

Abstract
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) is an adult onset cerebral small vessel disorder caused by the mutations of the neurogenic locus notch homolog protein 3 (NOTCH3) gene. The extracellular part of NOTCH3 is composed of 34 epidermal growth factor-like (EGF-like) repeat domains. Each EGF-like domain is rich of cysteine and glycine to produce three loops that are essential for high-affinity binding to its ligand. Nearly all reported CADASIL-associated mutations result in gain or loss of a cysteine residue within the EGF-like domains. Only a few cysteine-sparing NOTCH3 mutations have been documented in the patients with CADASIL to date. Here, we reported a Chinese CADASIL family with a cysteine-sparing NOTCH3 mutation. In this family, affected patients had dizziness, memory loss, gait instability, or hemiplegia. Brain magnetic resonance imaging (MRI) showed diffuse leukoencephalopathy with confluent signal abnormalities in the periventricular white matter, basal ganglia, and centrum semiovale bilaterally. By screening the entire coding region of NOTCH3, a novel missense mutation p.G149V (c.446G>T) was found. This mutation was not detected in 400 normal controls. Considering the critical position of glycine within the C-loop of EGF-like domain and its high conservation through evolution, p.G149V mutation could be a potential pathogenic cause for CADASIL.
Introduction
Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL, OMIM 125310) is an inherited small cerebral vessel disease characterized by migraine, recurrent stroke, mood disturbances, apathy, dementia, and premature death [1], [2]. CADASIL is caused by the mutations in the NOTCH3 gene (OMIM 600276), which encodes a single pass trans-membrane protein with an extracellular epidermal growth factor-like (EGF-like) repeat domain, a single trans-membrane domain, and an intracellular ankyrin domain [3], [4]. Each EGF-like domain contains six cysteine residues to form three loops, which are essential for the high-affinity for ligand binding. To date, more than 200 mutations of NOTCH3 have been reported in CADASIL patients. Among these, over 95% mutations are located in EGF-like repeat domain of NOTCH3 [data from HGMD]. The vast majority of mutations are missense and lead to either gain or lose a cysteine residue, resulting in an odd number of cysteine and further misfolding of the EGF-like repeat domain. This misfolding may alter the maturation, targeting, degradation and function of the NOTCH3 receptor, which is responsible for most phenotypes of CADASIL affected families. Interestingly, only several cysteine-sparing NOTCH3 mutations have been documented in the patients with CADASIL to date [5], [6].
In this study, we reported a novel cysteine-sparing NOTCH3 mutation at the nucleotide position 446 (c.446G>T; p.G149V) in a Chinese family with CADASIL.
Materials and Methods
The study was approved by the Institutional Review Board of Soochow University. Written informed consent forms from the family and 400 healthy blood donors were obtained. The clinical data on family members were obtained through interviews, physical examination, brain magnetic resonance imaging (MRI), and medical reports.
Genomic DNA from peripheral blood mononuclear cells of the proband and her relatives were isolated by the method of phenol-chloroform. All 33 exons and their flanking intronic sequences of gene NOTCH3 (NM_000435.2) were amplified by polymerase chain reaction (PCR) using DreamTaq Green PCR Master Mix (2X) (Fermentas) or TaKaRa LA Taq with GC buffer (Table 1). After the PCR products were directly sequenced by the Sanger method, DNA sequences were analyzed by comparing to the February 2009 human reference sequence (GRCh37/hg19) with the BLAT tool from the UCSC Genome Browser. The new mutation was checked with online database like HGMD (http://www.hgmd.org/) and the 1000 Genome project (http://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/).
Table 1. Information of primers used in PCR amplification for NOTCH3 gene.
| Primer Name | Sequence |
| E1F | GAATCTCTGGGTCCTCCAGGACTGG |
| E1R | AGGTGTCCGGCTCTGGGTGTGTAC |
| E2F | GAGGTTGCCCAAGCCACACACATG |
| E2R | CACCCTTACTTCCTGTCTCAGCACAC |
| E3-6F | CACTCAAGTCAGACTTCTTATTTGCCCTC |
| E3-6R | TGCGTGTTTCTTGCCTGTCTTGTG |
| E7-8F | GACCAAGCCCCCAGAATCACACAG |
| E7-8R | GGCAGAGCAGGAAGATCTGCCTATGAC |
| E9-10F | CTGTCACTGGGTCCTGCCTTGCTAC |
| E9-10R | AGTGTAGAGGTGGGGACGAAGGCTC |
| E11-2F | GCCAATGAGACAGCACAGACTCAGG |
| E11-2R | ATTGGTCCGAGGCCTCACTTGTG |
| E13-5F | GCTGACCCACACAGGCTGCATATC |
| E13-5R | GACTTCATGGAAGTTTGGGGACCAG |
| E16F | GCTCATTCCCTAACTCGTTCCATGG |
| E16R | CACTGATATGCAGCCTGTGTGGGTC |
| E17-8F | GTCTGGAGGGGAAGCACTCAGAGTC |
| E17-8R | TCCTGCCCTTGCCACATAGGTGAG |
| E19-20F | CCAGATCTACCCAGAGTGACACCCAC |
| E19-20R | AGTGCCAGGTGGGTGGAGTTACTGG |
| E21-3F | CAGAAACTCCCTTCCCTTCGATGTC |
| E21-3R | GTGCTGGGGTTCTTTGCGTCTTC |
| E24F | GACACGTGGGACACACCGATG |
| E24R | CCCCTCTCTCCCCTTGACTCTTC |
| E25F | CAAGCCACTTAGCAACCTGCCTCC |
| E25R | GGCATGGGTCCGTATATTCCAGTCC |
| E26-8F | CTGATCACGCCCATCATCCAC |
| E26-8R | AGATGGCGCTTCAGAGCCAGG |
| E29F | GGACCTTTGTCACTTCCAACCAAGAG |
| E29R | TTCCCAGGGCTCTGTGTGTATCTCC |
| E30-1F | TGAGGCACCCTAAGGACTAGTGGTG |
| E30-1R | CCCTGTGCCCTAGGAGTAGTTCTGTG |
| E32F | ACTGCTGACACCCAGTGGACCAAG |
| E32R | GCACACACATGGATCCAGACACAAG |
| E33F1 | AAGGCAAGGATGCAGGAGGGTTG |
| E33R1 | CTGACAGCTCGGTCACGCTGTC |
| E33F2 | CAAGCTGTGCCAGAGACACTGCAG |
| E33R2 | CCCAAGAGGCTGGAAGACTTTGCTAC |
| MscI mutF | CAGCTGCGGCCCTGGTgG |
| MscI mutR | ACTCACCCTGTCCTGGTCCCTCC |
All family members and unrelated 400 control individuals were screened by restriction enzyme FastDigest MscI (Fermentas, USA), which recognizes TGG/CCA sites generated with mutagenesis primers (Table 1) in wild type allele but not c.446G>T mutant allele of NOTCH3 gene.
Results
Patient history and clinical features
The proband of the family was a 43-year-old woman. At the 39 years of age, she complained progressive dizziness and memory impairment. She had no remarkable medical history. Cerebrovascular risk factors including hypertension, high glucose, hypercholesterolemia, and smoking were not found. Her neurological examination was unremarkable. Her performance on the mini-mental status examination (MMSE) was slightly impaired (a score of 25 out of 30). T2-weighted and fluid-attenuating inversion recovery (FLAIR) brain MRI (GE Healthcare Signa EXCITE 3.0T) revealed severe diffuse leukoencephalopathy with multiple confluent signal abnormalities in the periventricular white matter, basal ganglia, and centrum semiovale bilaterally (Fig. 1A–C and D–F). Diffusion-weighted imaging (DWI) sequence demonstrated acute ischemic stroke in the centrum semiovale (Fig. 1G). Cerebral magnetic resonance angiography (MRA) did not reveal segmental stenosis of the intracranial vessels, vascular malformation, or aneurysm (Fig. 1H). The patient was diagnosed as Binswanger’s disease. At the two-year follow-up, she still had dizziness and hypomnesia. MMSE score was 24 and MRI did not show any significant change in comparison with previous record. The patient reported that her elder sister had similar symptoms and MRI changes. Therefore, her family members were interviewed and clinical data were collected. On the basis of the family history and neuroimaging data, a diagnosis of CADASIL was suspected.
Figure 1. Brain MRI images of the proband showing characteristic diffuse leukoencephalopathy.
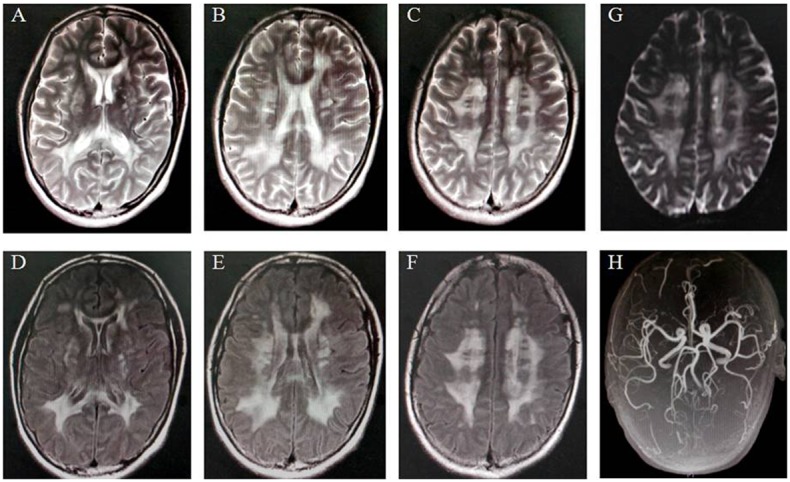
T2-weighted MRI images (A–C) showed diffuse hyperintensities in the periventricular white matter with a symmetrical distribution, the centrum semiovale, and the basal ganglia. Fluid-attenuated inversion recovery (FLAIR) T2 sequences (D–F) revealed confluent white-matter lesions in multiple brain regions. Diffusion-weighted imaging (DWI) sequence (G) demonstrated acute ischemic stroke in the centrum semiovale. MR angiography (H) revealed no intracranial arterial stenosis or occlusion.
The family pedigree was draw as in Fig. 2A. The elder sister of the proband (II1) initially presented dizziness, hypomnesia, and gait disability at 48 years of age. She was admitted for further examination at that time. She had no history of hypertension and diabetes mellitus. Her neurological examination was unremarkable except for bilateral positive Babinski sign, positive Romberg test, and mild memory disturbance (the MMSE score 23). Brain MRI indicated that multiple confluent hyperintensities were observed in the periventricular white matter, basal ganglia, and centrum semiovale. Her symptoms including memory loss were progressive. She gradually had the difficulty to chew, talk, and swallow. At the age of 52 years old, the patient was feed by a gastrostomy tube and was extremely thin so that she could not move. The MMSE score was 2. Neurological assessment revealed high muscle tone and brisk tendon reflexes in limbs.
Figure 2. A novel G149V mutation found in a CADASIL family.
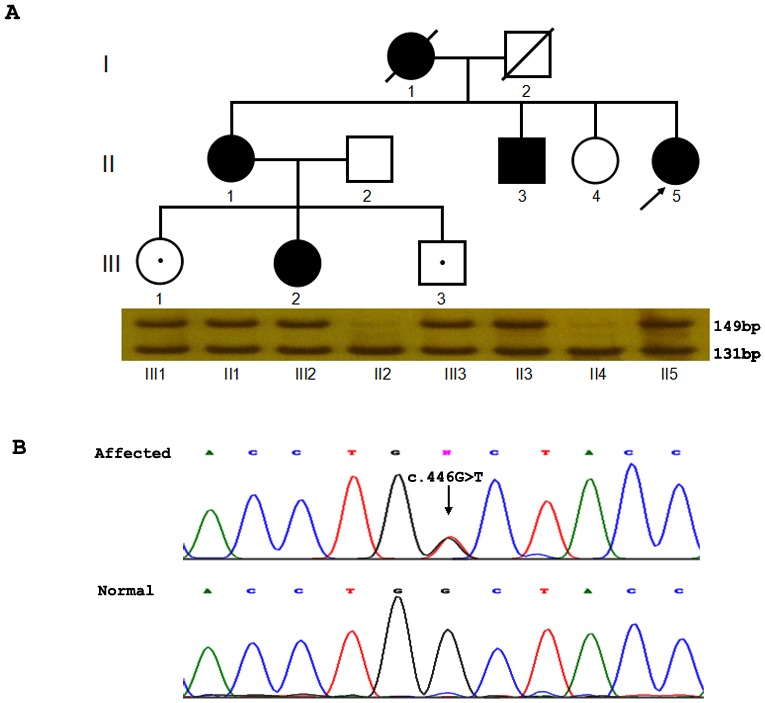
The pedigree of this Chinese CADASIL family was shown (A). The proband is indicated with an arrow. MscI digestion results corresponding to each individual were shown below the pedigree. The affected showed a digested band (131 bp) with an undigested band (149 bp), while the normal controls showed only one digested band at 131 bp. III1 and III3 carried the mutation but are asymptomatic so far. Electropherograms of the sequence showed the nucleotide change in the affected individual with respect to the normal control bp. III1 and III3 carried the mutation but are asymptomatic so far. Electropherograms of the sequence showed the nucleotide change in the affected individual with respect to the normal control (B). P.G149V (c.446G>T) was indicated with an arrow.
The elder brother of the proband (II3) was diagnosed as ischemic stroke for hemiplegia at 40 years of age. He did not have any vascular risk factors and was partially recovered within several months. The niece of the proband (III2) had dizziness at 26 years of age, but no neurological abnormality was found.
Genetic analysis
We first screened the exons 2–24, which encode 34 EGF-like repeat domains of NOTCH3 gene of the proband by direct sequencing. We found a novel G to T transversion at the nucleotide position 446 (c.446G>T), resulting in a change of glycine (GGC) to valine (GTC) at the amino acid position 149 in NOTCH3 (Fig. 2B). We further screened the remaining exons of NOTCH3 gene and no other potentially pathogenic nucleotide change was detected. The c.446G>T change causes a loss of MscI restriction enzyme site (TGG/CCA) in the mutant allele from the 149 bp PCR product amplified by the mutantgenesis primers which were designed to contain C at nucleotide position 448 instead of T. By MscI enzyme, the heterozygous c.446G>T mutation was found in all the affected individuals and two young asymptomatic individuals (III1 and III3) in the family (Fig. 2A). Additional 400 health controls have been screened by MscI digestion, however, this c.446G>T mutation was not found in these individuals. There was no report about this c.446G>T change in HGMD and the 1000 Genome project database.
The glycine at position 149 is located at the third EGF-like domain and lies between C5–C6 to form the C-loop of NOTCH3. It is corresponding to human EGF Gly36 and is highly conserved in nearly all the EGF-like repeat domains of NOTCH3 except domain 6, 25 and 32 (Fig. 3A). The glycine 149 lies in a center of a clusters reported mutations and shows high conservation through evolution from human to fly (Fig. 3B, 3C).
Figure 3. Alignment of EGF-like repeat domain of NOTCH3.

Alignment of C4–C6 in EGF-like domains of NOTCH3 was shown in the upper panel (A). Conserved G36 in human EGF is shown in the most EGF-like domains of NOTCH3. Alignment of C4–C6 in EGF-like repeat domain 3 homolog from different species was shown in the middle panel of the figure (B). Amino acid sequence alignment from various species showed the conservation of this glycine residue. The reported CADASIL mutations within this EGF-like repeat domain were indicated in the lower panel (C).
Discussion
In this study, we reported a novel NOTCH3 gene missense mutation (p.G149V) in a Chinese CADASIL family. The clinical features in this family included dizziness, cognitive deficits, gait instability, and ischemic stroke. All affected patients had no vascular risk factors. MRIs revealed T2-weighted hyperintensities in the periventricular and subcortical white matter. Based on family history, clinical symptoms, and typical MRI results, CADASIL was diagnosed.
CADASIL-causing protein NOTCH3 has 34 EGF-like repeat domains [7], [8]. Six highly conserved cysteine residues within each EGF-like domain form three intra-molecular disulfide bond pairs between C1–C3, C2–C4 and C5–C6. These three loops are essential for high-affinity binding to the ligand. When ligands bind to the extracellular EGF-like repeat domain, extracellular domain was released with a series of proteolytic cleavages, then translocates to the nucleus, and activates transcription. Mutations within EGF-like repeat domain alter the conformation of the receptor and lead to defects in NOTCH3 protein processing and trafficking, or interaction with the ligands [9]. As the second rich amino acid next to cysteine, glycine showed high degree of conservation at Gly18, Gly36, and Gly39 of human EGF (hEGF) domain. By genetic testing, we revealed a novel c.446G>T (p.G149V) mutation located in exon 4 of the NOTCH3 gene. The glycine at position 149 is located at the third EGF-like domain of NOTCH3 between C5–C6. This p.G149 is corresponding to the Gly36 of hEGF and is also highly conserved among almost all the EGF like domains of NOTCH3 except domain 6, 25 and 32 (Fig. 3A). Mutagenesis studies showed that the substitute of Gly36 by valine resulted in an apparent inability of EGF to fold into its native structure [10], which make it more easily to predict that G149V mutation might dysfunction the critical role of glycine and cause the phenotype of CADASIL. Similarly, the mutation c.445G>T (p.G149C) was reported in 5 individuals with CADASIL [11], which supported our hypothesis that the institution of glycine at position 149 of NOTCH3 is a potential pathogenic cause of CADASIL. In addition, this mutation was not found in 400 health controls and no other mutation of NOTCH3 gene is responsible for the phenotype of CADASIL, as further supported this notion.
To date, only a few cysteine-sparing missense mutations within EFG-like repeat domains have previously been reported as the cause of CADASIL. Four of them (R75P, Q151E, T577A, P685T) are located within C-loop of EGF-like domain (data from HMGD). It might be due to the hypothesis that C-loop is more likely to be responsible for the binding affinity since it is more conservative than the other two loops [12].
In summary, we found a novel cysteine-sparing NOTCH3 mutation G149V in a family with CADASIL. Considering its critical position of glycine within the C-loop of EGF-like domain, p.G149V mutation could be a potential pathogenic cause for CADASIL, which will be further confirmed by functional studies.
Acknowledgments
We would like to thank all the family members for their participation in this study. SM is a trainee in the International Collaborative Genetics Research Training Program (NIH grant D43 TW06176 to Jabs EW).
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work is supported by National Natural Science Foundation of China 30871355 and Ministry of Science and Technology of China 2013CB945400 (to MS). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Chabriat H, Vahedi K, Iba-Zizen MT, Joutel A, Nibbio A, et al. (1995) Clinical spectrum of CADASIL: a study of 7 families. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Lancet 346: 934–939. [DOI] [PubMed] [Google Scholar]
- 2. Dichgans M, Mayer M, Uttner I, Bruning R, Muller-Hocker J, et al. (1998) The phenotypic spectrum of CADASIL: clinical findings in 102 cases. Ann Neurol 44: 731–739. [DOI] [PubMed] [Google Scholar]
- 3. Tournier-Lasserve E, Joutel A, Melki J, Weissenbach J, Lathrop GM, et al. (1993) Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy maps to chromosome 19q12. Nat Genet 3: 256–259. [DOI] [PubMed] [Google Scholar]
- 4. Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, et al. (1996) Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature 383: 707–710. [DOI] [PubMed] [Google Scholar]
- 5. Bersano A, Ranieri M, Ciammola A, Cinnante C, Lanfranconi S, et al. (2012) Considerations on a mutation in the NOTCH3 gene sparing a cysteine residue: a rare polymorphism rather than a CADASIL variant. Funct Neurol 27: 247–252. [PMC free article] [PubMed] [Google Scholar]
- 6. Scheid R, Heinritz W, Leyhe T, Thal DR, Schober R, et al. (2008) Cysteine-sparing notch3 mutations: cadasil or cadasil variants? Neurology 71: 774–776. [DOI] [PubMed] [Google Scholar]
- 7. Domenga V, Fardoux P, Lacombe P, Monet M, Maciazek J, et al. (2004) Notch3 is required for arterial identity and maturation of vascular smooth muscle cells. Genes Dev 18: 2730–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Joutel A, Andreux F, Gaulis S, Domenga V, Cecillon M, et al. (2000) The ectodomain of the Notch3 receptor accumulates within the cerebrovasculature of CADASIL patients. J Clin Invest 105: 597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fouillade C, Monet-Lepretre M, Baron-Menguy C, Joutel A (2012) Notch signalling in smooth muscle cells during development and disease. Cardiovasc Res 95: 138–146. [DOI] [PubMed] [Google Scholar]
- 10. Campion SR, Niyogi SK (1994) Interaction of epidermal growth factor with its receptor. Prog Nucleic Acid Res Mol Biol 49: 353–383. [DOI] [PubMed] [Google Scholar]
- 11. Opherk C, Peters N, Herzog J, Luedtke R, Dichgans M (2004) Long-term prognosis and causes of death in CADASIL: a retrospective study in 411 patients. Brain 127: 2533–2539. [DOI] [PubMed] [Google Scholar]
- 12. Wouters MA, Rigoutsos I, Chu CK, Feng LL, Sparrow DB, et al. (2005) Evolution of distinct EGF domains with specific functions. Protein Sci 14: 1091–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.