 2009, 323, 1718–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkadi B.; Homolya L.; Szakács G.; Váradi A. Human Multidrug Resistance ABCB and ABCG Transporters: Participation in a Chemoimmunity Defense System. Physiol. Rev. 2006, 86, 1179–1236. [DOI] [PubMed] [Google Scholar]
- Zajchowski D. A.; Karlan B. Y.; Shawver L. K. Treatment-Related Protein Biomarker Expression Differs between Primary and Recurrent Ovarian Carcinomas. Mol. Cancer Ther. 2011, 11, 492–502. [DOI] [PubMed] [Google Scholar]
- Murray S.; Briasoulis E.; Linardou H.; Bafaloukos D.; Papadimitriou C. Taxane Resistance in Breast Cancer: Mechanisms, Predictive Biomarkers and Circumvention Strategies. Cancer Treat. Rev. 2012, 38, 890–903. [DOI] [PubMed] [Google Scholar]
- Yeh J. J.; Hsu W. H.; Wang J. J.; Ho S. T.; Kao A. Predicting Chemotherapy Response to Paclitaxel-Based Therapy in Advanced Non-Small-Cell Lung Cancer with P-Glycoprotein Expression. Respiration 2003, 70, 32–35. [DOI] [PubMed] [Google Scholar]
- Chiou J.-F.; Liang J.-A.; Hsu W.-H.; Wang J.-J.; Ho S.-T.; Kao A. Comparing the Relationship of Taxol-Based Chemotherapy Response with P-Glycoprotein and Lung Resistance-Related Protein Expression in Non-Small Cell Lung Cancer. Lung 2003, 181, 267–273. [DOI] [PubMed] [Google Scholar]
- Dean M.; Fojo T.; Bates S. Tumour Stem Cells and Drug Resistance. Nat. Rev. Cancer 2005, 5, 275–284. [DOI] [PubMed] [Google Scholar]
- Flesken-Nikitin A.; Hwang C.-I.; Cheng C.-Y.; Michurina T. V.; Enikolopov G.; Nikitin A. Y. Ovarian Surface Epithelium at the Junction Area Contains a Cancer-Prone Stem Cell Niche. Nature 2013, 495, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Li Y.; Yu T.-S.; McKay R. M.; Burns D. K.; Kernie S. G.; Parada L. F. A Restricted Cell Population Propagates Glioblastoma Growth after Chemotherapy. Nature 2012, 488, 522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schepers A. G.; Snippert H. J.; Stange D. E.; van den Born M.; van Es J. H.; van de Wetering M.; Clevers H. Lineage Tracing Reveals Lgr5+ Stem Cell Activity in Mouse Intestinal Adenomas. Science 2012, 337, 730–735. [DOI] [PubMed] [Google Scholar]
- Chaudhary P. M.; Roninson I. B. Expression and Activity of P-Glycoprotein, a Multidrug Efflux Pump, in Human Hematopoietic Stem Cells. Cell 1991, 66, 85–94. [DOI] [PubMed] [Google Scholar]
- Zinovyeva M. V.; Zijlmans J. M.; Fibbe W. E.; Visser J. W.; Belyavsky A. V. Analysis of Gene Expression in Subpopulations of Murine Hematopoietic Stem and Progenitor Cells. Exp. Hematol. 2000, 28, 318–334. [DOI] [PubMed] [Google Scholar]
- Zhou S.; Schuetz J. D.; Bunting K. D.; Colapietro A. M.; Sampath J.; Morris J. J.; Lagutina I.; Grosveld G. C.; Osawa M.; Nakauchi H.; Sorrentino B. P. The ABC Transporter Bcrp1/ABCG2 Is Expressed in a Wide Variety of Stem Cells and Is a Molecular Determinant of the Side-Population Phenotype. Nat. Med. 2001, 7, 1028–1034. [DOI] [PubMed] [Google Scholar]
- Marques D. S.; Sandrini J. Z.; Boyle R. T.; Marins L. F.; Trindade G. S. Relationships between Multidrug Resistance (MDR) and Stem Cell Markers in Human Chronic Myeloid Leukemia Cell Lines. Leuk. Res. 2010, 34, 757–762. [DOI] [PubMed] [Google Scholar]
- Martins-Neves S. R.; Lopes Á. O.; do Carmo A.; Paiva A. A.; Simões P. C.; Abrunhosa A. J.; Gomes C. M. F. Therapeutic Implications of an Enriched Cancer Stem-like Cell Population in a Human Osteosarcoma Cell Line. BMC Cancer 2012, 12, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston D. G. I.; Newman D. J. Taxoids: Cancer-Fighting Compounds from Nature. Curr. Opin. Drug Discovery Dev. 2007, 10, 130–144. [PubMed] [Google Scholar]
- Abidi A. Cabazitaxel: A Novel Taxane for Metastatic Castration-Resistant Prostate Cancer-Current Implications and Future Prospects. J. Pharmacol. Pharmacother. 2013, 4, 230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchet B. P.; Galmarini C. M. Cabazitaxel, a New Taxane with Favorable Properties. Drugs Today 2010, 46, 735–742. [DOI] [PubMed] [Google Scholar]
- Pivot X.; Koralewski P.; Hidalgo J. L.; Chan A.; Gonçalves A.; Schwartsmann G.; Assadourian S.; Lotz J. P. A Multicenter Phase II Study of XRP6258 Administered as a 1-H I.V. Infusion Every 3 Weeks in Taxane-Resistant Metastatic Breast Cancer Patients. Ann. Oncol. 2008, 19, 1547–1552. [DOI] [PubMed] [Google Scholar]
- Liu J.; Zhao Y.; Guo Q.; Wang Z.; Wang H.; Yang Y.; Huang Y. TAT-Modified Nanosilver for Combating Multidrug-Resistant Cancer. Biomaterials 2012, 33, 6155–6161. [DOI] [PubMed] [Google Scholar]
- Robert J.; Jarry C. Multidrug Resistance Reversal Agents. J. Med. Chem. 2003, 46, 4805–4817. [DOI] [PubMed] [Google Scholar]
- Nobili S.; Landini I.; Mazzei T.; Mini E. Overcoming Tumor Multidrug Resistance Using Drugs Able to Evade P-Glycoprotein or to Exploit Its Expression. Med. Res. Rev. 2012, 32, 1220–1262. [DOI] [PubMed] [Google Scholar]
- Shukla S.; Wu C.-P.; Ambudkar S. V. Development of Inhibitors of ATP-Binding Cassette Drug Transporters: Present Status and Challenges. Expert Opin. Drug Metab. Toxicol. 2008, 4, 205–223. [DOI] [PubMed] [Google Scholar]
- Xia C. Q.; Smith P. G. Drug Efflux Transporters and Multidrug Resistance in Acute Leukemia: Therapeutic Impact and Novel Approaches to Mediation. Mol. Pharmacol. 2012, 82, 1008–1021. [DOI] [PubMed] [Google Scholar]
- Tsuruo T.; Iida H.; Tsukagoshi S.; Sakurai Y. Overcoming of Vincristine Resistance in P388 Leukemia in Vivo and in Vitro through Enhanced Cytotoxicity of Vincristine and Vinblastine by Verapamil. Cancer Res. 1981, 41, 1967–1972. [PubMed] [Google Scholar]
- Cano-Gauci D. F.; Riorda J. R. Action of Calcium Antagonists on Multidrug Resistant Cells: Specific Cytotoxicity Independent of Increased Cancer Drug Accumulation. Biochem. Pharmacol. 1987, 36, 2115–2123. [DOI] [PubMed] [Google Scholar]
- Foxwell B. M.; Mackie A.; Ling V.; Ryffel B. Identification of the Multidrug Resistance-Related P-Glycoprotein as a Cyclosporine Binding Protein. Mol. Pharmacol. 1989, 36, 543–546. [PubMed] [Google Scholar]
- Solary E.; Velay I.; Chauffert B.; Caillot D.; Bidan J. M.; Dumas M.; Casasnovas O.; Guy H. Quinine Circumvents the Doxorubicin Resistance of a Multidrug Resistant Human Leukemic Cell-Line, K562/DXR. Nouv. Rev. Fr. Hématol. 1990, 32, 361–363. [PubMed] [Google Scholar]
- Stupp R.; Bauer J.; Pagani O.; Gerard B.; Cerny T.; Sessa C.; Bastian G.; Sarkany M.; Schläpfer J.; Giroux B.; Leyvraz S. Ventricular Arrhythmia and Torsade de Pointe: Dose Limiting Toxicities of the MDR-Modulator S9788 in a Phase I Trial. Ann. Oncol. 1998, 9, 1233–1242. [DOI] [PubMed] [Google Scholar]
- Fonseca S. B.; Kelley S. O. Peptide-Chlorambucil Conjugates Combat Pgp-Dependent Drug Efflux. ACS Med. Chem. Lett. 2011, 2, 419–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye S. B. Reversal of Drug Resistance in Ovarian Cancer: Where Do We Go from Here?. J. Clin. Oncol. 2008, 26, 2616–2618. [DOI] [PubMed] [Google Scholar]
- Abbasi M.; Lavasanifar A.; Uluda H. Recent Attempts at RNAi-Mediated P-Glycoprotein Downregulation for Reversal of Multidrug Resistance in Cancer. Med. Res. Rev. 2013, 33, 33–53. [DOI] [PubMed] [Google Scholar]
- Stege A.; Krühn A.; Lage H.. Overcoming Multidrug Resistance by RNA Interference. In Multi-Drug Resistance in Cancer; Zhou J., Ed.; Methods in Molecular Biology; Humana Press: New York, 2010; Vol. 596, pp 447–465. [DOI] [PubMed] [Google Scholar]
- Elbashir S. M.; Harborth J.; Lendeckel W.; Yalcin A.; Weber K.; Tuschl T. Duplexes of 21-Nucleotide RNAs Mediate RNA Interference in Cultured Mammalian Cells. Nature 2001, 411, 494–498. [DOI] [PubMed] [Google Scholar]
- Nieth C.; Priebsch A.; Stege A.; Lage H. Modulation of the Classical Multidrug Resistance (MDR) Phenotype by RNA Interference (RNAi). FEBS Lett. 2003, 545, 144–150. [DOI] [PubMed] [Google Scholar]
- Li L.; Xu J.; Min T.; Huang W. Reversal of MDR1 Gene-Dependent Multidrug Resistance Using Low Concentration of Endonuclease-Prepared Small Interference RNA. Eur. J. Pharmacol. 2006, 536, 93–97. [DOI] [PubMed] [Google Scholar]
- Stierlé V.; Laigle A.; Jollès B. Modulation of the Efficiency of a siRNA Directed against MDR1 Expression in MCF7-R Cells When Combined with a Second siRNA. Biochimie 2007, 89, 1033–1036. [DOI] [PubMed] [Google Scholar]
- Xiao H.; Wu Z.; Shen H.; Luo A.-L.; Yang Y.-F.; Li X.-B.; Zhu D.-Y. In Vivo Reversal of P-Glycoprotein-Mediated Multidrug Resistance by Efficient Delivery of Stealth RNAi. Basic Clin. Pharmacol. Toxicol. 2008, 103, 342–348. [DOI] [PubMed] [Google Scholar]
- Patil Y. B.; Swaminathan S. K.; Sadhukha T.; Ma L.; Panyam J. The Use of Nanoparticle-Mediated Targeted Gene Silencing and Drug Delivery to Overcome Tumor Drug Resistance. Biomaterials 2010, 31, 358–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamot C.; Drummond D. C.; Hong K.; Kirpotin D. B.; Park J. W. Liposome-Based Approaches to Overcome Anticancer Drug Resistance. Drug Resist. Updates 2003, 6, 271–279. [DOI] [PubMed] [Google Scholar]
- Št’astný M.; Strohalm J.; Plocová D.; Ulbrich K.; Říhová B. A Possibility to Overcome P-Glycoprotein (PGP)-Mediated Multidrug Resistance by Antibody-Targeted Drugs Conjugated to N-(2-Hydroxypropyl)methacrylamide (HPMA) Copolymer Carrier. Eur. J. Cancer 1999, 35, 459–466. [DOI] [PubMed] [Google Scholar]
- Fritzer M.; Szekeres T.; Szüts V.; Jarayam H. N.; Goldenberg H. Cytotoxic Effects of a Doxorubicin-Transferrin Conjugate in Multidrug-Resistant KB Cells. Biochem. Pharmacol. 1996, 51, 489–493. [DOI] [PubMed] [Google Scholar]
- Hrkach J.; Von Hoff D.; Ali M. M.; Andrianova E.; Auer J.; Campbell T.; De Witt D.; Figa M.; Figueiredo M.; Horhota A.; Low S.; McDonnell K.; Peeke E.; Retnarajan B.; Sabnis A.; Schnipper E.; Song J. J.; Song Y. H.; Summa J.; Tompsett D.; Troiano G.; Van Geen Hoven T.; Wright J.; LoRusso P.; Kantoff P. W.; Bander N. H.; Sweeney C.; Farokhzad O. C.; Langer R.; Zale S. Preclinical Development and Clinical Translation of a PSMA-Targeted Docetaxel Nanoparticle with a Differentiated Pharmacological Profile. Sci. Transl. Med. 2012, 4, 128ra39. [DOI] [PubMed] [Google Scholar]
- Wu J.; Lu Y.; Lee A.; Pan X.; Yang X.; Zhao X.; Lee R. J. Reversal of Multidrug Resistance by Transferrin-Conjugated Liposomes Co-Encapsulating Doxorubicin and Verapamil. J. Pharm. Pharm. Sci. 2007, 10, 350–357. [PubMed] [Google Scholar]
- Wender P. A.; Galliher W. C.; Goun E. A.; Jones L. R.; Pillow T. H. The Design of Guanidinium-Rich Transporters and Their Internalization Mechanisms. Adv. Drug Delivery Rev. 2008, 60, 452–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbard J. B.; Jessop T. C.; Lewis R. S.; Murray B. A.; Wender P. A. Role of Membrane Potential and Hydrogen Bonding in the Mechanism of Translocation of Guanidinium-Rich Peptides into Cells. J. Am. Chem. Soc. 2004, 126, 9506–9507. [DOI] [PubMed] [Google Scholar]
- Dubikovskaya E. A.; Thorne S. H.; Pillow T. H.; Contag C. H.; Wender P. A. Overcoming Multidrug Resistance of Small-Molecule Therapeutics through Conjugation with Releasable Octaarginine Transporters. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 12128–12133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wender P. A.; Galliher W. C.; Bhat N. M.; Pillow T. H.; Bieber M. M.; Teng N. N. H. Taxol-Oligoarginine Conjugates Overcome Drug Resistance in-Vitro in Human Ovarian Carcinoma. Gynecol. Oncol. 2012, 126, 118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Losic F.; Quinonero J.; Dubois V.; Alluis B.; Dechambre M.; Michel M.; Cailler F.; Fernandez A.-M.; Trouet A.; Kearsey J. Improved Therapeutic Efficacy of Doxorubicin through Conjugation with a Novel Peptide Drug Delivery Technology (Vectocell). J. Med. Chem. 2006, 49, 6908–6916. [DOI] [PubMed] [Google Scholar]
- Aroui S.; Brahim S.; Waard M. D.; Kenani A. Cytotoxicity, Intracellular Distribution and Uptake of Doxorubicin and Doxorubicin Coupled to Cell-Penetrating Peptides in Different Cell Lines: A Comparative Study. Biochem. Biophys. Res. Commun. 2010, 391, 419–425. [DOI] [PubMed] [Google Scholar]
- Lindgren M.; Rosenthal-Aizman K.; Saar K.; Eiríksdóttir E.; Jiang Y.; Sassian M.; Östlund P.; Hällbrink M.; Langel Ü. Overcoming Methotrexate Resistance in Breast Cancer Tumour Cells by the Use of a New Cell-Penetrating Peptide. Biochem. Pharmacol. 2006, 71, 416–425. [DOI] [PubMed] [Google Scholar]
- Stanzl E. G.; Trantow B. M.; Vargas J. R.; Wender P. A. Fifteen Years of Cell-Penetrating, Guanidinium-Rich Molecular Transporters: Basic Science, Research Tools, and Clinical Applications. Acc. Chem. Res. 2013, 46, 2944–2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wender P. A.; Cooley C. B.; Geihe E. I. Beyond Cell Penetrating Peptides: Designed Molecular Transporters. Drug Discovery Today Technol. 2012, 9, e49–e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torchilin V. P. Tat Peptide-Mediated Intracellular Delivery of Pharmaceutical Nanocarriers. Adv. Drug Delivery Rev. 2008, 60, 548–558. [DOI] [PubMed] [Google Scholar]
- Wender P. A.; Mitchell D. J.; Pattabiraman K.; Pelkey E. T.; Steinman L.; Rothbard J. B. The Design, Synthesis, and Evaluation of Molecules That Enable or Enhance Cellular Uptake: Peptoid Molecular Transporters. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 13003–13008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.-L.; Dubikovskaya E. A.; Hwang H.; Semyonov A. N.; Wang H.; Jones L. R.; Twieg R. J.; Moerner W. E.; Wender P. A. Single-Molecule Motions of Oligoarginine Transporter Conjugates on the Plasma Membrane of Chinese Hamster Ovary Cells. J. Am. Chem. Soc. 2008, 130, 9364–9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wender P. A.; Rothbard J. B.; Jessop T. C.; Kreider E. L.; Wylie B. L. Oligocarbamate Molecular Transporters: Design, Synthesis, and Biological Evaluation of a New Class of Transporters for Drug Delivery. J. Am. Chem. Soc. 2002, 124, 13382–13383. [DOI] [PubMed] [Google Scholar]
- Wender P. A.; Kreider E.; Pelkey E. T.; Rothbard J.; VanDeusen C. L. Dendrimeric Molecular Transporters: Synthesis and Evaluation of Tunable Polyguanidino Dendrimers That Facilitate Cellular Uptake. Org. Lett. 2005, 7, 4815–4818. [DOI] [PubMed] [Google Scholar]
- Chung H.-H.; Harms G.; Min Seong C.; Choi B. H.; Min C.; Taulane J. P.; Goodman M. Dendritic Oligoguanidines as Intracellular Translocators. Pept. Sci. 2004, 76, 83–96. [DOI] [PubMed] [Google Scholar]
- Cooley C. B.; Trantow B. M.; Nederberg F.; Kiesewetter M. K.; Hedrick J. L.; Waymouth R. M.; Wender P. A. Oligocarbonate Molecular Transporters: Oligomerization-Based Syntheses and Cell-Penetrating Studies. J. Am. Chem. Soc. 2009, 131, 16401–16403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiti K. K.; Jeon O.-Y.; Lee W. S.; Kim D.-C.; Kim K.-T.; Takeuchi T.; Futaki S.; Chung S.-K. Design, Synthesis, and Membrane-Translocation Studies of Inositol-Based Transporters. Angew. Chem., Int. Ed. 2006, 45, 2907–2912. [DOI] [PubMed] [Google Scholar]
- Mazel M.; Clair P.; Rousselle C.; Vidal P.; Scherrmann J. M.; Mathieu D.; Temsamani J. Doxorubicin-Peptide Conjugates Overcome Multidrug Resistance. Anticancer Drugs 2001, 12, 107–116. [DOI] [PubMed] [Google Scholar]
- Liang J. F.; Yang V. C. Synthesis of Doxorubicin-Peptide Conjugate with Multidrug Resistant Tumor Cell Killing Activity. Bioorg. Med. Chem. Lett. 2005, 15, 5071–5075. [DOI] [PubMed] [Google Scholar]
- Frankel A. D.; Pabo C. O. Cellular Uptake of the Tat Protein from Human Immunodeficiency Virus. Cell 1988, 55, 1189–1193. [DOI] [PubMed] [Google Scholar]
- Green M.; Loewenstein P. M. Autonomous Functional Domains of Chemically Synthesized Human Immunodeficiency Virus Tat Trans-Activator Protein. Cell 1988, 55, 1179–1188. [DOI] [PubMed] [Google Scholar]
- Vivès E.; Brodin P.; Lebleu B. A Truncated HIV-1 Tat Protein Basic Domain Rapidly Translocates through the Plasma Membrane and Accumulates in the Cell Nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [DOI] [PubMed] [Google Scholar]
- De Coupade C.; Fittipaldi A.; Chagnas V.; Michel M.; Carlier S.; Tasciotti E.; Darmon A.; Ravel D.; Kearsey J.; Giacca M.; Cailler F. Novel Human-Derived Cell-Penetrating Peptides for Specific Subcellular Delivery of Therapeutic Biomolecules. Biochem. J. 2005, 390, 407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones L. R.; Goun E. A.; Shinde R.; Rothbard J. B.; Contag C. H.; Wender P. A. Releasable Luciferin-Transporter Conjugates: Tools for the Real-Time Analysis of Cellular Uptake and Release. J. Am. Chem. Soc. 2006, 128, 6526–6527. [DOI] [PubMed] [Google Scholar]
- Saito G.; Swanson J. A.; Lee K.-D. Drug Delivery Strategy Utilizing Conjugation via Reversible Disulfide Linkages: Role and Site of Cellular Reducing Activities. Adv. Drug Delivery Rev. 2003, 55, 199–215. [DOI] [PubMed] [Google Scholar]
- Mulders T. M.; Keizer J.; Breimer D. D.; Mulder G. J. In Vivo Characterization and Modulation of the Glutathione/glutathione S-Transferase System in Cancer Patients. Drug Metab. Rev. 1995, 27, 191–229. [DOI] [PubMed] [Google Scholar]
- Wender P. A.; Goun E. A.; Jones L. R.; Pillow T. H.; Rothbard J. B.; Shinde R.; Contag C. H. Real-Time Analysis of Uptake and Bioactivatable Cleavage of Luciferin-Transporter Conjugates in Transgenic Reporter Mice. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 10340–10345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichler A.; Prior J. L.; Piwnica-Worms D. Imaging Reversal of Multidrug Resistance in Living Mice with Bioluminescence: MDR1 P-Glycoprotein Transports Coelenterazine. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 1702–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cragg G. M.; Kingston D. G. I.; Newman D. J.. Anticancer Agents from Natural Products, 2nd ed.; CRC Press: Boca Raton, FL, 2011. [Google Scholar]
- Elder M.World Market for Cancer Therapeutics, 4th ed.; Kalorama Information: Rockville, MD, 2012. [Google Scholar]
- Krishna R.; Mayer L. D. Multidrug Resistance (MDR) in Cancer: Mechanisms, Reversal Using Modulators of MDR and the Role of MDR Modulators in Influencing the Pharmacokinetics of Anticancer Drugs. Eur. J. Pharm. Sci. 2000, 11, 265–283. [DOI] [PubMed] [Google Scholar]
- Kirschberg T. A.; VanDeusen C. L.; Rothbard J. B.; Yang M.; Wender P. A. Arginine-Based Molecular Transporters: The Synthesis and Chemical Evaluation of Releasable Taxol-Transporter Conjugates. Org. Lett. 2003, 5, 3459–3462. [DOI] [PubMed] [Google Scholar]
- Gelderblom H.; Verweij J.; Nooter K.; Sparreboom A. Cremophor EL: The Drawbacks and Advantages of Vehicle Selection for Drug Formulation. Eur. J. Cancer 2001, 37, 1590–1598. [DOI] [PubMed] [Google Scholar]
- Lee F. Y.; Vessey A.; Rofstad E.; Siemann D. W.; Sutherland R. M. Heterogeneity of Glutathione Content in Human Ovarian Cancer. Cancer Res. 1989, 49, 5244–5248. [PubMed] [Google Scholar]
- Siegel R.; Naishadham D.; Jemal A. Cancer Statistics, 2012. CA: Cancer J. Clin. 2012, 62, 10–29. [DOI] [PubMed] [Google Scholar]
- NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines): Ovarian Cancer Including Fallopian Tube Cancer and Primary Peritoneal Cancer, version 1, 2013. [Google Scholar]
- Markman M. Phase III Randomized Trial of 12 Versus 3 Months of Maintenance Paclitaxel in Patients With Advanced Ovarian Cancer After Complete Response to Platinum and Paclitaxel-Based Chemotherapy: A Southwest Oncology Group and Gynecologic Oncology Group Trial. J. Clin. Oncol. 2003, 21, 2460–2465. [DOI] [PubMed] [Google Scholar]
- Bookman M. A.; Brady M. F.; McGuire W. P.; Harper P. G.; Alberts D. S.; Friedlander M.; Colombo N.; Fowler J. M.; Argenta P. A.; De Geest K.; Mutch D. G.; Burger R. A.; Swart A. M.; Trimble E. L.; Accario-Winslow C.; Roth L. M. Evaluation of New Platinum-Based Treatment Regimens in Advanced-Stage Ovarian Cancer: A Phase III Trial of the Gynecologic Cancer InterGroup. J. Clin. Oncol. 2009, 27, 1419–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong D. K.; Bundy B.; Wenzel L.; Huang H. Q.; Baergen R.; Lele S.; Copeland L. J.; Walker J. L.; Burger R. A. Intraperitoneal Cisplatin and Paclitaxel in Ovarian Cancer. N. Engl. J. Med. 2006, 354, 34–43. [DOI] [PubMed] [Google Scholar]
- Nakase I.; Konishi Y.; Ueda M.; Saji H.; Futaki S. Accumulation of Arginine-Rich Cell-Penetrating Peptides in Tumors and the Potential for Anticancer Drug Delivery in Vivo. J. Controlled Release 2012, 159, 181–188. [DOI] [PubMed] [Google Scholar]
- Kingston D. G. Recent Advances in the Chemistry of Taxol. J. Nat. Prod. 2000, 63, 726–734. [DOI] [PubMed] [Google Scholar]
- Fonseca S. B.; Pereira M. P.; Kelley S. O. Recent Advances in the Use of Cell-Penetrating Peptides for Medical and Biological Applications. Adv. Drug Delivery Rev. 2009, 61, 953–964. [DOI] [PubMed] [Google Scholar]
- Hampton T. Researchers Devise Innovative Method to Overcome Cancer-Related Drug Resistance. JAMA, J. Am. Med. Assoc. 2008, 300, 1507–1507. [DOI] [PubMed] [Google Scholar]
- Said Hassane F.; Saleh A. F.; Abes R.; Gait M. J.; Lebleu B. Cell Penetrating Peptides: Overview and Applications to the Delivery of Oligonucleotides. Cell. Mol. Life Sci. 2010, 67, 715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goun E. A.; Shinde R.; Dehnert K. W.; Adams-Bond A.; Wender P. A.; Contag C. H.; Franc B. L. Intracellular Cargo Delivery by an Octaarginine Transporter Adapted to Target Prostate Cancer Cells through Cell Surface Protease Activation. Bioconjugate Chem. 2006, 17, 787–796. [DOI] [PubMed] [Google Scholar]
- Jiang T.; Olson E. S.; Nguyen Q. T.; Roy M.; Jennings P. A.; Tsien R. Y. Tumor Imaging by Means of Proteolytic Activation of Cell-Penetrating Peptides. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 17867–17872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen Q. T.; Olson E. S.; Aguilera T. A.; Jiang T.; Scadeng M.; Ellies L. G.; Tsien R. Y. Surgery with Molecular Fluorescence Imaging Using Activatable Cell-Penetrating Peptides Decreases Residual Cancer and Improves Survival. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 4317–4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miele E.; Spinelli G. P.; Miele E.; Tomao F.; Tomao S. Albumin-Bound Formulation of Paclitaxel (Abraxane ABI-007) in the Treatment of Breast Cancer. Int. J. Nanomed. 2009, 4, 99–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukowska-Latallo J. F.; Candido K. A.; Cao Z.; Nigavekar S. S.; Majoros I. J.; Thomas T. P.; Balogh L. P.; Khan M. K.; Baker J. R. Jr. Nanoparticle Targeting of Anticancer Drug Improves Therapeutic Response in Animal Model of Human Epithelial Cancer. Cancer Res. 2005, 65, 5317–5324. [DOI] [PubMed] [Google Scholar]
- Shi N.-Q.; Gao W.; Xiang B.; Qi X. R. Enhancing Cellular Uptake of Activable Cell-Penetrating Peptide-Doxorubicin Conjugate by Enzymatic Cleavage. Int. J. Nanomed. 2012, 7, 1613–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]