

Abstract
Iron porphyrin carbenes (IPCs) are thought to be intermediates involved in the metabolism of various xenobiotics by cytochrome P450, as well as in chemical reactions catalyzed by metalloporphyrins and engineered P450s. While early work proposed IPCs to contain FeII, more recent work invokes a double bond description of the iron carbon bond, similar to that found in FeIV porphyrin oxenes. Here, we report the first quantum chemical investigation of IPC Mössbauer and NMR spectroscopic properties, as well as their electronic structures, together with comparisons to ferrous heme proteins and an FeIV oxene model. The results provide the first accurate predictions of the experimental spectroscopic observables as well as the first theoretical explanation of their electrophilic nature, as deduced from experiment. The preferred resonance structure is FeII←{:C(X)Y}0 and not FeIV={C(X)Y}2-, a result that will facilitate research on IPC reactivities in various chemical and biochemical systems.
Keywords: carbenes, catalysts, porphyrins, metalloenzymes, quantum chemistry
P450 cytochromes are a ubiquitous family of heme proteins that act as catalysts for numerous biochemical reactions with high valent ferryl species having been found to be important catalytic intermediates.[1] Biomimetic P450 metalloporphyrin models have also been found to be efficient catalysts for a broad range of organic reactions including C-H insertion, N-H insertion, cyclopropanation, as well as the olefination of aldehydes and ketones.[2] The active species responsible for such reactivities have been proposed to be metalloporphyrin carbene complexes. In particular, iron porphyrin carbene (IPC) complexes have been shown to undergo several of these reactions, including C-H insertions and cyclopropanations.[2f] IPC complexes were first observed several decades ago in the reactions of polyhalogenated methanes with porphyrins,[3] reactions similar to those observed in the metabolism by cytochrome P450 of various xenobiotics, including toxic polyhalogenated compounds as well as diverse drugs.[4] More recently, engineered cytochrome P450s have been used in synthetically important reactions not observed in nature,[2a] carbene transfers, with IPC complexes again being thought to be key catalytic intermediates.
Given the broad general interest in IPCs in chemistry and biochemistry, it is surprising that the electronic structures and associated reactivities of IPC complexes are poorly understood. For instance, whether IPCs are best described as FeII←{:C(X)Y}0 or FeIV={C(X)Y}2− (similar to FeIV=O2− in conventional P450 reactions), has not been resolved.[2f, 5] The presence of FeII was first proposed based on similarities with the UV-vis and NMR spectra of diamagnetic FeII porphyrins,[5c, d] and also used in an early extended Hückel theory study,[6] while in later work the presence of FeIV was proposed, based on similarities with the Mössbauer spectra of FeIV proteins and model systems.[2f, 5e] The double bond feature of the iron carbene bond in IPCs now seems generally accepted in the catalysis and biocatalysis areas,[2a-c, 2f, 2h, 5a] but has not, however, been investigated by ab initio quantum chemical methods. In addition, although IPCs were reported to effect electrophilic reactions via positive charge build-up on the substrates (Scheme 1),[2f, 2h, 5a, 7] there are no theoretical studies that explain this catalytic reactivity. Here, we use density functional theory (DFT) calculations to provide the first predictions of the Mössbauer and NMR spectroscopic observables of IPC complexes, as well as their geometries, and on the basis of these results together with a detailed investigation of molecular orbitals and charges, we determined the origins of their reactivity. In particular, the preferred resonance structure was found to be quite different to that currently used in experimental studies of IPCs.
Scheme 1.
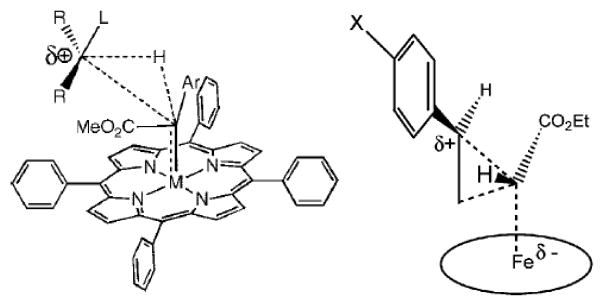
Experimentally proposed transition states in C-H insertion (left, Ref.2h) and cyclopropanation (right, Ref.5a) by IPCs.
We initially calculated the Mössbauer spectra of IPCs since the ability to predict spectroscopic observables can reasonably be expected to give some confidence in the quality of the calculations and hence, confidence in other computed properties. We first used X-ray structures[2f, 5c] to compute the Mössbauer spectra of “typical” FeIV and FeII complexes, 1 and 2, Figure 1. 1 contains an Fe=O bond and is the first FeIV complex with a reported X-ray structure,[8] while the heme site in carbomonoxymyogolobin (MbCO) is known to possess an FeII center.[9] We used the density functional theory (DFT) methods described previously. These enabled excellent predictions of the Mössbauer isomer shift (δFe) and quadrupole splitting (ΔEQ) values in >50 iron proteins and model systems encompassing the most common iron spin (S = 0, ½, 1, 3/2, 2, 5/2), oxidation (Fe0, FeII, FeIII, FeIV, FeVI), and coordination (2,3,4,5,6) states,[10] see Supporting Information for computational details of the methods used here.
Figure 1.

Molecular models used in this work for 1 [FeIV(O)(TMC)(NCCH3)]2+, TMC = 1,4,8,11-tetramethyl-1,4,8,11-tetraazacyclotetradecane; 2 MbCO active site; 3 [Fe(TPFPP)(CPh2)], TPFPP = meso-tetrakis(penta-fluorophenyl)porphyrinato dianion; 4 [Fe(TPFPP)(CPh2)(MeIm)], MeIm = N-methylimidazole; 5 [Fe(TPP)(C=C(C6H4Cl)2)], TPP = meso-tetraphenylporphyrinato dianion; 6 [Fe(TPP)(CCl2)]; 7 [Fe(TPP)(CCl(CH2OH))]; 8 [Fe(TPP)(CCl(CHMe2))]; 9 [Fe(TPP)(CCl(CHMe(OH))]; 10 [Fe(TPP)(CClCN)]; 11 [Fe(TPFPP)(C(Ph)CO2Et)]. Atom colors: N - blue, O - red, C - cyan, H - grey, Fe - black, Cl - green.
We first calculated the Mössbauer properties of 1 and 2 with all possible spin states and also evaluated the relative stability of other spin states with respect to the singlet state (ΔES=0). As shown in Table 1, the calculations correctly reproduced the experimental ground states of a triplet for 1 and a singlet for 2,[8, 9b] and only computed results obtained by using experimental spin states produced good predictions of both Mössbauer parameters, giving confidence in the methods used. We then investigated IPCs 3–5, Figure 1, intermediates in catalysis and the degradation of polyhalogenated compounds. All porphyrin meso-substituents were replaced with hydrogens to facilitate calculations. 3 is the only known IPC characterized by both X-ray crystallography and Mössbauer spectroscopy and the DFT results again reproduced the known singlet ground state, as well as providing good agreement between both of the experimental Mössbauer parameters and theory (Table 1). The predicted δFe/ΔEQ values of 0.19/−1.76 mm/s in 4 and 0.061/−1.94 mm/s in 5 were similar to those obtained with 3. So, overall, the Mössbauer isomer shifts in these IPC complexes are similar to the experimental results of ~0.09–0.22 mm/s in non-porphyrin iron carbene complexes.[11]
Table 1.
Mössbauer properties of FeIV, FeII and IPC complexes
| System | S | δFe(mm/s) | ΔEQ(mm/s) | ΔES=0(kcal/mol) | |
|---|---|---|---|---|---|
| [FeIV(O)(TMC)(NCCH3)]2+ | Expta) | 1 | 0.17 | 1.24 | |
| 1 | Calc | 0 | 0.11 | −3.22 | 0.00 |
| 1 | 0.13 | 1.25 | −28.74 | ||
| 2 | 0.097 | 0.53 | −13.28 | ||
| MbCO | Exptb) | 0 | 0.27 | 0.35 | |
| 2 | Calc | 0 | 0.29 | 0.27 | 0.00 |
| 1 | 0.38 | 0.33 | 30.99 | ||
| 2 | 0.48 | 2.87 | 58.40 | ||
| [Fe(TPFPP)(CPh2)] | Exptc) | 0 | 0.03 | (−)2.34 | |
| 3 | Calc | 0 | 0.10 | −2.37 | 0.00 |
| 1 | 0.053 | −2.97 | 20.61 | ||
| 2 | 0.056 | −2.90 | 69.16 |
In contrast to the rather small range of δFe values in IPCs, ΔEQ values cover a larger range since ΔEQ is related to the electric field gradient (EFG) tensor at the nucleus, which is more sensitive to the molecular environment than is δFe.[9b, 10d, 11] For instance, compared to the five-coordinate system 3, the six-coordinate complex 4 has a smaller absolute ΔEQ value, due to increased symmetry and thus, a reduced EFG. It is of interest to note that the δFe values in the IPCs are close to the experimental results of ~0.03–0.17 mm/s for FeIV species,[5e, 12] which was previously used to support the presence of an FeIV feature in IPCs[2f, 5e] since in general, δFe is a good indicator of iron oxidation state.[9b, 10c] However, exceptions were found due to strong ligand interactions,[13] and some non-porphyrin iron carbenes have been reported to be FeII systems.[11]
In contrast to δFe results (which show similarities to FeIV systems), the relative stability of the spin states for the five-coordinate IPC complex 3 are the same as the typical FeII complex, 2, and are quite different to those found in the typical FeIV complex 1, Table 1. The six-coordinate IPC complex 4 also exhibits the same trend with ΔEs=0 for the S=1 state being 17.70 kcal/mol higher, and for the S=2 state, 41.18 kcal/mol higher, indicating, again, similarities to FeII complexes.
To further investigate the FeII/FeIV bonding puzzle, we investigated the geometry optimized structures of IPCs. Since there are no prior reports of this kind of study, we evaluated a number of DFT methods including the commonly used hybrid functional B3LYP and another hybrid functional, mPW1PW91, as well as the more recently developed functionals M06, B97D, and ωB97XD, together with several basis sets (see Supporting Information for details). The key geometric parameters of interest are those around the carbene center: the iron carbene bond length RFeC, the average iron and porphyrin-nitrogen bond length RFeN, and the average length between the carbene carbon and its attached carbons, RCC. The best predictions are shown in Table 2 and have only a 0.012 Å mean absolute deviation (0.65% mean percentage deviation) for the singlet ground state. In contrast, results from using higher spin states (S=1 and 2) have significantly larger errors, with 0.069 and 0.133 Å mean absolute deviations, respectively. These data further support the energy results discussed above, and are consistent with the experimental NMR assignment of a diamagnetic ground state.[2f, 5c, d]
Table 2.
X-ray and computed geometric properties of IPCs
| System | S | RFeC(Å) | RFeN(Å) | RCC(Å) | |
|---|---|---|---|---|---|
| [Fe(TPFPP)(CPh2)(MeIm)] | Expta) | 0 | 1.827 | 1.973 | 1.495 |
| 4 | Calc | 0 | 1.828 | 2.002 | 1.478 |
| 1 | 2.018 | 1.993 | 1.458 | ||
| 2 | 2.048 | 2.093 | 1.458 | ||
| Fe(TFPP)(C(Ph)2) | Expta) | 0 | 1.767 | 1.967 | 1.483 |
| 3 | Calc | 0 | 1.764 | 1.991 | 1.478 |
| 1 | 1.969 | 1.980 | 1.454 | ||
| 2 | 2.030 | 2.112 | 1.453 | ||
| [Fe(TPP)(C=C(C6H4Cl)2)] | Exptb) | 0 | 1.690 | 1.985 | 1.487 |
| 5 | Calc | 0 | 1.675 | 1.992 | 1.484 |
| 1 | 1.751 | 2.050 | 1.485 | ||
| 2 | 2.058 | 2.000 | 1.489 |
We next investigated the 13C NMR shifts/shieldings in a series of IPCs in detail. As shown in Table 3, experimental solution 13C NMR chemical shifts of carbene carbons encompass a broad range, from 210 to 385 ppm downfield from TMS,[2c, 2f, 5d] which suggests that these NMR shifts may serve as sensitive probes of electronic structure. We used the geometry-optimized structures together with a series of DFT functionals and basis sets as well as the incorporation of solvent effects in the NMR chemical shielding calculations (see Supporting Information for computational details). The best results are shown in Table 3. As shown in Figure 2A, there is an excellent linear correlation between theory (σcalc) and experiment (δexpt) for the singlet state, with the correlation coefficient R2=0.982. The predicted shifts using this regression line (δpred’s) yielded a 6.71 ppm mean absolute deviation (2.34% mean percentage deviation), indicating that the broad range of 13C NMR chemical shifts in IPCs is well reproduced in our quantum chemical calculations. In contrast, results from using S=1 or S=2 states yielded ~9000 and ~30000 ppm mean absolute deviations, respectively, due to large hyperfine shifts, Table 3. These results provide strong additional evidence for a diamagnetic ground state in these IPC complexes, quite different to the paramagnetic states seen in FeIV species in heme/non-heme proteins and model systems. [1, 8, 14] In addition, there were essentially no differences between the theory-experiment correlations when solvent effects were included (R2 = 0.980) using the polarizable continuum method, see Table S6.
Table 3.
13C NMR chemical shifts/shieldings and charges
| δexpt a)(ppm) | σcalc (S=0)(ppm) | δpred (S=0)(ppm) | δpred (S=1)(ppm) | δpred (S=2)(ppm) | QC(e) | |
|---|---|---|---|---|---|---|
| 4 | 385.44 | −198.88 | 380.27 | 2857.51 | 15809.38 | 0.324 |
| 3 | 358.98 | −178.38 | 349.57 | 800.64 | 9408.93 | 0.326 |
| 11 | 327.47 | −163.06 | 326.61 | 1075.39 | 12579.69 | 0.229 |
| 8 | 324.00 | −165.80 | 330.73 | 22695.04 | 23522.84 | 0.275 |
| 9 | 312.00 | −155.75 | 315.66 | 21584.70 | 21712.35 | 0.241 |
| 7 | 302.70 | −155.77 | 315.69 | 21592.17 | 28689.45 | 0.200 |
| 6 | 224.70 | −97.29 | 228.08 | 3421.81 | 74530.39 | 0.053 |
| 10 | 210.00 | −77.57 | 198.53 | 1386.70 | 51894.04 | 0.056 |
Figure 2.
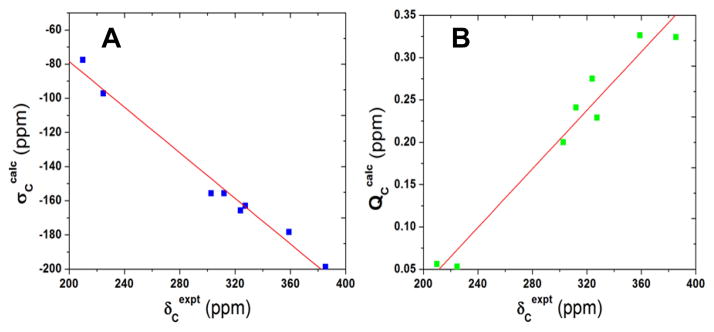
Plots of (A) computed 13C NMR chemical shieldings and (B) charges versus experimental 13C NMR chemical shifts in IPCs.
Given the success in predicting the spectroscopic observables, we next investigated the atomic charges of the carbene carbons in the IPC systems. As shown in Table 3, these charges (QC’s) cover a large range, ~0.3 e. The charges are all positive, with the charges in the known catalysts 3, 4, and 11 being particularly large, suggesting a physical basis for their electrophilic reactivity, as proposed experimentally (Scheme 1).[2f, 2h, 5a, 7] A good correlation between these charges and the experimental NMR chemical shifts was also found, as illustrated in Figure 2B (R2=0.958), suggesting that 13C NMR spectroscopy may be used as a probe of the reactivities of other IPC complexes.
The positive charges also suggest that the dominant feature in the metal-carbene bond involves carbene-to-metal donation. That is, carbenes with more electron-withdrawing substituents (e.g. CCl2 as opposed to CPh2) are associated with less positive charge (see Table 3), because electron-withdrawing substituents hinder the carbene’s electron donation ability. These results thus support the importance of a FeII←{:C(X)Y}0 resonance structure over FeIV={C(X)Y}2− since the latter is associated with dominant metal-to-carbene back-donation, and thus partial negative charges on the carbene carbon, inconsistent with the electrophilic reactivities seen experimentally.
To further investigate the electronic structures of IPCs we examined the molecular orbitals (MOs) of the three IPC complexes having reported X-ray structures (3–5) and compared the MO results obtained with those of typical FeIV and FeII systems (1,2). Although the IPCs have different structural features (five-coordinate, six-coordinate, as well as a vinylidene carbene), the electronic configurations of the frontier metal orbitals (FMOs) are the same: (dxy)2(dxz)2(dyz)2(dz2)0(dx2–y2)0, as illustrated for 3 in the left-hand column of Figure 3. This is consistent with a d6 FeII configuration, basically as found in the typical low spin FeII system 2 reported previously[10c] and other non-catalyst IPC models.[6] In contrast, as seen in the right-hand column in Figure 3 for the typical FeIV system 1 (in the S=0 state, to be more readily compared with the diamagnetic IPCs), the FMOs are (dxy)2(dxz)2(dyz)0(dx2–y2)0(dz2)0, consistent with the expected d4 FeIV configuration. This has the same energy order as the FMOs in the triplet ground state: (dxy)2(dxz)1(dyz)1(dx2–y2)0(dz2)0 reported previously,[15] and is clearly different to that seen in the IPC complexes. It is also interesting to note that compared to the two Fe dπ and O pπ interactions in the ground state of the FeIV complex 1,[15] there is only one Fe dπ and C pπ interaction in the ground state of IPC complex 3, as shown in Figure 3. The overlap-weighted natural atomic orbital bond order for Fe-O in 1, 0.797, is ca. 50% larger than that for Fe-C in 3, 0.548, which is further evidence for the difference between Fe-O bonding in the FeIV system and Fe-C bonding in the IPCs.
Figure 3.

Frontier metal MO diagrams of IPC complex 3 (left) and 1 (right) with S=0.
To further evaluate the possibility of the FeIV state in IPC complexes, calculations of 3 with a deliberate initial setup of FeIV and (CPh2)2− and even with a different occupied Fe 3d orbital sequence (dxy > dxz/dyz) were performed. After convergence all systems yielded the same results as shown in Figure 3, additional evidence that the FeIV state is not stable in these IPC complexes.
So, the MO results together with the charges, spin state trends and NMR shifts described above are all very similar to those found in FeII systems, rather than those seen in FeIV species. The predominant resonance structure is thus FeII←{:C(X)Y}0, quite different to the double-bond picture (FeIV={C(X)Y}2-, analogous to FeIV=O2− ) used in much current literature,[2a–c, 2f, 2h, 5a] but is consistent with earlier spectroscopic suggestions[5c, d] and their electrophilic reactivities.[2f, 2h, 5a, 7] The less favorable FeIV state in IPC complexes as compared to FeIV=O2− intermediates is, however, reasonable, since carbon has a lower electronegativity than oxygen.
The results described above are of broad general interest since they represent the first successful predictions of the Mössbauer spectra (δFe, ΔEQ), the 13C NMR chemical shifts/shieldings as well as the local geometries of IPC complexes. They also provide the first theoretical basis for the origin of their electrophilic catalytic nature and also indicate that 13C NMR spectroscopy may be a useful probe of IPC reactivity. Unlike the frequently used picture of a double bond between iron and carbon[2a-c, 2f, 2h, 5a] as being analogous to the catalytic intermediates in conventional P450 reactions (FeIV=O2-), IPC complexes are best described as involving FeII←{:C(X)Y}0, a result that should facilitate future studies of IPC systems in catalysis, and in bioinorganic chemistry.
Supplementary Material
Footnotes
This work was supported by NSF grant CHE-1300912 to YZ and NIH grants GM085774 toYZ and GM065307 to EO.
Supporting information for this article is available on the WWW under http://www.angewandte.org.
Contributor Information
Rahul L. Khade, Department of Chemistry, Chemical Biology, and Biomedical Engineering, Stevens Institute of Technology, Castle Point on Hudson, Hoboken NJ 07030 (USA), Fax: (+) 1-201-216-8240
Wenchao Fan, Department of Chemistry, Chemical Biology, and Biomedical Engineering, Stevens Institute of Technology, Castle Point on Hudson, Hoboken NJ 07030 (USA), Fax: (+) 1-201-216-8240.
Dr. Yan Ling, Department of Chemistry and Biochemistry, University of Southern Mississippi, 118 College Drive #5043, Hattiesburg, MS 39406 (USA)
Liu Yang, Department of Chemistry, Chemical Biology, and Biomedical Engineering, Stevens Institute of Technology, Castle Point on Hudson, Hoboken NJ 07030 (USA), Fax: (+) 1-201-216-8240.
Prof. Dr. Eric Oldfield, Department of Chemistry, University of Illinois at Urbana-Champaign, 600 South Matthews Avenue, Urbana, IL 61801 (USA)
Prof. Dr. Yong Zhang, Email: yong.zhang@stevens.edu, Department of Chemistry, Chemical Biology, and Biomedical Engineering, Stevens Institute of Technology, Castle Point on Hudson, Hoboken NJ 07030 (USA), Fax: (+) 1-201-216-8240
References
- 1.a) Meunier B, de Visser SP, Shaik S. Chem Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. [DOI] [PubMed] [Google Scholar]; b) Bernhardt R. J Biotech. 2006;124:128–145. doi: 10.1016/j.jbiotec.2006.01.026. [DOI] [PubMed] [Google Scholar]; c) de Visser SP, Kumar D, Cohen S, Shacham R, Shaik S. J Am Chem Soc. 2004;126:8362–8363. doi: 10.1021/ja048528h. [DOI] [PubMed] [Google Scholar]; d) Irigaray P, Belpomme D. Carcinogenesis. 2010;31:135–148. doi: 10.1093/carcin/bgp252. [DOI] [PubMed] [Google Scholar]
- 2.a) Coehlo PS, Brustad EM, Kannan A, Arnold FH. Science. 2013;339:307–310. doi: 10.1126/science.1231434. [DOI] [PubMed] [Google Scholar]; b) Che CM, Lo VKY, Zhou CY, Huang JS. Chem Soc Rev. 2011;40:1950–1975. doi: 10.1039/c0cs00142b. [DOI] [PubMed] [Google Scholar]; c) Che CM, Zhou CY, Wong ELM. Top Organomet Chem. 2011;33:111–138. [Google Scholar]; d) Lu H, Dzik WI, Xu X, Wojtas L, de Bruin B, Zhang XP. J Am Chem Soc. 2011;133:8518–8521. doi: 10.1021/ja203434c. [DOI] [PubMed] [Google Scholar]; e) Chan KS, Lau CM, Yeung SK, Lai TH. Organometallics. 2007;26:1981–1985. [Google Scholar]; f) Li Y, Huang JS, Zhou ZY, Che CM, You XZ. J Am Chem Soc. 2002;124:13185–13193. doi: 10.1021/ja020391c. [DOI] [PubMed] [Google Scholar]; g) Thu HY, Tong GSM, Huang JS, Chan SLF, Deng QH, Che CM. Angew Chem-Int Edit. 2008;47:9747–9751. doi: 10.1002/anie.200803157. [DOI] [PubMed] [Google Scholar]; h) Mbuvi HA, Woo LK. Organometallics. 2008;27:637–645. [Google Scholar]; i) Wang JC, Xu ZJ, Guo Z, Deng QH, Zhou CY, Wan XL, Che CM. Chem Comm. 2012;48:4299–4301. doi: 10.1039/c2cc30441d. [DOI] [PubMed] [Google Scholar]; j) Li Y, Huang JS, Zhou ZY, Che CM. J Am Chem Soc. 2001;123:4843–4844. doi: 10.1021/ja003184q. [DOI] [PubMed] [Google Scholar]; k) Chen Y, Huang LY, Zhang XP. Org Lett. 2003;5:2493–2496. doi: 10.1021/ol0347444. [DOI] [PubMed] [Google Scholar]; l) Baumann LK, Mbuvi HM, Du G, Woo LK. Organometallics. 2007;26:3995–4002. [Google Scholar]
- 3.Mansuy D, Lange M, Chottard JC, Guerin P, Morliere P, Brault D, Rougee M. J Chem Soc, Chem Comm. 1977:648–649. [Google Scholar]
- 4.a) Simonneaux G, Le Maux P. Top Organomet Chem. 2006:83–122. [Google Scholar]; b) Tolando R, Ferrara R, Eldirdiri NI, Albores A, King LJ, Manno M. Xenobiotica. 1996;26:425–435. doi: 10.3109/00498259609046721. [DOI] [PubMed] [Google Scholar]; c) Mansuy D, Battioni JP, Chottard JC, Ullrich V. J Am Chem Soc. 1979;101:3971–3973. [Google Scholar]; d) Groves JT, Avaria-Neisser GE, Fish KM, Imachi M, Kuczkowski RL. J Am Chem Soc. 1986;108:3837–3838. [Google Scholar]; e) Lafite P, Dijols S, Zeldin DC, Dansette PM, Mansuy D. Arch Biochem Biophys. 2007;464:155–168. doi: 10.1016/j.abb.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Taxak N, Patel B, Bharatam PV. Inorg Chem. 2013;52:5097–5109. doi: 10.1021/ic400010d. [DOI] [PubMed] [Google Scholar]
- 5.a) Lai TS, Chan FY, So PK, Ma DL, Wong KY, Che CM. Dalton Trans. 2006:4845–4851. doi: 10.1039/b606757c. [DOI] [PubMed] [Google Scholar]; b) Mansuy D, Lange M, Chottard JC, Bartoli JF, Chevrier B, Weiss R. Angew Chem Int Ed. 1978;17:781–782. [Google Scholar]; c) Mansuy D, Battioni JP, Lavallee DK, Fischer J, Weiss R. Inorg Chem. 1988;27:1052–1056. [Google Scholar]; d) Guerin P, Battioni JP, Chottard JC, Mansuy D. J Organomet Chem. 1981;218:201–209. [Google Scholar]; e) English DR, Hendrickson DN, Suslick KS. Inorg Chem. 1983;22:367–368. [Google Scholar]
- 6.Tatsumi T, Hoffmann R. Inorg Chem. 1981;20:3771–3784. [Google Scholar]
- 7.Wolf JR, Hamaker CG, Djukic JP, Kodadek T, Woo LK. J Am Chem Soc. 1995;117:9194–9199. [Google Scholar]
- 8.Rohde JU, In JH, Lim MH, Brennessel WW, Bukowski MR, Stubna A, Munck E, Nam W, Que L. Science. 2003;299:1037–1039. doi: 10.1126/science.299.5609.1037. [DOI] [PubMed] [Google Scholar]
- 9.a) Kachalova GS, Popov AN, Bartunik HD. Science. 1999;284:473–476. doi: 10.1126/science.284.5413.473. [DOI] [PubMed] [Google Scholar]; b) Debrunner PG. In: Iron Porphyrins. Lever ABP, Gray HB, editors. Vol. 3. VCH Publishers; New York: 1989. pp. 139–234. [Google Scholar]
- 10.a) Ling Y, Zhang Y. J Am Chem Soc. 2009;131:6386–6388. doi: 10.1021/ja9006723. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Katigbak J, Zhang Y. J Phys Chem Lett. 2012;3:3503–3508. doi: 10.1021/jz301689b. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang Y, Mao JH, Oldfield E. J Am Chem Soc. 2002;124:7829–7839. doi: 10.1021/ja011583v. [DOI] [PubMed] [Google Scholar]; d) Zhang Y, Mao JH, Godbout N, Oldfield E. J Am Chem Soc. 2002;124:13921–13930. doi: 10.1021/ja020298o. [DOI] [PubMed] [Google Scholar]
- 11.Guillaume V, Thominot P, Coat F, Mari A, Lapinte C. J Organomet Chem. 1998;565:75–80. [Google Scholar]
- 12.Ling Y, Davidson VL, Zhang Y. J Phys Chem Lett. 2010;1:2936–2939. doi: 10.1021/jz101159x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Andres H, Bominaar EL, Smith JM, Eckert NA, Holland PL, Munck E. J Am Chem Soc. 2002;124:3012–3025. doi: 10.1021/ja012327l. [DOI] [PubMed] [Google Scholar]; b) MacDonnell FM, Ruhlandt-Senge K, Ellison JJ, Holm RH, Power PP. Inorg Chem. 1995;34:1815–1822. [Google Scholar]; c) Zhang Y, Oldfield E. J Phys Chem B. 2003;107:7180–7188. [Google Scholar]
- 14.a) Costas M, Mehn MP, Jensen MP, Que L. Chem Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]; b) Ikezaki A, Ohgo Y, Nakamura M. Coord Chem Rev. 2009;253:2056–2069. [Google Scholar]
- 15.Zhang Y, Oldfield E. J Am Chem Soc. 2004;126:4470–4471. doi: 10.1021/ja030664j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.