

Abstract
Primary ciliary dyskinesia is a genetically heterogeneous autosomal recessive disease in which mutations disrupt ciliary function, leading to impaired mucociliary clearance and life-long lung disease. Mouse tracheal cells with a targeted deletion in the axonemal dynein intermediated chain gene Dnaic1 differentiate normally in culture but lack ciliary activity. Gene transfer to undifferentiated cultures of mouse Dnaic1−/− cells with a lentiviral vector pseudotyped with avian influenza hemagglutinin restored Dnaic1 expression and ciliary activity. Importantly, apical treatment of well-differentiated cultures of mouse Dnaic1−/− with lentiviral vector also restored ciliary activity, demonstrating successful gene transfer from the apical surface. Treatment of Dnaic1flox/flox mice expressing an estrogen responsive Cre recombinase with different doses of tamoxifen indicated that restoration of ~20% of ciliary activity may be sufficient to prevent the development of rhinosinusitis. However, while administration of a β-galactosidase expressing vector to control mice demonstrated efficient gene transfer to the nasal epithelium, treatment of Dnaic1−/− mice resulted in a low level of gene transfer, demonstrating that the severe rhinitis present in these animals impedes gene transfer. The results demonstrate that gene replacement therapy may be a viable treatment option for primary ciliary dyskinesia, but further improvements in the efficiency of gene transfer are necessary.
Keywords: cilia, airway, lentiviral, bronchiectasis, rhinosinusitis, DNAI1
Introduction
Primary ciliary dyskinesia (PCD) is a rare genetic disease in which mutations in critical genes impair the function of motile cilia resulting in reduced or absent mucociliary clearance (MCC) 1, 2. Patients with PCD, as a result of the defect in MCC, suffer from neonatal respiratory distress, chronic rhinosinusitis, otitis media, and repeated airway infections, resulting in bronchiectasis 1, 2. Additionally, abnormalities of situs occur with increased frequency in this population 3, 4, and males with PCD are typically infertile 1, 2. Current treatments are designed to reduce the incidence and severity of respiratory tract infections, as there are no corrective treatments available 5, 6.
Recent genetic studies have identified some of the genes mutated in PCD, including components of the dynein motors (DNAI1, DNAI2, DNAH5, DNAH11), radial spokes (RSPH4A, RSPH9) and other genes involved in ciliary assembly (KTU, LRRC50) 7. Because of the genetically heterogeneous nature of the disease, which makes it unlikely that a single drug can be identified to treat/correct each causative mutation, gene transfer may be a viable therapeutic option for PCD. For example, a drug that corrects a specific defect in one axonemal protein would be unlikely to correct mutations in a different axonemal protein. In contrast, if a suitable gene transfer vector were available, by simply replacing the cDNA the vector could be used to correct most, if not all, PCD causing mutations. Heterozygotes have apparently normal ciliary function, indicating that dominant negative mutations are not common, and the conducting airways, which are the site of the most serious pathology, are relatively accessible to administration of a viral vector 1, 2.
Much prior effort has been devoted to developing gene transfer as a therapy for other airway diseases, most notably, cystic fibrosis. These studies have identified several challenges to successful gene therapy, including the resistance of well-differentiated (polarized) airway epithelium to apical transduction by viral vectors, the difficulties of obtaining long-term expression, and the development of an immune response to many vector components 8-10. Although gene therapy of airway diseases has not yet been successful, researchers are continuously making advances in vector design and production. For example, several groups have identified viral envelope proteins useful for targeting lentiviral vectors to the apical membrane of differentiated airway epithelial cells 11-14. Our group has recently demonstrated that pseudotyping lentiviral vectors with an avian influenza hemagglutinin (HA) protein allows efficient apical transduction of differentiated airway epithelial cells 15, 16. The incorporation of endogenous cellular promoters and the use of integrating lentiviral vectors to transduce airway epithelium in animal models have resulted in stable, long-term expression 11-14, 17.
To test the hypothesis that gene transfer could restore normal ciliary activity to PCD cells, we utilized an inducible mouse model of PCD we have previously characterized 18. In this model, activation of an estrogen responsive Cre recombinase (CreER) by tamoxifen treatment causes the deletion of two exons flanked by loxP sites in the axonemal dynein intermediate chain 1 (Dnaic1) gene. The mouse Dnaic1 protein is 82% identical to the human DNAI1 protein, and mutations in DNAI1 cause about 10% of PCD cases 19-22. The deletion disrupts the structure of critical WD40 domains in the Dnaic1 protein and prevents the assembly of the outer dynein arm 23, resulting in immotile cilia and a PCD phenotype. Unlike traditional knock-out models, the use of an inducible system avoids the complications of neonatal hydrocephalus 24, 25, heart defects 26, 27, and other situs abnormalities that frequently occur in PCD mice, allowing us to study adult PCD animals18.
In this report, we have tested the ability of an HA-pseudotyped lentiviral vector to restore ciliary activity to both undifferentiated and differentiated PCD cells in vitro. We also utilized our inducible mouse model to estimate the level of gene transfer required to prevent upper airway disease and to investigate the turnover of a ciliary protein, two important aspects that will have to be considered when designing clinical trials for PCD. Finally, we examined the effect of preexisting rhinosinusitis on the ability of the HA-pseudotyped lentiviral vector to transduce the nasal epithelium of PCD animals.
Results
Construction of lentiviral vectors
A full-length mouse cDNA for Dnaic1 was cloned into a lentivirus gene transfer vector (SIN6.1CB-W) based on the equine infectious anemia virus (EIAV, 15) and the construct was verified by direct sequencing (Fig. 1a). The Dnaic1 cDNA was under control of hybrid promoter consisting of the human CMV enhancer followed by the chicken β-actin promoter (CB) that is ubiquitously expressed 16. Additional vectors expressing the reporter genes EGFP, firefly luciferase, or β-galactosidase (β-gal) from the same construct were utilized as controls in these studies 16. Vectors were also constructed that contained an internal ribosomal entry site (IRES) after the Dnaic1 cDNA followed by a cDNA encoding EGFP, however, these vectors were found to be inefficient. Viral particles pseudotyped with influenza hemagglutinin (HA) from fowl plague virus were produced by transfection of 293T cells as previously described 16. Transduction of 293 cells with Dnaic1-encoding lentivirus resulted in expression of a protein of the correct size that reacted with a purified monoclonal antibody against human DNAI1 on Western blotting (Fig. 1b, 18) confirming the vector was expressing full-length Dnaic1.
Figure 1.
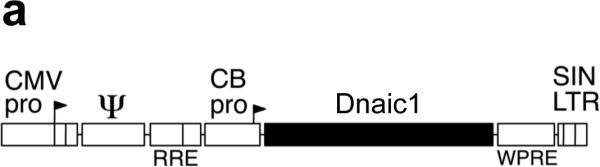
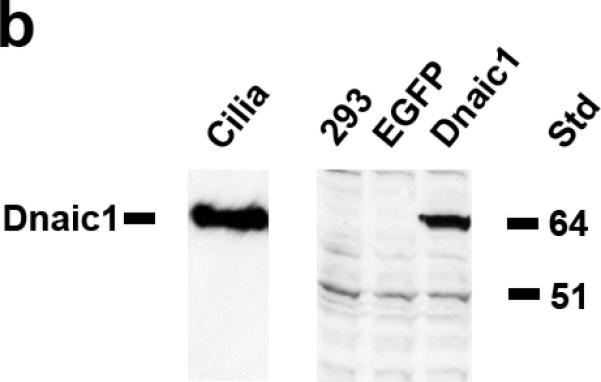
a) Diagram of the lentiviral gene transfer construct used in these studies. The mouse Dnaic1 cDNA was expressed from a hybrid CMV enhancer/chicken β-actin promoter (CB). The vector also contains an upstream CMV enhancer/promoter fused to the R and U5 domains of the EIAV long terminal repeat (LTR) sequence, the viral Rev Responsive Element (RRE), RNA encapsidation sequence (Ψ), the woodchuck hepatitis virus post-transcriptional regulatory element (WPRE) and a 3’ self-inactivating (SIN) LTR sequence. b) Western blot demonstrating expression of full-length Dnaic1 protein. Cultures of 293 cells were untreated (negative control) or transduced with lentiviral vectors expressing EGFP (EGFP) or Dnaic1 (Dnaic1) from the CB promoter. Purified mouse cilia (cilia) isolated from tracheal cell cultures 32, 33 served as a positive control. Molecular size markers are shown (Std.).
Gene transfer to undifferentiated PCD cells restores ciliary activity
To test the hypothesis that exogenous expression of Dnaic1 could restore ciliary activity to Dnaic1 −/− (PCD) cells, mouse tracheal epithelial (MTE) cells from Dnaic1 flox/flox mice that also express CreER were cultured at an air/liquid interface (ALI) on porous inserts 18. Cells were treated with tamoxifen to activate CreER and induce the deletion of Dnaic1 and generate PCD cells as previously described 18. After the cells reached confluence, but before ciliated cell differentiation was visible (day 5 of culture), cultures were treated apically with Dnaic1-encoding HA-EIAV lentivirus and then monitored for the appearance of ciliary activity. As a negative control, cultures received either no vector or were transduced with a lentiviral vector expressing EGFP from the CB promoter. Each experiment also included cultures that were not treated with tamoxifen or cultures from heterozygous mice (Dnaic1 flox/wt ,CreER+ ) treated with tamoxifen as a positive control for ciliogenesis. As expected, the percentage of the culture surface area demonstrating active ciliary beating in the PCD cells and PCD cells transduced with the EGFP expressing control vector was extremely low, averaging <1% in 4 separate experiments (Supplemental Video S1). Transduction of PCD cells with a Dnaic1 expressing lentivirus resulted in a clearly visible increase in ciliary activity, seemingly in parallel with the development of ciliary activity in the positive control cultures. In four separate experiments, treatment of undifferentiated cultures of PCD cells with lentivirus expressing Dnaic1 increased the level of ciliary activity significantly over untreated PCD cultures. At the time when ciliary activity was maximal in the vector-treated cultures, the level of ciliary activity averaged ~10% of the positive control cultures, compared to ~0.4% for the PCD cultures (p= 0.04 by student's t-test, n=4; Figure 2a). Measurement of ciliary beat frequency (CBF) using the SAVA system demonstrated that there was no significant difference in CBF between the positive control cultures and the virally transduced cultures (heterozygote=18.4 Hz; experimental=18.2 Hz; p=0.42 by students t-test, n=9; Supplemental Video S2, S3). Figure 2b illustrates the typical time course of the development of ciliary activity in an experiment in which treatment with a lentiviral vector expressing Dnaic1 from the CB promoter increased ciliary activity from 0.1% to 8.5%. Ciliary surface activity in the positive control cultures typically reached 40-50%, depending on the culture and the time of measurement. Ciliary activity was maintained in the virally transduced cultures for the remainder of the experiment, usually 2-3 weeks, at which time ciliary activity in the positive control cultures was also diminishing due to the limited lifespan of primary cultures.
Figure 2.
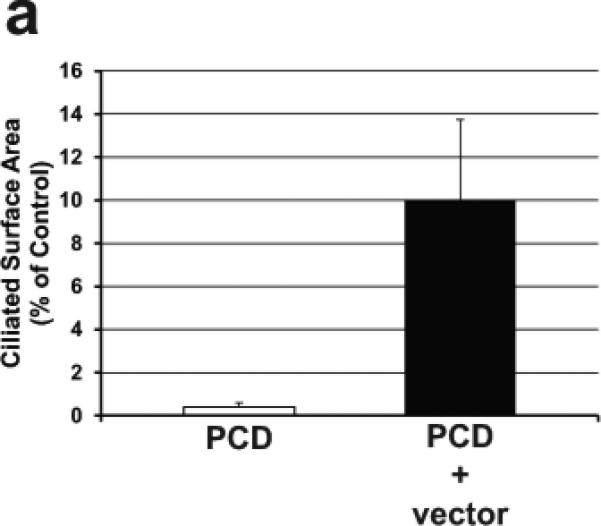
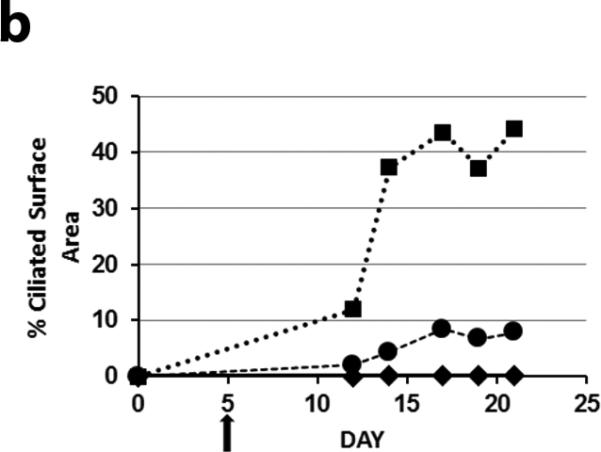
a) Ciliary activity as a percentage of surface area in PCD cultures and undifferentiated PCD cultures transduced with a lentiviral vector expressing Dnaic1. Data are expressed as the percentage of parallel positive control cultures and represents the average ± S.E.M. of 4 separate experiments. b) Representative experiment showing the increase in ciliary activity vs. time in culture of positive control cells (squares), PCD cells (diamonds), or in undifferentiated PCD cells transduced apically with the vector (circles). Vector was administered on day 5 (arrow).
At the end of these experiments, individual cultures were harvested for various additional analyses. Qualitative analysis of cellular DNA was performed by PCR amplification using primers specific for deleted exons of Dnaic1 followed by gel electrophoresis. Because the primers span the intron between exons 17 and 18, it is possible to distinguish products derived from the endogenous genomic Dnaic1 (333 bp) and products derived from the vector encoded cDNA (223 bp). These experiments confirmed the essentially complete deletion of the endogenous Dnaic1 in cultures treated with tamoxifen (as shown previously; Fig. 3a in ref. 18) and the presence of the integrated viral genome in cultures treated with vector (2/2 cultures; data not shown). Treatment of PCD cultures with Dnaic1 expressing lentivirus also resulted in easily detectable levels of Dnaic1 RNA. Quantitative RT-PCR using exon 17-18 specific primers demonstrated that virally transduced cultures expressed Dnaic1 RNA at levels between 7 and ~140-fold higher than the PCD cultures when analyzed 4 weeks after transduction (n=3; data not shown).
Figure 3.
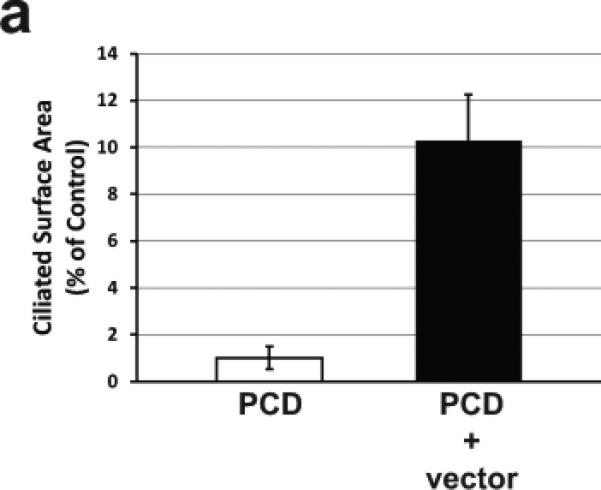
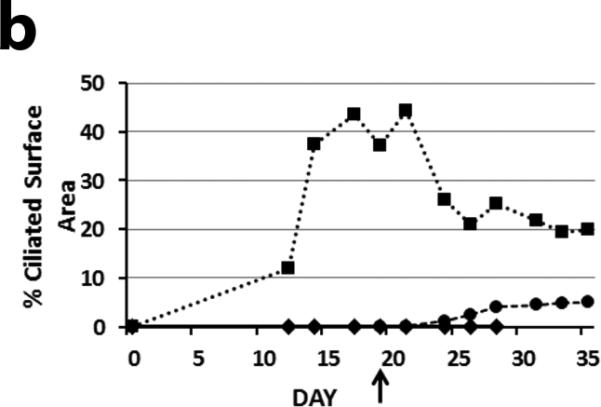
a) Ciliary activity as a percentage of surface area in PCD cultures and differentiated PCD cultures transduced apically with a lentiviral vector expressing Dnaic1. Data are expressed as the percentage of parallel positive control cultures and represents the average ± S.E.M. of 4 separate experiments. b) Same experiment as in 2b showing the increase in ciliary activity vs. time in culture of positive control cells (squares), PCD cells (diamonds) or in differentiated PCD cells transduced apically with the vector (circles). Vector was administered on day 19 (arrow).
Apical gene transfer to differentiated PCD cells restores ciliary activity
Gene transfer to differentiated airway epithelial cells from the apical surface has been shown to be a major obstacle to successful gene therapy of lung diseases, including cystic fibrosis. To determine if apical delivery of an HA-EIAV lentivirus encoding wild-type Dnaic1 to fully-differentiated PCD cells could restore ciliary activity, tamoxifen-treated MTE cells from Dnaic1 flox/flox /CreER+ animals were cultured at an ALI for 19 days. At this time, all positive control (non-PCD) cultures were well-differentiated and displayed easily visible ciliary activity covering 35-65% of the culture surface, while the PCD cultures had few or no actively beating ciliated cells. Apical treatment of the PCD cultures with Dnaic1-expressing lentiviral vectors again resulted in an increase in ciliary activity. The percentage of the surface area of the vector-treated cultures increased to levels as high as 8.8%, while the PCD cultures again averaged less than 1.0%. The average ciliary activity in treated cultures was 10-fold higher than that in the PCD cultures (6.1% vs 0.6%; p=0.02 by student's t-test, n=4). Compared to the positive control cultures that had an average of 56% of their surface covered by active ciliated cells, treatment with the lentiviral vector restored ~10% of the normal level of ciliary activity (Fig. 3a). Active ciliated cells appeared in the cultures 5-6 days after vector application (Fig. 3b), and increased in number over the next several days. RT-PCR analysis demonstrated full-length Dnaic1 RNA in differentiated PCD cultures treated with vector, and qRT-PCR with primers specific for exons 17-18 demonstrated levels several thousand-fold that observed in the PCD cultures (n=3; data not shown). The amount of intact Dnaic1 RNA in the cultures transduced after differentiation appeared greater than that found in cultures transduced early in culture ; this most likely reflects the increased cell turnover in the cultures transduced early due to the longer time interval between the viral treatment and the RNA isolation in the two groups. Similarly, Western blot analysis showed the levels of intact Dnaic1 protein to be higher in the virally transduced differentiated cultures than in the virally transduced undifferentiated cultures. Dnaic1 protein was expressed in the late cultures at almost 60% of the level in positive control cultures, while no full-length Dnaic1 protein was detectable in the cultures that were transduced at earlier time points (data not shown).
In contrast to the cultures treated with vector before differentiation, in which the appearance of ciliary activity roughly paralleled the development of ciliary activity in the positive controls, we noticed a delay of 5-6 days before ciliary activity was observed in the already differentiated PCD cultures following vector treatment (compare Figure 2b and 3b which are from the same experiment). One hypothesis for the delay in restoration of ciliary activity to the differentiated PCD cells is that the incorporation of the vector-expressed wild-type Dnaic1 protein into the already fully assembled ciliary axonemes may be rate-limiting. Very little is known about the time required for assembly or turnover of axonemal proteins in mammalian systems, and this may be an important consideration when designing or testing treatments for PCD. Therefore we took advantage of the inducibility of our system to study the turnover of the intact Dnaic1 protein in cultured tracheal epithelial cells. In these studies, MTE cells from Dnaic1 flox/flox /CreER+ mice were cultured at an ALI in the absence of tamoxifen until ciliated cell differentiation was well-established (14-21 days). The percentage of the culture surface area covered with actively beating cilia was measured and the cultures were then treated with tamoxifen (1 μM tamoxifen in media on day 0, 2, and 4) to induce the deletion of exons 17-18 from Dnaic1. Cultures were harvested at various times during treatment and the amount of intact Dnaic1 gene and the level of Dnaic1 protein were measured, along with the amount of ciliary activity remaining. As expected, treatment with tamoxifen rapidly induced the deletion of the floxed Dnaic1, decreasing the percentage of intact Dnaic1 to ~60% 48 hrs after tamoxifen addition and to < 25% after 5 days of tamoxifen treatment (Fig. 4). Similarly, the amount of intact Dnaic1 protein in the cellular (soluble) fraction of total cell lysates decreased rapidly over the first 5 days of treatment, while the level of deleted Dnaic1 protein increased (Fig. 4 and supplemental Fig. S1). In contrast, the level of intact Dnaic1 protein in the axonemal fraction (pellet) decreased at a slower rate, with over 50% of the untreated control level remaining 5 days after initiation of treatment and almost 30% remaining 14 days after initiation of treatment (Fig. 4 and supplemental Fig. S1). The high levels of intact Dnaic1 protein remaining in the axonemal fraction were reflected in even higher levels of ciliary activity, with over 90% of the activity of untreated control cultures maintained at 5 days, and >70% maintained at 14 days (Fig. 4). These results suggest that Dnaic1, and likely other ciliary proteins, are very stable once they are incorporated into the axonemal structure. Further, these results suggest that the delay in the restoration of ciliary activity in well-differentiated PCD cultures may be due to the relatively slow incorporation of the newly synthesized Dnaic1 protein into pre-existing axonemes.
Figure 4.
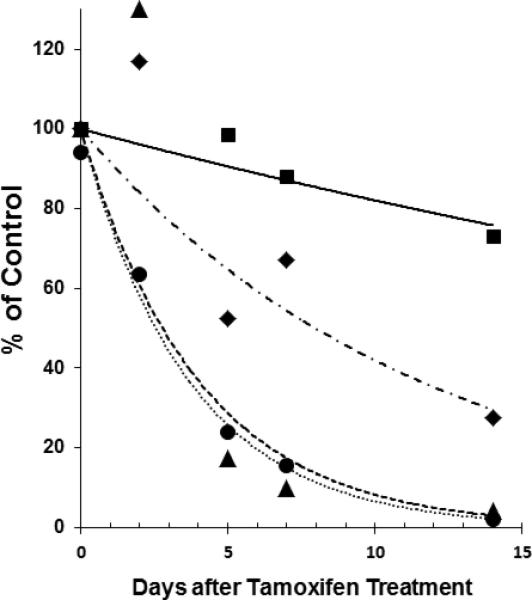
Turnover of Dnaic1 protein in ciliated cells. Differentiated cultures of Dnaic1 flox/flox CreER+− cells were treated with tamoxifen (day 0, 2, and 4) and the percentage of intact Dnaic1 DNA (circles), cytoplasmic protein (triangles), and axonemal protein (diamonds) were measured over time, along with the level of ciliary activity (squares). Measurements were taken before treatment (day 0) and at various times after treatment with tamoxifen was begun. Data shown are from a single experiment. See text for details.
The above studies clearly demonstrate that apical treatment with a lentiviral vector can restore ciliary activity to PCD cells in vitro, however the percentage of cells corrected was only 10-15% of the positive control cultures. To determine if this level of transfer might be sufficient to improve the outcome of PCD patients, we treated Dnaic1 flox/flox /CreER+ mice with different doses of tamoxifen to induce different levels of Dnaic1 gene deletion. We also included several Dnaic1 WT/flox / CreER+ animals that were not treated with tamoxifen, but because of low levels of leakiness (i.e. CreER activation and deletion of the target gene in the absence of tamoxifen treatment 18), also had varying levels of genomic Dnaic1 deletion. Mucociliary clearance was measured in the nasopharynx as previously described18. Following the measurement of MCC, the nasal cavity was fixed for histology and the level of intact genomic Dnaic1 remaining was determined in excised tracheal tissue by qPCR. As shown in Fig. 5a, the rate of mucociliary clearance was directly correlated with the level of intact genomic Dnaic1 remaining (R2 =0.7). As noted in our previous study 18, most animals with < 20% of intact genomic Dnaic1 remaining had essentially no mucociliary clearance, although 2 animals with 16% and 19% Dnaic1 had MCC rates of ~2 mm/min and 1 animal with 22% of the wild-type level of intact genomic Dnaic1 had a MCC rate of 3.8 mm/min. Examination of the nasal cavity of these animals revealed essentially normal histology in 2 while the third showed severe mucus accumulation and sinusitis (Fig. 5b). In contrast, all animals with lower levels of intact genomic Dnaic1 (<15%) had extensive mucus accumulation in the nasal cavity, in agreement with previous studies of these PCD mice. Together, these studies indicate that successful gene transfer of as little as 20% of the normal level of a ciliary gene may be able to restore a low level of MCC and significantly lessen the severity of disease.
Figure 5.

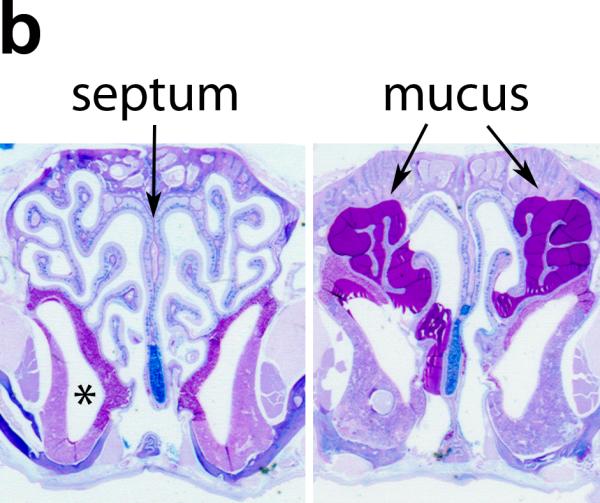
a) Relationship between level of intact genomic Dnaic1 and MCC. Most animals with >20% Dnaic1 remaining had MCC rates > 2 mm/min and no evidence of disease. b) Histology of the nasal cavity of animals with 16% (left) and 19% (right) of the wild-type level of Dnaic1 showing the absence (left) or presence (right; arrow) of rhinosinusitis. The maxillary sinus (*) and nasal septum (arrow) are indicated (left). Sections were stained with alcian blue-periodic acid Schiff.
To determine if the HA-pseudotyped lentiviral vector could efficiently transduce PCD airway epithelium in vivo, we administered vector to groups of PCD and control animals by nasal inhalation. To measure both the level of gene transduction and the stability of expression, we first utilized a vector expressing both firefly luciferase and β-gal from a single CB promoter construct 16. Four days after vector administration, animals were anesthetized and luciferase activity was measured in the nasal cavity as described in methods. Clear evidence of gene transfer to the control animals was observed, with an average luciferase activity ~8-fold higher than the untreated (no virus) negative control animal (1.6 × 106 ± 2.9 × 105 photons/sec/cm2 (avg. ± S.E.M., n=6) vs. 2.1 × 105 ± 8.0 × 104 photons/sec/cm2 (avg. ± S.E.M. of 3 repeat measures); Fig. 6). In contrast, only one of the PCD animals exhibited a substantial level of luciferase activity (1.0 × 106photons/sec/cm2), with the PCD animals together averaging 4.6 × 105 ± 1.5x 105 photons/sec/cm2 (avg ± S.E.M., n=5; significantly different from controls with p< 0.005 by student's t-test) The level of luciferase activity was increased in the control animals at 7 and 12 days after treatment, while the level of activity in the PCD animals, although increased, remained significantly lower than the controls. At day 7, the control animals averaged 1.3 x107 ± 2 × 106 compared to 1.7 × 106 ± 8.4 × 105 for the PCD animals; at day 12 the control animals averaged 2.4 × 107 ± 5.9 × 106 while the PCD animals averaged 2.2 × 106 ± 1.2 × 106 (significantly different at both days with p< 0.007). After one month, the level of luciferase activity declined in both groups, and was no longer significantly different between them, although the average activity remained higher in the controls (Fig. 6). To examine the cellular distribution of viral mediated gene transfer, animals were euthanized and tissues were processed for detection of β-gal. Sections were prepared and β-gal positive cells were enumerated in the nasal cavity and trachea. In agreement with the luciferase data, there were significantly fewer β-gal positive cells in the nasal cavity of the PCD mice compared to the control animals (2.2 ± 2 vs 116 ± 54, mean ± S.E.M.in 3 sections/ animal; n=5; p=0.03 by student's t-test). In contrast, there was no significant difference in the numbers of β-gal positive mice in sections of the tracheas from PCD mice compared to the control animals (174 ± 107 vs 30 ±24; n=5; p>0.11 by student's t-test), indicating that the inhibition of gene transfer was limited to the site of disease pathology, i.e., the nasal cavity. To further examine the distribution of virally transduced cells in the PCD animals compared to controls, additional groups of PCD and control animals were treated with an HA-pseudotyped lentivirus expressing only β-gal. Animals were again administered vector by nasal inhalation, but were euthanized and examined for positive gene transfer about the time of maximum luciferase expression (Fig. 6; 12 days after treatment). In confirmation of our earlier study 16, control animals displayed a high level of gene transfer to the ciliated epithelium of the nasal cavity. β-gal positive cells were easily identified along the septum, nasal turbinates, and throughout the nasopharynx, while olfactory tissue was routinely negative (Fig. 7a, c). PCD animals displayed little or no gene transfer to the nasal epithelium (Fig. 7b, d). While control animals averaged over 500 β-gal positive cells, PCD animals had significantly fewer positive cells (532 ± 42; n=7 vs. 62 ± 31; n=9; p=1.3 x10-7 by student's t-test). In contrast, transfer to the trachea was again similar between the two groups (control=285 ± 31, PCD =298 ± 39; p> 0.4 by student's t-test, Fig 7e, f, g, h). These data clearly demonstrate that the accumulated mucus and/or inflammation in the nasal cavity of the PCD animals prevent efficient gene transfer.
Figure 6.
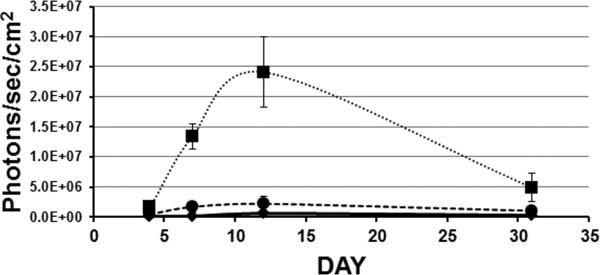
Time course of luciferase activity in the nasal cavity of control (squares; n=6) and PCD (circles; n=5) mice transduced with a lentiviral vector. Data = average ± S.E.M. Untreated (no virus) negative control (diamonds) exhibited <1 × 106 photons/sec/cm2 at all time points.
Figure 7.

Gene transfer in the nasal cavity (a, b, c, d) and trachea (e, f, g, h) of control (a, c, e, g) and PCD (b, d, f, h) mice. HA-pseudotyped lentivirus expressing β-gal was administered to mice by nasal inhalation and 12 days later tissues were stained for the expression of β-gal and paraffin sections were evaluated. Both the nasal cavity and the trachea exhibited positive β-gal staining in the control mice. In PCD animals the level of gene transfer in the nasal cavity was greatly reduced (note the mucus (*) in b, d; compare with Figure 5), while gene transfer to the trachea was not impeded. Nasal sections (a, b, c, d) are from ~level 23 36. Arrowheads pointing at the nasal septum in a, b, indicate the location of the higher magnification images in c, d. Sections were counter-stained with neutral fast red. Scale bars =50 μm in c, d, e, f, g, h.
Discussion
Primary ciliary dyskinesia is a genetically heterogeneous disease that occurs with an incidence of approximately 1:16,000, and is therefore classified as a “rare” or “orphan” disease. Currently there are no specific treatments available for PCD, and individuals with PCD suffer from repeated respiratory tract infections eventually leading to bronchiectasis 2, 5. Our laboratories have been investigating the development of gene replacement therapy as a treatment for PCD and other respiratory diseases, including cystic fibrosis. To investigate the pathogenesis and treatment of PCD, we have previously developed an inducible mouse model of PCD that does not develop hydrocephalus or developmental (heart) defects 18. In this work, we have utilized this model to investigate several aspects of gene therapy as a treatment for PCD.
Our initial studies demonstrated that expression of the wild-type Dnaic1 from a lentiviral vector successfully restored ciliary activity to cultures of undifferentiated PCD cells. Measurements of ciliary beat frequency were not significantly different between the virally transduced cultures and the controls, suggesting that expression of the wild-type Dnaic1 was sufficient to restore normal ciliary activity to the PCD cells. This is consistent with an earlier study in which a VSV-g pseudotyped lentiviral vector was used to express DNAI1 in human PCD cells 28. While it may be expected that expression of the wild-type protein will also lead to the correct orientation and regulation of ciliary activity required for efficient mucociliary clearance, further studies are required to test this hypothesis.
Importantly, for delivery of a gene transfer vector to the airways by inhalation, the vector must be able to transduce the airway epithelium via the apical surface. We have recently reported that influenza HA-pseudotyped vectors can mediate efficient gene transfer to murine airways 16. In this study, we found that apical administration of influenza HA-pseudotyped vector expressing Dnaic1 to fully differentiated cultures of PCD cells restored ciliary activity to a similar level as did treatment of undifferentiated cells. In cultures that were examined at the end of the experiment, we observed higher levels of Dnaic1 RNA and protein in cultures that were transduced late in culture compared to those transduced early. Although we think it is likely this difference is due to the longer time period between transduction and analysis allowing for greater turnover (loss) of the transduced cells in the cultures treated early, other possibilities need to be considered. For example, it is possible that in the late stage cultures the PCR analysis was detecting residual vector RNA. In both conditions, the maximum level of correction achieved was approximately 10%; it is unclear at this time what the rate-limiting step for correction is. Interestingly, the appearance of ciliary activity in apically transduced differentiated cultures required several days, whereas expression of a reporter protein (e.g., EGFP) is typically observed in <72 hrs (e.g.,15). This observation prompted us to examine the stability the Dnaic1 protein by taking advantage of the inducible nature of our model. Unlike Chlamydomonas, an organism that rapidly sheds and regrows its flagella 29, there is little known about the turnover of respiratory ciliary proteins. Our studies indicated that although the Cre-mediated deletion of Dnaic1 and the decline in the level of cytoplasmic Dnaic1 protein occurred rapidly after tamoxifen treatment, the level of axonemal Dnaic1 protein and ciliary activity declined at a much slower rate. These studies demonstrate that once ciliary proteins are assembled into axonemal structures they exhibit an increased stability. These results also suggest that the process of Dnaic1 assembly into outer dynein arms (ODA) and the incorporation of sufficient ODA into the existing ciliary axonemes to restore motility requires several days. Alternatively, it is possible that the vector is transducing undifferentiated or preciliated cells that are then undergoing ciliated cell differentiation. In our previous study, 80% of the cells transduced by the HA-pseudotyped vector were identified as ciliated cells, making this seem less likely 15. Additional studies using a vector expressing both a reporter and the Dnaic1 protein are necessary to distinguish between these two possibilities.
Although the studies reported here demonstrate that ciliary activity can be restored to individual PCD cells by gene transfer, it is not known what level of gene transfer will be necessary to prevent or reduce the disease symptoms caused by PCD. Efficient mucociliary clearance in the airways requires the continuous transport of mucus, from the bronchioles to the larynx, and so the distribution as well as the number of cells transduced will be important determinants of success. To investigate the level of gene transfer required, we again took advantage of the inducible nature of our model system to generate individual animals with varying levels of Dnaic1 deletion. Measurement of MCC rates in the nasopharynx and examination of nasal histology showed that animals with ~ 20% of the wild-type level of intact genomic Dnaic1 remaining had some MCC remaining and most were free of obvious disease. Although this number (20%) is clearly an approximation, we have successfully transduced up to 40% of the surface epithelial cells in the trachea and large airways of mice using HA-pseudotyped lentiviral vector 16, suggesting that improvements in vector design may make gene therapy for PCD possible.
An additional barrier to gene transfer to the airways of adult individuals with PCD, cystic fibrosis, or other diseases is the existence of disease-associated pathology, including the presence of excess mucus and/or the presence of inflammatory cells and mediators that may reduce or prevent gene transfer. Mice with a deletion of Dnaic1 invariably develop rhinosinusitis with an accumulation of mucus and inflammatory cells in the nasal cavity and sinuses. Administration of an HA-pseudotyped vector to groups of control and PCD mice by nasal inhalation revealed a profound inhibition of gene transfer to the nasal cavity due to the presence of rhinosinusitis. In contrast, gene transfer to the trachea and lower airways of PCD mice was not significantly affected, demonstrating that the vector was able to successfully transduce regions of the airways lacking the excess mucus and inflammation present in the nasal cavity. However, additional strategies will be required for successful gene transfer to the airways of individuals with pre-existing disease. These may include pre-treatment to reduce the level of mucus accumulation and/or inflammation prior to administration of the vector. Alternatively, administration of vector to affected individuals early in the course of the disease, perhaps prenataly 30, may allow successful gene transfer before the development of severe symptoms.
In summary, the results presented here demonstrate that restoration of ciliary activity to PCD cells by gene transfer to the apical surface of airway epithelial cells is possible using an HA-pseudotyped lentiviral vector, and that successful transfer of ~20% of the normal endogenous level of the mutated gene may be sufficient to prevent or mitigate the consequences of disease. Using an inducible mouse model of PCD that allows studies of adult animals, these studies have also demonstrated a significant inhibition of gene transfer to the nasal cavity as a result of pre-existing rhinosinusitis.
Methods
Generation of animals
The generation and breeding of the Dnaic1flox/flox mice expressing CreER from the ROSA promoter has been described 18. These mice are maintained on a mixed background (C57Bl6/129) and when treated with tamoxifen provide a long-lived animal model of PCD. All procedures using animals were approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill, and were conducted in accordance with policies for the ethical treatment of animals established by the National Institutes of Health.
Cloning of Dnaic1
The cloning of mouse Dnaic1 into the lentiviral vector was performed using standard molecular biology procedures and reagents. RNA was isolated from cultured mouse tracheal epithelial cells and a full-length cDNA for mouse Dnaic1 was amplified using PfuUltra polymerase (Stratagene, Santa Clara, CA) and primers ACCaagcttGCAATGCCCTCGAAACAG (sense) and TTTtctagaCTGGGCCATACTCTCAGGTTTT (anti-sense). The product was cloned into the TOPO vector (TOPO-TA cloning kit; Invitrogen) and sequenced. A clone with the correct sequence was used to amplify the Dnaic1 cDNA using Fidelity Fast Start polymerase (Roche Diagnostics, Indianapolis, IN) with primers incorporating Sal1 and Not1 restriction sites (AATTgtcgaccgGCAATGCCCTCGAAACAGat; TTTgcggccgCTGGGCCATACTCTCAGGTTTT). The product was digested, ligated into the lentiviral vector SIN6.1CB-W, sequenced, and used for production of vector.
Production of Lentiviral vectors
Recombinant EIAV-based lentiviral vector stocks were generated by transient transfection of 293T-based TAB22 cells as previously described 16, 31. Briefly, lentiviral plasmids were transfected into 293T cells using the standard calcium phosphate method. After a 48 h incubation cell supernatant containing lentiviral vectors was harvested, clarified by low speed centrifugation (100 g, 10 min), and filtered through 0.2 μm PES membranes. Vector was concentrated by high speed centrifugation at 5000 g (Beckman Avanti J-E centrifuge, JS-5.3 rotor) for 21 h at 8°C. Vector pellets were suspended in vector formulation buffer (5 mM Hepes (pH 7.4), 37 mM NaCl and 40 mM lactose) and stored at −80°C. Recombinant lentiviral vector stocks were titered on 293T cells by serial dilution as previously described 16. Titers for these experiments ranged from 6-10 × 107 infectious units per ml (IFU).
Western blotting of Dnaic1
Detection of Dnaic1 protein by Western blotting was performed using a mouse monoclonal antibody generated against a synthetic peptide from the human DNAI1 protein 18 using standard procedures. For analysis of mouse tracheal epithelial cells, total cell lysates were prepared in Mammalian Protein Extraction Reagent (Pierce, Rockford, IL). Ciliary axonemes were isolated from cultured mouse tracheal epithelial cells as previously described 32 and prepared in gel-loading buffer. For studies of protein turnover, both the soluble (cytoplasmic) and pellet (axonemal) fractions from individual cultures were analyzed. Protein samples were fractionated on 4 to 12% Bis-Tris gradient gels (Invitrogen, Carlsbad, CA), transferred to nitrocellulose membranes, and probed using Amersham ECL Plus reagents (GE Healthcare, Buckinghamshire, UK), all according to manufacturer's instructions. For normalization of loading, Western blots of cytoplasmic extracts were reprobed with an antibody to GAPDH (Sigma, St. Louis, MO), and axonemal pellets were reprobed with mouse anti-acetylated α-tubulin (Invitrogen). Quantification of signals was performed on an Odyssey Imaging System (Licor, Lincoln, NB).
Culture, tamoxifen treatment, and transduction of MTE cells
Procedures for the isolation and culture of mouse tracheal epithelial (MTE) cells have been described in detail previously 18, 33. To generate PCD cells for gene transfer studies, MTE cells from Dnaic1flox/flox; CreER+/− mice were seeded on collagen coated Millicell Cell Culture Inserts (12 mm; 0.4 μm, Millipore) and treated with 1 μM tamoxifen for the first 5-7 days of culture. As we have observed no difference between heterozygous and wild-type cultures, mice from both genotypes were used as controls. Lentiviral vectors with a titer of 6-10 × 107 infectious units (IFU) per ml (33 μL) were applied to the apical surface of washed cultures (day 5 or 19) and allowed to incubate for 3-4 hours before the supernatant was removed. Cultures were washed and refed 48 hrs after infection.
For Dnaic1 turnover studies, differentiated cultures of MTE cells from Dnaic1flox/flox;CreER±mice were cultured in the absence of tamoxifen until ciliated cell differentiation was apparent (day 14-21). Experimental cultures were then treated with 1 μM tamoxifen in media on day 0, 2, and 4, while positive control cultures received only media. Ciliary activity was measured and parallel cultures were harvested for DNA, RNA, and protein analysis. Four separate experiments were performed with similar results, although not all data were collected from each experiment.
Measurement of ciliary activity
The extent of ciliary activity and measurements of ciliary beat frequency (CBF) were performed essentially as previously described 18 using a Nikon Eclipse TE2000 microscope and the SAVA software package 34. Briefly, for measurements of the percentage of active ciliated surface area, nine different low-magnification fields (one central, eight peripheral) from each insert were measured. Typically, 2-3 inserts were measured and averaged at each time point. Because the absolute level of ciliogenesis varied between cultures, data is also expressed as the percentage of positive control cultures. For comparison between vector-treated and untreated cultures, the data shown in Figures 2a and 3a is taken from the time-point when vector-treated cultures showed maximal ciliary activity as a percentage of positive control. For determining CBF, positive control (heterozygous) cultures and PCD cultures treated with vector on day 5 were washed free of mucus and debris, equilibrated at 37° C, and 9 random fields were analyzed on day 25 (20 days after vector treatment).
Measurement of Dnaic1 RNA and DNA
RNA and DNA were isolated from MTE cell cultures or tail snips using RNeasy or DNeasy reagents and kits (Qiagen Inc., Valencia, CA) according to the manufactures protocols. DNA and RNA concentrations were determined by spectrophotometry. Qualitative PCR analysis of samples was performed by using primers (TGCTCCAAATCCTACTCCAG and AACACAGTGGAAGAGTACGG) that are internal to the deleted region of Dnaic1 to amplify cellular DNA followed by gel electrophoresis using standard procedures. The primers produce a 323 bp product with intact genomic DNA, no product from the deleted genomic DNA (tamoxifen-treated), and a 223 bp product from the vector encoded cDNA. Quantitative PCR to determine the amount of intact Dnaic1 in various samples was performed using LightCycler SYBR Green reagents (Roche Applied Science, Mannheim, Germany) and a Roche LightCycler using the same primers as previously published18. Each sample was analyzed in duplicate. Relative changes in the level of Dnaic1 were calculated from the efficiency of the PCR reaction and the cross-point deviation between samples. For experiments in which the level of Dnaic1 was being quantified, signals were normalized to the single-copy reference gene β-ENaC (Scnn1b)18; for the estimation of levels of Dnaic1 mRNA, signals were normalized to the level of the ciliated cell-specific gene dynein axonemal heavy chain 5 (Dnahc5) using primers GTCTGGATGGGCGGCATGACTA and ATCCTGTAGCCCCTCGAGCTCA.
Tamoxifen treatment of mice
To generate PCD mice, Dnaic1flox/flox; CreER+− mice, 6-9 weeks old, were treated with tamoxifen (five intraperitoneal injections of 75 μg/g body weight18). For the experiment shown in Fig. 5, animals were treated with 2 or 3 injections of tamoxifen to generate mice with varying levels of intact genomic Dnaic1 remaining. Additional Dnaic1flox/wt; CreER+− mice with varying levels of intact genomic Dnaic1 (due to the spontaneous activation of CreER) and Dnaic1wt/wt mice were included in the study. After allowing sufficient time for ciliated cell turnover (>6 mos; 18), mucociliary clearance was measured in the nasopharynx, the nasal cavity was fixed in formalin for histology, and the percentage of intact genomic Dnaic1 remaining was determined in tracheal tissue.
Measurement of MCC
Measurement of mucociliary clearance in the nasopharynx was performed as previously described in detail 18. In this procedure, the transport of endogenous particles is recorded with a video camera after exposing the nasopharynx by dissection immediately after euthanizing the animal. The time required for the particles to move a calibrated distance (0.5-1 mm) is measured and the MCC rate reported for each animal is the average speed of 5-10 particles.
Treatment of Mice with lentiviral vectors
Lentiviral vectors were administered to Dnaic1−/−; CreER+− (PCD) and control mice by nasal inhalation 16. Control animals included both Dnaic1+− and Dnaic1+/+ littermates; both were treated with tamoxifen. Mice were anesthetized with isoflurane and the concentrated vector solution (7 × 108 IFU) was administered dropwise to the tip of the nose in a small volume (20-25 μl), where it was spontaneously inhaled into the nasal cavity.
Quantification of luciferase
For live animal imaging of nasal luciferase expression 16, mice were anesthetized with isoflurane and D-luciferin (Gold Biotechnology, St Louis, MO, USA) substrate (20 μl of 25mg/ml D-luciferin in PBS) was delivered by intranasal administration 35. Mice were imaged using the IVIS Lumina optical imaging system (Caliper Life Sciences, Hopkinton,MA, USA) and images of luciferase activity were collected using a charge-coupled device camera interfaced to a computer. Data is presented as the average ± S.E.M.in photons/sec/cm2.
Quantification of β-galactosidase
Tissue from the lungs, trachea and nasal cavity of treated animals were prepared and stained with X-Gal Solution as previously described 16. For quantification, sections were scanned at high resolution (2400 dpi) and stained (blue) cells were counted. To quantify transduction in the nasal cavity, multiple sections (3 or 4) at different levels of the nasal cavity were counted for each animal and the total number of stained cells were compared. In the first experiment, sections were prepared from levels 9, 11, and 23; in the second experiment, sections were prepared from levels 5, 9, 11, and 23, based on the system of Mery et al. 36. For quantification in the trachea, a single section the length of the trachea was evaluated; both sides of the longitudinal section through the trachea were counted. Data is reported as mean ± S.E.M.
Statistical Analyses
Results between PCD and vector-treated groups were compared using a student's t-test with a p-value of ≤ 0.05 accepted as significant. In experiments where more than two groups were being compared, an ANOVA was first performed to determine significance. For the data shown in Figure 5a, the coefficient of determination (R2) was determined by linear regression.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. P. Sears for technical assistance with the recording and analysis of ciliary activity for this project. This research was funded by NIH grant 1RC1HL10081 to L.E.O.. Research Support was also provided by Oxford BioMedica Ltd to J.C.O.
Footnotes
Conflict of interest
Dr. J.C. Olsen is a named inventor on patents involving EIAV-based gene transfer technology and has received royalties for the use of EIAV-based vectors. All other authors declare no potential conflict of interest.
Supplementary information
Supplementary information is available at Gene Therapy's website.
References
- 1.Bush A, Chodhari R, Collins N, Copeland F, Hall P, Harcourt J, et al. Primary ciliary dyskinesia: current state of the art. Archives of disease in childhood. 2007;92(12):1136–40. doi: 10.1136/adc.2006.096958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leigh MW, Pittman JE, Carson JL, Ferkol TW, Dell SD, Davis SD, et al. Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genet Med. 2009;11(7):473–87. doi: 10.1097/GIM.0b013e3181a53562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kennedy MP, Omran H, Leigh MW, Dell S, Morgan L, Molina PL, et al. Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation. 2007;115(22):2814–21. doi: 10.1161/CIRCULATIONAHA.106.649038. [DOI] [PubMed] [Google Scholar]
- 4.Nakhleh N, Francis R, Giese RA, Tian X, Li Y, Zariwala MA, et al. High prevalence of respiratory ciliary dysfunction in congenital heart disease patients with heterotaxy. Circulation. 2012;125(18):2232–42. doi: 10.1161/CIRCULATIONAHA.111.079780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knowles MR, Daniels LA, Davis SD, Zariwala MA, Leigh MW. Primary ciliary dyskinesia. Recent advances in diagnostics, genetics, and characterization of clinical disease. Am J Respir Crit Care Med. 2013;188(8):913–22. doi: 10.1164/rccm.201301-0059CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stillwell PC, Wartchow EP, Sagel SD. Primary Ciliary Dyskinesia in Children: A Review for Pediatricians, Allergists, and Pediatric Pulmonologists. Pediatr Allergy Immunol Pulmonol. 2011;24(4):191–196. doi: 10.1089/ped.2011.0099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zariwala MA, Omran H, Ferkol TW. The emerging genetics of primary ciliary dyskinesia. Proc.Am.Thorac.Soc. 2011;8(5):430–3. doi: 10.1513/pats.201103-023SD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griesenbach U, Alton EW. Cystic fibrosis gene therapy: successes, failures and hopes for the future. Expert Rev Respir Med. 2009;3(4):363–71. doi: 10.1586/ers.09.25. [DOI] [PubMed] [Google Scholar]
- 9.Parsons DW. Airway gene therapy and cystic fibrosis. J Paediatr Child Health. 2005;41(3):94–6. doi: 10.1111/j.1440-1754.2005.00556.x. [DOI] [PubMed] [Google Scholar]
- 10.Pickles RJ. Physical and biological barriers to viral vector-mediated delivery of genes to the airway epithelium. Proc Am Thorac Soc. 2004;1(4):302–8. doi: 10.1513/pats.200403-024MS. [DOI] [PubMed] [Google Scholar]
- 11.Buckley SM, Howe SJ, Sheard V, Ward NJ, Coutelle C, Thrasher AJ, et al. Lentiviral transduction of the murine lung provides efficient pseudotype and developmental stage-dependent cell-specific transgene expression. Gene Ther. 2008;15(16):1167–75. doi: 10.1038/gt.2008.74. [DOI] [PubMed] [Google Scholar]
- 12.Medina MF, Kobinger GP, Rux J, Gasmi M, Looney DJ, Bates P, et al. Lentiviral vectors pseudotyped with minimal filovirus envelopes increased gene transfer in murine lung. Mol Ther. 2003;8(5):777–89. doi: 10.1016/j.ymthe.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 13.Mitomo K, Griesenbach U, Inoue M, Somerton L, Meng C, Akiba E, et al. Toward gene therapy for cystic fibrosis using a lentivirus pseudotyped with Sendai virus envelopes. Mol Ther. 2010;18(6):1173–82. doi: 10.1038/mt.2010.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sinn PL, Burnight ER, Hickey MA, Blissard GW, McCray PB., Jr Persistent gene expression in mouse nasal epithelia following feline immunodeficiency virus-based vector gene transfer. J Virol. 2005;79(20):12818–27. doi: 10.1128/JVI.79.20.12818-12827.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McKay T, Patel M, Pickles RJ, Johnson LG, Olsen JC. Influenza M2 envelope protein augments avian influenza hemagglutinin pseudotyping of lentiviral vectors. Gene Ther. 2006;13(8):715–24. doi: 10.1038/sj.gt.3302715. [DOI] [PubMed] [Google Scholar]
- 16.Patel M, Giddings AM, Sechelski J, Olsen JC. High efficiency gene transfer to airways of mice using influenza hemagglutinin pseudotyped lentiviral vectors. J Gene Med. 2013;15(1):51–62. doi: 10.1002/jgm.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stocker AG, Kremer KL, Koldej R, Miller DS, Anson DS, Parsons DW. Single-dose lentiviral gene transfer for lifetime airway gene expression. J Gene Med. 2009;11(10):861–7. doi: 10.1002/jgm.1368. [DOI] [PubMed] [Google Scholar]
- 18.Ostrowski LE, Yin W, Rogers TD, Busalacchi KB, Chua M, O'Neal WK, et al. Conditional deletion of dnaic1 in a murine model of primary ciliary dyskinesia causes chronic rhinosinusitis. Am J Respir Cell Mol Biol. 2010;43(1):55–63. doi: 10.1165/rcmb.2009-0118OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guichard C, Harricane MC, Lafitte JJ, Godard P, Zaegel M, Tack V, et al. Axonemal dynein intermediate-chain gene (DNAI1) mutations result in situs inversus and primary ciliary dyskinesia (Kartagener syndrome). Am J Hum Genet. 2001;68(4):1030–5. doi: 10.1086/319511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pennarun G, Escudier E, Chapelin C, Bridoux AM, Cacheux V, Roger G, et al. Loss-of-function mutations in a human gene related to Chlamydomonas reinhardtii dynein IC78 result in primary ciliary dyskinesia. Am J Hum Genet. 1999;65(6):1508–19. doi: 10.1086/302683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zariwala M, Noone PG, Sannuti A, Minnix S, Zhou Z, Leigh MW, et al. Germline mutations in an intermediate chain dynein cause primary ciliary dyskinesia. American Journal of Respiratory Cell and Molecular Biology. 2001;25(5):577–83. doi: 10.1165/ajrcmb.25.5.4619. [DOI] [PubMed] [Google Scholar]
- 22.Zariwala MA, Leigh MW, Ceppa F, Kennedy MP, Noone PG, Carson JL, et al. Mutations of DNAI1 in primary ciliary dyskinesia: evidence of founder effect in a common mutation. Am J Respir Crit Care Med. 2006;174(8):858–66. doi: 10.1164/rccm.200603-370OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilkerson CG, King SM, Koutoulis A, Pazour GJ, Witman GB. The 78,000 M(r) intermediate chain of Chlamydomonas outer arm dynein is a WD-repeat protein required for arm assembly. J Cell Biol. 1995;129(1):169–78. doi: 10.1083/jcb.129.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee L. Riding the wave of ependymal cilia: Genetic susceptibility to hydrocephalus in primary ciliary dyskinesia. J Neurosci Res. 2013 doi: 10.1002/jnr.23238. [DOI] [PubMed] [Google Scholar]
- 25.Vogel P, Read RW, Hansen GM, Payne BJ, Small D, Sands AT, et al. Congenital hydrocephalus in genetically engineered mice. Vet Pathol. 2012;49(1):166–81. doi: 10.1177/0300985811415708. [DOI] [PubMed] [Google Scholar]
- 26.Francis RJ, Christopher A, Devine WA, Ostrowski L, Lo C. Congenital heart disease and the specification of left-right asymmetry. Am J Physiol Heart Circ Physiol. 2012;302(10):H2102–11. doi: 10.1152/ajpheart.01118.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tan SY, Rosenthal J, Zhao XQ, Francis RJ, Chatterjee B, Sabol SL, et al. Heterotaxy and complex structural heart defects in a mutant mouse model of primary ciliary dyskinesia. J Clin Invest. 2007;117(12):3742–52. doi: 10.1172/JCI33284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chhin B, Negre D, Merrot O, Pham J, Tourneur Y, Ressnikoff D, et al. Ciliary beating recovery in deficient human airway epithelial cells after lentivirus ex vivo gene therapy. PLoS Genet. 2009;5(3):e1000422. doi: 10.1371/journal.pgen.1000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lefebvre PA, Nordstrom SA, Moulder JE, Rosenbaum JL. Flagellar elongation and shortening in Chlamydomonas. IV. Effects of flagellar detachment, regeneration, and resorption on the induction of flagellar protein synthesis. J Cell Biol. 1978;78(1):8–27. doi: 10.1083/jcb.78.1.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.David AL, Peebles D. Gene therapy for the fetus: is there a future? Best Pract Res Clin Obstet Gynaecol. 2008;22(1):203–18. doi: 10.1016/j.bpobgyn.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 31.Johnson LG, Mewshaw JP, Ni H, Friedmann T, Boucher RC, Olsen JC. Effect of host modification and age on airway epithelial gene transfer mediated by a murine leukemia virus-derived vector. J. Virol. 1998;72:8861–72. doi: 10.1128/jvi.72.11.8861-8872.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ostrowski LE, Blackburn K, Radde KM, Moyer MB, Schlatzer DM, Moseley A, et al. A proteomic analysis of human cilia: identification of novel components. Mol Cell Proteomics. 2002;1(6):451–65. doi: 10.1074/mcp.m200037-mcp200. [DOI] [PubMed] [Google Scholar]
- 33.You Y, Richer EJ, Huang T, Brody SL. Growth and differentiation of mouse tracheal epithelial cells: selection of a proliferative population. Am J Physiol Lung Cell Mol Physiol. 2002;283(6):L1315–21. doi: 10.1152/ajplung.00169.2002. [DOI] [PubMed] [Google Scholar]
- 34.Sisson JH, Stoner JA, Ammons BA, Wyatt TA. All-digital image capture and whole-field analysis of ciliary beat frequency. J Microsc. 2003;211(Pt 2):103–11. doi: 10.1046/j.1365-2818.2003.01209.x. [DOI] [PubMed] [Google Scholar]
- 35.Buckley SM, Howe SJ, Rahim AA, Buning H, McIntosh J, Wong SP, et al. Luciferin detection after intranasal vector delivery is improved by intranasal rather than intraperitoneal luciferin administration. Hum Gene Ther. 2008;19(10):1050–6. doi: 10.1089/hum.2008.023. [DOI] [PubMed] [Google Scholar]
- 36.Mery S, Gross EA, Joyner DR, Godo M, Morgan KT. Nasal diagrams: a tool for recording the distribution of nasal lesions in rats and mice. Toxicol Pathol. 1994;22(4):353–72. doi: 10.1177/019262339402200402. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.