

Abstract
Endocannabinoids, including anandamide (AEA), have been implicated in neuroprotective on-demand responses. Related to such a response to injury, an excitotoxic kainic acid (KA) injection (i.p.) was found to increase AEA levels in the brain. To modulate the endocannabinoid response during events of excitotoxicity in vitro and in vivo, we utilized a new generation compound (AM5206) that selectively inhibits the AEA deactivating enzyme fatty acid amide hydrolase (FAAH). KA caused calpain-mediated spectrin breakdown, declines in synaptic markers, and disruption of neuronal integrity in cultured hippocampal slices. FAAH inhibition with AM5206 protected against the neurodegenerative cascade assessed in the slice model 24 h postinsult. In vivo, KA administration induced seizures and the same neurodegenerative events exhibited in vitro. When AM5206 was injected immediately after KA in rats, the seizure scores were markedly reduced as were levels of cytoskeletal damage and synaptic protein decline. The pre- and postsynaptic proteins were protected by the FAAH inhibitor to levels comparable to those found in healthy control brains. These data support the idea that endocannabinoids are released and converge on pro-survival pathways that prevent excitotoxic progression.
Keywords: AM5206, Excitotoxicity, Endocannabinoid system, Hippocampus, Neuroprotection
Introduction
The endogenous cannabinoid system is involved in a myriad of physiological processes, including nociception, memory, feeding behavior, lipid metabolism, and gastrointestinal motility (Rodriguez de Fonseca et al. 2005; Viveros et al. 2008; Kunos et al. 2009; Izzo and Sharkey 2010). The endocannabinoids anandamide (AEA) and 2-arachidonoyl glycerol (2-AG) have also been linked to on-demand responses that protect against seizure damage, stroke/ischemia, traumatic brain injury, and multiple sclerosis (see reviews, Bahr et al. 2006; Fernandez-Ruiz et al. 2010; Hwang et al. 2010; Sagar et al. 2010). The endocannabinoids may mediate their protective effects through several pathways linked to CB1 receptors, including triggering potassium channel opening, inhibition of calcium currents, and eliciting signaling element responses for neuronal maintenance (Deadwyler et al. 1995; Gomez del Pulgar et al. 2000; Galve-Roperh et al. 2002; Karanian et al. 2005b, 2007; Molina-Holgado et al. 2005). Correspondingly, activated CB1 receptors have been reported to reduce neurotransmitter release and in turn, suppress excitability and potential excitotoxic progression (Hajos et al. 2000; Kreitzer and Regehr 2001; Ohno-Shosaku et al. 2001; Wilson et al. 2001). From the many studies, promoting cannabinoid responses is being pursued as a protection strategy. Blocking endocannabinoid inactivation mechanisms, in particular, is an ideal target for neuroprotective modulation of cannabinergic signaling.
Enhanced cannabinoid signaling can be achieved by preventing AEA hydrolysis/inactivation by fatty acid amide hydrolase (FAAH). FAAH is highly expressed by neurons in the rat hippocampus, neocortex, and cerebellum (Egertova et al. 1998), and is responsible for degrading AEA after its release from neurons (Basavarajappa 2007;Di Marzo 2008). Studies with FAAH inhibitors and FAAH-knockout mice have described the outcome of elevated signaling through CB1 receptors (Cravatt et al. 2001; Kathuria et al. 2003; Karanian et al. 2005b, 2007). Targeted FAAH inhibition successfully enhances endocannabinoid signaling in the hippocampus, mediated predominantly via CB1 receptors (Deadwyler et al. 1993; Onaivi 2009). It has also been suggested in different animal models of epilepsy that high concentrations of CB1 receptors in the hippocampal formation reduce seizure activity by protecting neurons against excessive glutamatergic activity (Arida et al. 2005; Araujo et al. 2010). These findings suggest a key role played by endocannabinoids in excitotoxic protection.
FAAH inhibition is a potential avenue for the attenuation of neural overexcitation and excitotoxic progression (Cippitelli et al. 2008; Janero et al. 2009; Straiker and Mackie 2009). Several studies have documented the neuroprotective capabilities of FAAH inhibitors, showing reduced hippocampal excitability (Coomber et al. 2008) and seizure severity (Karanian et al. 2005b, 2007). FAAH inhibitors are particularly advantageous to promote CB1 signaling without the adverse psychotropic effects usually associated with direct activation of the CB1 receptor (Karanian et al. 2007; Janero et al. 2009). In the present study, we investigated the neuroprotective effects of the new generation, reversible FAAH inhibitor AM5206 in vitro and in vivo. Our results indicate that after excitotoxic insults, FAAH inhibition with AM5206 protects against neural compromise in hippocampal slices and reduces seizure activity and seizure-associated damage in rats.
Materials and Methods
Animals
Animal use protocols were approved by the Institutional Animal Care and Use Committees of the University of Connecticut and the University of North Carolina Pembroke. Groups of male rats for in vivo work and litters of rat pups (11 to 12 days postnatal) for hippocampal slice work were obtained from Charles River Laboratories (Wilmington, MA). The animals were housed according to guidelines from the National Institutes of Health and had food and water available ad libitum.
Chemicals and Antibodies
The reversible FAAH inhibitor was synthesized following a distinct reaction pathway to make the trifluoromethylketophenoxypentyl analog AM5206. Kainic acid (KA) was obtained from Tocris (Ellisville, MO). Affinity purification of antibodies to the calpain-mediated spectrin fragment BDPN and to α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptor subunit glutamate receptor 1 (GluR1, also referred to as GluA1 or GluR-A) was conducted using previously described methods (Bahr 1995;Bahr et al. 2002). Anti-synapsin II was purchased from CalbioChem (San Diego, CA) and anti-actin from Sigma (St. Louis, MO). All other reagents were from Sigma.
FAAH and MAGL Fluorometric Assays
Recombinant rat FAAH and recombinant hexa-histidine-tagged human monoacylglycerol lipase (MAGL) were expressed in E. coli as previously described (Patricelli et al. 1998; Ramarao et al. 2005; Zvonok et al. 2008). FAAH and MAGL activities were performed in 96-well plates using the fluorogenic substrates arachidonoyl 7-amino-4-methylcoumarin amide for FAAH, and arachidonoyl, 7-hydroxy-6-methoxy-4-methylcoumarin ester for MAGL. Fluorescence was recorded in 20-min intervals at emission wavelengths of 460 nm. Concentration-response curves across increasing concentrations of AM5206 (Graphpad Prism, San Diego, CA), and IC50 values were determined.
Endocannabinoid and AM5206 Detection
Sprague–Dawley rats were injected i.p. with phosphate-buffered saline (PBS), 9.5 mg/kg KA, or 60 mg/kg AM5206. After 2–3 h, the cortex and other tissues were rapidly dissected, flash-frozen and stored at −80°C. From the samples, protein content was precipitated with ice-cold acetone, followed by liquid–liquid phase extraction of endocannabinoids or AM5206 using methanol and chloroform. For endocannabinoid analysis, subsequent liquid chromatography-mass spectrometry analyses were conducted as previously described (Williams et al. 2007; Wood et al. 2008; 2010). Chromatographic separation was achieved using an Agilent Zorbax SB-CN column (2.1×50 mm) on a Finnigan TSQ Quantum Ultra triple quad mass spectrometer (Thermo Electron Corporation, Waltham, MA) with an Agilent 1100 HPLC on the front end (Agilent Technologies, Palo Alto, CA) in positive APCI mode. The mobile phase consisted of 10 mM ammonium acetate (pH 7.3) and methanol. AM5206 levels were assessed as eluted from a Phenomenex Gemini C18 column (2×50 mm, 5 μm pore size) with a C18 guard column, using 0.1% formic acid in water and 0.1% formic acid in methanol, ionized with negative electrospray ionization, and detected in selected reaction monitoring mode.
Organotypic Hippocampal Slice Cultures
Brain tissue from 11- to 12-day-old postnatal Sprague–Dawley rat pups was rapidly removed to prepare slices as previously described (Bahr 1995; Karanian et al. 2005b, 2007). Transverse slices of hippocampus (400 μm) were prepared and placed on Millicell-CM insert membranes (Millipore Corporation, Bedford, MA) with culture medium consisting of 50% basal medium Eagle, 25% Earle’s balanced salts, 25% horse serum, and defined supplements (Bahr et al. 1994). The slices were maintained in culture at 37°C in 5% CO2-enriched atmosphere for a 15–20-day maturation period before experimental use.
Model of Excitotoxicity in Slice Cultures
Cultured hippocampal slices were pretreated for 60 min with serum-free media plus 0.1% DMSO, in the absence of presence of 10–30 μM AM5206. The media was aspirated and the wells washed once with fresh medium. Slices were subsequently treated with vehicle or 60 μM KA for 2 h in the absence or presence of 10–30 μM of AM5206. The media was then removed, and the slices washed and incubated with or without AM5206 for 24 h. At that time, the slices were harvested for histology and immunoblotting.
Histology
Cultured hippocampal slices were fixed in formalin overnight at 4°C, incubated in PBS, and mounted onto Superfrost slides (Fisher Scientific, Pittsburgh, PA) for Nissl staining.
Immunoblot Analysis
Groups of six to eight hippocampal slices each were harvested with a soft brush in ice-cold harvest buffer composed of 0.32 M sucrose, 5 mM HEPES (pH 7.5), 1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM ethylene glycol tetraacetic acid (EGTA), and the protease inhibitors antipain, 4-(2-aminoethyl) benzenesulfonyl fluoride, pepstatin A, E-64, bestatin, leupeptin, and aprotinin (2 μg/ml each). The slices were sonicated in lysis buffer of 6 mM Tris (pH 8.1), 0.2 mM EDTA, 0.2 mM EGTA, and protease inhibitors. Protein content was determined in the homogenized samples with a bovine serum albumin standard. Aliquots (75 μg) of the protein samples were denatured in SDS at 100°C, separated by electrophoresis on 4–15% SDS-PAGE, and then blotted to nitrocellulose. Immunodetection used antibodies against calpain-mediated spectrin BDPN, synapsin II, the AMPA receptor subunit GluR1, and the protein load control actin. Anti-IgG-alkaline phosphatase conjugates were used for secondary antibody incubation. Development of immunoreactive species used the 5-bromo-4-chloro-3-indolyl phosphate and nitroblue tetrazolium substrate system and was terminated prior to maximum intensity in order to avoid saturation. Integrated optical density of the bands was determined with image analysis at high resolution with BIOQUANT software (R & M Biometrics; Nashville, TN). Mean integrated densities for various antigens are expressed as mean values ± SEM.
Lactate Dehydrogenase Assay
Lactate dehydrogenase (LDH) activity in the culture medium was measured using the Cytotox 96 Non-Radioactive Cyto-toxicity Kit (Promega, Madison, WI). Thirty microliters of media were transferred to wells of a 96-well plate in triplicate, mixed with assay reagent and allowed to react for 30 min. Stop solution was then added to each well and absorbances immediately recorded at 490 nm using a Molecular Devices Plate Reader (Sunnyvale, CA).
Model of Excitotoxicity in Vivo
Sprague–Dawley rats were injected i.p. with PBS or 9 mg/kg KA. Immediately following the KA injection, the rats were administered a subsequent injection (i.p.) with either vehicle or 8 mg/kg AM5206. The animals were then monitored and scored for seizure activity (see below) by observers blinded to the treatment groups. At 48 h postinjection, brains were rapidly removed with ice-cold buffer containing a protease inhibitor cocktail. Hippocampi were homogenized in lysis buffer with protease inhibitors. Protein content was determined and samples assessed for BDPN, GluR1, and synapsin II.
Seizure Scoring
For the excitotoxicity study, animals were observed for 4 h following injections. The seizure score was recorded every 15 min, representative of the seizure expression during that period. The seizure rating scale consisted of the following seven stages: stage 0, normal behavior; stage 1, freezing, staring, or mouth/facial movements; stage 2, rigid posture, head nodding or isolated twitches; stage 3, tail extension, unilateral–bilateral forelimb clonus, or repetitive scratching; stage 4, rearing with one or both forepaws extended; stage 5, clonic seizures with loss of posture, jumping, and falling; stage 6, severe tonic seizures.
Plasma Analysis
Trunk blood from rats treated with AM5206 was collected in heparinized tubes, centrifuged at 5,000 rpm for 5 min and the plasma drawn off for analyses. Alanine amino-transferase (ALT) activity was measured with Infinity ALT Liquid Stable reagent, and blood urea nitrogen (BUN) levels were assessed using Infinity Urea Liquid Stable reagent (Thermotrace, Melbourne, Australia).
Statistical Analyses
Data were analyzed using unpaired t tests or one-way ANOVA followed by the Tukey’s post hoc tests.
Results
We monitored the endocannabinoid response in rats injected with an excitotoxic level of KA (9.5 mg/kg). Mass spectrometry was used to test whether enhancement of brain AEA levels occurred in the KA-treated animals (Fig. 1b) as compared to control rats (Fig. 1a). A 2.7-fold increase in AEA was evident in the cortical samples from KA rats (n=6; Fig. 1c). No change in AEA was evident in plasma samples after the KA treatment (96±15% of vehicle-injected controls) nor in mesencephalic tissue (92±8% of control) or hindbrain (87±13%). KA also increased cortical 2-AG levels from 91,320±7,020 to 362,200±45,140 ng/g (mean ± SEM; P<0.0001), without any change in plasma 2-AG levels (28.1±1.6 ng/μl in vehicle rats; 26.2±2.7 ng/μl in KA rats). Thus, as previously reported, the on-demand cannabinergic response after an excitotoxic insult elevates endocannabinoid levels in the brain (Hansen et al. 2001; Marsicano et al. 2003; Karanian et al. 2007).
Fig. 1.

Exposure to the excitotoxin KA increases AEA levels in the brain. Vehicle (a) or 9.5 mg/kg KA (b) was systemically administered to rats. At the 2 h postinjection time, cortical tissue was rapidly dissected, flash-frozen in liquid nitrogen, and assayed for AEA levels by mass spectrometry as described in the “Materials and Methods” section. Time of elution (minutes) from the reverse phase column is represented by the X-axes and is noted for reference peaks and AEA (shaded). AEA levels (nanograms per gram of wet tissue weight) were determined using an AEA standard curve and means ± SEM are shown (c). Unpaired t test; triple asterisk, P<0.001
To test whether FAAH inhibition promotes the protective nature of the endocannabinoid system, we used the new generation FAAH inhibitor AM5206. In separate FAAH and MAGL assays, AM5206 caused a concentration-dependent inhibition of both activities, but more potently inhibited FAAH over MAGL (Fig. 2). Similar results were found across three separate experiments, with AM5206 being 280- to 480-fold more selective for FAAH (42±6 nM, IC50±SD) compared to MAGL (15,130±2,800 nM).
Fig. 2.

AM5206 selectively inhibits FAAH activity. Recombinant FAAH and MAGL were treated with varying concentrations of AM5206 in triplicate, and the two hydrolytic activities were determined using specific fluorogenic substrates. Data were normalized to 100% fluorescence activity in the absence of drug and are presented as means ± SEM
Next, we tested AM5206 for its neuroprotective ability by assessing the cellular and molecular events of excitotoxicity in the hippocampal slice model (Vanderklish and Bahr 2000; Bahr et al. 2002; Karanian et al. 2005b). Histological analysis of cellular integrity was performed by Nissl stain, showing that slice cultures possess native neuronal organization of hippocampal subfields (Fig. 3a). Compared to vehicle-treated slices (Fig. 3b), neurons of the KA-treated tissue showed pronounced pyknotic changes with a severe loss of pyramidal neurons (Fig. 3c). AM5206 protected against KA-induced neurodegeneration by preserving neuronal structure, as confirmed by an absence of pathogenic manifestations (Fig. 3d).
Fig. 3.

AM5206 affords neuronal protection in the hippocampus after KA-induced excitotoxicity in vitro. Organotypic hippocampal slice cultures were used, and a low-power photomicrograph shows their characteristic maintenance of native cellular organization (a). Nissl staining was also used to assess cellular integrity across treatment groups. CA3 pyramidal zones are shown from slices treated consistently with vehicle (b), those pretreated with vehicle before a 2-h exposure to 60 μM KA (c), and slices pretreated with 10 μM AM5206 be fore the KA insult (d). After a washout step, the cultured slices were then incubated with vehicle or AM5206 for 24 h before the tissue was fixed, sectioned, and stained. The KA insult resulted in neuronal loss and obvious pyknotic changes that were reduced by the FAAH inhibitor. DG, dentate gyrus; so, stratum oriens; sp, stratum pyramidale; sr, stratum radiatum. Scale bar: a, 400 μm; b–d, 45 μm
For further detection of excitotoxic neurodegeneration in the rat hippocampus, we immunoblotted the slice samples to assess BDPN, a marker of cytoskeletal breakdown, and for the synaptic proteins synapsin II and AMPA-R subunit glutamate receptor 1 (GluR1; also referred to as GluA1 or GluR-A). The excitotoxic insult caused the characteristic production of N-terminal spectrin breakdown product BDPN mediated by calpain and a corresponding loss of synaptic components (Fig. 4a). The neuroprotective potential of AM5206 is evidenced by the nearly complete elimination of BDPN 24 h postinsult (Fig. 4b). In addition, the postsynaptic marker GluR1 exhibited significant protection by the FAAH inhibitor (Fig. 4a, c), and similar results were found concerning the presynaptic synapsin II (see Fig. 4a). Assessment of LDH released from damaged neurons into the media also indicated significant cellular protection by AM5206 (Fig. 4d). The FAAH inhibitor treatment lowered KA-induced LDH levels by 60%.
Fig. 4.

Treatment with AM5206 elicits several indicators of neuroprotection in the hippocampal slice model. The slices were treated consistently with vehicle (veh), pretreated with vehicle before a 2-h KA exposure, or pretreated with AM5206 before the KA insult. After the KA exposure and a washout step, slices were returned to their prior condition of the absence or presence of AM5206. Slices were harvested 24 h postinsult in groups of six to eight and assessed by immunoblotting for calpain-mediated spectrin breakdown product BDPN, synaptic markers GluR1 and synapsin II (syn II), and protein load control actin (see blots in a). Integrated optical densities for BDPN (b) and GluR1 (c) across the treatment groups are compared as means ± SEM. Unpaired t test compared to KA+vehicle data, single asterisk P<0.05; double asterisk P<0.01. Treated slices (25–27 per condition) divided into three groups were also assessed for LDH activity released into the culture medium (d). LDH release is expressed as the colorimetric assay absorbance (mean ± SEM) corrected for background activity present in fresh medium. AM5206 effect: single asterisk, P=0.026
Extending the work from the in vitro slice model required assessment of AM5206 in the intact animal. To first confirm that AM5206 crosses the blood–brain barrier, a high level of AM5206 (60 mg/kg) was injected i.p. into rats and was subsequently detected in the cortex at a concentration of 2–6 ng per gram of brain tissue at 2 h postinjection. After 3 h, AM5206 was still detected in the cortex (0.25–0.73 ng/g) as well as in the hippocampus (0.39–1.3 ng/g). The average plasma level that AM5206 reached was 27.1±3.9 ng/μl(n=6 rats). As evidence that the FAAH inhibitor modulated the endocannabinoid system, the baseline AEA to 2-AG ratio (1.6±0.14) was increased 44% in the AM5206 animals to 2.3±0.15 (n=8; P<0.01).
To further evaluate neuroprotection in vivo, we tested whether AM5206 injected i.p. into rats provided protection against KA-induced seizures. For the work with theexcitotoxic rat model, the study included three treatment groups of animals: (1) those that received two vehicle i.p. injections, (2) those that received KA (9.5 mg/kg) followed by an immediate vehicle injection, or (3) those that received KA followed immediately by an injection of 8 mg/kg AM5206. Seizures were scored, and AM5206 demonstrated a 79% reduction in seizure severity compared to animals that only received vehicle after the KA treatment (ANOVA, P<0.0001) (Fig. 5a). To determine if a correspondence exists between seizure protection and molecular indicators of excitotoxicity, hippocampi were removed from the three treatment groups and immunoblotted against BDPN, GluR1, and synapsin II. The hippocampal samples from KA alone animals showed strong immunoreactivity for BDPN, and this was associated with a decline in GluR1 and synapsin II (Fig. 5b). Hippocampi from rats treated with KA and AM5206, however, showed reduced BDPN immunoreactivity and preserved staining for the synaptic markers (see Fig. 5b, c). Thus, systemically administered AM5206 attenuates seizure severity and elicits cytoskeletal and synaptic protection.
Fig. 5.

AM5206 affords seizure and neuronal protection after KA-induced excitotoxicity in vivo. Seizures were induced by i.p. injection of 9.5 mg/kg KA (n=12 rats), and following the KA administration animals were immediately injected with either vehicle or 8 mg/kg AM5206. Vehicle-treated control rats (veh, n=11) did not receive KA or AM5206. Seizures were scored by blinded raters over a 4-h period (a) and mean scores ± SEM are shown (ANOVA, P<0.0001). At 48 h postinjection, hippocampal tissue was rapidly dissected, homogenized, and equal protein aliquots assessed by immunoblot for BDPN, GluR1, synapsin II (syn II), and actin (b). Mean integrated optical densities for GluR1 (c) are shown (±SEM; ANOVA, P<0.001). Post hoc tests compared to KA+ vehicle data, single asterisk P<0.05; triple asterisk, P<0.001
Finally, AM5206 was evaluated to provide an initial safety assessment. Sprague–Dawley rats were injected i.p. daily for 14 days with 0–15 mg/kg AM5206, thus covering a dosage range above that used for neuroprotection. Across the 14-day period, no obvious behavioral abnormalities were observed, including seizure events. In the AM5206 rats, scores given on treatment days 12–14 remained at the typical low baseline level characteristic of vehicle-treated controls (Fig. 6a). Mean scores from the three treatment groups were not different than zero according to one-sample t tests. In plasma samples collected from the same animals, ALT activity (Fig. 6b) and BUN levels (Fig. 6c) were also unchanged by the repeated administration of AM5206. Injecting a hepatotoxin to induce organ toxicity, such positive-control rats indeed exhibited much elevated ALT activity (280±50 U/L) and BUN (130±14 mg/dl) levels in plasma samples. These initial safety results show that the FAAH inhibitor AM5206 does not cause adverse effects on behavior or major organ functions.
Fig. 6.
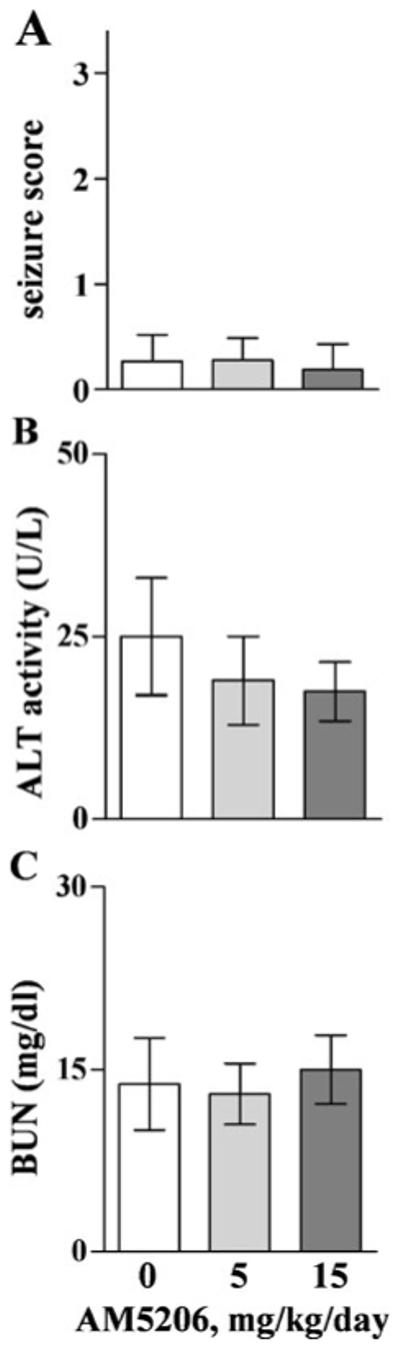
Repeated administrations of AM5206 were assessed for signs of toxicity in rats. Animals in groups of four were injected i.p. with vehicle (0 mg/kg) or 5–15 mg/kg AM5206 daily for 14 days. They were evaluated by seizure scorers during the 2-h postinjection periods on the last 3 days of treatment (a); overall mean scores ± SD are shown. Blood was subsequently collected for measures of ALT activity (b) and BUN levels (c) in plasma, which are plotted as means ± SEM
Discussion
Substantial evidence indicates that the endocannabinoid system plays a protective role against excitotoxic events linked to seizure activity and associated neurodegeneration. The proconvulsant KA was found to increase cortical AEA in the present study, adding to the evidence that endocannabinoid levels are elevated as part of internal repair responses to injury (Panikashvili et al. 2001; Maccarrone et al. 2003; Marsicano et al. 2003; Pavlopoulos et al. 2006). Blocking cannabinergic inactivation is an avenue to promote such responses, and the hydrolase FAAH is a primary mediator of breaking down released endocannabinoids (Tsou et al. 1999; Cravatt et al. 2001; Egertova et al. 2003; Kathuria et al. 2003; Morozov et al. 2004). Exploiting FAAH as a key regulatory site of endocannabinoid signaling, we have shown that the new generation, reversible FAAH inhibitor AM5206 modulates the endocannabinoid system and provides protection against excitotoxicity in vitro and in vivo. Protection was evident with respect to all parameters measured, including histological assessment, cytoskeletal damage, pre- and postsynaptic markers, LDH release, and seizure scores.
KA is a seizure-inducing excitotoxin, and we found that it elicits a pathogenic cascade in cultured hippocampal slices. The cascade includes calpain-mediated cytoskeletal damage, a typical concomitant loss of pre- and postsynaptic markers, abundant pyknotic neurons within a seizure-prone brain region and a corresponding reduction in neuronal density. Each element of the KA-induced cascade was attenuated by AM5206. The protection results were similar to those against AMPA-induced excitotoxicity in hippocampal slices produced by an irreversible FAAH inhibitor as well as by an endocannabinoid transport inhibitor (Karanian et al. 2005b). The organotypic tissue model, in conjunction with specific antibodies to synaptic and cytoskeletal markers, makes for a sensitive method to evaluate neuroprotection. Neuronal morphology and organized strata across subfields were stably maintained for months in culture. The hippocampal slice cultures are widely used to model various neuropathologies due to their expression of similar signaling, genetic, and cellular responses to pathogenic insults as found in vivo (Vornov et al. 1994; Bahr 1995; Caba and Bahr 2004; Bonde et al. 2005; Karanian et al. 2005b; Ryzhikov and Bahr 2008; Jourdi et al. 2009). The slice model of excitotoxicity is particularly valuable for characterizing neuroprotective modulators of the endocannabinoid system. The cytoskeletal and synaptic protection described here suggests that AM5206 protects against KA-induced neuronal atrophy by reducing calpain-mediated spectrin breakdown and preserving synaptic signaling.
The AM5206-mediated neuroprotection in the slice model translated to protective results in an in vivo model of excitotoxicity. KA is commonly used to induce seizures in order to understand neurodegenerative mechanisms (Wang et al. 2005). KA is known to elicit seizure damage characteristic of epilepsy, and the seizure scores exhibited in the KA-treated rats were significantly reduced by the FAAH inhibitor. Reduced seizure severity was evident across the seizure scoring period, providing evidence that AM5206 has an extended influence on excitotoxic progression. Pharmacologically controlled modulation of the endocannabinoid system through the safe targeting of FAAH is important since complete inhibition, considering FAAH-knockout mice for instance, was found to produce susceptibility to spastic and proconvulsant activity (Cravatt et al. 2001; Clement et al. 2003). Features of the excitotoxic cascade found in the slice model were present in vivo, and AM5206 also prevented the cytoskeletal damage and synaptic decline measured in the rat model. The ability of AM5206 to cross the blood–brain barrier and enhance the protective nature of the cannabinoid system without adverse effects demonstrates that endocannabinoid modulation is a potential avenue to treat excitotoxic injuries, including those stemming from seizure events.
Basal activity of the cannabinergic network in the brain plays an important part in the maintenance of neural connectivity and cell survival. Several studies have shown that blocking endocannabinoid signals causes synaptic disruption, increases excitotoxic vulnerability, and decreases survival responses (Parmentier-Batteur et al. 2002; Marsicano et al. 2003; Khaspekov et al. 2004; Karanian et al. 2005a). Correspondingly, enhancing endocannabinoid signaling leads to improved neuronal survival (Marsicano et al. 2003; Karanian et al. 2005b; Fernandez-Ruiz et al. 2010; Wolf et al. 2010). Much focus has been put on FAAH inhibition for an avenue to modulate levels of AEA as a major component of on-demand responses (Hwang et al. 2010). Selective inhibition of FAAH was achieved with the new generation inhibitor AM5206, which exhibits FAAH selectivity that approaches three orders of magnitude as compared to its action on MAGL. AM5206’s degree of protection against KA-induced seizures and excitotoxic damage is comparable to that produced by the irreversible FAAH inhibitor AM374 (Karanian et al. 2007). It is also comparable to the excitotoxic protection produced in vitro and in vivo by dual modulation of the endocannabinoid system (Karanian et al. 2005b), elicited by AM374 as well as through the inhibition of endocannabinoid transport that has individually resulted in protective effects (Lastres-Becker et al. 2002; Marsicano et al. 2003; Karanian et al. 2005b). Thus, selective FAAH inhibition may be an ideal strategy with which to protect against consequences ascribed to excitotoxic events, including seizures, stroke, and traumatic brain injury. Complete understanding of the effects of enhancing endocannabinoid signaling in the CNS and thorough safety evaluations for select FAAH inhibitors are important future goals.
Acknowledgments
The authors thank Robert Kwon, Bharti Patel, Ana Charalambides, Joanna Cooper, Sarah Barnes, and Professor José Manautou for their assistance on different parts of this project. The work was supported by the National Institutes of Health (grants DA07215, R44 DA023737, and R25 GM077634) and the Oliver Smithies Grant from the North Carolina Biotechnology Center (Research Triangle Park, NC). The funding agencies had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Vinogran Naidoo, Spyros P. Nikas, and David A. Karanian contributed equally to this work.
References
- Araujo BH, Torres LB, Cossa AC, Naffah-Mazzacoratti Mda G, Cavalheiro EA. Hippocampal expression and distribution of CB1 receptors in the Amazonian rodent Proechimys: an animal model of resistance to epilepsy. Brain Res. 2010;1335:35–40. doi: 10.1016/j.brainres.2010.03.031. [DOI] [PubMed] [Google Scholar]
- Arida RM, Scorza FA, de Amorim CR, Cavalheiro EA. Proechimys guyannensis: an animal model of resistance to epilepsy. Epilepsia. 2005;46(Suppl 5):189–197. doi: 10.1111/j.1528-1167.2005.01065.x. [DOI] [PubMed] [Google Scholar]
- Bahr BA. Long-term hippocampal slices: a model system for investigating synaptic mechanisms and pathologic processes. J Neurosci Res. 1995;42:294–305. doi: 10.1002/jnr.490420303. [DOI] [PubMed] [Google Scholar]
- Bahr BA, Abai B, Gall CM, Vanderklish PW, Hoffman KB, Lynch G. Induction of β-amyloid-containing polypeptides in hippocampus: evidence for a concomitant loss of synaptic proteins and interactions with an excitotoxin. Exp Neurol. 1994;129:81–94. doi: 10.1006/exnr.1994.1149. [DOI] [PubMed] [Google Scholar]
- Bahr BA, Bendiske J, Brown QB, Munirathinam S, Caba E, Rudin M, Urwyler S, Sauter A, Rogers G. Survival signaling and selective neuroprotection through glutamatergic transmission. Exp Neurol. 2002;174:37–47. doi: 10.1006/exnr.2001.7852. [DOI] [PubMed] [Google Scholar]
- Bahr BA, Karanian DA, Makanji SS, Makriyannis A. Targeting the endocannabinoid system in treating brain disorders. Expert Opin Investig Drugs. 2006;15:351–365. doi: 10.1517/13543784.15.4.351. [DOI] [PubMed] [Google Scholar]
- Basavarajappa BS. Critical enzymes involved in endocannabinoid metabolism. Protein Pept Lett. 2007;14:237–246. doi: 10.2174/092986607780090829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonde C, Noraberg J, Noer H, Zimmer J. Ionotropic glutamate receptors and glutamate transporters are involved in necrotic neuronal cell death induced by oxygen–glucose deprivation of hippocampal slice cultures. Neuroscience. 2005;136:779–794. doi: 10.1016/j.neuroscience.2005.07.020. [DOI] [PubMed] [Google Scholar]
- Caba E, Bahr BA. Biphasic NF-κB activation in the excitotoxic hippocampus. Acta Neuropathol. 2004;108:173–182. doi: 10.1007/s00401-004-0876-5. [DOI] [PubMed] [Google Scholar]
- Cippitelli A, Cannella N, Braconi S, Duranti A, Tontini A, Bilbao A, Defonseca FR, Piomelli D, Ciccocioppo R. Increase of brain endocannabinoid anandamide levels by FAAH inhibition and alcohol abuse behaviours in the rat. Psychopharmacology (Berl) 2008;198:449–460. doi: 10.1007/s00213-008-1104-0. [DOI] [PubMed] [Google Scholar]
- Clement AB, Hawkins EG, Lichtman AH, Cravatt BF. Increased seizure susceptibility and proconvulsant activity of anandamide in mice lacking fatty acid amide hydrolase. J Neurosci. 2003;23:3916–3923. doi: 10.1523/JNEUROSCI.23-09-03916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coomber B, O’Donoghue MF, Mason R. Inhibition of endocannabinoid metabolism attenuates enhanced hippocampal neuronal activity induced by kainic acid. Synapse. 2008;62:746–755. doi: 10.1002/syn.20547. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci USA. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deadwyler SA, Hampson RE, Bennett BA, Edwards TA, Mu J, Pacheco MA, Ward SJ, Childers SR. Cannabinoids modulate potassium current in cultured hippocampal neurons. Recept Channels. 1993;1:121–134. [PubMed] [Google Scholar]
- Deadwyler SA, Hampson RE, Mu J, Whyte A, Childers S. Cannabinoids modulate voltage sensitive potassium A-current in hippocampal neurons via a cAMP-dependent process. J Pharmacol Exp Ther. 1995;273:734–743. [PubMed] [Google Scholar]
- Di Marzo V. Endocannabinoids: synthesis and degradation. Rev Physiol Biochem Pharmacol. 2008;160:1–24. doi: 10.1007/112_0505. [DOI] [PubMed] [Google Scholar]
- Egertova M, Giang DK, Cravatt BF, Elphick MR. A new perspective on cannabinoid signalling: complementary localization of fatty acid amide hydrolase and the CB1 receptor in rat brain. Proc Biol Sci. 1998;265:2081–2085. doi: 10.1098/rspb.1998.0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egertova M, Cravatt BF, Elphick MR. Comparative analysis of fatty acid amide hydrolase and cb1 cannabinoid receptor expression in the mouse brain: evidence of a widespread role for fatty acid amide hydrolase in regulation of endocannabinoid signaling. Neuroscience. 2003;119:481–496. doi: 10.1016/s0306-4522(03)00145-3. [DOI] [PubMed] [Google Scholar]
- Fernandez-Ruiz J, Garcia C, Sagredo O, Gomez-Ruiz M, de Lago E. The endocannabinoid system as a target for the treatment of neuronal damage. Expert Opin Ther Targets. 2010;14:387–404. doi: 10.1517/14728221003709792. [DOI] [PubMed] [Google Scholar]
- Galve-Roperh I, Rueda D, Gomez del Pulgar T, Velasco G, Guzman M. Mechanism of extracellular signal-regulated kinase activation by the CB(1) cannabinoid receptor. Mol Pharmacol. 2002;62:1385–1392. doi: 10.1124/mol.62.6.1385. [DOI] [PubMed] [Google Scholar]
- Gomez del Pulgar T, Velasco G, Guzman M. The CB1 cannabinoid receptor is coupled to the activation of protein kinase B/Akt. Biochem J. 2000;347:369–373. doi: 10.1042/0264-6021:3470369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajos N, Katona I, Naiem SS, MacKie K, Ledent C, Mody I, Freund TF. Cannabinoids inhibit hippocampal GABAergic transmission and network oscillations. Eur J Neurosci. 2000;12:3239–3249. doi: 10.1046/j.1460-9568.2000.00217.x. [DOI] [PubMed] [Google Scholar]
- Hansen HH, Schmid PC, Bittigau P, Lastres-Becker I, Berrendero F, Manzanares J, Ikonomidou C, Schmid HH, Fernandez-Ruiz JJ, Hansen HS. Anandamide, but not 2-arachidonoylglycerol, accumulates during in vivo neurodegeneration. J Neurochem. 2001;78:1415–1427. doi: 10.1046/j.1471-4159.2001.00542.x. [DOI] [PubMed] [Google Scholar]
- Hwang J, Adamson C, Butler D, Janero DR, Makriyannis A, Bahr BA. Enhancement of endocannabinoid signaling by fatty acid amide hydrolase inhibition: a neuroprotective therapeutic modality. Life Sci. 2010;86:615–623. doi: 10.1016/j.lfs.2009.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izzo AA, Sharkey KA. Cannabinoids and the gut: new developments and emerging concepts. Pharmacol Ther. 2010;126:21–38. doi: 10.1016/j.pharmthera.2009.12.005. [DOI] [PubMed] [Google Scholar]
- Janero DR, Vadivel SK, Makriyannis A. Pharmacotherapeutic modulation of the endocannabinoid signalling system in psychiatric disorders: drug-discovery strategies. Int Rev Psychiatry. 2009;21:122–133. doi: 10.1080/09540260902782778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdi H, Hamo L, Oka T, Seegan A, Baudry M. BDNF mediates the neuroprotective effects of positive AMPA receptor modulators against MPP+-induced toxicity in cultured hippocampal and mesencephalic slices. Neuropharmacology. 2009;56:876–885. doi: 10.1016/j.neuropharm.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanian DA, Brown QB, Makriyannis A, Bahr BA. Blocking cannabinoid activation of FAK and ERK1/2 compromises synaptic integrity in hippocampus. Eur J Pharmacol. 2005a;508:47–56. doi: 10.1016/j.ejphar.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Karanian DA, Brown QB, Makriyannis A, Kosten TA, Bahr BA. Dual modulation of endocannabinoid transport and fatty acid amide hydrolase protects against excitotoxicity. J Neurosci. 2005b;25:7813–7820. doi: 10.1523/JNEUROSCI.2347-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanian DA, Karim SL, Wood JT, Williams JS, Lin S, Makriyannis A, Bahr BA. Endocannabinoid enhancement protects against kainic acid-induced seizures and associated brain damage. J Pharmacol Exp Ther. 2007;322:1059–1066. doi: 10.1124/jpet.107.120147. [DOI] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Khaspekov LG, Brenz Verca MS, Frumkina LE, Hermann H, Marsicano G, Lutz B. Involvement of brain-derived neurotrophic factor in cannabinoid receptor-dependent protection against excitotoxicity. Eur J Neurosci. 2004;19:1691–1698. doi: 10.1111/j.1460-9568.2004.03285.x. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- Kunos G, Osei-Hyiaman D, Batkai S, Sharkey KA, Makriyannis A. Should peripheral CB(1) cannabinoid receptors be selectively targeted for therapeutic gain? Trends Pharmacol Sci. 2009;30:1–7. doi: 10.1016/j.tips.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lastres-Becker I, Hansen HH, Berrendero F, De Miguel R, Perez-Rosado A, Manzanares J, Ramos JA, Fernandez-Ruiz J. Alleviation of motor hyperactivity and neurochemical deficits by endocannabinoid uptake inhibition in a rat model of Huntington’s disease. Synapse. 2002;44:23–35. doi: 10.1002/syn.10054. [DOI] [PubMed] [Google Scholar]
- Maccarrone M, Gubellini P, Bari M, Picconi B, Battista N, Centonze D, Bernardi G, Finazzi-Agro A, Calabresi P. Levodopa treatment reverses endocannabinoid system abnormalities in experimental Parkinsonism. J Neurochem. 2003;85:1018–1025. doi: 10.1046/j.1471-4159.2003.01759.x. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutierrez SO, van der Stelt M, Lopez-Rodriguez ML, Casanova E, Schutz G, Zieglgansberger W, Di Marzo V, Behl C, Lutz B. CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science. 2003;302:84–88. doi: 10.1126/science.1088208. [DOI] [PubMed] [Google Scholar]
- Molina-Holgado F, Pinteaux E, Heenan L, Moore JD, Rothwell NJ, Gibson RM. Neuroprotective effects of the synthetic cannabinoid HU-210 in primary cortical neurons are mediated by phosphatidylinositol 3-kinase/AKT signaling. Mol Cell Neurosci. 2005;28:189–194. doi: 10.1016/j.mcn.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Morozov YM, Ben-Ari Y, Freund TF. The spatial and temporal pattern of fatty acid amide hydrolase expression in rat hippocampus during postnatal development. Eur J Neurosci. 2004;20:459–466. doi: 10.1111/j.1460-9568.2004.03507.x. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabi-noids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron. 2001;29:729–738. doi: 10.1016/s0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Onaivi ES. Cannabinoid receptors in brain: pharmacogenetics, neuropharmacology, neurotoxicology, and potential therapeutic applications. Int Rev Neurobiol. 2009;88:335–369. doi: 10.1016/S0074-7742(09)88012-4. [DOI] [PubMed] [Google Scholar]
- Panikashvili D, Simeonidou C, Ben-Shabat S, Hanus L, Breuer A, Mechoulam R, Shohami E. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature. 2001;413:527–531. doi: 10.1038/35097089. [DOI] [PubMed] [Google Scholar]
- Parmentier-Batteur S, Jin K, Mao XO, Xie L, Greenberg DA. Increased severity of stroke in CB1 cannabinoid receptor knock-out mice. J Neurosci. 2002;22:9771–9775. doi: 10.1523/JNEUROSCI.22-22-09771.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patricelli MP, Lashuel HA, Giang DK, Kelly JW, Cravatt BF. Comparative characterization of a wild type and transmembrane domain-deleted fatty acid amide hydrolase: identification of the transmembrane domain as a site for oligomerization. Biochemistry. 1998;37:15177–15187. doi: 10.1021/bi981733n. [DOI] [PubMed] [Google Scholar]
- Pavlopoulos S, Thakur GA, Nikas SP, Makriyannis A. Cannabinoid receptors as therapeutic targets. Curr Pharm Res. 2006;12:1751–1769. doi: 10.2174/138161206776873743. [DOI] [PubMed] [Google Scholar]
- Ramarao MK, Murphy EA, Shen MW, Wang Y, Bushell KN, Huang N, Pan N, Williams C, Clark JD. A fluorescence-based assay for fatty acid amide hydrolase compatible with high-throughput screening. Anal Biochem. 2005;343:143–151. doi: 10.1016/j.ab.2005.04.032. [DOI] [PubMed] [Google Scholar]
- Rodriguez de Fonseca F, Del Arco I, Bermudez-Silva FJ, Bilbao A, Cippitelli A, Navarro M. The endocannabinoid system: physiology and pharmacology. Alcohol Alcohol. 2005;40:2–14. doi: 10.1093/alcalc/agh110. [DOI] [PubMed] [Google Scholar]
- Ryzhikov S, Bahr BA. Gephyrin alterations due to protein accumulation stress are reduced by the lysosomal modulator Z-Phe-Ala-diazomethylketone. J Mol Neurosci. 2008;34:131–139. doi: 10.1007/s12031-007-9009-7. [DOI] [PubMed] [Google Scholar]
- Sagar DR, Jhaveri MD, Richardson D, Gray RA, de Lago E, Fernandez-Ruiz J, Barrett DA, Kendall DA, Chapman V. Endocannabinoid regulation of spinal nociceptive processing in a model of neuropathic pain. Eur J Neurosci. 2010;31:1414–1422. doi: 10.1111/j.1460-9568.2010.07162.x. [DOI] [PubMed] [Google Scholar]
- Straiker A, Mackie K. Cannabinoid signaling in inhibitory autaptic hippocampal neurons. Neuroscience. 2009;163:190–201. doi: 10.1016/j.neuroscience.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou K, Mackie K, Sanudo-Pena MC, Walker JM. Cannabinoid CB1 receptors are localized primarily on cholecystokinin-containing GABAergic interneurons in the rat hippocampal formation. Neuroscience. 1999;93:969–975. doi: 10.1016/s0306-4522(99)00086-x. [DOI] [PubMed] [Google Scholar]
- Vanderklish PW, Bahr BA. The pathogenic activation of calpain: a marker and mediator of cellular toxicity and disease states. Int J Exp Pathol. 2000;81:323–339. doi: 10.1111/j.1365-2613.2000.00169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viveros MP, de Fonseca FR, Bermudez-Silva FJ, McPartland JM. Critical role of the endocannabinoid system in the regulation of food intake and energy metabolism, with phylogenetic, developmental, and pathophysiological implications. Endocr Metab Immune Disord Drug Targets. 2008;8:220–230. doi: 10.2174/187153008785700082. [DOI] [PubMed] [Google Scholar]
- Vornov JJ, Tasker RC, Coyle JT. Delayed protection by MK-801 and tetrodotoxin in a rat organotypic hippocampal culture model of ischemia. Stroke. 1994;25:457–464. doi: 10.1161/01.str.25.2.457. [DOI] [PubMed] [Google Scholar]
- Wang Q, Yu S, Simonyi A, Sun GY, Sun AY. Kainic acid-mediated excitotoxicity as a model for neurodegeneration. Mol Neurobiol. 2005;31:3–16. doi: 10.1385/MN:31:1-3:003. [DOI] [PubMed] [Google Scholar]
- Williams J, Wood J, Pandarinathan L, Karanian DA, Bahr BA, Vouros P, Makriyannis A. Quantitative method for the profiling of the endocannabinoid metabolome by LC-atmospheric pressure chemical ionization-MS. Anal Chem. 2007;79:5582–5593. doi: 10.1021/ac0624086. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Kunos G, Nicoll RA. Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron. 2001;31:453–462. doi: 10.1016/s0896-6273(01)00372-5. [DOI] [PubMed] [Google Scholar]
- Wolf SA, Bick-Sander A, Fabel K, Leal-Galicia P, Tauber S, Ramirez-Rodriguez G, Muller A, Melnik A, Waltinger TP, Ullrich O, Kempermann G. Cannabinoid receptor CB1 mediates baseline and activity-induced survival of new neurons in adult hippocampal neurogenesis. Cell Commun Signal. 2010;8:1–14. doi: 10.1186/1478-811X-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JT, Williams JS, Pandarinathan L, Courville A, Keplinger MR, Janero DR, Vouros P, Makriyannis A, Lammi-Keefe CJ. Comprehensive profiling of the human circulating endocannabinoid metabolome: clinical sampling and sample storage parameters. Clin Chem Lab Med. 2008;46:1289–1295. doi: 10.1515/CCLM.2008.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JT, Williams JS, Pandarinathan L, Janero DR, Lammi-Keefe CJ, Makriyannis A. Dietary docosahexaenoic acid supplementation alters select physiological endocannabinoid-system metabolites in brain and plasma. J Lipid Res. 2010;51:1416–1423. doi: 10.1194/jlr.M002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zvonok N, Williams J, Johnston M, Pandarinathan L, Janero DR, Li J, Krishnan SC, Makriyannis A. Full mass spectrometric characterization of human monoacylglycerol lipase generated by large-scale expression and single-step purification. J Proteome Res. 2008;7:2158–2164. doi: 10.1021/pr700839z. [DOI] [PMC free article] [PubMed] [Google Scholar]