

Abstract
DNA–protein interactions play critical roles in the control of genome expression and other fundamental processes. An essential element in understanding how these systems function is to identify their molecular components. We present here a novel strategy, Hybridization Capture of Chromatin Associated Proteins for Proteomics (HyCCAPP), to identify proteins that are interacting with any given region of the genome. This technology identifies and quantifies the proteins that are specifically interacting with a genomic region of interest by sequence-specific hybridization capture of the target region from in vivo cross-linked chromatin, followed by mass spectrometric identification and quantification of associated proteins. We demonstrate the utility of HyCCAPP by identifying proteins associated with three multicopy and one single-copy loci in yeast. In each case, a locus-specific pattern of target-associated proteins was revealed. The binding of previously unknown proteins was confirmed by ChIP in 11 of 17 cases. The identification of many previously known proteins at each locus provides strong support for the ability of HyCCAPP to correctly identify DNA-associated proteins in a sequence-specific manner, while the discovery of previously unknown proteins provides new biological insights into transcriptional and regulatory processes at the target locus.
Keywords: chromatin, DNA−protein interactions, mass spectrometry, proteomics, hybridization, ribosome biogenesis, DNA-binding proteins, chromatin immunoprecipitation, ChIP, transcription factors, transcriptional regulation, GENECAPP, HyCCAPP, rDNA, X-element
Introduction
DNA–protein interactions are fundamental to the control of genome expression and play critical roles in mediating DNA replication,1 chromatin organization/segregation,2 and the transcription of genes and noncoding RNAs.3 Some proteins associate with DNA in a site- and sequence-specific manner, such as transcription factors that recognize specific cis regulatory elements to modulate the transcription of nearby genes. Other proteins, such as histones and cohesins, bind much of the genome, often with periodic binding patterns.4
Numerous technologies exist to study these interactions, including DNase footprinting,5 formaldehyde-assisted isolation of regulatory elements (FAIRE),6 and chromatin immunoprecipitation (ChIP).7 DNase footprinting and FAIRE provide information on protein occupancy by revealing sites of the genome protected by or depleted of bound proteins.5,6 ChIP-seq is used extensively to examine sites across the genome that are bound by DNA-associated proteins.8 However, a major limitation is that ChIP is protein-centric, in that a candidate protein must be chosen as a potential DNA binder. Therefore, while ChIP-seq reveals the specific binding patterns of a given protein within the genome, it does not provide information about other proteins bound to those DNA regions. In the absence of a means to discover proteins bound to specific chromosomal loci in living cells, much of how the genome is replicated, protected, and expressed will remain obscure.
To address these limitations, we describe here a corollary technology to ChIP that is DNA-centric; that is, rather than isolating DNA–protein complexes through capture of the protein, we capture a DNA locus and identify the proteins interacting with that DNA sequence. Knowledge of these DNA-associated proteins is essential to understanding the physiological role and functional properties of the locus. Importantly, this technology is not only able to identify already known DNA interactors, such as those that are studied using ChIP, but also is able to reveal new and previously unsuspected proteins.
Conceptually this is very similar to ChIP analysis, differing only in the nature of the affinity capture step (DNA hybridization as opposed to antibody binding). In practice, however, it is much more difficult. First, in contrast to ChIP, where captured DNA is amplified by the polymerase chain reaction (PCR) for subsequent analysis, no amplification is possible for captured protein, and thus much less material is available for mass spectrometric (MS) analysis. Second, the expression levels of different proteins within cells vary by many orders of magnitude, and in some cases important proteins are present at exceedingly low levels. This dramatically complicates their separation and MS analysis.
Despite these difficulties there have been two reports of DNA-centric capture approaches. Déjardin and Kingston described the “Proteomics of Isolated Chromatin segments” (PICh) strategy to identify proteins bound to human or Drosophila telomeric regions using locked nucleic acid (LNA) probes.9,10 Byrum et al. developed an approach referred to as “Chromatin Affinity Purification with Mass Spectrometry” (ChAP-MS), in which a transcription factor (LexA) binding site is engineered into the yeast genome at one specific locus and affinity captured via binding of a LexA–Protein A conjugate.11 They employed this methodology to identify proteins associated with the yeast GAL1 gene promoter.11 While constituting an important early proof-of-principle, the Kingston approach has thus far been limited to the capture of very high copy number short repetitive sequences,9,10 and the ChAP-MS approach requires either engineering of the target genome to introduce a protein binding site11 or introduction of a transcription activator-like (TAL) fusion protein.12
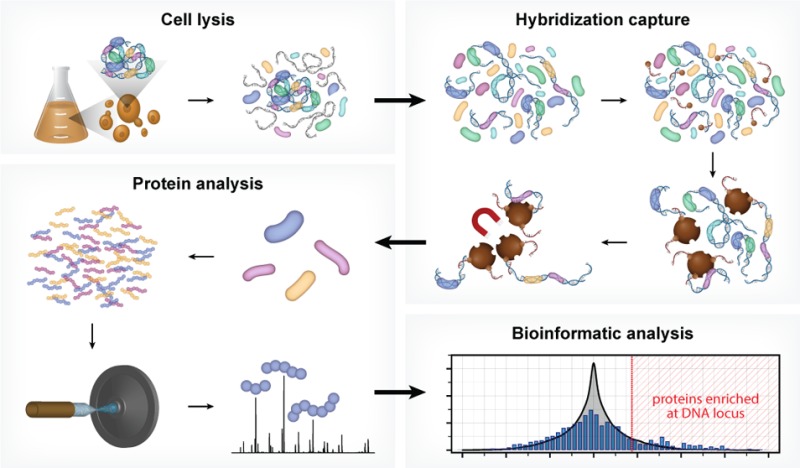
In the present work we address these limitations, with development of a new technology, Hybridization Capture of Chromatin Associated Proteins for Proteomics (HyCCAPP), which combines (i) sequence-specific hybridization capture of DNA fragments of interest directly from a cleared yeast lysate, (ii) state-of-the-art mass spectrometric analysis, and (iii) a bioinformatics analysis pipeline to statistically differentiate between real and background signal (Figure 1). We validated HyCCAPP by capturing and interrogating four genomic regions in Saccharomyces cerevisiae: two regions within the rDNA locus, 25S and 5S (∼150–200 copies/cell), the X-element adjacent to the telomeres (∼35 copies/cell) and the GAL1-10 promoter (single copy/cell) (Figure 2). The different loci have different functions, which are reflected in the proteins that interact with them. These differences were evident in the results from HyCCAPP analysis, which produced distinct sets of proteins for each of the four loci, validating the specificity of the technology. These locus-specific protein lists include many, although not all, previously identified protein interactors, as well as numerous previously unknown interactors that expand our understanding of the biology at these loci. Eleven of 17 proteins identified in our proteomic analysis to interact with the various loci were validated by chromatin-immunoprecipitation from TAP-tagged yeast strains, confirming the ability of the technology to discover new interactors. HyCCAPP is a robust technology that can be used to study any DNA fragment of interest in the yeast genome.
Figure 1.

HyCCAPP is a multistep process that uses sequence-specific hybridization to capture DNA loci of interest from formaldehyde cross-linked cells. The overall procedure involves (1) cross-linking cells with formaldehyde followed by cell lysis, (2) hybridization capture from sonicated and RNaseA-treated lysate using desthiobiotin oligonucleotide capture probes and streptavidin-coated magnetic beads, (3) mass spectrometric analysis of captured proteins using LC–MS/MS, and (4) bioinformatic analysis of mass spectrometric data to determine the proteins enriched at the captured DNA locus of interest relative to non-enriched yeast lysate.
Figure 2.
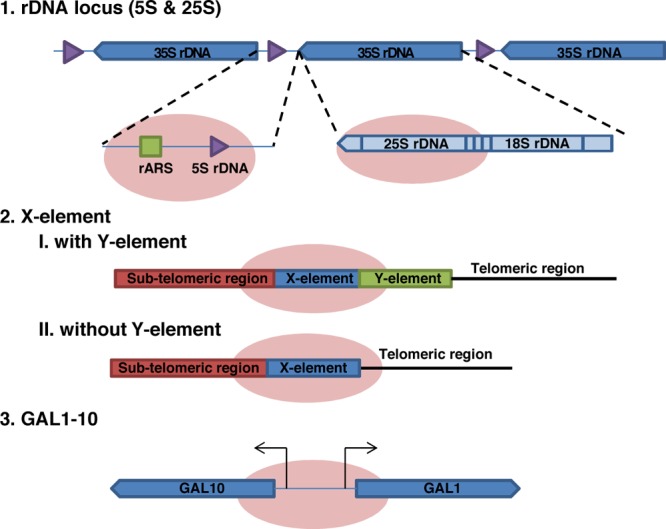
Four genomic loci analyzed by HyCCAPP. The four DNA regions studied are depicted by the pink ovals. These include the 25S and 5S regions of the rDNA locus (a 150 copy 9 kb tandem repeat on chromosome XII), the X-element (a 35 copy 462 bp sequence near the telomeric repeats), and the single copy GAL1-10 promoter region. The 25S rDNA locus is transcribed as part of the 35S rRNA by RNA polymerase I, while the 5S rDNA locus is transcribed by RNA polymerase III. The X-element is located either directly adjacent to the telomeric repeats or separated by one or more copies of the Y′ element. The GAL1-10 promoter is a 660 bp region containing the upstream activating sequence (UAS) for divergent control of both the GAL1 and GAL10 genes.
Materials and Methods
Cell Growth and Cross-Linking for HyCCAPP
Saccharomyces cerevisiae strain Y1788 cells were grown in yeast extract peptone dextrose (YPD) purchased from Sigma-Aldrich (Y1375). Small scale inoculations of 5 mL were shaken overnight at 30 °C at 200 rpm in an Amerex 747 shaker incubator until saturation. Four large flasks (4 L) containing 1.5 L of YPD were each inoculated with 5 mL of saturated culture and shaken at 30 °C at 200 rpm until the OD600 was between 1.75 and 2.00 measured with an Agilent 8453 UV–vis spectrometer. This cell density is greater than that typically employed in ChIP experiments (OD600 of 0.5–1), in order to reduce the volumes of yeast culture and amounts of formaldehyde required. Flasks were removed from the shaker incubator and placed on stir plates with a 2-in. stir bar added to each flask. Formaldehyde (37% solution Sigma-Aldrich F38775) was added to each flask to a final concentration of just under 3% (122 mL). The cells were stirred at room temperature for 30 min. Excess formaldehyde was then quenched with the addition of 5 M Tris pH = 8 (Teknova T5581) to a final concentration of 714 mM, or 250 mL per 1.5 L of cells. The quenching reaction was stirred for an additional 10 min at room temperature. The cross-linked cells were then centrifuged at 4 °C at 5,000g for 20 min using an Avanti J-25I centrifuge. The cell pellet was washed once with 1X PBS (10X PSB stock Teknova P0191) and centrifuged again at 4 °C at 3,000g for 30 min using an Allegra 6KR centrifuge. Cell pellets were then stored at −80 °C and were stable for at least 2 months.
Cell Lysis and Chromatin Sonication/Solubilization
Cell pellets were resuspended in lysis buffer made fresh each time containing 200 mM NaCl (Teknova S0250), 20 mM EDTA (Teknova E0307-06), 50 mM Tris pH = 7 (Teknova T1070) and protease inhibitors diluted 200X from the Sigma-Aldrich cocktail in DMSO (P8215). A standard HyCCAPP experiment requires 1 L of lysis buffer. Cell pellets from 3 L of cross-linked cells were resuspended in 200 mL of lysis buffer. The cell suspension was lysed using Constant Systems TS Series Cell Disruptor at 30 kpsi. After lysis, SDS was added to the cell lysate to a final concentration of 1% from a 20% solution (Bio-Rad 161-0418) and incubated in a 60 °C water bath for 8 min. The cell lysate was then sonicated in 50 mL aliquots using a MisoniX Ultrasonic Processor S4000 at 20 V for 3 min 30 sec with 4 s on/off intervals. The solution was kept in an ice bath for the duration of the sonication. It is critical that the solution does not heat up during the sonication step, as this will reverse the formaldehyde cross-links. Insoluble cellular debris was cleared from solution with a 12 min, 4 °C, 8,000g centrifugation. The pellet was then removed, and the supernatant was diluted into lysis buffer such that the final concentration of SDS was 0.2% (5-fold dilution). RNaseA (Life Technologies 12091-039) was then added to a final concentration of 60 μg/mL. For 1L of cell lysate, this corresponds to 3 mL from the 20 mg/mL stock. The lysate was incubated at 37 °C for 1 h at 150 rpm in the Amerex shaker. After RNaseA treatment, the lysate was centrifuged at 15,000g for 15 min at room temperature to remove insoluble particulates. Sera-Mag streptavidin particles (Thermo Fisher Scientific 30152104010350) were washed once with wash buffer (200 mM NaCl, 0.2% SDS, 50 mM Tris pH = 8), resuspended in wash buffer, and added to the lysate to remove endogenously biotinylated moieties. For each 1 L of cell lysate from 3 L of cross-linked cells, 4 mL of Sera-Mag particles was used for this step. After washing, the beads were resuspended in ∼5 mL of wash buffer. The solution was shaken at 150 rpm at room temperature for 1 h. The streptavidin particles were then removed using the DynaMag-50 magnet (Life Technologies 12302D) in 50 mL aliquots. The removal of magnetic particles from each 50 mL aliquot of lysate should take between 2 and 4 min. It is important to use a magnet of this strength in order to limit the overall preparation time.
Hybridization/Capture/Washing/Elution
The amount of DNA in the lysate was measured using PicoGreen (Life Technologies P7581). From a 3 L cell lysate preparation, typically between 1 and 3 pmol of chromatin was present. For hybridization, the amount of capture oligonucleotide added depends on copy number of the target region and number of capture oligonucleotides used. For each of the multicopy loci, 100 pmol of capture oligonucleotide was added to about 300 mL of lysate from a 3 L cell preparation. For the single-copy locus, 20 pmol of each of the 10 capture probes was added to a total of 1 L of lysate from a 3 L cell preparation. The volume of capture oligonucleotide added to each lysate sample was negligible relative to the volume of the lysate (1–5 μL into ∼300–1000 mL). The lysate samples with added capture oligonucleotides were shaken at 150 rpm and incubated at 37 °C for 3 h. After this time, the sample was removed from heat and cooled to room temperature (∼15 min). Sera-Mag streptavidin particles were washed and resuspended in a volume of wash buffer equal to their volume prior to washing. For each 100 pmol of capture oligonucleotide, 1 mL of bead slurry was added. The bead slurry was then added to the lysate sample and shaken at 150 rpm at room temperature for 1 h. The beads were then isolated in a 50 mL conical tube held against a DynaMag-50 magnet, by repeatedly adding aliquots of the lysate sample to the tube and allowing the beads to be drawn to the magnet. For the single-copy locus, the beads were collected into two 50 mL conical tubes. After removal of the final aliquot of lysate material, the beads were washed with wash buffer four times. A 5 mL aliquot of the lysate material was set aside at this step and stored at 4 °C. For each washing step, the wash buffer was gently added to the beads to minimize the formation of bubbles from the detergent. To resuspend the beads, the 50 mL conical centrifuge tube was gently inverted multiple times and then placed on a rocker to gently and continually mix the solution but prevent formation of large amounts of bubbles. The samples were rocked at room temperature for 5 min in between each washing step. After the fourth wash, the beads were concentrated into 3 mL of wash buffer and collected against a smaller magnet, the Magna-Sep (Life Technologies K1585-01), into low retention 1.7 mL tubes. For each multicopy locus, the entire amount of beads was concentrated into a single tube. For the single-copy locus, because twice as many beads were added, the beads were split between two tubes. The beads were washed twice in this smaller volume and incubated gently on a rocker for 1 h at room temperature. After the final wash, the wash buffer was replaced with 1.2 mL of release buffer for each tube (200 mM NaCl, 0.2% SDS, 50 mM Tris pH = 8, 10 mM d-Biotin (Life Technologies B-20656)). The samples were gently rocked for 2 h at room temperature to release all of the captured material from the beads. At the end of this incubation, the beads were collected against the magnet, and the solution was removed from each tube, leaving the beads behind. Trichloroacetic acid (350 μL) was then added to each of the samples, which were gently vortexed and placed on ice for 10 min. At this stage, the same amount of TCA was added to two 1.2 mL aliquots of cell lysate, which were then also placed on ice. The samples were then centrifuged at maximum speed at 4 °C for 20 min using a Eppendorf 5417R centrifuge. The supernatant was removed, and the pellet was washed twice with chilled acetone with 5 min centrifugations after each wash. The pellet was then heated at 95 °C to remove excess acetone. At this step, the samples can be stored at −20 °C for future processing. To solubilize the DNA–protein complexes in the pellet and reverse the formaldehyde cross-links, 100 μL of 200 mM Tris pH = 8 containing 1 mg of RapiGest13 (commercially available from Waters Corporation, Milford, MA), kindly provided by Professor Neil Kelleher, was added to each tube. The sample was gently pipetted, centrifuged briefly, and then heated at 95 °C for 25 min. The sample was gently vortexed every 5 min to aid in solubilization of the pellet. After heating, the samples were allowed to cool and were processed for either qPCR analysis or mass spectrometry analysis.
qPCR Analysis
Samples for qPCR analysis were diluted at least 10-fold from the cross-link reversal sample into 1X TE buffer (10 mM Tris pH = 7 1 mM EDTA). This step is necessary to dilute the SDS. Taqman assays were designed for each of the loci studied and ordered from IDT. A standard curve was made using dilutions of purified yeast genomic DNA. Each sample was analyzed in duplicate on a 96-well plate (Roche 04729692001). Each well contained 5 μL of sample, 10 μL of LightCycler 480 probes master (Roche 04707494001), 4.5 μL water, and 0.5 μL 40X primer probe mix. Each plate was centrifuged for 2 min at 2,000g after pipetting and analyzed using the Roche 480 LightCycler. Each qPCR run included a 5 min preincubation step at 95 °C, amplification cycles, and a 2 min cooling step at 40 °C. Each amplification cycle included a 10 s 95 °C incubation with a temperature ramp of 4.4 °C/s, a 30 s incubation at 60 °C with a temperature ramp of 2.2 °C/s, and a third 1 s incubation at 72 °C with a temperature ramp of 4.4 °C/s. Detection of the FAM fluorophore was performed during the 72 °C incubation using a 483–533 filter set. Analysis of the resultant qPCR curves and calculation of Cp values was performed using the Roche 480 LightCycler software and the 2nd quant/2nd derivative function.
Mass Spectrometry Analysis
After the reverse cross-linking step, 10 μL of 300 mM iodoacetamide (Sigma-Aldrich I1149) was added to each sample and incubated for 30 min at room temperature. Next, 15 μL of 300 mM DL-dithiothreitol (Sigma-Aldrich D9779) was added, and the samples were incubated again for 30 min at room temperature. The sample was diluted with 900 μL of 25 mM ammonium bicarbonate (Teknova A2012), and trypsin (Promega V5111) was added to a ratio of 5–10 μg sample per μg trypsin. The sample was incubated at 37 °C overnight with gentle rotation. Trifluoroacetic acid (TFA) (Sigma-Aldrich T6508) was added to a final concentration of 0.5% TFA in each of the trypsin digest tubes (50 μL in 1000 μL solution). The tubes were incubated at 37 °C for 30 min and then centrifuged at 13,000g for 10 min. The RapiGest byproducts are water-immiscible, so some precipitation may be observed. The supernatant was transferred to another tube. The samples were then desalted using a C18 solid-phase extraction pipet tip (OMIX C18, 100 μL, Agilent Technologies). The tip was conditioned in 70% ACN, 0.1% TFA, and equilibrated in 0.1% TFA. The peptides were bound by pipetting the entire volume over to a low-binding tube (in 200 μL increments). The sample was then pipetted back-and-forth between the two tubes three times. The OMIX tip was then rinsed in 0.1% TFA five times. The bound peptides were eluted into 150 μL of 70% acetonitrile (ACN), 0.1% TFA. The samples were then dried in a Savant SVC-100H SpeedVac Concentrator (about 30 min). After removing all liquid, the samples were reconstituted in 25 μL of 95:5 H2O/ACN, 0.1% formic acid and then vortexed several times. The samples were then spun in a benchtop centrifuge for 2 min, and 24 μL of the supernatant was loaded into HPLC sample vials. Care should be taken to avoid bubbles. This sample allows for 3 technical replicate injections of 8 μL each. Samples were analyzed by HPLC-ESI-MS/MS using a system consisting of a high performance liquid chromatograph (nanoAcquity, Waters) connected to an electrospray ionization (ESI) Orbitrap mass spectrometer (LTQ Velos, Thermo Fisher Scientific). HPLC separation employed a 100 × 365 μm fused silica capillary microcolumn packed with 20 cm of 3 μm diameter, 100 Å pore size, C18 beads (Magic C18, Bruker), with an emitter tip pulled to approximately 2 μm using a laser puller (Sutter Instruments). Peptides were loaded on-column at a flow rate of 500 nL/min for 30 min and then eluted over 120 min at a flow rate of 300 nL/min with a gradient of 2% to 30% ACN, in 0.1% formic acid. Full-mass scans were performed in the FT orbitrap between 300 and 1500 m/z at a resolution of 60,000, followed by 10 MS/MS HCD scans of the 10 highest intensity parent ions at 42% relative collision energy and 7,500 resolution, with a mass range starting at 100 m/z. Dynamic exclusion was enabled with a repeat count of two over the duration of 30 s and an exclusion window of 120 s.
Mass Spectrometry Data Analysis
The acquired precursor MS and MS/MS spectra were searched against an S. cerevisiae protein database (Uniprot reviewed database, containing 6,883 sequences) using SEQUEST, within the Proteome Discoverer 1.3.0.339 software package (Thermo Fisher Scientific). Masses for both precursor and fragment ions were treated as monoisotopic. Oxidized methionine (+15.995 Da) and carbamidomethylated cysteines (+57.021 Da) were allowed as dynamic modifications. The database search permitted for up to two missed trypsin cleavages and ion masses were matched with a mass tolerance of 10 ppm for precursor masses and 0.1 Da for HCD fragments. The output from the SEQUEST search algorithm was validated using the Percolator algorithm. The data were filtered using a 5% false discovery rate, based on q-values. All raw data files and searched data files are available at PeptideAtlas.org under the name “HyCCAPPyeast”.
Bioinformatics Analysis
Delta-Rank Distribution
The result of the mass spectrometric data searching was exported into Microsoft Excel. The numbers of peptide spectral matches (PSMs) were summed across the three technical replicates from each biological replicate. Here, biological replicates are HyCCAPP experiments from different cell growths, while technical replicates are different MS runs from the same biological replicate sample. The total number of PSMs per technical replicate were normalized across all runs to account for variability in protein amount. The total number of normalized PSMs was then summed across the three technical replicates for all HyCCAPP experiments and yeast lysate controls. Proteins not identified in a given mass spectrometric run, but identified in at least one of the other mass spectrometric runs from the HyCCAPP experiments or yeast lysate samples, were given a PSM value of 0.0417 (1/total number of biological replications (1/24)) in place of the zero. Although the ensuing analysis involves rank order, imputing a small number in place of zeros is necessary in order to approximate fold change. The four biological replicates for the HyCCAPP experiments against each of the four loci were averaged. There were eight biological replicates of the yeast lysate control sample run on the mass spectrometer: four of the samples were from each of the four multicopy HyCCAPP experiments, and four corresponded to the GAL1-10 HyCCAPP experiment. To compare the proteins identified in the HyCCAPP experiments to those in the yeast lysate, we used the set of four corresponding yeast lysate samples for each of the loci studied. Each of the lists of proteins were then sorted relative to the number of PSMs, and the proteins were ranked; the protein with the most PSMs was given a rank of 1, and the second most abundant protein a rank of two, etc. The lists from each of the four loci studied were then compared with corresponding yeast lysate samples. Differences in rank were calculated for each protein identified between the HyCCAPP sample and the yeast lysate sample, where a positive delta-rank indicated enrichment in the HyCCAPP experiment. The resultant delta-rank distribution was used to filter true hits from background contribution.
Delta-Rank Threshold Calculation
To determine the delta-rank threshold to filter out true hits from the remainder of identified proteins for each of the four loci, we compared different biological replicates of yeast lysate samples to each other. In the HyCCAPP analysis, the most enriched proteins captured on loci of interest have the largest delta-rank. Comparing different biological replicates of yeast lysate to one other will provide a delta-rank distribution that results from a sample where only biological and technical fluctuations are responsible for differences across samples. Among the eight biological replicates of yeast lysate, there are 70 different possible comparisons when averaging four biological replicates and comparing two such averages (since 70 = 8 choose 4 = 8!/(4!4!)). We combined the yeast lysate samples in all 70 combinations and determined the delta-rank corresponding to different thresholds. For each yeast lysate delta-rank comparison, the number of proteins corresponding to 1% of the sample, 2.5%, 5%, and 10% were determined, and the corresponding delta-rank of each of these proteins was extracted. These ranks were then averaged and used as the threshold levels for the HyCCAPP delta-rank distributions for each of the four loci. False discovery rates (FDRs) were calculated for each of the loci for the four thresholds by dividing the threshold level (e.g., 10%) by the number of proteins above the corresponding delta-rank in the gene:yeast lysate delta-rank distribution normalized to the total number of proteins in each distribution. For example, there were 1831 total delta-ranks from the 25S rDNA gene:yeast lysate comparison. From this list, 393 proteins were above the 10% threshold. Therefore, the FDR was determined to be 10%/(393/1831) or 46.6%.
Chromatin Immunoprecipitation
TAP-tagged strains were obtained from the Thermo Scientific Yeast TAP Tagged ORF library (YSC1177). Small scale (5 mL) inoculations were grown in YPD overnight to saturation and diluted into 500 mL of YPD. Once cells reached an OD of 0.75, formaldehyde was added to a final concentration of 1% and cross-linked for 20 min at room temperature with a stir bar added and stirred at medium speed. Tris was then added to a final concentration of 500 mM to inactivate unreacted formaldehyde,14 and the sample was stirred at room temperature for an additional 10 min. Cells were isolated through centrifugation at 5,000g at 4 °C for 20 min. The pellet was washed once with 1X PBS, centrifuged at 3,000g for 30 min at 4 °C, and stored at −80 °C. Cells from 125 mL of cell culture were resuspended in about 1.5 mL of lysis buffer (50 mM Tris pH = 7, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% Na-deoxycholate with 1X protease inhibitor cocktail (Halt Protease inhibitor cocktail Thermo Fisher Scientific 78429)). About 400 μL of glass beads (Sigma-Aldrich G8772) was added to a 2 mL screw-top tube (Sarstedt, 72.693), and the cell suspension was pipetted over the beads. The cells were then disrupted using a bead beater (BioSpec 3110BX) at standard settings for 4 × 50 s cycles. In between cycles, the samples were kept on ice. The solution was then pipetted to a new tube, leaving behind the beads, and sonicated using a microtip sonicator (Fisher Scientific, 550 Sonic Dismembrator) at setting 5 for 3 min total time with 4 s on/off cycles. During sonication, the samples were kept in an ice bath. The samples were then centrifuged at 14,000g for 5 min to remove insoluble debris. The supernatant was then removed and split in half into two tubes. A 50 μL aliquot of this input solution was diluted into 300 μL of 1XTE and stored at −20 °C. Five microliters of the TAP Tag antibody (Thermo Fisher Scientific CAB1001) was added to one tube of each sample. The other half of the lysate sample served as the no-antibody control. The tubes were then incubated overnight at 4 °C with constant rotation. The samples were then equilibrated to room temperature, and 50 μL of Dynabeads Protein G beads (Life Technologies 100.04D) was added to each tube and rotated at room temperature for 1 h. The beads were then collected against a magnet and washed seven times: twice with lysis buffer, twice with lysis buffer containing 500 mM NaCl, twice with wash buffer (10 mM Tris pH = 7, 250 mM LiCl, 0.25% NP-40, 0.25% Na-deoxycholate and 1 mM EDTA), and once with stringent wash buffer (10 mM Tris pH = 7, 1 mM EDTA, 140 mM NaCl, 0.5% SDS). The samples were then eluted into 95 μL of elution buffer (50 mM Tris pH = 7, 1 mM EDTA and 0.5% SDS) by heating for 30 min at 65 °C. The beads were then isolated against a magnet, and the solution was diluted into 300 μL of 1X TE. Proteinase K (2 μL, New England BioLabs P8107S) was then added to each tube, including the input material samples, and incubated at 42 °C for 1 h. The samples were then transferred to 65 °C and incubated for 4–5 h. Phenol/chloroform/isoamyl alcohol mixture (0.4 mL, Sigma-Aldrich 77617) was then added to each sample, followed by vortexing and centrifugation to separate the immiscible layers. The aqueous layer from each sample was isolated and diluted into 1.3 mL of 100% ethanol and incubated at −20 °C overnight. The DNA was then isolated by centrifugation (20 min, 4 °C, 15,000g), and the pellet was washed once with 70% ethanol. The concentration of DNA was measured using a NanoDrop (Thermo Scientific NanoDrop 2000c UV–vis spectrophotometer). Equal amounts of DNA were used for qPCR analysis. Each sample was diluted in 1X TE for qPCR analysis, as described above.
Results
HyCCAPP Procedure
Each step in the HyCCAPP process is critical to success, and was explored and optimized in order to arrive at the overall protocol employed here. Brief descriptions of issues and solutions are provided below, and a detailed overall protocol is provided in Materials and Methods.
Cell Number, Cross-Linking, and Lysis
A typical detection limit for the mass spectrometric identification of peptides in a complex background is in the low femtomole (1 fmol = 10–15 mol = 6 × 108 molecules) range. Assuming a 1% overall efficiency of the HYCAPP process (see below), 6 × 1011 cells are needed to obtain 10 fmol of a single copy/cell protein. We performed HyCCAPP at a scale sufficient to see most proteins (1011 yeast cells per experiment). Because many DNA–protein interactions are dynamic and transient in nature, formaldehyde was utilized to stabilize DNA–protein interactions. Formaldehyde is a widely used zero-length cross-linking reagent that reacts rapidly with proteins and DNA, and the resultant cross-links can be reversed if desired.15 The concentration (3%) and reaction time (30 min at room temperature) were optimized to maximize DNA-related protein IDs (see Materials and Methods). After testing multiple lysis methods (including bead beating, conventional French press, lysozyme digestion, and high-pressure cell disruptors), we obtained the best reproducibility and efficiency (90% cell lysis, based upon total DNA and protein recovery) with a commercially available cell disruptor system (unpublished).
Chromatin Solubilization and Fragmentation
Hybridization capture requires that the cross-linked chromatin be effectively solubilized and efficiently fragmented. We employed high concentrations of detergent (1% sodium dodecyl sulfate (SDS)) and a short 60 °C incubation for chromatin solubilization. Including this step in the procedure greatly improved overall protein and DNA recovery. The size distribution of chromatin fragments is also a critical variable in HyCCAPP. Longer DNA fragments carry more associated protein molecules, increasing MS signal intensities, but the binding location of those proteins is necessarily less well-defined, compromising spatial resolution. After exploring both enzymatic (restriction enzymes and micrococcal nuclease) and physical fragmentation of the cross-linked chromatin, we found that sonication provided the most reproducible results, generating chromatin fragments ∼1 kb on average. In ChIP procedures the chromatin is generally fragmented more extensively than this, in order to produce shorter DNA fragments, which in turn yield greater resolution. However, in HyCCAPP, there are two significant advantages to employing longer chromatin fragments: first, as mentioned above, the longer fragments carry more associated proteins, facilitating MS detection, and second, the probability that DNA molecules are fragmented within the capture sequence (thereby reducing capture yield) is lower for longer fragments.
Capture Oligonucleotide Design and Hybridization
We designed 30 nt long capture oligonucleotides, with calculated melting temperatures from 50 to 70 °C (Supplementary Table 1). Binding to streptavidin-conjugated beads is a useful approach to capture cross-linked chromatin that has been hybridized to biotinylated oligonucleotides; however, we found that elution of the captured material with heat produced unacceptably high levels of background proteins, compromising our ability to identify low-abundance captured proteins by MS. We therefore employed capture oligonucleotides that included a 5′-desthiobiotin moiety.16 Desthiobiotin is a biotin analogue that binds with 100-fold lower affinity to streptavidin than biotin, allowing elution by competitive displacement with free biotin under mild conditions. The selective elution of the capture oligonucleotides/hybridized chromatin from support particles greatly reduced background levels from nonspecifically bound proteins, as revealed by MS analysis of the eluted material. For each of the multicopy loci, only a single capture oligonucleotide was used; however, for the single-copy GAL1-10 locus, 10 capture oligonucleotides were utilized in parallel in order to increase overall capture efficiency. The optimum molar ratio of capture oligonucleotide to targeted chromatin locus was 100 for the multicopy loci and 20 for each of the 10 capture oligonucleotides for the single-copy GAL1-10 locus. Hybridization of capture oligonucleotides to the cross-linked and fragmented chromatin was carried out in RNase A-treated lysate at 37 °C for 3 h, followed by capture of the resultant complexes on magnetic beads. The beads were washed and then incubated in a biotin-containing buffer to elute bound material.
Efficiency and Specificity of Hybridization Capture for Multicopy Loci
Hybridization capture efficiency and specificity for multicopy loci were evaluated using qPCR to determine the number of copies of target DNA captured, and PicoGreen binding was used to determine the total amount of DNA captured (target region plus nonspecifically captured DNA).17 See Supplementary Table 2 for qPCR primer sequences. qPCR showed a capture efficiency (copies of target DNA captured divided by input copies) of 0.6–1.2% for each of the three multicopy loci. The capture specificity (copies of target DNA captured using the target capture oligonucleotide divided by that captured using an off-target oligonucleotide) ranged from 77- to 812-fold relative to the other multicopy loci (Figure 3). This specificity measurement reveals the amount of the targeted DNA captured relative to the two other regions; however, it does not reveal the total amount of off-target DNA captured. To measure this, we used PicoGreen binding to compare the total DNA obtained in specific capture experiments with the total DNA obtained in control capture experiments using a scrambled sequence oligonucleotide that is not significantly complementary to any region within the yeast genome. The difference in total DNA obtained in the specific and control capture experiments is an approximation of the amount of DNA captured through off-target hybridization. For each of the three multicopy loci, the ratio of specifically captured to nonspecifically captured total DNA is at least 2 (Supplementary Figure 1), meaning that at least 50% of the total DNA captured in each HyCCAPP experiment corresponds to the targeted region. Although this value may seem low, it is important to note that, assuming the contaminating DNA represents sequences distributed widely across the genome, the captured fragment greatly outnumbers any other specific sequence, and thus nonspecific capture is not expected to recover particular associated proteins with high enrichment.
Figure 3.

Multicopy locus hybridization capture efficiency and specificity. The fractions of target DNA captured by HyCCAPP from the 25S rDNA locus, 5S rDNA locus, and X-element, as measured by locus-specific qPCR assays (Supporting Information) are shown in panels A, B, and C, respectively. Error bars represent the standard deviation of duplicate measurements. The first, second, and third bars in each plot show the fractions of DNA captured using the 25S rDNA capture oligonucleotides, the 5S rDNA capture oligonucleotides, and the X-element capture oligonucleotides, respectively. Numerical values in each case are shown.
Efficiency and Specificity of Hybridization Capture for the Single Copy GAL1-10 Promoter
In order to increase hybridization capture efficiency for the single-copy divergent GAL1-10 promoter, we employed 10 capture oligonucleotides across a ∼1400 bp region (Figure 4a). Due to the greater length of this region, compared to the multicopy loci examined, we designed two qPCR assays targeting regions approximately 1 kb apart. These assays were employed to measure the capture efficiency, capture specificity (by comparison to capture with a scrambled oligonucleotide control), and the approximate length of the captured fragments. We combined capture oligonucleotides 1–5 to target the GAL10 end of the capture region and capture oligonucleotides 6–10 to target the GAL1 end of the capture region. These combinations were used in separate hybridization capture experiments. As shown in Figure 4b, oligonucleotides 1–5 captured ∼0.4% of the GAL10 region and ∼0.1% of the GAL1 region, while oligonucleotides 6–10 captured ∼0.6% of the GAL1 region and ∼0.1% of the GAL10 region. This corresponds to a total capture efficiency of ∼1.2%, comparable to the results obtained for the multicopy loci using only a single capture sequence. The capture specificity compared to a scrambled oligonucleotide control was approximately 100-fold. The differences in the two qPCR assay signals for each set of capture oligonucleotides reflects and is consistent with the distribution of chromatin fragment sizes produced by the sonication (∼1 kb average length, data not shown); more signal is obtained from the qPCR assay closest to the capture oligonucleotide sequences.
Figure 4.

GAL1-10 locus hybridization capture efficiency and specificity. Ten capture oligonucleotides (30 nt) were designed for the GAL1-10 promoter region spanning a total length of about 1400 bp (a). The capture probes alternated spacing of 100 bp and 200 bp to avoid synchronicity with nucleosome repetition. Two qPCR assays were designed, one targeting a region near the start of the GAL10 gene (dark gray) and one targeting a region near the start of the GAL1 gene (light gray). HyCCAPP was performed using either capture oligonucleotides 1–5, capture oligonucleotides 6–10, or a scrambled oligonucleotide control (b). Error bars represent the standard deviation of duplicate measurements.
MS Analysis
MS analysis of protein samples was performed by standard methods (see Materials and Methods). The raw MS data were processed using rank order statistics and false discovery rate (FDR) criteria to distinguish true binders from false positive binders. This process will be presented in detail above.
Validation of HyCCAPP through Analysis of Four Genomic Loci in Yeast
To develop and test the HyCCAPP procedure, we analyzed four genomic loci in yeast, each with distinct expectations in terms of bound proteins (see below): the 25S rDNA (∼150 copies/cell), 5S rDNA (∼150–200 copies/cell), X-element (∼35 copies/cell), and the GAL1-10 promoter (single copy/cell) (Figure 2). Capture oligonucleotides were designed as described in Materials and Methods, and the sequences and calculated melting temperatures are provided in Supplementary Table 1. Each experimental run employed 1011 yeast cells. This experimental scale was used for either a single HyCCAPP experiment on the GAL1-10 region or for three HyCCAPP experiments on any of the multicopy loci. An equal volume of yeast lysate was removed from each cell preparation for separate MS analysis and comparison to captured material. Four biological replicates were performed for each of the four loci.
Bioinformatic Analysis
More than 1000 proteins were identified in HyCCAPP experiments for each of the four loci (with four biological replicates each). Although these numbers are large, they are comparable to the numbers of proteins identified in affinity purification and mass spectrometry (AP-MS) experiments,18 which are conceptually similar to the DNA capture experiments presented here. As has been shown in AP-MS studies, these large lists include both true protein–protein interactions and nonspecific protein binders, and the challenge is designing proper control experiments and accurately identifying the true binders.19 Similarly, the protein lists obtained in HyCCAPP experiments include both the desired proteins that truly interact with the target region (henceforth “true hits”) and false positive proteins that are not actually specifically interacting with the target region (henceforth “false positives”). False positives often correspond to the most highly abundant proteins present; alternatively, characteristics such as a general but nonspecific affinity for nucleic acids (e.g., any basic and thus positively charged protein) or for the capture beads or associated streptavidin can also lead to artifactual binding.
The goal of the bioinformatics data analysis is to reduce the incidence of false positives as much as possible, without also eliminating the true hits. We implemented a rank order methodology to identify the proteins that were most enriched in the captured samples relative to yeast lysate. Relative protein abundances were estimated from their corresponding numbers of peptide spectral matches (PSMs). Because we are operating close to the limit of protein detection, many true positives may not be reproducibly identified in the MS analysis (even if they are reproducibly captured). We therefore opted to combine data from biological replicates, to maximize the breadth of our analysis, as follows. For both yeast lysate and captured samples, proteins were sorted by abundance and ranked correspondingly. For each captured sample (four biological replicates for each of four loci), the average difference in rank between the sample and its paired lysate was calculated for all proteins, yielding a “delta-rank” distribution for each of the four loci (see Figure 5 and Materials and Methods). The proteins that were most enriched in the captured samples have large positive delta-ranks and thus correspond to the right end of the delta-rank distributions; conversely, the proteins that were most abundant in the yeast lysate and were not captured in the HyCCAPP process have large negative delta-ranks and are thus represented at the left end of the delta-rank distributions. The distributions are centered around a delta-rank value of zero, consistent with substantial recovery of abundant, nonspecifically bound proteins. Importantly, each of the distributions shows a significant tail on the right side (large positive delta-rank). Many of these proteins are likely to be the true hits.
Figure 5.

Delta-rank histograms obtained from bioinformatic analysis of HyCCAPP MS results for each of the four loci studied. Delta-rank values are plotted along the x-axis, with positive delta-ranks indicating enrichment in the HyCCAPP sample and negative delta-ranks indicating enrichment in the yeast lysate sample. The y-axis values are the numbers of observations of each delta-rank, divided by the total number of delta-rank observations (density). Delta-rank values obtained in gene vs lysate comparisons are shown in red (fit to an area density plot), and delta-rank values obtained from lysate vs lysate comparisons are shown in blue. The vertical dashed black line on each plot indicates the 10% threshold value employed (see text).
We sought to identify a threshold of delta-rank values that would maximize true hits while minimizing false positives, essentially setting a false discovery rate (FDR). To determine the threshold differentiating the most enriched proteins from background proteins, we repeated the delta-rank analysis for 70 pairs of yeast lysate samples, generating a background distribution of delta-ranks (Supplementary Figure 2). This background distribution captures the natural fluctuations in delta-rank between lysate samples, in the absence of any capture enrichment. We then identified delta-rank thresholds for the top 1%, 5%, or 10% of lysate delta-ranks, with associated conservative FDR values (false discovery corresponds here to nonspecifically captured proteins) of <30–50% (see Materials and Methods). The true FDR is likely lower than these values, because of how missing proteins not observed in the lysate are treated. We settled on a threshold (10%) that identified the greatest fraction of known binding proteins in our analysis (vertical lines, Figure 5).
Expected Proteins Are Enriched via HyCCAPP
Common Proteins Found at All Loci
The filtered lists of proteins at the chosen threshold for each locus contain from 393 to 610 proteins (see Supporting Information). These lists contain proteins that are unique to each locus, proteins that are shared by some of the four loci, and proteins common to all four loci. There were 117 proteins found at all loci. These proteins would be expected to include both ubiquitous DNA-binding proteins and proteins that bind nonspecifically to chromatin. We evaluated these proteins by GO analysis using the web-based tool FunSpec.20 GO term enrichment was found for ubiquitous DNA binding functionalities, including DNA helicases (p = 8.1 × 10–6), proteins modulating DNA topology (p = 9.0 × 10–4), and proteins localized to the nucleus (p = 6.8 × 10–3). Other enriched GO terms include spindle pole components (p = 2.3 × 10–6), COPI vesicle (p = 1.3 × 10–10), ER to Golgi transport (p = 1.8 × 10–4), and cell-wall proteins (p = 2.2 × 10–4). While some of these proteins may indeed correspond to true hits,for example, spindle pole bodies that are likely attached to chromatin through protein–protein interactions,21 others likely result from nonspecific protein binding. For further analysis of locus-specific binders, we therefore removed proteins from the list found at all four loci; while this removes commonly bound DNA binders, it is also likely to remove contaminating proteins that have high affinity for DNA.
Locus-Specific Protein Lists
We sought to compile protein lists specific for each of the four loci, to capture the unique physiology of each region. It is known that some proteins bind at both the telomeric and rDNA regions (e.g., silencing proteins22); we therefore included proteins that were found at one or more of these loci (5s rDNA, 25S rDNA, X-element) but excluded those that were either present at all four loci or present at three of the four if one of the three was GAL1-10. For the single-copy GAL1-10 locus we included proteins if they were also identified at no more than one of the multicopy loci. These four locus-specific protein lists are provided in the Supporting Information.
We expect these locus-specific protein lists to contain proteins that reflect the function of the gene and largely exclude contaminant proteins. To confirm this, we first combined the locus-specific lists to verify that the majority of proteins are in fact DNA-associated. This analysis revealed a striking enrichment for DNA-binding processes. The seven GO terms enriched with a Bonferroni-corrected p-value of less than 10–4 include DNA binding, transcriptional control, and both nucleus and nucleolus localization (as expected due to the representation of nucleolar-localized rDNA loci, Table 1). The group was also enriched for genes that produce benomyl sensitivity when deleted, as expected for proteins involved in chromosome segregation since microtubule-disrupting benomyl interferes with chromosome dynamics.23
Table 1. GO Term Enrichment of Locus-Specific Proteinsa.
| GO term | p-value |
|---|---|
| Nucleus | 8.66 × 10–13 |
| DNA-dependent transcription | 3.88 × 10–7 |
| Transcriptional control | 7.12 × 10–6 |
| Nucleolus | 1.30 × 10–5 |
| DNA conformation modification | 1.66 × 10–5 |
| Benomyl sensitivity | 4.54 × 10–5 |
The locus-specific protein lists obtained for each of the four loci were combined and analyzed for GO enrichment using FunSpec. The results with a Bonferroni-corrected p-value of <10–4 are shown.
Functional Groups Missing from Capture Experiments
For comparison, we also performed GO term enrichment analysis on the pooled set of proteins from the opposite end of the delta-rank distributions (left side of the delta-rank distributions of Figure 5). As mentioned above, these proteins are likely to correspond to abundant proteins present in the lysate that were not captured in the HyCCAPP experiments. Table 2 shows the over-represented GO terms, which are very different from those obtained for the captured proteins and correspond primarily to cytosolic processes such as enzymatic activities, metabolic processes, and biosynthetic and degradative pathways. The striking differences in GO term enrichment for proteins from the two ends of the distributions provides a strong indication that the HyCCAPP technology is truly isolating DNA–protein complexes representative of normal interactions in vivo.
Table 2. GO Term Enrichment of Yeast Lysate Enriched Proteinsa.
| GO term | p-value |
|---|---|
| Oxidoreductase activity | <1 × 10–14 |
| Catalytic activity | <1 × 10–14 |
| Oxidation–reduction process | <1 × 10–14 |
| Cytoplasm | <1 × 10–14 |
| Mitochondria | <1 × 10–14 |
| Metabolic process | 1.84 × 10–14 |
| Cellular amino acid biosynthetic process | 3.9 × 10–12 |
| Proteasome complex | 4.76 × 10–12 |
| Electron transport | 4.78 × 10–12 |
| ER | 4.5 × 10–11 |
| Nucleotide binding | 1.14 × 10–9 |
| Protein processing (proteolytic) | 1.24 × 10–9 |
| Proteasome storage granule | 5.47 × 10–9 |
| ATP binding | 2.71 × 10–8 |
| Binding | 8.82 × 10–8 |
| Transferase activity | 9.42 × 10–8 |
| ER membrane | 2.33 × 10–7 |
| Pyridoxal phosphate binding | 3.15 × 10–7 |
The locus-specific protein lists for each of the four loci were partially selected on the basis of a delta-rank threshold of 314. To generate a similar list of proteins that are most enriched in the yeast lysate relative to each of the HyCCAPP samples, proteins with a delta-rank of −314 or below were combined from each of the four HyCCAPP experiments. This list was then analyzed for GO enrichment. There were nearly 40 GO terms enriched with a Bonferroni-corrected p-value of <10–4. About half of the terms are listed here, which are representative of the total list and is devoid of redundant GO terms (see Supplementary Table 3 for complete list).
Validation of HyCCAPP Capture by Chromatin Immunoprecipitation
As discussed in detail below, we chose 17 proteins bound to various loci to validate by traditional chromatin immunoprecipitation (ChIP) and qPCR (Figure 6). We chose proteins for which there was some corroborating evidence that they may represent new binders (see below). Eleven of the 17 proteins (65%) were enriched by ChIP at their respective loci, but not at the control INO1 promoter (with the exception of some chromatin factors; see Supplementary Table 4). These results highlight our ability to identify new binding proteins at these regions, validating the HyCCAPP procedure. Below we discuss each locus specifically in terms of known binders and new proteins implicated as bound to the regions.
Figure 6.

ChIP-qPCR confirmation assays for selected proteins identified in HyCCAPP. ChIP-qPCR was performed for 17 TAP-tagged yeast strains expressing proteins found in HyCCAPP experiments at the GAL1-10 promoter (a) and the 25S rDNA locus (b–d). Recovered DNA for immunoprecipitation experiments and no-antibody controls was quantified by qPCR using the primers developed against the GAL10 promoter region (see Figure 4a and Supporting Information) and 25S rDNA region (Supporting Information). The qPCR signal from the ChIP samples is an average of two biological replicates and is depicted as the left (dark gray) bar in each chart, while the qPCR signal from the no antibody control sample is shown as the right (light gray) bar in each chart. For each TAP-tagged strain, the qPCR signal was normalized to input DNA. ChIP enrichment was considered significant if both replicates showed at least 2-fold enrichment compared to the no-antibody control (indicated with *), since the t test has lower power on duplicates (see Supplementary Table 4).
Known Proteins and New Biology Captured by HyCCAPP
X-Element
Chromosome ends in yeast are composed of telomeric and subtelomeric regions, including X-elements and Y′-elements that occur in varying combinations.24 It is well-known that telomeric repeats are involved in genome stability, affect chromosome maintenance, and play a role in transcriptional silencing.24 The subtelomeric regions are less well characterized but are also thought to play a role in chromosome stability.24 The 462 bp X-element core is the only sequence motif found on every chromosome end. This region contains an autonomously replicating sequence (ARS), which is bound by the origin recognition complex (ORC).1,24 The proximity of the X-element to the telomeric region varies depending upon the presence of one or more adjacent Y′-elements. In HyCCAPP experiments for the X-element, we thus expect to identify proteins not only associated with the X-element but also associated with telomeric repeats and Y′ elements.
After applying the bioinformatics filtering and removal of common proteins, there were 232 locus-specific proteins found at the X-element. The list was enriched for both single-stranded and double-stranded telomeric DNA binding proteins (p = 2.1 × 10–3, 3.6 × 10–3), nuclear proteins (p = 3.0 × 10–4) and chromosome-associated proteins (p = 2.0 × 10–3), all functions expected for this DNA locus based on its physiological role. Included on the list were several expected proteins, including Orc4 that binds to the ARS,1 Sir4 involved in telomeric silencing,25 and Cdc13,26 Stm1,27 Top1,28 Dot5,29 and Ebs130 that are all known to bind to the telomeres. We also identified over 20 other proteins that are involved in DNA and chromosome maintenance and nearly 50 other proteins that are known to localize to the nucleus. To garner support for other possible telomere-associated proteins, we compared the X-element locus-specific list to proteins identified in screens for mutants with defects in telomere maintenance. Eleven proteins were identified that had previously been shown to affect telomere length when mutated;31 several of these proteins were not previously known to associate directly with the X-element. We also assessed if novel proteins identified at this locus interact genetically or physically with other captured proteins, under the assumption that true binders on our list may be more likely to interact than random. Indeed, we observed 430 genetic interactions32 (GIs) among genes encoding the X-element-bound proteins, more than expected by chance (p = 0.002). We also observed 157 protein–protein interactions32 (PPIs) (p = 0.138), which although not statistically significant still demonstrates that many of the captured proteins are known to be physically associated with each other. Organizing the X-element captured proteins based on these interactions identified specific complexes or subgroups of functionally enriched proteins that further highlight the physiological processes at or near the X-elements (Figure 7). In all, nearly 40% of the proteins captured at the X-element are known to directly associate with telomeres, aid in their maintenance, bind DNA, or reside in the nucleus. Together these results provide further strong validation that HyCCAPP is indeed identifying locus-specific protein binders.
Figure 7.

Interaction network and GO Slim enrichment for locus-specific proteins at X-element locus. Between the 232 locus-specific proteins at the X-element, we observed 430 GIs and 157 PPIs, represented by the blue and gray lines, respectively, in the network. The protein interaction network was organized and analyzed for GO-slim enrichment using the GOlorize plugin for Cytoscape 2.4.62 Seven GO categories were enriched with a Benjamini & Hochberg False Discovery Rate correction of less than 0.05, with proteins in significantly enriched categories colored according to the key. Refined functional enrichment was performed for each protein cluster (encircled) using the program FunSpec.20
Given the confidence garnered for X-element captured proteins, we looked for new functions implicated in telomere biology. Of note is the capture of Pfa4, a palmitoyltransferase required for protein secretion and reported to reside in the endoplasmic reticulum (ER) membrane.33 Pfa4 was identified in a screen for proteins required for heterochromatin silencing34 and later shown to palmitoylate the telomere binding protein Rif1.35 Pfa4 is required for Rif1 localization to attachment sites of telomeres to nuclear membrane (which is contiguous with the ER membrane). The gene encoding Pfa4 has interactions with genes encoding several other X-element captured proteins, including the nuclear envelope protein Mps3 required for telomere organization during cell division.36 Our results strongly suggest that Pfa4 is physically bound to telomeres, likely at the sites of their attachment to the nuclear membrane (this hypothesis has not yet been tested due to lack of availability of either a TAP-tagged Pfa4 strain or a ChIP-compatible anti-Pfa4 antibody).
GAL1-10
The GAL1-10 promoter region is a single-copy locus in the yeast genome that has been extensively studied.37 As a single-copy region, its analysis by HyCCAPP is more challenging than the analysis of multicopy loci, as less DNA and thus less associated protein for MS analysis will be captured (see Discussion). This 660 bp intergenic region contains an upstream activating sequence (UAS) that controls expression of the divergent GAL1 and GAL10 genes, as well as short noncoding RNAs that may regulate expression of the flanking genes.38
After applying the bioinformatics filtering and removal of common proteins, there were 415 locus-specific proteins found at the GAL1-10 locus. Numerous proteins known to be associated with this region were identified as well as many DNA-binding proteins and other DNA-related proteins. These include Rsc3 and Sth1, both members of the RSC complex39 that is known to bind at the GAL UAS. Additionally, we identified three members of the Kin28 complex (Kin28, Cdc28, and Ccl1), which is also known to bind to the GAL1-10 region.40 The capture of these proteins is especially interesting, since the downstream open reading frames should be transcriptionally silenced under our growth conditions. Also surprising was the capture of RNA Pol II subunits, which is expected for the locus under inducing conditions. Together, the capture of these proteins may reflect a poised transcription complex or could indicate the expression of noncoding regulatory RNAs in the region.38 In all, over 40 known DNA-related proteins were present on the GAL1-10 locus-specific list. Interestingly, we also found a significant enrichment of proteins required for normal growth on galactose (including transcriptional regulator Bdf1, Spt20, RSC subunit Sth1, and Zeo1, a protein reported to reside at the plasma membrane, p = 9.1 × 10–3), supporting their association with the GAL1-10 promoter. Using a traditional ChIP-qPCR assay with strains expressing TAP-tagged proteins, we confirmed that two of the four proteins (Bdf1, Sth1, and possibly Zeo1) bind the GAL1-10 locus under these conditions (Figure 6a).
25S and 5S rDNA
The rDNA locus in yeast is a 9 kb tandem repeat that physically localizes to the nucleolus and encodes rRNA along with an autonomously replicating sequence (ARS) near the 5S gene.41 Roughly half of the rDNA repeats are transcribed at any given moment, while the others are transcriptionally silenced.42 Each rDNA unit encodes two transcripts: the 35S transcribed by RNA polymerase (Pol) I and the 5S transcribed by RNA Pol III,43 separated by intergenic spacers that can be conditionally transcribed by Pol II.44 The 35S transcript is processed into 18S, 5.8S, and 25S fragments that are chemically modified via methylation and pseudouridylation, folded around ribosomal proteins (RPs), and exported to the cytosol where they mature into functional complexes.42 Errors in the complicated assembly process are monitored by a surveillance system that may be coupled to nuclear export and triggers rapid degradation of misfolded complexes.42 Proper ribosome assembly requires precise stoichiometry of rRNA and RPs, although how their synthesis is coordinated remains unclear. Both rRNA and RP production are tightly controlled according to the translational demands of the cell and regulated in an unknown manner via the growth-regulating signaling pathways Ras/PKA and TOR.45 Due to its repetitive nature, the rDNA locus is subject to high levels of DNA breaks and is therefore a hotspot for DNA damage repair.46
Through the HyCCAPP procedure, we identified 216 locus-specific proteins enriched at the region encoding the 25S rRNA (referred to as the “25S locus”) and 316 proteins at the 5S region, with 78 proteins shared by both loci. Both lists were independently enriched (p < 1 × 10–3) for proteins involved in DNA/nucleotide binding and proteins localized to the nucleus. The 25S list was also weakly enriched for expected functions including chromatin regulators (p = 1 × 10–3), proteins involved in transcription-coupled repair (p = 4 × 10–4), nucleolar proteins (p = 1 × 10–4), and RNA helicases (p = 5 × 10–3), while the 5S list was enriched for RNA polymerase subunits (p = 8 × 10–6) and kinases (p = 1 × 10–4). We found 46 and 37 proteins on the 25S and 5S lists, respectively, that participate in processes expected to occur at these loci (including chromatin regulators, DNA damage-repair proteins, proteins annotated as involved in rRNA synthesis41 or known to be required for the ribosome biogenesis42). We validated four of the DNA damage proteins found at the 25S locus by ChIP-qPCR (Figure 6b). Our pipeline identified an additional 24 and 26 proteins at the 25S and 5S loci, respectively, that were identified in screens for rRNA processing/ribosome biogenesis factors,47,48 for a total of 39 proteins across the two lists. Both lists were enriched above chance for proteins that display genetic interactions (GIs) (p = 0.002 for the 5S list and p = 0.015 for the 25S list), consistent with the functional relationships of proteins on the lists (Figure 8). In all we identified 111 proteins with support for binding at one or both loci and an additional 156 proteins that interacted genetically or physically with the supported binders, together representing 60% of the proteins enriched at these loci.
Figure 8.

Interaction network and GO Slim enrichment for locus-specific proteins at 25S and 5S rDNA. As shown in Figure 7 but for the combined 215 and 315 locus-specific proteins captured at the 25S and/or 5S regions.
We therefore delved deeper into the striking diversity of biological processes captured by HyCCAPP of the rDNA locus (Figure 8). Nearly 50 proteins were involved in chromatin regulation, chromosome segregation/recombination, or DNA damage repair, consistent with the high frequency of DNA breaks and recombination errors at the rDNA repeats. We also captured two replication proteins (Mcm2, Orc5) at the 5S locus, consistent with the known ARS in the region. Both rDNA loci were bound by polymerase subunits, including subunits specific to Pol III (Tfc6 at 5S) and Pol II (at both regions), as well as subunits common to all three polymerases (at both loci). One component of the UAF transcription complex, Rrn5, that works with Pol I to transcribe the 35S transcript49 was also captured at the 25S region.
The extensive processing of the 35S rRNA was long thought to occur post-transcriptionally, but recent evidence suggests that processing can occur on the nascent transcript as transcription is occurring.50 We found a striking number of rRNA processing enzymes and helicases, several of which we validated by ChIP (Figure 6c). A number of additional proteins identified at the rDNA locus are not characterized in terms of ribosome biogenesis but were identified in a screen for genes required for rRNA processing;47 our finding that they are localized to the rDNA locus supports a role in rRNA processing. In addition to rRNA processing factors, we captured several proteins involved in rRNA methylation (Bmt2 and Rcm1), also in agreement with the recent realization of cotranscriptional rRNA modification.50,51
Preribosome complexes must be exported to the cytosol for maturation; it has been proposed that export could serve as a surveillance mechanism for proper assembly, yet how this occurs is not known.52 HyCCAPP identified several proteins involved in nuclear transport, including nuclear pore subunit Nup145,53 preribosome export chaperones Ltv154 and Srp40,55 and members of the THO (Tho2, Hrp1) and TREX-2 (Thp1, Sac3, Tex2) complexes. The THO/TREX-2 complex is thought to provide surveillance for proper pre-mRNA production, by coupling transcription, processing, and nuclear export.56 Capture of the THO/TREX-2 complex is especially intriguing, since the complex is not known to monitor rRNA complexes. TREX-2 subunit Sac3 is required for normal ribosome biogenesis,48 further supporting the role of this complex in rRNA surveillance and/or export.
Somewhat surprisingly, HyCCAPP identified at the rDNA loci a number of signaling proteins that are central to growth-rate regulation and RP synthesis. Ifh1, a transcription factor controlling RP expression,45b,57 was captured at the 25S region and validated by ChIP (Figure 6d), raising the possibility that Ifh1 plays a role in coordinating rRNA transcription and RP synthesis, perhaps via transcriptional regulation or through its known association with processing factors.58 We identified several upstream regulators of cellular growth, including members of the TOR pathway (including Tor-complex subunits Lst8, Tsc11 and downstream kinase Sch9), the RAS/PKA pathway (RAS guanine-nucleotide exchange factor Cdc25, heterotrimeric G-protein Gpg1, and stress-activated cAMP phosphodiesterase, Pde2), Sak1 (a kinase regulating the AMPK kinase, Snf159), and others. We validated the rDNA binding of Pde2, Cdc25, and Sak1 by ChIP (Figure 6d). Binding of these regulators to the rDNA locus is reminiscent of the known binding of Tor1 to this region, which is required for proper rDNA transcriptional regulation in response to nutrient cues.60 While further dissection will be required, our results suggest that other regulators of yeast growth control reside at the rDNA locus and may coordinate ribosome production with translational requirements.
Discussion
We describe here a new technology, HyCCAPP, for the identification of proteins bound to specific genomic loci in yeast. The approach utilizes in vivo formaldehyde cross-linking to covalently attach DNA-associated proteins to the genomic DNA, chromatin fragmentation and sequence-specific DNA hybridization to capture the region of interest, mass spectrometry to identify the associated proteins, and a rank order-based bioinformatic filtering process to enrich true binding proteins over nonspecific background binding proteins. The technology was demonstrated in the analysis of four genomic regions in yeast: the multicopy 5S rRNA, 25S rRNA, and X-element loci, and the single-copy GAL1-10 promoter locus. In each case a locus-specific pattern of target-associated proteins was revealed, which includes both previously known and previously unknown target-associated proteins. The binding of the previously unknown proteins was confirmed by immunoprecipitation from TAP-tagged yeast strains and qPCR in 11 of 17 cases, an ∼65% rate of validation. The identification of many previously known proteins at each locus provides strong support for the ability of HyCCAPP to correctly identify DNA-associated proteins in a sequence-specific manner, while the discovery of previously unknown proteins provides new biological insights into transcriptional and regulatory processes at the target locus. We present below a brief discussion of several interesting aspects of the HyCCAPP technology.
Proteins Expected but Not Observed
While it is gratifying that many proteins previously identified at the target regions are observed in our analysis, there are also many binding proteins identified in the literature that were not identified in this HyCCAPP analysis. For example, we initially chose the GAL1-10 region for analysis because it is one of the best-characterized promoters of the yeast genome, with many known protein interactors. However, while we did detect Rsc3, Sth1, Kin28, among other known binders,39,40 we did not observe promoter binders Gal4, Gal80,37b Mig1,37e or Reb1.37a Interestingly, these known binding proteins were also missed in the recent ChAP-MS study of the same region,11 which employed an engineered construct of the GAL1-10 promoter that enabled capture using antibody binding. The absence of some known binding proteins in the captured material was also observed in the work of Déjardin and Kingston on telomeric regions.9 Although further work will be required to understand the basis for such discrepancies, several possibilities are worthy of mention.
First, it is important to understand that HyCCAPP is fundamentally limited by the sensitivity of the MS analysis. MS detection limits for different peptides and proteins vary widely, depending upon factors such as their ionization efficiency, solubility, and polarity. If none of the tryptic peptides corresponding to a target protein are present at levels greater than their detection limit, they will not be observed. Typical peptide MS detection limits for discovery proteomics in complex mixtures are in the 1–10 fmol (6 × 108 to 6 × 109 copies) range, but in many cases they may lie outside that range. Thus, if for any reason, the amount of the peptides from a given protein lies below the detection limit for those peptides, the peptides will not be observed.
Second, it is possible that the occupancy of the promoter region is low; in other words, if only a fraction of the genomes in the cell culture are bound at the moment of cross-linking, that may cause the MS signal obtained for that protein to be below the detection limit of the analysis. This limitation does not affect standard ChIP protocols, because of the very high levels of PCR amplification they employ, which can reveal even just a few copies of the target DNA region.
Third, HyCCAPP employs formaldehyde as a cross-linker. Advantages of formaldehyde for this application include that it is inexpensive, rapidly crosses the cell wall and cell membrane, reacts rapidly, produces reversible cross-links, and has been very widely used and validated in ChIP protocols. However, it is a very short “zero-length” cross-linker, which means that if the formaldehyde-reactive sites on the DNA and protein are not physically close enough to one another, they will not be cross-linked. It is thus possible that the efficiency of cross-linking is extremely low, which is not a problem in ChIP studies due to the high levels of amplification employed but may be too low for HyCCAPP, as the protein that is captured must be directly analyzed without any amplification possible.
Single-Stranded DNA Regions Are Accessible in Cross-Linked Chromatin
A central issue in the development of HyCCAPP was the ability to hybridize-capture probes effectively to the double-stranded genomic DNA present in chromatin. Déjardin and Kingston employed LNA probes to address this issue, with the idea that the increased thermodynamic stability of LNA:DNA duplexes would enable strand invasion and displacement, allowing hybridization capture.9 The HyCCAPP procedure described here is an evolution of a strategy we described previously, referred to as GENECAPP.61 In GENECAPP, which was employed successfully for in vitro studies on a model system, restriction enzyme digestion was used to fragment chromatin at defined sites, followed by exonuclease digestion to produce single-stranded regions suitable for hybridization capture with ordinary capture oligonucleotides.61 However, control experiments in which no restriction enzyme or exonuclease digestions were employed revealed to our surprise that hybridization occurred equally well in their absence. This observation led to the development of HyCCAPP, in which neither restriction digestion nor exonuclease digestion are employed, greatly reducing the expense and complexity of the process. Interestingly, if the same protocol is applied to yeast genomic DNA, rather than chromatin, either with or without cross-linking, no DNA is captured. It is likely that this has bearing upon the observed capture efficiency of ∼1% in HyCCAPP, as only accessible single-stranded regions can be captured. Further exploration of the mechanism underlying this behavior will be necessary to fully understand this aspect of HyCCAPP.
Utility of HyCCAPP in Discovering New Biology
Our initial results here highlight the power of HyCCAPP to discover new biology, through the identification of novel proteins bound to any particular region of a genome. In this sense, the diverse processes that occur at the rDNA locus provide a perfect showcase for the procedure. We captured proteins involved in chromosome functions (including DNA replication, repair, and segregation), transcription (spanning RNA Pol subunits, Mediator components, and site-specific transcription factors), and cotranscriptional processes (such as rRNA processing, modification, export, and surveillance). Several of the novel captured proteins were reported to reside in other subcellular compartments but were identified in screens affecting rRNA biogenesis or (or telomere silencing in the case of Pfa4), strongly supporting a real functional connection. Furthermore, the capture and ChIP validation of upstream signaling proteins bound to the rDNA locus underscores the power to formulate new hypotheses and seed future study.
Conclusion and Future Directions
In conclusion, HyCCAPP offers a powerful new approach to the study of locus-specific chromatin-associated proteins in yeast. We are currently in the process of extending the strategy to the analysis of mammalian genomes, which are ∼300-fold more complex than the yeast genome, and implementing a multiplex strategy to permit many loci to be captured in parallel, thereby increasing throughput while reducing cost and labor. As these and other improvements to the technology are made, the capabilities of HyCCAPP will continue to develop, providing an increasingly powerful new tool for the study of genomic processes.
Acknowledgments
We acknowledge A. J. Bureta and Gloria Sheynkman for assistance with figure preparation. This work was supported by the NIH Center of Excellence in Genomics Sciences grant 1 P50 HG004952.
Supporting Information Available
Additional figures and tables containing DNA oligonucleotide sequence information and mass spectrometry data and analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
A.P.G., B.L.F., J.K.D., L.M.S., M.O., and M.R.S. conceived of experiments. J.K.D. and H.G.A. developed and executed biochemical methods of HyCCAPP. M.S. analyzed captured proteins on MS. A.P.G., C.K., J.K.D., L.M.S., M.O., and M.R.S. developed methods for analysis of MS data. A.P.G. and J.K.D. provided biological insight into captured proteins at the four loci. A.P.G., J.K.D., and L.M.S. prepared figures and manuscript. L.M.S., M.O., and B.L.F. edited and reviewed manuscript.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Sun J. Y.; Kong D. C. DNA replication origins, ORC/DNA interaction, and assembly of pre-replication complex in eukaryotes. Acta Biochim. Biophys. Sin. 2010, 427433–439. [DOI] [PubMed] [Google Scholar]
- Goshima G.; Saitoh S.; Yanagida M. Proper metaphase spindle length is determined by centromere proteins Mis12 and Mis6 required for faithful chromosome segregation. Genes Dev. 1999, 13131664–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Martens J. A.; Laprade L.; Winston F. Intergenic transcription is required to repress the Saccharomyces cerevisiae SER3 gene. Nature 2004, 4296991571–574. [DOI] [PubMed] [Google Scholar]; b Xu Z. Y.; Wei W.; Gagneur J.; Perocchi F.; Clauder-Munster S.; Camblong J.; Guffanti E.; Stutz F.; Huber W.; Steinmetz L. M. Bidirectional promoters generate pervasive transcription in yeast. Nature 2009, 45772321033–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Glynn E. F.; Megee P. C.; Yu H. G.; Mistrot C.; Unal E.; Koshland D. E.; DeRisi J. L.; Gerton J. L. Genome-wide mapping of the cohesin complex in the yeast Saccharomyces cerevisiae. PLoL Biol. 2004, 291325–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Arya G.; Maitra A.; Grigoryev S. A. A structural perspective on the where, how, why, and what of nucleosome positioning. J. Biomol. Struct. Dyn. 2010, 276803–820. [DOI] [PubMed] [Google Scholar]
- Galas D. J.; Schmitz A. DNAse footprinting - simple method for detection of protein-DNA binding specificity. Nucleic Acids Res. 1978, 593157–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon J. M.; Giresi P. G.; Davis I. J.; Lieb J. D. Using formaldehyde-assisted isolation of regulatory elements (FAIRE) to isolate active regulatory DNA. Nat. Protoc. 2012, 72256–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon M. J.; Larsen P. L.; Varshavsky A. Mapping protein DNA interactions in vivo with formaldehyde - evidence that histone-H4 is retained on a highly transcribed gene. Cell 1988, 536937–947. [DOI] [PubMed] [Google Scholar]
- a Robertson G.; Hirst M.; Bainbridge M.; Bilenky M.; Zhao Y. J.; Zeng T.; Euskirchen G.; Bernier B.; Varhol R.; Delaney A.; Thiessen N.; Griffith O. L.; He A.; Marra M.; Snyder M.; Jones S. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods 2007, 48651–657. [DOI] [PubMed] [Google Scholar]; b Chen X.; Xu H.; Yuan P.; Fang F.; Huss M.; Vega V. B.; Wong E.; Orlov Y. L.; Zhang W. W.; Jiang J. M.; Loh Y. H.; Yeo H. C.; Yeo Z. X.; Narang V.; Govindarajan K. R.; Leong B.; Shahab A.; Ruan Y. J.; Bourque G.; Sung W. K.; Clarke N. D.; Wei C. L.; Ng H. H. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell 2008, 13361106–1117. [DOI] [PubMed] [Google Scholar]
- Dejardin J.; Kingston R. E. Purification of proteins associated with specific genomic loci. Cell 2009, 1361175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antao J. M.; Mason J. M.; Dejardin J.; Kingston R. E. Protein landscape at Drosophila melanogaster telomere-associated sequence repeats. Mol. Cell. Biol. 2012, 32122170–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrum S. D.; Raman A.; Taverna S. D.; Tackett A. J. ChAP-MS: A method for identification of proteins and histone posttranslational modifications at a single genomic locus. Cell Rep. 2012, 21198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrum S. D.; Taverna S. D.; Tackett A. J. Purification of a specific native genomic locus for proteomic analysis. Nucleic Acids Res. 2013, 41206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H. Z.; Nichols A.; Liu D. J. Direct identification and quantification of aspartyl succinimide in an IgG2 mAb by RapiGest assisted digestion. Anal. Chem. 2009, 8141686–1692. [DOI] [PubMed] [Google Scholar]
- Sutherland B. W.; Toews J.; Kast J. Utility of formaldehyde cross-linking and mass spectrometry in the study of protein-protein interactions. J. Mass Spectrom. 2008, 436699–715. [DOI] [PubMed] [Google Scholar]
- Orlando V. Mapping chromosomal proteins in vivo by formaldehyde-crosslinked-chromatin immunoprecipitation. Trends Biochem. Sci. 2000, 25399–104. [DOI] [PubMed] [Google Scholar]
- Hirsch J. D.; Eslamizar L.; Filanoski B. J.; Malekzadeh N.; Haugland R. P.; Beechem J. M.; Haugland R. P. Easily reversible desthiobiotin binding to streptavidin, avidin, and other biotin-binding proteins: uses for protein labeling, detection, and isolation. Anal. Biochem. 2002, 3082343–357. [DOI] [PubMed] [Google Scholar]
- Dragan A. I.; Bishop E. S.; Casas-Finet J. R.; Strouse R. J.; Schenerman M. A.; Geddes C. D. Metal-enhanced PicoGreen fluorescence: application to fast and ultra-sensitive pg/mL DNA quantitation. J. Immunol. Methods 2010, 3621–295–100. [DOI] [PubMed] [Google Scholar]
- Gavin A. C.; Aloy P.; Grandi P.; Krause R.; Boesche M.; Marzioch M.; Rau C.; Jensen L. J.; Bastuck S.; Dumpelfeld B.; Edelmann A.; Heurtier M. A.; Hoffman V.; Hoefert C.; Klein K.; Hudak M.; Michon A. M.; Schelder M.; Schirle M.; Remor M.; Rudi T.; Hooper S.; Bauer A.; Bouwmeester T.; Casari G.; Drewes G.; Neubauer G.; Rick J. M.; Kuster B.; Bork P.; Russell R. B.; Superti-Furga G. Proteome survey reveals modularity of the yeast cell machinery. Nature 2006, 4407084631–636. [DOI] [PubMed] [Google Scholar]
- Gingras A. C.; Gstaiger M.; Raught B.; Aebersold R. Analysis of protein complexes using mass spectrometry. Nat. Rev. Mol. Cell Biol. 2007, 88645–654. [DOI] [PubMed] [Google Scholar]
- Robinson M. D.; Grigull J.; Mohammad N.; Hughes T. R., Funspec: A web-based cluster interpreter for yeast. BMC Bioinform. 2002, 3 (35). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout M. P.; Kilmartin J. V. Components of the yeast spindle and spindle pole body. J. Cell Biol. 1990, 11151913–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. S.; Boeke J. D. An unusual form of transcriptional silencing in yeast ribosomal DNA. Genes Dev. 1997, 112241–254. [DOI] [PubMed] [Google Scholar]
- Hoyt M. A.; Stearns T.; Botstein D. Chromosome instability mutants of Saccharomyces cerevisiae that are defective in microtubule-mediated processes. Mol. Cell. Biol. 1990, 101223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis E. J. The chromosome ends of Saccharomyces cerevisiae. Yeast 1995, 11161553–1573. [DOI] [PubMed] [Google Scholar]
- Gotta M.; Laroche T.; Formenton A.; Maillet L.; Scherthan H.; Gasser S. M. The clustering of telomeres and colocalization with Rap1, Sir3, and Sir4 proteins in wild-type Saccharomyces cerevisiae. J. Cell Biol. 1996, 13461349–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier B.; Driller L.; Jaklin S.; Feldmann H. M. New function of CDC13 in positive telomere length regulation. Mol. Cell. Biol. 2001, 21134233–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dyke M. W.; Nelson L. D.; Weilbaecher R. G.; Mehta D. V. Stm1p, a G4 quadruplex and purine motif triplex nucleic acid-binding protein, interacts with ribosomes and subtelomeric y′ DNA in Saccharomyces cerevisiae. J. Biol. Chem. 2004, 2792324323–24333. [DOI] [PubMed] [Google Scholar]
- Lotito L.; Russo A.; Chillemi G.; Bueno S.; Cavalieri D.; Capranico G. Global transcription regulation by DNA topoisomerase I in exponentially growing Saccharomyces cerevisiae cells: Activation of telomere-proximal genes by TOP1 deletion. J. Mol. Biol. 2008, 3772311–322. [DOI] [PubMed] [Google Scholar]
- Izawa S.; Kuroki N.; Inoue Y. Nuclear thioredoxin peroxidase Dot5 in Saccharomyces cerevisiae: Roles in oxidative stress response and disruption of telomeric silencing. Appl. Microbiol. Biotechnol. 2004, 641120–124. [DOI] [PubMed] [Google Scholar]
- Zhou J. L.; Hidaka K.; Futcher B. The Est1 subunit of yeast telomerase binds the Tlc1 telomerase RNA. Mol. Cell. Biol. 2000, 2061947–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askree S. H.; Yehuda T.; Smolikov S.; Gurevich R.; Hawk J.; Coker C.; Krauskopf A.; Kupiec M.; McEachern M. J. M. A genome-wide screen for Saccharomyces cerevisiae deletion mutants that affect telomere length. Proc. Natl. Acad. Sci. U.S.A. 2004, 101238658–8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M.; Baryshnikova A.; Bellay J.; Kim Y.; Spear E. D.; Sevier C. S.; Ding H. M.; Koh J. L. Y.; Toufighi K.; Mostafavi S.; Prinz J.; Onge R. P. S.; VanderSluis B.; Makhnevych T.; Vizeacoumar F. J.; Alizadeh S.; Bahr S.; Brost R. L.; Chen Y. Q.; Cokol M.; Deshpande R.; Li Z. J.; Lin Z. Y.; Liang W. D.; Marback M.; Paw J.; Luis B. J. S.; Shuteriqi E.; Tong A. H. Y.; van Dyk N.; Wallace I. M.; Whitney J. A.; Weirauch M. T.; Zhong G. Q.; Zhu H. W.; Houry W. A.; Brudno M.; Ragibizadeh S.; Papp B.; Pal C.; Roth F. P.; Giaever G.; Nislow C.; Troyanskaya O. G.; Bussey H.; Bader G. D.; Gingras A. C.; Morris Q. D.; Kim P. M.; Kaiser C. A.; Myers C. L.; Andrews B. J.; Boone C. The genetic landscape of a cell. Science 2010, 3275964425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam K. K. Y.; Davey M.; Sun B. M.; Roth A. F.; Davis N. G.; Conibear E. Palmitoylation by the DHHC protein Pfa4 regulates the ER exit of Chs3. J. Cell Biol. 2006, 174119–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson E. E.; Fox C. A. The Ku complex in silencing the cryptic mating-type loci of Saccharomyces cerevisiae. Genetics 2008, 1802771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S.; Patterson E. E.; Cobb J.; Audhya A.; Gartenberg M. R.; Fox C. A. Palmitoylation controls the dynamics of budding-yeast heterochromatin via the telomere-binding protein Rif1. Proc. Natl. Acad. Sci. U.S.A. 2011, 1083514572–14577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober H.; Ferreira H.; Kalck R.; Gehlen L. R.; Gasser S. M. Yeast telomerase and the SUN domain protein Mps3 anchor telomeres and repress subtelomeric recombination. Genes Dev. 2009, 238928–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Reagan M. S.; Majors J. E. The chromatin structure of the GAL1 promoter forms independently of Reb1p in Saccharomyces cerevisiae. Mol. Gen. Genet. 1998, 2592142–149. [DOI] [PubMed] [Google Scholar]; b Lue N. F.; Chasman D. I.; Buchman A. R.; Kornberg R. D. Interaction of GAL4 and GAL80 gene regulatory proteins in vitro. Mol. Cell. Biol. 1987, 7103446–3451. [DOI] [PMC free article] [PubMed] [Google Scholar]; c West R. W.; Chen S. M.; Putz H.; Butler G.; Banerjee M. GAL1-GAL10 divergent promoter region of Saccharomyces-cerevisiae contains negative control elements in addition to functionally separate and possibly overlapping upstream activating sequences. Genes Dev. 1987, 1101118–1131. [DOI] [PubMed] [Google Scholar]; d West R. W.; Yocum R. R.; Ptashne M. Saccharomyces-cerevisiae GAL1-GAL10 divergent promoter region - location and function of the upstream activating sequence UASG. Mol. Cell. Biol. 1984, 4112467–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Johnston M.; Flick J. S.; Pexton T. Multiple mechanisms provide rapid and stringent glucose repression of GAL gene-expression in Saccharomyces-cerevisiae. Mol. Cell. Biol. 1994, 1463834–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseley J.; Rubbi L.; Grunstein M.; Tollervey D.; Vogelauer M. A ncRNA modulates histone modification and mRNA induction in the yeast GAL gene cluster. Mol. Cell 2008, 325685–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floer M.; Wang X.; Prabhu V.; Berrozpe G.; Narayan S.; Spagna D.; Alvarez D.; Kendall J.; Krasnitz A.; Stepansky A.; Hicks J.; Bryant G. O.; Ptashne M. A RSC/nucleosome complex determines chromatin architecture and facilitates activator binding. Cell 2010, 1413407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder S. C.; Schwer B.; Shuman S.; Bentley D. Dynamic association of capping enzymes with transcribing RNA polymerase II. Genes Dev. 2000, 14192435–2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry J. M.; Hong E. L.; Amundsen C.; Balakrishnan R.; Binkley G.; Chan E. T.; Christie K. R.; Costanzo M. C.; Dwight S. S.; Engel S. R.; Fisk D. G.; Hirschman J. E.; Hitz B. C.; Karra K.; Krieger C. J.; Miyasato S. R.; Nash R. S.; Park J.; Skrzypek M. S.; Simison M.; Weng S.; Wong E. D. Saccharomyces genome database: The genomics resource of budding yeast. Nucleic Acids Res. 2012, 40D1D700–D705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolford J. L.; Baserga S. J. Ribosome biogenesis in the yeast Saccharomyces cerevisiae. Genetics 2013, 1953643–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T. Regulation of ribosomal RNA gene copy number and its role in modulating genome integrity and evolutionary adaptability in yeast. Cell. Mol. Life Sci. 2011, 6881395–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. H.; Mueller J. E.; Bryk M. Sir2 represses endogenous polymerase II transcription units in the ribosomal DNA nontranscribed spacer. Mol. Biol. Cell 2006, 1793848–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Xiao L. J.; Grove A. Coordination of ribosomal protein and ribosomal RNA gene expression in response to TOR signaling. Curr. Genomics 2009, 103198–205. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Martin D. E.; Soulard A.; Hall M. N. TOR regulates ribosomal protein gene expression via PKA and the forkhead transcription factor FHL1. Cell 2004, 1197969–979. [DOI] [PubMed] [Google Scholar]; c Powers T.; Walter P. Regulation of ribosome biogenesis by the rapamycin-sensitive TOR-signaling pathway in Saccharomyces cerevisiae. Mol. Biol. Cell 1999, 104987–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ramachandran V.; Herman P. K. Antagonistic interactions between the cAMP-dependent protein kinase and TOR signaling pathways modulate cell growth in Saccharomyces cerevisiae. Genetics 2011, 1872441–454. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Neumansilberberg F. S.; Bhattacharya S.; Broach J. R. Nutrient availability and the RAS/cyclic AMP pathway both induce expression of ribosomal-protein genes in Saccharomyces-cerevisiae but by different mechanisms. Mol. Cell. Biol. 1995, 1563187–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conconi A. The yeast rDNA locus: A model system to study DNA repair in chromatin. DNA Repair 2005, 48897–908. [DOI] [PubMed] [Google Scholar]
- Peng W. T.; Robinson M. D.; Mnaimneh S.; Krogan N. J.; Cagney G.; Morris Q.; Davierwala A. P.; Grigull J.; Yang X. Q.; Zhang W.; Mitsakakis N.; Ryan O. W.; Datta N.; Jojic V.; Pal C.; Canadien V.; Richards D.; Beattie B.; Wu L. F.; Altschuler S. J.; Roweis S.; Frey B. J.; Emili A.; Greenblatt J. F.; Hughes T. R. Panoramic view of yeast noncoding RNA processing. Cell 2003, 1137919–933. [DOI] [PubMed] [Google Scholar]
- Li Z. H.; Lee I.; Moradi E.; Hung N. J.; Johnson A. W.; Marcotte E. M. Rational extension of the ribosome biogenesis pathway using network-guided genetics. PLoS Biol. 2009, 710e1000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keys D. A.; Lee B. S.; Dodd J. A.; Nguyen T. T.; Vu L.; Fantino E.; Burson L. M.; Nogi Y.; Nomura M. Multiprotein transcription factor UAF interacts with the upstream element of the yeast RNA polymerase I promoter and forms a stable preinitiation complex. Genes Dev. 1996, 107887–903. [DOI] [PubMed] [Google Scholar]
- Kos M.; Tollervey D. Yeast pre-rRNA processing and modification occur cotranscriptionally. Mol. Cell 2010, 376809–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osheim Y. N.; French S. L.; Keck K. M.; Champion E. A.; Spasov K.; Dragon F.; Baserga S. J.; Beyer A. L. Pre-18S ribosomal RNA is structurally compacted into the SSU processome prior to being cleaved from nascent transcripts in Saccharomyces cerevisiae. Mol. Cell 2004, 166943–954. [DOI] [PubMed] [Google Scholar]
- Kressler D.; Hurt E.; Bassler J. Driving ribosome assembly. Biochim. Biophys. Acta, Mol. Cell Res. 2010, 18036673–683. [DOI] [PubMed] [Google Scholar]
- Davis L. I. The nuclear-pore complex. Annu. Rev. Biochem. 1995, 64, 865–896. [DOI] [PubMed] [Google Scholar]
- Seiser R. M.; Sundberg A. E.; Wollam B. J.; Zobel-Thropp P.; Baldwin K.; Spector M. D.; Lycan D. E. Ltv1 is required for efficient nuclear export of the ribosomal small subunit in Saccharomyces cerevisiae. Genetics 2006, 1742679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikonomova R.; Sommer T.; Kepes F. The Srp40 protein plays a dose-sensitive role in preribosome assembly or transport and depends on its carboxy-terminal domain for proper localization to the yeast nucleoskeleton. DNA Cell Biol. 1997, 16101161–1173. [DOI] [PubMed] [Google Scholar]
- Luna R.; Rondon A. G.; Aguilera A. New clues to understand the role of THO and other functionally related factors in mRNP biogenesis. Biochim. Biophys. Acta, Gene Regul. Mech. 2012, 18196514–520. [DOI] [PubMed] [Google Scholar]
- Schawalder S. B.; Kabani M.; Howald I.; Choudhury U.; Werner M.; Shore D. Growth-regulated recruitment of the essential yeast ribosomal protein gene activator Ifh1. Nature 2004, 43270201058–1061. [DOI] [PubMed] [Google Scholar]
- Rudra D.; Mallick J.; Zhao Y.; Warner J. R. Potential interface between ribosomal protein production and pre-rRNA processing. Mol. Cell. Biol. 2007, 27134815–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbing K.; McCartney R. R.; Schmidt M. C. Purification and characterization of the three Snf1-activating kinases of Saccharomyces cerevisiae. Biochem. J. 2006, 393, 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck T.; Hall M. N. The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors. Nature 1999, 4026762689–692. [DOI] [PubMed] [Google Scholar]
- Wu C.-H.; Chen S.; Shortreed M. R.; Kreitinger G. M.; Yuan Y.; Frey B. L.; Zhang Y.; Mirza S.; Cirillo L. A.; Olivier M.; Smith L. M. Sequence-specific capture of protein-DNA complexes for mass spectrometric protein identification. PLoS One 2011, 610e26217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia O.; Saveanu C.; Cline M.; Fromont-Racine M.; Jacquier A.; Schwikowski B.; Aittokallio T. GOlorize: A cytoscape plug-in for network visualization with gene ontology-based layout and coloring. Bioinformatics 2007, 233394–396. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.