

Abstract
Background
Curvularia lunata is an important maize foliar fungal pathogen that distributes widely in maize growing area in China. Genome sequencing of the pathogen will provide important information for globally understanding its virulence mechanism.
Results
We report the genome sequences of a highly virulent C. lunata strain. Phylogenomic analysis indicates that C. lunata was evolved from Bipolaris maydis (Cochliobolus heterostrophus). The highly virulent strain has a high potential to evolve into other pathogenic stains based on analyses on transposases and repeat-induced point mutations. C. lunata has a smaller proportion of secreted proteins as well as B. maydis than entomopathogenic fungi. C. lunata and B. maydis have a similar proportion of protein-encoding genes highly homologous to experimentally proven pathogenic genes from pathogen-host interaction database. However, relative to B. maydis, C. lunata possesses not only many expanded protein families including MFS transporters, G-protein coupled receptors, protein kinases and proteases for transport, signal transduction or degradation, but also many contracted families including cytochrome P450, lipases, glycoside hydrolases and polyketide synthases for detoxification, hydrolysis or secondary metabolites biosynthesis, which are expected to be crucial for the fungal survival in varied stress environments. Comparative transcriptome analysis between a lowly virulent C. lunata strain and its virulence-increased variant induced by resistant host selection reveals that the virulence increase of the pathogen is related to pathways of toxin and melanin biosynthesis in stress environments, and that the two pathways probably have some overlaps.
Conclusions
The data will facilitate a full revelation of pathogenic mechanism and a better understanding of virulence differentiation of C. lunata.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-15-627) contains supplementary material, which is available to authorized users.
Keywords: Curvularia lunata, Genome sequencing, RNA-seq, Pathogenicity variation
Background
The maize leaf spot caused by C. lunata have ever made tremendous yield loss of maize in 11 provinces of maize growing areas in China since the 1990s. Especially in northern China, for instance, it occurred over 192000 hm2 and led to 8 million kg yield loss in Liaoning province in 1996 [1–3]. Many research projects have been designed to discover the disease occurrence pattern, resistance breeding and integrated control of the disease. As the application of resistance varieties containing tropic and sub-tropic germplasms in large growing areas, the incidence of disease infection and its severity were declined massively and less damage was observed in field. However, in recent years, the disease has bounced back again and caused serious damages in some maize growing areas such as Liaoning, Anhui and Henan province etc. The main cause of the disease recurrent was expected to link to large area of monoculture with same or similar resistant germplasm which then become a matrix to induce pathogen virulence variation.
C. lunata is a major organism causing the leaf spot disease in maize [4], but Cochliobolus lunatus as teleomorph of C. lunata is expected to form when the pathogen suffer stress condition in maize field in most cases. The teleomorph is not a major form to cause the foliar disease. C. lunata has 5 pathogenic types from high to low virulence in China. The distribution of pathogenic types varies in different maize growing areas. C. lunata has broad host range including maize, wheat, barley and sorghum and other grasses. Meanwhile a multiple virulence factors have been demonstrated to be involved in pathogen infection to maize, such as cellulose [5], non-host specific toxin (methyl 5-(hydroxymethyl)-furan-2-carboxylate) [6], melanin [7]. It is worth mentioning that some of virulence related genes have been successfully cloned in previous work such as clt-1 regulating non-host specific toxin production, brn1 being required for DHN melanin synthesis [8], two mitogen-activated protein kinases (MAPK) encoding genes (clk1 and clm1) [9, 10]. Previous study preliminarily showed that brn1 was not only involved in melanin synthesis, but also associated with toxin biosynthesis. Whereas, the role of these genes in collaborated regulation of toxins and melanin production remains unknown. In addition, several novel virulence-associated genes and large suites of enzymes such as polyketide synthases (PKS) being involved in secondary metabolism were also associated with pathogenicity in most fungal pathogens [11, 12]. However, there is no detailed knowledge to reveal the genetically synergistic regulation mechanism of those virulence-associated genes in the C. lunata so far.
C. heterostrophus (agamotype: B. maydis) causing a widely distributed maize fungal disease, southern leaf blight, belongs to Cochliobolus as well as C. lunatus (agamotype: C. lunata). As the genome of B. maydis has been accomplished in recent years, it provides a great deal of bioinformation for globally understanding the detailed infection mechanism of C. lunata and its interaction mechanism with maize. Based on previous work, it was found that C. lunata showed high homology with B. maydis in genetic evolution, thus C. lunata could be profiled in genome wide through comparative genome analysis. However, until now, very little information is known about developmental and pathogenic process of C. lunata from a global view, even though a normalized full-length cDNA library of C. lunata was constructed in the previous study [13]. As we mentioned, the production of melanin and non-host specific toxin may share the common gene regulation network or overlap some node genes responsible for cross-talking between both regulation network systems. Three velvet-like protein-encoding genes being interactive with brn1 gene were successfully screened in C. lunata by yeast two hybrid [14]. Further work showed that velvet-like protein was involved in the regulation of pathogen pathogenicity [15].
PAMP (pathogen associated molecular pattern) model is widely applied in a group of crops to illustrate basic immunity or mechanism of C. lunata-maize interaction. Previous study showed that planting resistant maize varieties for long term in a certain areas would induce virulence variation of C. lunata and some genes served as hallmarks displaying the virulence variation, most of which are secreted protein-encoding genes. Similarly a large amount of secreted proteins have been identified in B. maydis (C. heterostrophus), which are responsible for specific interaction of B. maydis with maize germplasm [16]. Taken together, it is expected that multiple virulence-associated genes control pathogen infection in host plants. Although recently the functions of virulence-associated genes were characterized, the details on the relations among the expression modes of these genes still remain unclear, owing to lack of global understanding of C. lunata genome sequence. However, the global analysis on genome sequence of the pathogen C. lunata will be imperative and facilitate more rapid identification of genes responsible for pathogenicity and pathogen-plant interactions. Hence, in this work, firstly the draft genome sequence of C. lunata CX-3 with high virulence was presented, and comparative analyses of genome repertoire among C. lunata CX-3 isolated from maize, C. lunata m118 isolated from sorghum and B. maydis C5 were conducted in pathogen proliferation and development, virulence and genetic variation, secondary metabolism, plant-pathogen interactions, signaling pathway and detoxification. Secondly, RNA-seq was applied to comparatively analyze transcriptional profiles of C. lunata WS18 with weak virulence and its virulence-increased variant WS18-Pob21-11. This work will provide abundant information for globally uncovering the infection mechanism of C. lunata to maize, thereby the new insight to the pathogen development and infection mechanism would be generated in genome wide. Last but not least comparative genomic and transcriptome analysis for virulence variation of C. lunata will contribute to better understanding what is required to develop flexible strategies to control the pathogen infection.
Results
Genome sequencing and general features
The genome of C. lunata CX-3 was sequenced (65× coverage) with Illumina sequence technology; the sequenced reads were assembled into 340 scaffolds (N50, 788 kb) with a total size of 35.5 Mb, similar to B. maydis C5 (36.5 Mb) (GenBank: AIDY00000000), but bigger than C. lunata m118 (31.2 Mb) (JGI: 403758) (Table 1). By mapping 23390 unigenes to the scaffolds of C. lunata CX-3, the completeness of C. lunata CX-3 genome was estimated to be > 99%. By prediction, C. lunata CX-3 genome encoded a total of 11234 protein genes similar to C. lunata m118 and fewer than B. maydis C5 (Table 1). Similarly, the proportion of genes encoding secreted proteins in C. lunata CX-3 was 7.5% (840 proteins), similar to 7.6% in C. lunata m118 (834), but higher than 6.7% in B. maydis C5 (886) (Table 1). Although the proportions of secreted proteins in the two plant pathogens were close to 7-10% in other sequenced plant pathogens such as Magnaporthe oryzae [17, 18], they were far lower than that in three insect pathogenic fungi (16.2% in Cordyceps militaris, 17.6% in Metarhizium anisopliae and 15.1% in Metarhizium acridum) [19, 20].
Table 1.
Comparison of genome features between C. lunata and B. maydis
| Features | C. lunataCX-3 | C. lunatam118 | B. maydisC5 |
|---|---|---|---|
| Assembly size (Mb) | 35.5 | 31.2 | 36.5 |
| Scaffolds | 340 | 171 | 89 |
| GC (%) | 50.22 | 50.9 | 50.0 |
| Repeated sequences (%) | 2.44 | 1.07 | 1.33 |
| Protein-coding genes | 11,234 | 11,004 | 13,316 |
| Gene density (genes per Mb) | 314.7 | 353.8 | 280 |
| Mean gene length (Bp) | 1,448 | 1,429 | 1,284 |
| Secreted proteins | 840 | 834 | 886 |
B. maydis = C. heterostrophus.
Approximately 55% of C. lunata CX-3 genes have >90% of amino acid sequence identity with C. lunata m118 (Figure 1A). Although the sexual stages of both C. lunata and B. maydis are Cochliobolus species, only about 30% of C. lunata CX-3 genes have >90% of amino acid sequence identity with B. maydis C5, and the proportion is far lower that between M. anisopliae and M. acridum (more than 50%) [19]. Comparative genomic analysis showed that C. lunata CX-3, C. lunata m118 and B. maydis C5 have a large number of species-specific and strains-specific genes (Figure 1B). 10082, 9835 and 10476 orthologous core genes and 820, 864 and 1924 species-specific genes were identified in C. lunata CX-3, C. lunata m118 and B. maydis C5, respectively. The proportion of species-specific genes in C. lunata CX-3 (about 7.3%) was similar to C. lunata m118 (7.9%), but far lower than B. maydis C5 (14.4%) (Figure 1B), showing that C. lunata CX-3 has more differences with B. maydis C5 followed by C. lunata m118. In addition, 859 and 837 strains-specific genes were also identified in C. lunata CX-3 and C. lunata m118, respectively (Figure 1B), showing that two different C. lunata strains have distinct difference in genome. Genomic islands (GIs) are part of a genome and contain at least three contiguous gene-encoding proteins that do not exist in the reference genome [21]. GIs have many functions, especially they are involved in symbiosis or pathogenesis and may help organism’s adaptation. GIs are classed into many sub-classes based on their function. For example, GIs associated with pathogenesis are often called as pathogenicity islands (PAIs), and GIs containing many antibiotic resistant genes are referred to as antibiotic resistance islands (ARIs). It was found by reciprocal analysis that C. lunata CX-3 has 40 and 16 GIs separately in comparison to C. lunata m118 and B. maydis C5, which probably play key roles in virulence.
Figure 1.
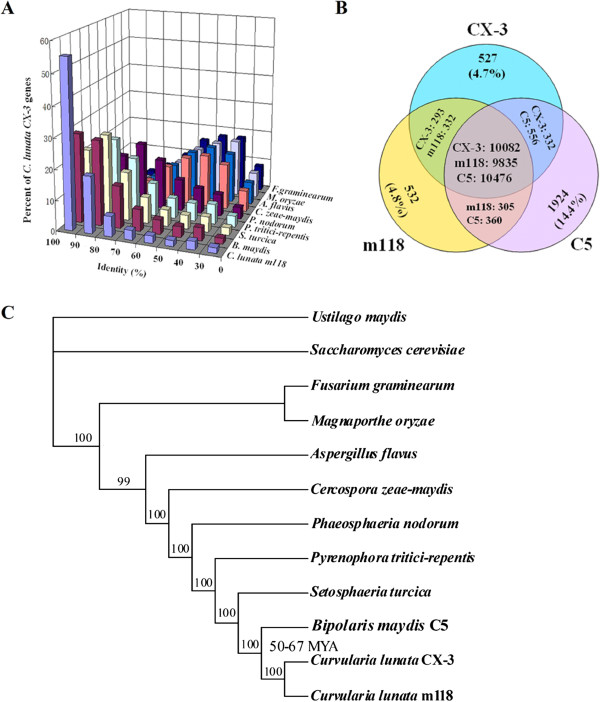
Comparative genomics and evolutionary analysis of C. lunata. (A) Amino acid sequence identity of C. lunata CX-3 with other fungi. (B) Reciprocal Blast analysis of the protein sequences among the three pathogenic fungi C. lunata CX-3, C. lunata m118 and B. maydis C5 with a cut-off E value of 1e-5. CX-3, C. lunata CX-3; m118, C. lunata m118; C5, B. maydis C5. In relative to B. maydis C5, C. lunata CX-3 and C. lunata m118 have 820 (7.3%) and 864 (7.9%) species-specific genes; In relative to C. lunata, B. maydis C5 has 1924 (14.4%) species-specific genes. C. lunata CX-3 and C. lunata m118 have 859 and 837 strains-specific genes, respectively. (C) A phylogenetic tree constructed with the Dayhoff amino acid substitution model representing the evolutionary relationships of C. lunata and other fungi. MYA, million years ago. B. maydis = C. heterostrophus.
Identity analysis between orthologous proteins shows that C. lunata CX-3 has an average about 85.9% amino acid identity with C. lunata m118, higher than with other plant pathogenic fungi such as B. maydis C5 (78.6%), Setosphaeria turcica (JGI: 401988) (75.8%) and Pyrenophora tritici-repentis (GenBank: AAXI00000000) (70.9%), especially far higher than with M. oryzae (GenBank: AACU00000000) orthologs (47.9%) (Table 2). Therefore, C. lunata and B. maydis are more close than three Aspergillus species (A. oryzae, A. nidulans and A. fumigatus) with an average of 68% protein sequence identity [21], but more diverged than two Metarhizium spp. (M. anisopliae and M. acridum) with an average of 89.8% protein sequence identity [20]. A phylogenomic analysis revealed that C. lunata CX-3 and C. lunata m118 diverged 24.3–50.4 million years ago (MYA) and diverged from their most closely related maize pathogen B. maydis 50.4–67.0 MYA (Figure 1C). In additionally, B. maydis diverged from S. turcica 67.1-93.0 MYA, which diverged from P. tritici-repentis 93–136 MYA. Therefore, the lineage leading to C. lunata (C. lunatus) diverged from B. maydis (C. heterostrophus) around Cretaceous Extinction (65 MYA), and the Cochliobolus lineage diverged from other plant pathogenic fungi after the Triassic-Jurassic Event (200 MYA) [22]. It was also showed in Figure 1C that M. oryzae and Fusarium graminearum diverged 274–403 MYA (before the Triassic-Jurassic Event), which was much earlier than the divergence time between Cochliobolus lineage and other plant pathogenic fungi.
Table 2.
Genome-wide analysis of C. lunata CX-3 gene sets
| Characteristics | CX-3 | CCC core a | CX-3-m118 specific b | CX-3-C5 specific c | CX-3 specific |
|---|---|---|---|---|---|
| Protein-encoding genes | 11234 | 10082 | 293 | 332 | 527 |
| Protein families | 2830 | 2780 | 71 | 66 | 48 |
| Secreted proteins | 840 | 736 | 41 | 23 | 40 |
| PHI genesd | 1904 | 1880 | 2 | 10 | 12 |
| Transposases | 161 | 78 | 5 | 34 | 44 |
| Proteases | 516 | 502 | 5 | 4 | 5 |
| Glycoside hydrolases | 219 | 216 | 0 | 1 | 2 |
| MFS transporters | 252 | 252 | 0 | 0 | 0 |
| ABC transporters | 47 | 47 | 0 | 0 | 0 |
| P450s | 147 | 147 | 0 | 0 | 0 |
| GPCRs | 129 | 128 | 1 | 0 | 0 |
| Pth11-like GPCRs | 37 | 0 | 0 | 0 | 0 |
| Protein kinases | 153 | 153 | 0 | 0 | 0 |
| Fungal specific transcription factors | 222 | 209 | 9 | 2 | 2 |
| Backbone genes for secondary metabolism | 34 | 34 | 0 | 0 | 0 |
| Orthologs in C. lunata m118 | 10375 | 10082 | 293 | NA | NA |
| Orthologs in B. maydis C5 | 10414 | 10082 | NA | 332 | NA |
| Orthologs in S. turcica | 10268 | 9833 | 125 | 238 | 72 |
| Orthologs in P. tritici-repentis | 9979 | 9656 | 111 | 144 | 68 |
| Orthologs in M. oryzae | 8462 | 8320 | 56 | 62 | 24 |
| Identity to C. lunata m118 orthologs | 85.9% | 86.4% | 68.8% | NA | NA |
| Identity to B. maydis C5 orthologs | 78.6% | 79.1% | NA | 64.2% | NA |
| Identity to S. turcica orthologs | 75.8% | 76.5% | 53.3% | 62.7% | 57.2% |
| Identity to P. tritici-repentis orthologs | 70.9% | 71.6% | 50.4% | 52.8% | 47.0% |
| Identity to M. oryzae orthologs | 47.9% | 48.0% | 38.4% | 39.3% | 36.9% |
CX-3: C. lunata CX-3. aCCC core: C. lunata CX-3, C. lunata m118 and B. maydis C5 genes grouped with a cutoff E value of 1e-5 during reciprocal Blast analysis. bCX-3-m118 specific: C. lunata CX-3 and C. lunata m118 specific genes in relative to B. maydis C5 grouped with a cutoff E value of 1e-5. cCX-3-C5 specific: C. lunata CX-3 and B. maydis C5 specific genes in relative to C. lunata m118 grouped with a cutoff E value of 1e-5. dPHI genes, pathogen-host interaction genes identified by Blast analysis against the PHI database with a cutoff E value of 1e-5. Identity was estimated using amino acid sequences. GPCR, G-protein-coupled receptor; MFS, major facilitator superfamily; NA, not available. B. maydis = C. heterostrophus.
Pfam matches were performed to identify 2830 conserved protein families containing 8471 proteins in C. lunata CX-3 genome close to C. lunata m118 with 2827 families containing 8181 proteins and slightly lower than B. maydis C5 with 2860 families containing 8886 proteins. In phytopathogenic fungi, family expansions were identified in glycoside hydrolases (GHs), cutinases and pectin lyases compare to insect pathogenic fungi [19], showing the important roles of GHs, cutinases and pectin lyases in pathogenicity of phytopathogenic fungi. As a plant pathogen, C. lunata CX-3 has a large number of GHs, cutinases and pectin lyases (Additional file 1: Table S1). In comparison with C. lunata m118, C. lunata CX-3 has family expansions in transposase, fungal specific transcription factors, major facilitator superfamily, ABC superfamily, cytochrome P450, protein kinase, serine protease, subtilisin and glucosidase, and family constrictions in G-protein coupled receptor and heterokaryon incompatibility (Additional file 1: Table S1 and S2), which are expected to be crucial for the fungal survival in varied stress environments.
To excavate potential pathogenic genes, a Blast search of the C. lunata CX-3 genome was performed against the pathogen-host interaction (PHI) database, which collected the experimentally proven genes affecting the outcome of pathogen-host interactions of fungi, bacteria and oomycetes [23]. 17.0% of predicted genes in C. lunata CX-3 (1904), identified by a Blast search of C. lunata CX-3 genome against the PHI database, are putatively related to pathogen-host interaction. The percentage of PHI genes in C. lunata CX-3 (17.0%) is close to other plant pathogenic fungi (16.6% in C. lunata m118, 15.0% in B. maydis C5, 17.0% in F. graminearum and 17.7% in Aspergillus flavus) (Additional file 1: Table S2).
Transposases and repeat-induced point mutation
Like rice pathogen Magnaporthe grisea, C. lunata has a high degree of genetic variability and prone to form novel pathogenic variants to infect the resistant host. Transposases and repetitive elements make a greater contribution to genetic instability and pathogenic variation [24]. The transposable elements are closely associated with the virulence of most ascomycetes including C. lunata. Thus, an understanding of the change of transposons and repeat elements in C. lunata not only provides an insight into their impact on genome evolution and also contributes to shedding light on mechanisms of pathogenic variation.
The number of transposases in C. lunata CX-3 (161) was far more than C. lunata m118 (24) and B. maydis C5 (97) (Additional file 1: Table S3). Out of 161 transposases in C. lunata CX-3, 83 were DNA transposases, 54 LTR transposases and 24 LINE transposases. However, C. lunata m118 genome lacks LINE transposases. Most of these transposases in C. lunata CX-3 are rich in DNA (79), LINE (24) and LTR (54) transposases, of which subclasses such as TcMar (70), Tad1 (22), Copia (18) and Gypsy (28) are much more abundant than other subclasses (Additional file 1: Table S3). Transcriptome analysis also showed that 3 transposase genes (CL00030, CL10102 and CL00391) were up-regulated in C. lunata with higher virulence.
Repeat-induced point mutation (RIP) in ascomycete fungi is a genome defense, which hyper-mutates repetitive DNA and limit the accumulation of transposable elements [25]. RIP preferentially introduces CA/G dinucleoutides to TA during the sexual cycle in many fungi, such as Metarhizium spp. and Trichoderma spp. etc. 68 paired C. lunata CX-3 paralogous genes showing >80% nucleotide sequence identities were used to estimate nucleotide mutation frequencies. The results showed that there was strong mutation bias: C:G to T:A (Figure 2), which is introduced by repeat induced point mutations (RIP) [26]. The C. lunata CX-3 genome contains many transposable elements, suggesting that they can escape damage by RIP.
Figure 2.
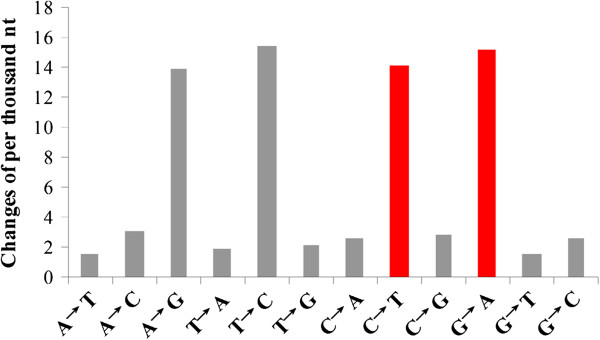
Nucleotide mutation rates in C. lunata CX-3 paralogous genes showing >80% identity in paired protein sequences. For calculations, the gene of each pair with >90% nucleotide sequence similarities to the ortholog from S. turcica was used as the reference.
Protein families involved in degrading plant cell wall and cuticle
C. lunata as a plant pathogen need to break through the passive defense (cell wall, cuticle) of plant for successful infection [27]. Therefore, it would be expected to produce and secrete large numbers of extracellular degrading enzymes to degrade the plant cell wall and cuticle such as glycoside hydrolases, pectinase and cutinase, in which glycoside hydrolase (GH) is one of the representative. The number of GH genes in C. lunata CX-3 (219) is close to C. lunata m118 (214) and the average in plant pathogenic fungi (211) and less than B. maydis C5 (241) (Additional file 1: Table S4), but far larger than that in three insect pathogenic fungi (159 in M. anisopliae, 130 in M. acridum, 135 in C. militaris) [19, 20]. There is no distinct difference in the number of GH genes for each family except for GH18 and GH43 family among C. lunata CX-3, C. lunata m118 and B. maydis C5. 31.9% of GH genes in C. lunata CX-3 (75) are identified to be putative PHI genes, 34.1% in C. lunata m118 (83) and 34.0% in B. maydis C5 (82), which are close to the average in plant pathogenic fungi (31.5%) but higher than that in insect fungi Metarhizium spp. (about 20%) [20].
Plant pathogenic fungi contain almost all GH families of insect pathogenic fungi (Metarhizium spp. and C. militaris), but some GH families existing in C. lunata and other plant pathogenic fungi are not present in insect fungi, including GH6, GH7, GH45 and GH61 cellulases, GH10 xylanases, GH13 amylases, GH30 glucosylceramidases, GH62 and GH78 rhamnosidases, GH79 and GH115 glucuronidases, GH125 mannosidases [19]. The GH families of cellulases are well presented in C. lunata CX-3 (55), C. lunata m118 (53) and B. maydis C5 (56), including GH3, GH6, GH7, GH45 and GH61 cellulases. Among the GH families the GH3 and GH61 cellulases are the majority, but lack of GH5 cellulases. GH18 chitinases are involved in degrading insect chitin, so predictably GH18 genes are less in C. lunata and other plant pathogenic fungi (11 in C. lunata CX-3, 14 in C. lunata m118, 9 to 19 in other phytopathogenic fungi) than in insect pathogenic fungi (30 in M. anisopliae and 21 in M. acridum) [19]. Additionally, GH16 family of xyloglucosyl transferases for xyloglucan catabolism play an important role in digesting plant cell walls, and they are well presented in plant pathogenic fungi including C. lunata CX-3 (19), C. lunata m118 (19) and B. maydis C5 (21).
Protein families for transportation
In fungal transporters, ATP-binding cassette (ABC) transporters and the major facilitator superfamily (MFS) are the two biggest superfamilies. The ABC transporters are multi-component primary active transporters, which transport both small molecules and macromolecules under ATP hydrolysis, whereas the MFS transporters are single-polypeptide secondary carriers, capable of transporting small solutes in response to chemiosmotic ion gradients [28, 29]. Drug transporters of the ABC and MFS transporters in plant pathogenic fungi can function roles not only in the secretion of endogenous fungal pathogenic factors such as toxins but also in protecting against exogenous plant defense compounds such as phytoalexins, thereby playing an important role in virulence to plant [19, 30]. The C. lunata CX-3 genome encodes a great quantity of transporters (590) which is close to C. lunata m118 (594) and B. maydis C5 (593) (Additional file 1: Table S6). C. lunata CX-3 has 47 ABC transporters, close to 45 in C. lunata m118 and 46 in B. maydis C5. MFS transporters account for the highest proportion (252/590 in C. lunata CX-3, 251/594 in C. lunata m118 and 233/593 in B. maydis C5) (Additional file 1: Tables S1 and S6). 60-70% of MFS transporters (167/252 in CX-3, 171/251 in m118 and 158/233 in C5) and 80-85% of ABC transporters (40/47 in CX-3, 38/45 in m118 and 38/46 in C5) were identified as putative PHI genes in C. lunata CX-3, C. lunata m118, B. maydis C5 (Additional file 1: Table S2).
In the ABC transporters, the multidrug resistance (MDR) and the pleiotropic drug resistance (PDR) subfamilies function in resisting antifungal agents [31]. C. lunata CX-3 has 9 MDR subfamily of transporters, more than C. lunata m118 (6), B. maydis C5 (7) and the average of plant pathogens (8) (Additional file 1: Table S7). However, the number of PDR transporters in C. lunata CX-3 (10) is in line with C. lunata m118 (10), B. maydis C5 (10) and the average of plant pathogens (10). The two drug:H+ antiporter (DHA) subfamilies (DHA1 with 12 spanner and DHA2 with 14 spanner) of MFS transporter can make toxic compounds be secreted into outer environment [31]. DHA1 subfamily of transporters well exist in the C. lunata CX-3 genome (49 in CX-3 versus 52 in m118, 51 in C5 and an average of 39 in plant pathogenic fungi) (Additional file 1: Table S7). In comparison to DHA1, little DHA2 subfamily of transporters are present in C. lunata CX-3 (26) as well as C. lunata m118 (27) and B. maydis C5 (15), however which is higher than the average of plant pathogenic fungi. These results show that there are differences in gene family expansions occurred in DHA1, DHA2 and MDR among C. lunata CX-3, C. lunata m118 and B. maydis C5. Transcriptome sequencing of both the low virulent C. lunata strain and its virulence-increased variant showed that 16 transporters were up-regulated in the virulence-increased variant response to the selective pressure of host in the pathogen-plant interactions. Half of these transporters (8) were MFS transporters including CL01123, CL02605, CL03193, CL04682, CL04921, CL05805, CL08969 and CL09075, of which CL01123 and CL05805 were DHA1 subfamily of MFS transporters and CL04682 and CL04921 were DHA2 subfamily of MFS transporters. In the 16 transporters, there was only one MDR subfamily of ABC transporter (CL06556).
Protein families for detoxification
Cytochrome P450 enzymes (CYPs) have a function with the conversion of hydrophobic intermediates of metabolisms and the detoxification of natural and environmental pollutants [32]. The genome sequencing of C. lunata facilitates the excavation of novel CYPs from C. lunata. Through the genome search against cytochrome P450 database [33], 137 P450 genes were identified in C. lunata CX-3, more than 106 in C. lunata m118, but less than 162 in B. maydis C5 (Additional file 1: Tables S1 and S8). 23 subfamilies of CYPs are presented in C. lunata CX-3 and/or C. lunata m118 but absent in B. maydis C5, and 13 subfamilies of CYPs are absent in both C. lunata CX-3 and C. lunata m118 but present in B. maydis C5, thus showing the significant differences in CYP families expansions in these genomes. Interestingly, there are a high percentage of PHI genes (about 70% or higher) in the CYPs of plant pathogenic fungi, for example, 112/137 (81%) in C. lunata CX-3, 95/107 (88%) in C. lunata m118 and 114/162 (70%) in B. maydis C5 (Additional file 1: Table S2), showing that most of CYPs are involved in the pathogen-plant interactions. CYP65 is the biggest subfamily of P450s both in C. lunata CX-3 (17), C. lunata m118 (12) and B. maydis C5 (21) (Additional file 1: Table S8). Additionally, CYP505 subfamily of P450s are present in these genomes (3 in CX-3, 2 in m118 and 4 in C5). It was reported that CYP65 catalyzed the epoxidation reaction in the mycotoxin trichothecene biosynthesis of F. graminearum [34], and CYP505 as well as CYP65 were involved in the fumonisin biosynthesis of a maize pathogen F. verticillioides [35, 36], suggesting that CYP505s and CYP65s in C. lunata and B. maydis probably be related to the toxin biosynthesis and thus play roles in fungal virulence.
Virulence-associated signaling pathways
In fungal pathogenic lifestyle, G-protein-coupled receptors (GPCRs) are essential for plant recognition and pheromone/nutrient sensing [19]. They can transduce environmental signals by means of heterotrimeric G proteins into secondary messengers to regulate gene expression and ultimately cellular responses [37]. One GPCR of M. grisea, Pth11, mediates appressorium formation and virulence [38]. The C. lunata CX-3 genome contains a large number of GPCR-like genes with 129 in total and 51 putative Pth11-like GPCRs, which is similar to 139/54 in C. lunata m118 and but more than 102/30 in B. maydis C5 (Additional file 1: Tables S1 and S9). G-protein alpha subunit is a component of heterotrimeric G protein [39], and activates distinct downstream effectors and influence the pathogenicity [40, 41]. Notably, C. lunata CX-3 contains four G-protein alpha subunits (CL02298, CL05431, CL05929 and CL06671) that are PHI genes, and it was similarly found in C. lunata m118 and B. maydis C5 (Additional file 1: Table S2). The four G-protein alpha subunits showed 97% (CL02298), 92% (CL05431), 99% (CL05929) and 61% (CL06671) amino acid identities with heterotrimeric G protein alpha subunit subtype 2 of S. turcica (GenBank: AEJ38171), guanine nucleotide-binding protein alpha-3 subunit protein of Neofusicoccum parvum (GenBank: EOD45444), G-alpha subunit of Phaeosphaeria nodorum (GenBank: XP_001800368) and guanine nucleotide-binding protein alpha-3 subunit of P. tritici-repentis (GenBank: XP_001931840), respectively.
Likewise, three main signaling pathways (MAPK, cAMP and Ca), being mediated by protein kinases, control virulence-associated development of the fungal pathogen. Fungal protein kinases are classed into 9 groups, of which STE and CMGC (MAPK family) kinases are involved in MAPK pathway, AGC (PKA family) kinases in cAMP pathway, and CMGC (CLK and RCK family), CAMK and AGC (PKC family) kinases in Ca signaling pathway. The C. lunata CX-3 genome contains 153 protein kinases and it is slightly more than 147 in C. lunata m118 and 140 in B. maydis C5 and close to the average (154) of plant pathogenic fungi (Additional file 1: Tables S1 and S10). Because signal transduction plays a critical role in fungal development and infection [20], most of these protein kinases are highly homologous to gene-encoded proteins from PHI database (127/153 in CX-3, 124/147 in m118 and 114/140 in C5) (Additional file 1: Tables S1 and S2). Although there are no distinct differences in the number of protein kinases in the three genomes, the function of signal pathways regulated by protein kinases in different organisms are distinct. For example, Pmk1 MAPK pathway in M. grisea is involved in pathogenesis and regulates appressorium formation, however its homologous MAPK pathway in S. cerevisiae is related to both the pheromone signaling and filamentation.
S. cerevisiae has five MAPK pathways such as Fus3, Kss1, Hog1, Mpk1 and Smk1, which are involved in pheromone response, filamentous growth, osmoregulation, cell wall integrity and spore wall assembly, respectively [42]. M. grisea has three MAPK pathways such as Pmk1, Mps1 and Osm1, which are homologous to Fus3 or Kss1, Mpk1 and Hog1 pathways of S. cerevisiae, respectively [18]. Pmk1 pathway regulates appressorium formation, penetration and colonization; MPS1 pathway invasive growth, conidiation and penetration; OSM1 osmoregulation and stress response [18]. Blastp search analysis reveals that the homologues of MAPK pathways of S. cerevisiae and M. grisea were identified in the C. lunata CX-3 genome (Additional file 1: Table S11). The homologues of pmk1 and mps1, clk1 (CL06419) and clm1 (CL11087) of C. lunata, were cloned and characterized in our previous study. Clk1 gene influenced conidiospore formation, growth, cell degrading enzymes activity and virulence of C. lunata [9], which supported a result that homologues of pmk1 were essential for fungal pathogenicity in all plant pathogens [43]. Clm1 gene regulated cell-wall integrity, conidiospore formation, infection, cell degrading enzymes activity [10].
Cyclic AMP (cAMP) signaling pathway was involved in the induction of appressorium formation and turgor-driven process, therefore leading to plant infection [18]. cAMP can activate downstream effectors such as cAMP-dependent protein kinase (PKA) [44]. Catalytic subunit (CPKA) of PKA was required for appressorium maturation of M. grisea [45]. The C. lunata CX-3 genome contains two gene-encoded PKA catalytic subunits (CL03564 and CL01641), therefore proper studies of these genes will contribute to elaborating the cAMP signaling mediating appressorium formation.
Histidine kinase (HK) phosphorelay signaling as a major mechanism is used by some organisms (bacteria, slime molds, plants and fungi) to sense and adapt to their environment [20, 46]. In fungi, HK signaling mediates multi biological processes such as secondary metabolite biosynthesis, stress response, virulence and differentiation [46–48]. The pathway is two-component signaling pathway containing a sensor HK and a response regulator (RR). HKs well exist in C. lunata CX-3 (19), C. lunata m118 (19) and B. maydis C5 (20) and other plant pathogenic fungi (9 to 21) (Additional file 1: Table S10). It’s worth noting that all HKs are PHI genes.
Virulence-associated secondary metabolite genes
One primary goal of studying the genome of a fungal pathogen is to identify secondary metabolites that are served as virulence factors such as host specific toxins (HST), non host specific toxin (NHST) and melanin [16]. The impact of HST/NHST on plant hosts was understood early, since they make the producing fungi be highly virulent to crops. C. lunata CX-3 is capable of producing diverse secondary metabolites such as NHST and melanin that aid in niche exploitation and pathogenicity. Although toxin and melanin are two key pathogenic factors in C. lunata, the core genes for their biosynthesis are not identified in C. lunata yet. The C. lunata CX-3 genome provides the feasibility to identify the core genes for melanin and toxin such as non-ribosomal peptide synthetase (NRPS) and polyketide synthase (PKS). It was reported previously that PKS1 and PKS2, two PKS of B. maydis C5, functioned as the biosynthesis of T-toxin, and HST1, one NPRS of Bipolaris zeicola played a key role in the HC-toxin biosynthesis. The number of core genes for secondary metabolites in C. lunata CX-3 (36) is close to the average of other plant pathogenic fungi (41) (Table 3). The C. lunata CX-3 genome encodes similar amounts of NRPS (6), PKS (16) and NRPS-PKS hybrid genes (HYBRID) (2) with C. lunata m118 (5 NRPS, 14 PKS and 2 HYBRID). While, the numbers of NRPS and PKS in the two C. lunata genomes are slightly less than B. maydis C5 (9 NRPS and 19 PKS), and the latter has no HYBRID. These results show that the differences in the number of NPRS and PKS between strains or species are consistent with their adapting to the diverse environments and hosts.
Table 3.
Numbers of backbone-genes for the biosynthesis of secondary metabolites in different pathogenic fungi
| Classifications | Fungal species | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CX-3 | m118 | C5 | CZM | PN | ST | PTR | MO | AF | FG | |
| DMAT | 1 | 2 | 3 | 1 | 2 | 2 | 0 | 3 | 8 | 0 |
| HYBRID | 2 | 2 | 0 | 1 | 1 | 2 | 1 | 5 | 2 | 0 |
| NRPS | 6 | 5 | 9 | 7 | 9 | 9 | 12 | 8 | 18 | 10 |
| NRPS-Like | 10 | 9 | 7 | 8 | 5 | 7 | 6 | 6 | 14 | 10 |
| PKS | 16 | 14 | 22 | 11 | 12 | 23 | 14 | 23 | 25 | 12 |
| PKS-Like | 1 | 1 | 3 | 2 | 9 | 3 | 6 | 2 | 3 | 2 |
| Total | 36 | 33 | 44 | 30 | 38 | 46 | 39 | 47 | 70 | 34 |
Classifications: DMAT, dimethylallyl tryptophan synthase; HYBRID, hybrid PKS-NRPS enzyme; NRPS, non-ribosomal peptide synthetase; PKS, polyketide synthetase;. Fungal species: CX-3, C. lunata CX-3; m118, C. lunata m118; C5, B. maydis C5; CZM, C. zeae-maydis; PN, Phaeosphaeria nodorum, ST, S. turcica; PTR, P. tritici-repentis, MO, M. oryzae; AF, A. flavus; FG, F. graminearum. B. maydis = C. heterostrophus.
C. lunata CX-3 PKSs were divided into different clusters based on phylogenetic analysis of the ketoacyl CoA synthase (KS) domain of PKSs with other known PKS for melanin or toxin biosynthesis as references (Figure 3A). There were differences in the domains of different PKSs in C. lunata CX-3 based on domain analysis, but the domains of C. lunata CX-3 PKSs were similar with known PKSs. Interestingly, PKSs were grouped into two kinds based on PKS domain components. One kind was reducing PKSs with KS, acyltransferase (AT) and dehydratase (DH) domains at least, including 12 C. lunata CX-3 PKSs and 6 other known PKSs being involved in different kinds of toxin biosynthesis such as Alternaria alternata ACT-toxin, Gibberella zeae zearalenone, Gibberella moniliformis fumonisin, Aspergillus ochraceus ochratoxin and C. heterostrophus T-toxin (Figure 3C). The other kind was non-reducing PKSs without DH domain, including 2 C. lunata CX-3 PKSs and 9 other known PKSs for melanin biosynthesis. It was suggested that C. lunata CX-3 has conserved PKSs related to the biosynthesis of toxin and melanin. Notably CL04686 and CL09981 both encoding PKSs were up-regulated in virulence-enhanced strain. However, phylogenetic and modular analyses suggested that the protein structures of C. lunata CX-3 NRPSs were obviously different from other known NRPSs being involved in the biosynthesis of mycotoxins such as HC-toxin of Cochliobolus carbonum, AM-toxin of A. alternate, gliotoxin of Aspergillus fumigatus and enniatin of Fusarium equiseti (Additional file 2: Figure S1).
Figure 3.
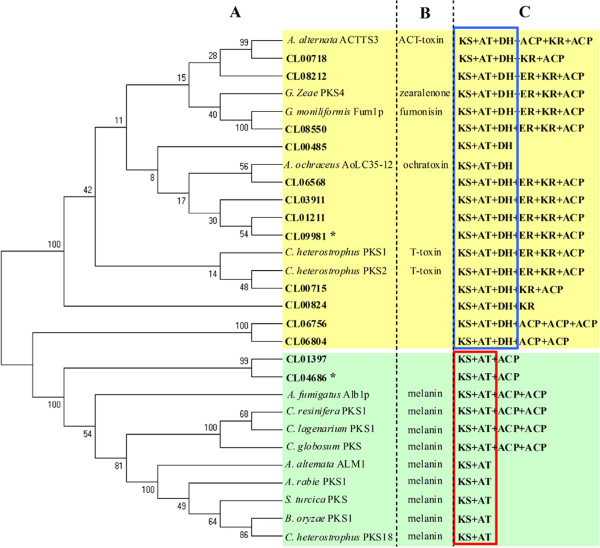
Phylogenetic and domain analyses of C. lunata CX-3 and other fungal polyketide synthases (PKS). (A) A neighbor-joining tree of ketoacyl CoA synthase (KS) domain sequences among fungi. (B) Toxin and melanin related to PKS in other fungi. (C) Domain analysis of the PKS. Domain definitions: KS, ketoacyl CoA synthase; AT, acyltransferase domain; DH, dehydratase domain; ER, enoyl reductase domain; KR, ketoreductase domain; ACP, acyl carrier protein domain; The accessions of other fungi PKSs are shown in the Materials and methods. *CL04686 and CL09981 are up-regulated in virulence-enhanced strain. C. heterostrophus = B. maydis.
Small, cysteine-rich peptides and effector proteins
The small cysteine-rich proteins (SCRPs) were secreted directly into host plant cells and perform multiple biological functions such as host recognition or colonization [49, 50], the induction of host HR [51–53], pathogenicity [54], and antimicrobiosis [55, 56]. Some SCRPs as virulence effectors showed carbohydrate binding activity that not only facilitated fungal virulence by perturbing host cell signaling or interfering with host recognition of the pathogen or suppressing pathogen-associated molecular pattern (PAMP)-triggered immunity (PTI) [18, 57], but also induced effector-triggered immunity (ETI) governed by a gene-for-gene system in plants containing homologous resistance (R) proteins in the pathogen-host interaction [58]. A recently identified class of conserved effectors in fungi are LysM effectors that contain lysine motifs (LysMs) other than recognizable protein domains [59]. It is feasible to under-predict putative SCRPs in the C. lunata CX-3 genome due to the short length (<150 amino acid residues) of SCRPs, which contribute to extending the annotation of the C. lunata CX-3 genome with a specific search (see Materials and Methods). 76 potential SCRPs were found in the genome, ranging in size from 72 to 150 residues (Additional file 1: Table S12). Among these 76 predicted SCRPs in C. lunata CX-3, CL08250 (147 residues) with a LysM showed 34% of amino acid identity with Ecp6 protein of Cladosporium fulvum, which was a LysM-containing effector and virulence factor [60]. CL08356 (92 residues) containing an antifungal protein domain showed 48% of amino acid identity with an antifungal protein (GenBank: CAR79018) of Fusarium avenaceum. CL11021 (125 residues) containing a hydrophobin domain showed 45% of amino acid identity with a hydrophobin (GenBkan: ABY48863) from B. maydis. Several secreted hydrophobins as fungal effectors were examined for their roles in virulence such as Hum3 and Rsp1 of U. maydis [58, 61], fungal development and plant colonization such as Mhp1 of M. grisea [62]. Additionally, although >150 residues in size of a secreted protein (CL01513, 234 residues), it contains a LysM (Additional file 1: Table S12) and shows 42% of amino acid identity with Ecp6 (GenBank: ACF19427) of Passalora fulva. As described above, cysteine-rich polypeptides such as CL01513, CL08250, CL08356 and CL11021 were served as potential candidates for pathogen effectors in C. lunata CX-3.
Comparative transcriptome analysis for pathogenicity variation
In the previous works, under the successive selection pressure of resistant maize germplasm, C. lunata virulence was enhanced [4]. Although some hallmarks related to virulence variation were screened, the variation mechanism at the transcriptional level was not deeply understood yet. In order to further shed light on the molecular regulation mechanism for virulence differentiation of C. lunata, high-throughput RNA-Seq was performed to compare the transcriptional differences between C. lunata WS18 with low virulence and its virulence-enhanced variant WS18-Pob21-11 and to further screen the crux genes that involved in the virulence variation of C. lunata under the host selection pressures.
>9.7 million tags were sequenced for each strain and 76.2% and 74.6% of predicted C. lunata CX-3 genes were expressed in WS18 and WS18-Pob21-11 respectively. However, the transcriptional profile of WS18-Pob21-11 was different from WS18 after successive host direction selection (Figure 4). A total of 200 genes including 32 putative PHI genes were significantly (PDR ≤ 0.001,) up-regulated and 164 genes including 35 putative PHI genes down-regulated in the virulence-enhanced strain (Additional file 1: Table S13), showing that C. lunata presented the obvious change at transcriptional regulation under the continued selective pressure from resistant host. In general, differential genes were involved in transport, oxidation-reduction process, metabolic process, mycelium development, response to stress, pigment biosynthetic process and protein metabolic process, etc., most of which were significantly up-regulated under the successive selection pressure from host. In contrast, almost all differentially expressed genes being involved in the carbohydrate metabolism, protein modification and cellular component organization were significantly down-regulated (Figure 5).
Figure 4.
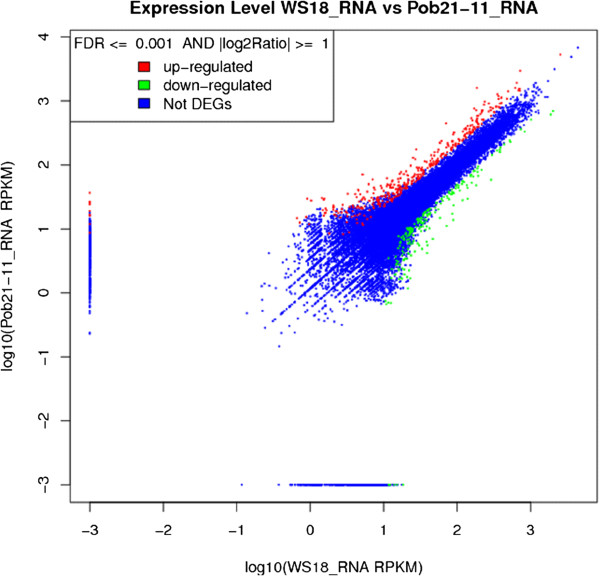
Differentially expressed genes in the virulence-enhanced strain compared to the wild type. The red and green color separately mean up-regulated and down-regulated unigenes, and the blue color means no differentially expressed unigenes. A total of 200 and 164 genes were significantly (PDR ≤ 0.001,) up-regulated and down-regulated in the virulence-enhanced strain WS18-Pob21-11.
Figure 5.
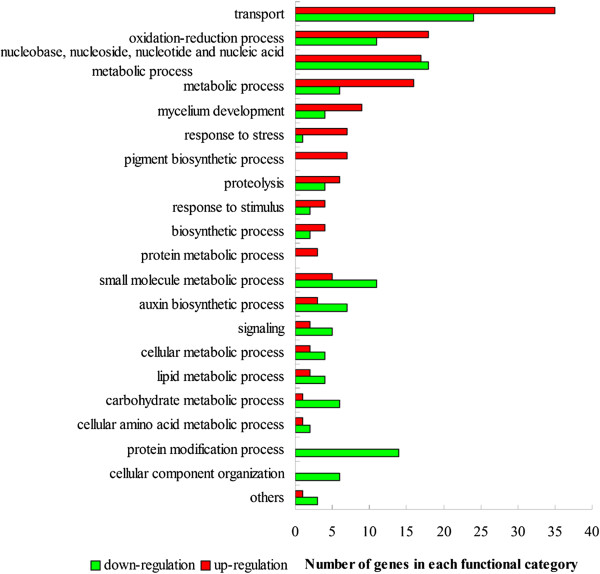
Functional enrichment of differentially expressed genes in C. lunata WS18-Pob21-11 compared to WS18.
Like other plant pathogenic fungi, C. lunata could produce 1,8-dihydroxynaphthalene (DHN) melanin and toxin as two important pathogenic factors to make plant infected. In this study, it was found that the virulence-enhanced strain produced more melanin than the wild-type strain (data not shown), showing that the differential genes related to the biosynthesis of DHN-melanin were at lest partly responsible for the virulence variation under the selective pressures from host [4]. Responses to the selective pressure of host, CL04685 encoding scytalone dehydratase (SCD) were up-regulated PHI genes and it was involved in melanin synthesis [63, 64]. Therefore, it was proved that the pathway of melanin synthesis was related to virulence differentiation of C. lunata. Interestingly, it was found by Blastp of differential genes against the proteins sequences of the C. lunata CX-3 genome that 12 flanking genes of CL04685 (scd) in genome were up-regulated in WS18-Pob21-11, including CL04673 (alpha-1,6-mannosyltransferase subunit), CL04676 (epimerase), CL04678 (Pc16g10800), CL04680 (epimerase), CL04681 (NADP(+)-dependent dehydrogenase), CL04682 (MFS transporter), CL04686 (polyketide synthase), CL04687 (metallo-beta-lactamase), CL04688 (17-beta-hydroxysteroid dehydrogenase), CL04689 (epimerase), CL04690 (monoxygenase) and CL04692 (methyltransferase). Among the 12 flanking genes, the CL04682, CL04688 and CL04690 were PHI genes (Additional file 1: Table S13). Genes being involved in the biosynthesis of secondary metabolites were usually clustered [65], and the combination of CL04685 and its 12 flanking genes was similar to a sirodesmin biosynthesis-associated gene cluster of Leptosphaeria maculans [66] and a cercosporin-associated gene cluster of Cercospora nicotianae [67]. Thus, the 13 genes belonged to a gene cluster for the synthesis of secondary metabolites in C. lunata (Figure 6). In this cluster, CL04683 with no expression change has 27% of amino acid sequence identity with aflatoxin regulatory protein AflR (GenBank: AAM03003) which was involved in the regulation of aflatoxin clusters in A. flavus and Aspergillus parasiticus and sterigmatocystin cluster in Aspergillus nidulans, and 24% of amino acid sequence identity with cercosporin regulatory protein CTB8 (GenBank: DQ991510) of C. nicotianae. CL04682, a MFS transporter, has 61% of amino acid sequence identity with aflatoxin efflux pump (GenBank: XP_002844735) of Arthroderma otae. It was suggested that this cluster in C. lunata is a putative gene cluster for toxin biosynthesis of C. lunata. Therefore, the up-regulation of the cluster genes showed that they were involved in pathogenicity variation.
Figure 6.
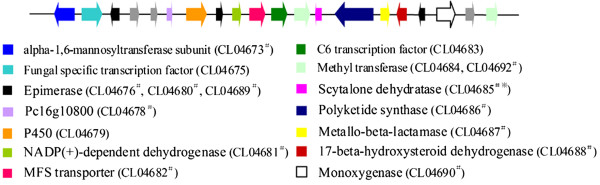
Gene cluster for toxin synthesis in the C. lunata CX-3 genome. ※Scytalone dehydratase involves in melanin synthesis. #Genes were up-regulated in highly virulent C. lunata strain.
A complex network of regulatory and signaling components involved in regulation of morphogenesis and virulence [68, 69]. Therefore, although a gene cluster related to toxin and melanin biosynthesis played roles in the pathogenicity variation, other differently expressed genes were probably relative to the change, particularly up-regulated genes (Figure 7).
Figure 7.

Part of up-regulated genes related to the virulence variation in highly virulent C. lunata strain. A: WS18, B: WS18-Pob21-11.
Discussion
In this study, we reported the first genome sequence of C. lunata, an important pathogen of maize. Genome sequencing of the pathogen will make more contribution to the understanding of its evolutionary relationship with other pathogenic fungi, and efficiently screening of pathogenicity-associated genes as well as detection of its virulence variation under its interaction with host. It was showed that C. lunata CX-3 has significant differences from C. lunata m118 and B. maydis C5 in the number and sequence of genes-encoded proteins, although their evolutionary relationships are very close. As a plant pathogen, the C. lunata CX-3 genome like B. maydis contains about thousands of putative PHI genes (17.0% of all the genes) which are well known to be closely involved in the interaction with plants. Additionally, it was also found that the proportion of secretary proteins in C. lunata CX-3 was close to that in C. lunata m118 and B. maydis C5. Secreted proteins of pathogens have some crucial effectors responsible for mediating plant-pathogen interaction, some of which may play a role in the induction/suppression of plant resistance against pathogen infection [70, 71]. Transcriptional profile deriving from RNA-seq showed that the virulence variation of C. lunata was not only related to the up-regulation of genes probably being involved in the biosynthesis pathway of toxin and melanin, it also related to the up-regulation of other pathogenicity associated genes. In other words, multiple regulatory networks involved in the virulence differentiation of C. lunata response to the selective pressures from resistant hosts.
It was found by the phylogenomic analysis that the Cochliobolus lineage was diverged from other well known plant pathogenic fungi including S. turcica, P. tritici-repentis and M. oryzae, which was consistent with Leonard’s result [72]. Comparatively, Cochliobolus lineage was more similar to S. turcica and P. tritici-repentis than to M. oryzae in protein sequences. A great deal of expressed sequence tags (ESTs) sourced from the suppression subtractive hybridization (SSH) library of C. lunata revealed a similar trend that C. lunata has high sequence similarities with S. turcica and P. tritici-repentis [4], which would allow us more efficiently and more easily to find novel functional factors or mechanisms in C. lunata for attacking host plants. Furthermore the similar resistant genes could also be searched between mentioned pathogenic fungi and applied in a way to share homologous gene sources.
A wide range of plant pathogenic fungi exhibit a high degree of genetic variability through varied ways [4, 18], based on the PAMP models (pathogen–associated molecular pattern), in which a numerous effectors are involved in compatible or incompatible interaction between pathogen and host plants. In our case, small cysteine-rich proteins (SCRPs), for example, were speculated to be contributor to host immunity response if incompatible interaction of pathogen and maize resistant varieties happen, which could serve as effectors responsible for inducing host resistance response such as PTI and ETI [70].
Transposases encoded by transposons played a key role in the evolution of eukaryotic gene regulatory network, and coordinated and regulated eukaryotic gene expression [73]. Therefore serious pathogenic variation of C. lunata was attributed to the presence of a large amount of transposases in C. lunata [4]. The amount of transposases in C. lunata CX-3 is much larger than not only in C. lunata m118 and B. maydis C5, also in other Ascomycetes [20]. The phenomenon could be explained by high C to T and G to A transitions mediated by RIP in the C. lunata CX-3 genome during its sexual cycle [26], as also proved in many Ascomycetes [17, 19, 20, 74, 75]. Thus, it was speculated that shift of C:G to T:A might make the pathogen more frequently mutate in virulence differentiation of C. lunata, at least partly, suggesting that C. lunata CX-3 has a strong potential to evolve into other pathogenic types of strains.
C. lunata successively undergoes asexual and sexual stages in his life cycle like other fungi. So far, the asexual stage of C. lunata is well known, but its sexual stage is still poorly understood. In sexual stage, C. lunata probably survived in soil in the shape of ascospores [76], but this stage was infrequent in nature. The asexual stage of C. lunata is key in causing disease to host plant. During the interaction of pathogen-plant, C. lunata and other pathogenic fungi undergo several complex and crucial steps, including attachment to the plant surface, germination on the plant surface and formation of infection structures, penetration of the host and colonization of the host tissue, which are crucial to cause disease [77]. Pathogenic factors, including toxin, melanin, cell wall degrading enzymes (pectinase, cellulase, hemicellulase, protease, amylase and phospholipase), cutinase and hormone are essential for completing these processes. Furthermore, many gene-encoded proteins such as GPCRs, kinases, core genes for the biosynthesis of secondary metabolite, CYPs, ABC transporters and MFS transporters are directly or indirectly involved in these processes. Excitingly, a great many of these proteins had been identified in the C. lunata CX-3 genome by bioinformatics and most of these protein families were involved in the pathogen-host interaction (Additional file 1: Table S2), which facilitated the understanding of pathogenic mechanism of C. lunata. In the previous study, the chemical structure of non-host-specific toxin in C. lunata was identified successfully, and it was speculated based on the toxin structure that genes related to toxin biosynthesis may exist in genome with clustering status. It was found through the pathogen genome and transcriptome sequencing that alpha-1,6-mannosyltransferase, epimerase, P450, MFS transcripter, methyl transferase, polyketide synthase, monooxygenase and 17-beta-hydroxysteroid dehydrogenase etc. might be members of a gene cluster and responsible for the toxin synthesis, which was a very great progress on the identification of genes related to toxin biosynthesis, even though there was no idea that those genes how to work in synergistic way so far.
Although C. lunata has greatly close relation to B. maydis and both they are maize pathogens, there are distinct differences in pathogenicity-associated families. MFS transporters, G-protein coupled receptors, protein kinases and proteases families in C. lunata being involved in transport, signal transduction or degradation are expanded in relative to B. maydis. Cytochrome P450, lipases, glycoside hydrolases and polyketide synthases families for detoxification, hydrolysis or secondary metabolites biosynthesis are contracted compared to B. maydis, which are expected to be crucial for the fungal survival in varied stress environments.
Comparative transcriptional studies for the wild type strain with low virulence and its variant with high virulence provided a global realization of gene expression response to the directly selective pressures of resistant host plants, which would contribute to better understand the virulence differentiation of C. lunata and clarify the biosynthesis pathways of secondary metabolites such as toxin and melanin. Results suggested that toxin and melanin may share some biosynthesis related genes or regulation genes, and gene clusters for the toxin and melanin biosynthesis may overlap. Nevertheless toxin and melanin were synergistic pathogenic factors to maize, which might take place in virulence differentiation induced by host selective pressures. In other words, an unknown crossovers regulation mode of toxin and melanin may govern global virulence performance. For example, brn1 (CL07173)-encoded 1,3,8-trihydroxynaphthalene reductase is not just involved in melanin biosynthesis but also mediates toxin biosynthesis in C. lunata [78].
Although the identification of PHI genes in the genome wide and pathogenicity variation analysis in transcriptome level were performed, there are still some limitations in this work. The identification of PHI genes and the family classification were based on Blastp search against corresponding databases. Nevertheless the function of homologous genes in different organisms may be different, thus the specific function of the interested genes especially PHI genes in C. lunata should be further confirmed experimentally.
Conclusions
In conclusion, we report the genome sequence and comparative genome analysis, and conduct transcriptional regulation of pathogenicity variation in the plant pathogenic fungus C. lunata. The genome and transcriptome data should facilitate the identification of candidate genes and accelerate the molecular studies on biology, pathogenic mechanism and virulence differentiation of C. lunata. The candidate genes for plant-pathogen interaction are worthy of interest in C. lunata.
Methods
Fungal strains
C. lunata CX-3 strain was selected for genome sequence since it is highly virulent to maize and caused typical lesion on the maize leaves. Its colony morphology on PDA plate at 28°C is less aerial hyphae [9]. The strain is stored in our laboratory and used for the study of pathogenicity mechanism of the pathogen for ten years. The strain is maintained on potato dextrose agar (PDA) medium at 4°C or silicone beads at −20°C.
Genome sequencing and assembly
The whole genome of C. lunata CX-3 strain was de novo sequenced using Illumina, the next generation sequencing technology. Two DNA libraries with 700 bp and 5 kb insert fragments were constructed and Paired-End sequenced with the Illumina Genome Analyzer at the Beijing Genomics Institute (BGI, Shenzhen, China). The genome sequence was assembled by SOAPdenovo software (BGI) [79].
Gene prediction and annotation
The genes structures of the C. lunata CX-3 genome were predicted using Augustus software [80], with the annotated gene information of M. grisea as a reference. The predicted genes were inspected manually to maximize the accuracy of gene prediction. All questionable open reading frames were individually searched against the NCBI (reference proteins) refseq_protein database by Blastp [20]. Repetitive sequences were predicted using RepeatMasker 3.3.0 [81], and the database version and the search engine were 20110920 and rmblastn 2,2,27+, respectively. The potential secreted proteins were screened by combining Target 1.1 [82], SignalP 4.1 Server [83], TMHMM Server v. 2.0 [84] and Big-GPI softwares [85]. Small cystein-rich proteins (SCRPs) were identified from predicted scecreted proteins based on their expected sequence characteristics [86]. Secreted proteins with 20 to 150 amino acids and at least four cysteins were served as putative SCRPs.
Orthology and phylogenomic analysis
Orthologous protein domains in fungi were used for phylogenomic analysis by the Inparanoid 7.0 database [87]. In total, 1328 orthologous proteins were screened out between used fungi with a cutoff E value of 1e-20 and aligned using Clustal W 2.1 [88]. The concatenated amino acid sequences was used to created a neighbor-joining tree using with TREE-PUZZLE program with the Dayhoff model [89]. The divergence time between species was estimated using r8s software with the Langley-Fitch medel [90] by calibrating with the origin of the Ascomycota at 500–650 million years ago [91].
Protein family classifications
The protein families of C. lunata CX-3 genome were classified by Pfam analysis (http://pfam.sanger.ac.uk/). To identify virulence-associated genes, Blastp searches of the C. lunata CX-3 genome were performed against protein sequences in the pathogen-host interaction database (version 3.2, http://www.phi-base.org/) with a cutoff E value of 1e-5. G-protein-coupled receptors were screened through local Blastp against GPCRDB database (http://www.gpcr.org/7tm/) with best hits and further confirmed by searching seven transmembrane helices. Pth11-like GPCRs in C. lunata were identified by local Blastp searching against Pth11 with a cutoff E value of 1e-10 [74]. Carbohydrate-active enzymes were classified using local Blastp searching against the CAZy database (http://www.cazy.org/). Protease families and kinases were identified by local Blastp analysis respectively against the MEROPS peptidase database with a cutoff E value of 1e-20 and KinBase database with a cutoff E value of 1e-10 [92]. Transporters were analyzed through local Blastp against Transporter Classification Database with a cutoff E value of 1e-40 [93]. Cytochrome P450s were classified based on Blastp alignment against P450 database with a cutoff E value of 1e-10 (http://drnelson.uthsc.edu/CytochromeP450.html).
Analysis of genes involved in secondary metabolism
PKS, NRPS and NRPS-PKS were identified through submitting the whole genome data to SMURF system [94]. Different PKS or NRPS proteins were submitted to SBSPKS database to modulate and extract conserved domains by Blast search [95]. Phylogenetic analysis for the conserved domains was conducted by aligning the domain sequences and creating the Neighbor-joined tree with MEGE 5.0 using a Dayhoff model with 1,000 of bootstrap method. PKSs in C. lunata CX-3 and B. maydis were used in this analysis. In addition, toxin-associated PKSs used in the analysis included Gibberella zeae PKS4 (GenBank: ABB90283) for the biosynthesis of zearalenones, Aspergillus ochraceus PKS (GenBank: AAT92023) for ochratoxin, Gibberella moniliformis Fum1p (GenBank: AAD43562) for fumonisin, Alternaria alternate ACTTS3 (GenBank: BAJ14522) for ACT-toxin, B. maydis PKS1 (GenBank: AAB08104) and PKS2 (GenBank: AAR90257) for T-toxin. The melanin-associated PKSs used in the analysis included B. maydis PKS18 (GenBank: AAR90272), A. fumigatus Alb1p (GenBank: ACJ13039), Ceratocystis resinifera PKS1 (GenBank: AAO60166), Colletotrichum lagenarium PKS1 (GenBank: BAA18956), Chaetomium globosum PKS (GenBank: AFP82905), A. alternate ALM1 (GenBank: BAK64048), Ascochyta rabie PKS1 (GenBank: ACS74449), S. turcica PKS (GenBank: AEE68981), Bipolaris oryzae PKS1 (GenBank: BAD22832). The mycotoxin-associated NRPSs used for phylogenetic and modular analyses included Cochliobolus carbonum NRPS (GenBank: AAA33023) for HC-toxin [96], Fusarium equiseti NRPS (GenBank: CAA79245) for enniatin [97], Tolypocladium inflatum NRPS (GenBank: CAA82227) for cyclosporin [98], A. fumigatus Glip (GenBank: AAW03307) for gliotoxin [99], and A. alternate NRPS (GenBank: AAF01762) for AM-toxin [100].
Transcriptome analysis
C. lunata stains WS18 and its variant WS18-Pob21-11 were selected for transcriptome analysis using RNA-seq technology. Transcriptomes of WS18-Pob21-11 and WS18 were served as test group and control. C. lunata WS18 and its variant WS18-Pob21-11 obtained by the successive induction of resistant maize population Pob21 were lowly and highly virulent to Pob21, respectively [4]. To extract total RNA of WS18 and WS18-Pob21-11, their mycelia were cultured in potato-dextrose (PD) liquid medium with 140 rpm of shaking at 28°C for 96 h. The incubated medium was harvested by filtration with sterile cheese cloth, washed using sterilized Milli-Q and it was grinded in liquid nitrogen. Then, total RNA was extracted using Trizol™ Reagent (Invitrogen, USA) and treated with DNase I (Takara, Japan) for removing genome DNA with the recommended method by the manufacturer. Messenger RNA was extracted, cut off randomly and then reverse transcribed into cDNA.
A sequencing library was constructed by PCR amplification of cDNA for tag preparation. The tags were Paired-End sequenced using de novo sequencing technique with Illumina HiSeq™ 2000. Tags only containing adapter sequences were omitted, then the remaining tags were mapped to the C. lunata CX-3 genome or annotated genes [19]. For comparing expression of each mapped gene between samples, the abundance of each tag was transformed to the value of transcripts per million tags (TPM) [19]. The mapped genes with a cutoff of P ≤ 0.05 and FDR ≤0.001 were identified as significantly differential genes [101].
Availability of supporting data
This Whole Genome Shotgun project has been deposited at DDBJ/EMBL/GenBank under the accession JFHG00000000 for C. lunata CX-3. The version described in this paper is version JFHG01000000. Phylogenetic data (alignments and phylogenetic trees) can be available from the Dryad Digital Repository: http://doi.org/10.5061/dryad.22v18.
Electronic supplementary material
Additional file 1: Comparative genomics analysis of C. lunata. The file is composed by 13 tables in separate excel sheets, in which the additional information refer to comparative genomic properties and gene family analysis of C. lunata with other pathogenic fungi. Table S1. The number of genes for selected gene families in C. lunata and other Ascomycetes. Table S2. Major protein families involved in pathogen-host interaction in C. lunata and other Ascomycetes. Table S3. Putative transposase-encoding genes. Table S4. Glycoside hydrolase in C. lunata and other fungi. Table S5. Protease genes in different fungal genomes, classed by MEROPS families. Table S6. Transporters in C. lunata CX-3, C. lunata m118 and B. maydis C5. Table S7. Drug transporters of C. lunata and other fungi. Table S8. – Cytochrome P450 genes in different fungal genomes, classed by CYP families. Table S9. G-protein-coupled receptors in different fungal genomes. Table S10. The number of protein kinases in different fungal genomes. Table S11. Proteins for MAP Kinase pathway in the C. lunata CX-3 genome. Table S12. The pupative small, cysteine-rich peptides encoding genes in C. lunata CX-3 genome. Table S13. Differentially expressed genes in WS18-Pob21-11 compared to WS18. (XLS 146 KB)
Additional file 2: Figure S1: Phylogenetic and domain analyses of C. lunata CX-3 NRPSs compared with known NRPS from other fungi for mycotoxin biosynthesis. (DOC 2 MB)
Acknowledgements
We are grateful to Chengshu Wang research group in Institute of Plant Physiology and Ecology, Chinese Academy of Sciences for their help in phylogenomic analysis, protein family classification, RIP analysis and the identification of core genes for secondary metabolites. We are also grateful to other members of this research group including Lili Fan, Chuanjin Yu, Meng Wang, Saravanakumar Kandasamy, Zhiquan Cao and Jianan Sun for their help in data analysis and manuscript revise. This work was supported by grants from China Agriculture Research System (CARS-02), and the National Natural Science Foundation of China (Nos. 31171798).
Abbreviations
- MAPK
Mitogen activated protein kinase
- PKS
Polyketide synthases
- CX-3
C. lunata CX-3
- m118
C. lunata m118
- C5
B. maydis C5
- GI
Genomic island
- PAI
Pathogenicity island
- ARI
Antibiotic resistance island
- MYA
Million years ago
- GH
Glycoside hydrolase
- ABC
ATP-binding cassette
- RIP
Repeat-induced point mutation
- MFS
Major facilitator superfamily
- MDR
Multidrug resistance
- PDR
Pleiotropic drug resistance
- DHA
Drug:H+ antiporter
- CYP
Cytochrome P450 enzyme
- GPCR
G-protein-coupled receptor
- cAMP
Cyclic AMP
- HK
Histidine kinase
- HST
Host specific toxin
- NHST
Non host specific toxin
- NRPS
Non-ribosomal peptide synthetase
- HYBRID
NRPS-PKS hybrid
- SCRP
Small cysteine-rich protein
- LysM
Lysin motif
- PHI
Pathogen-host interaction
- PAMP
Pathogen associated molecular pattern
- PTI
PAMP-triggered immunity
- ETI
Effector-triggered immunity
- TPM
Transcripts per million tags.
Footnotes
Competing interests
The authors declare no competing financial interests.
Authors’ contributions
SG carried out DNA and RNA extraction, phylogenetic analysis, gene annotation, protein family classification and transcriptome analysis, and drafted the manuscript. YQL conducted RIP analysis, participated in phylogenetic analysis, transcriptome analysis and the design of the study, and drafted the manuscript. JG carried out the identification of secreted proteins and drafted the manuscript. YS participated in nucleic acid extraction, gene annotation and transcriptome analysis. KF participated in gene annotation and transcriptome analysis. YYL revised the manuscript. JC conceived of the study, participated in its design and helped to draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
Shigang Gao, Email: shggao@163.com.
Yaqian Li, Email: yaqianli2008@163.com.
Jinxin Gao, Email: jinxingao@yeah.net.
Yujuan Suo, Email: yujsuo@163.com.
Kehe Fu, Email: fkh0112@sina.com.
Yingying Li, Email: liyingying16@163.com.
Jie Chen, Email: jiechen59@sjtu.edu.cn.
References
- 1.Dai FC, Gao WD, Wu RJ, Jin XH. A noticeable corn disease: Curvularia leaf spot. ACTA Phytopathologica Sinica. 1995;25:330. [Google Scholar]
- 2.Dai FC, Wang XM, Zhu ZD, Gao WD, Huo NX. Curvularia leaf spot of maize: pathogens and varietal resistance. ACTA Phytopathologica Sinica. 1998;2:123–129. [Google Scholar]
- 3.Macri F, Lenna P. Leaf corn blight incited by Curvularia lunata (Wakk.) Boed. J Plant Pathol. 1974;10:27–35. [Google Scholar]
- 4.Gao S, Liu T, Li Y, Wu Q, Fu K, Chen J. Understanding resistant germplasm-induced virulence variation through analysis of proteomics and suppression subtractive hybridization in a maize pathogen Curvularia lunata. Proteomics. 2012;12:3524–3535. doi: 10.1002/pmic.201200105. [DOI] [PubMed] [Google Scholar]
- 5.Feng J, Gao Z, Xue C, Zhuang J, Chen J, Bai S. The pathogenesis of the cell-degrading enzymes produced by Curvularia lunata. Rain Fed Crops. 2002;22:164–167. [Google Scholar]
- 6.Liu T, Liu L, Jiang X, Huang X, Chen J. A new furanoid toxin produced by Curvularia lunata, the causal agent of maize Curvularia leaf spot. Can J Plant Pathol. 2009;31:22–27. doi: 10.1080/07060660909507568. [DOI] [Google Scholar]
- 7.Xu SF, Chen J, Liu LX, Wang XF, Huang XL, Zhai YH. Proteomics associated with virulence differentiation of Curvularia lunata in maize in China. J Integr Plant Biol. 2007;49:487–496. doi: 10.1111/j.1744-7909.2007.00469.x. [DOI] [Google Scholar]
- 8.Liu T, Xu S, Liu L, Zhou F, Hou J, Chen J. Cloning and characteristics of Brn1 gene in Curvularia lunata causing leaf spot in maize. Eur J Plant Pathol. 2011;131:211–219. doi: 10.1007/s10658-011-9800-8. [DOI] [Google Scholar]
- 9.Gao SG, Zhou FH, Liu T, Li YY, Chen J. A MAP kinase gene, Clk1, is required for conidiation and pathogenicity in the phytopathogenic fungus Curvularia lunata. J Basic Microbiol. 2012;53:214–223. doi: 10.1002/jobm.201100518. [DOI] [PubMed] [Google Scholar]
- 10.Wang JY, Chen J. Cloning and functional analysis of Clm1 in Curvularia lunata. Acta Phytopathologica Sinica. 2011;41:464–472. [Google Scholar]
- 11.Biggins JB, Ternei MA, Brady SF. Malleilactone, a polyketide synthase-derived virulence factor encoded by the cryptic secondary metabolome of Burkholderia pseudomallei group pathogens. J Am Chem Soc. 2012;134:13192–13195. doi: 10.1021/ja3052156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baker SE, Kroken S, Inderbitzin P, Asvarak T, Li BY, Shi L, Yoder OC, Turgeon BG. Two polyketide synthase-encoding genes are required for biosynthesis of the polyketide virulence factor, T-toxin, by Cochliobolus heterostrophus. Mol Plant Microbe Interact. 2006;19:139–149. doi: 10.1094/MPMI-19-0139. [DOI] [PubMed] [Google Scholar]
- 13.Liu T, Liu LX, Liu ZC, Hou JM, Gao SG, Zhou FH, Chen J. Construction and characterization of a norm a lized full-length cDNA library of Curvularia lunata. Acta Phytopathol Sinica. 2010;40:250–257. [Google Scholar]
- 14.Jing J, Liu T, Chen J. Construction and characterization of yeast two hybrid cDNA library of Curvularia lunata. Acta Phytopathol Sinica. 2012;42:93–96. [Google Scholar]
- 15.Bayram O, Krappmann S, Ni M, Bok JW, Helmstaedt K, Valerius O, Braus-Stromeyer S, Kwon NJ, Keller NP, Yu JH, Braus GH. VelB/VeA/LaeA complex coordinates light signal with fungal development and secondary metabolism. Science. 2008;320:1504–1506. doi: 10.1126/science.1155888. [DOI] [PubMed] [Google Scholar]
- 16.Condon BJ, Leng Y, Wu D, Bushley KE, Ohm RA, Otillar R, Martin J, Schackwitz W, Grimwood J, MohdZainudin N. Comparative genome structure, secondary metabolite, and effector coding capacity across Cochliobolus pathogens. PLoS Genet. 2013;9:e1003233. doi: 10.1371/journal.pgen.1003233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cuomo CA, Guldener U, Xu JR, Trail F, Turgeon BG, Di Pietro A, Walton JD, Ma LJ, Baker SE, Rep M, Adam G, Antoniw J, Baldwin T, Calvo S, Chang YL, Decaprio D, Gale LR, Gnerre S, Goswami RS, Hammond-Kosack K, Harris LJ, Hilburn K, Kennell JC, Kroken S, Magnuson JK, Mannhaupt G, Mauceli E, Mewes HW, Mitterbauer R, Muehlbauer G, et al. The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science. 2007;317:1400–1402. doi: 10.1126/science.1143708. [DOI] [PubMed] [Google Scholar]
- 18.Dean RA, Talbot NJ, Ebbole DJ, Farman ML, Mitchell TK, Orbach MJ, Thon M, Kulkarni R, Xu JR, Pan H, Read ND, Lee YH, Carbone I, Brown D, Oh YY, Donofrio N, Jeong JS, Soanes DM, Djonovic S, Kolomiets E, Rehmeyer C, Li W, Harding M, Kim S, Lebrun MH, Bohnert H, Coughlan S, Butler J, Calvo S, Ma LJ. The genome sequence of the rice blast fungus Magnaporthe grisea. Nature. 2005;434:980–986. doi: 10.1038/nature03449. [DOI] [PubMed] [Google Scholar]
- 19.Zheng P, Xia Y, Xiao G, Xiong C, Hu X, Zhang S, Zheng H, Huang Y, Zhou Y, Wang S, Zhao GP, Liu X, St Leger RJ, Wang C. Genome sequence of the insect pathogenic fungus Cordyceps militaris, a valued traditional Chinese medicine. Genome Biol. 2011;12:R116. doi: 10.1186/gb-2011-12-11-r116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao Q, Jin K, Ying SH, Zhang Y, Xiao G, Shang Y, Duan Z, Hu X, Xie XQ, Zhou G, Peng G, Luo Z, Huang W, Wang B, Fang W, Wang S, Zhong Y, Ma LJ, St Leger RJ, Zhao GP, Pei Y, Feng MG, Xia Y, Wang C. Genome sequencing and comparative transcriptomics of the model entomopathogenic fungi Metarhizium anisopliae and M. acridum. PLoS Genet. 2011;7:e1001264. doi: 10.1371/journal.pgen.1001264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fedorova ND, Khaldi N, Joardar VS, Maiti R, Amedeo P, Anderson MJ, Crabtree J, Silva JC, Badger JH, Albarraq A, Angiuoli S, Bussey H, Bowyer P, Cotty PJ, Dyer PS, Egan A, Galens K, Fraser-Liggett CM, Haas BJ, Inman JM, Kent R, Lemieux S, Malavazi I, Orvis J, Roemer T, Ronning CM, Sundaram JP, Sutton G, Turner G, Venter JC, et al. Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genet. 2008;4:e1000046. doi: 10.1371/journal.pgen.1000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McElwain JC, Punyasena SW. Mass extinction events and the plant fossil record. Trends Ecol Evol. 2007;22:548–557. doi: 10.1016/j.tree.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 23.Winnenburg R, Baldwin TK, Urban M, Rawlings C, Kohler J, Hammond-Kosack KE. PHI-base: a new database for pathogen host interactions. Nucleic Acids Res. 2006;34:D459–D464. doi: 10.1093/nar/gkj047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kang S, Lebrun MH, Farrall L, Valent B. Gain of virulence caused by insertion of a Pot3 transposon in a Magnaporthe grisea avirulence gene. Mol Plant Microbe Interact. 2001;14:671–674. doi: 10.1094/MPMI.2001.14.5.671. [DOI] [PubMed] [Google Scholar]
- 25.Hood ME, Katawczik M, Giraud T. Repeat-induced point mutation and the population structure of transposable elements in Microbotryum violaceum. Genetics. 2005;170:1081–1089. doi: 10.1534/genetics.105.042564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Galagan JE, Selker EU. RIP: the evolutionary cost of genome defense. Trends Genet. 2004;20:417–423. doi: 10.1016/j.tig.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 27.Dangl JL, Jones JD. Plant pathogens and integrated defence responses to infection. Nature. 2001;411:826–833. doi: 10.1038/35081161. [DOI] [PubMed] [Google Scholar]
- 28.Dimroth P. Primary sodium ion translocating enzymes. Biochim Biophys Acta. 1997;1318:11–51. doi: 10.1016/S0005-2728(96)00127-2. [DOI] [PubMed] [Google Scholar]
- 29.Paulsen IT, Brown MH, Skurray RA. Proton-dependent multidrug efflux systems. Microbiol Rev. 1996;60:575–608. doi: 10.1128/mr.60.4.575-608.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Waard MA. Significance of ABC transporters in fungicide sensitivity and resistance. Pestic Sci. 1997;51:271–275. doi: 10.1002/(SICI)1096-9063(199711)51:3<271::AID-PS642>3.0.CO;2-#. [DOI] [Google Scholar]
- 31.Roohparvar R, De Waard MA, Kema GH, Zwiers LH. MgMfs1, a major facilitator superfamily transporter from the fungal wheat pathogen Mycosphaerella graminicola, is a strong protectant against natural toxic compounds and fungicides. Fungal Genet Biol. 2007;44:378–388. doi: 10.1016/j.fgb.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 32.Crešnar B, Petrič S. Cytochrome P450 enzymes in the fungal kingdom. Biochimica et Biophysica Acta (BBA)-Proteins & Proteomics. 2011;1814:29–35. doi: 10.1016/j.bbapap.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 33.Nelson DR. The cytochrome p450 homepage. Hum Genomics. 2009;4:59–65. doi: 10.1186/1479-7364-4-1-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kimura M, Tokai T, Takahashi-Ando N, Ohsato S, Fujimura M. Molecular and genetic studies of fusarium trichothecene biosynthesis: pathways, genes, and evolution. Biosci Biotechnol Biochem. 2007;71:2105–2123. doi: 10.1271/bbb.70183. [DOI] [PubMed] [Google Scholar]
- 35.Butchko RA, Plattner RD, Proctor RH. Deletion analysis of FUM genes involved in tricarballylic ester formation during fumonisin biosynthesis. J Agric Food Chem. 2006;54:9398–9404. doi: 10.1021/jf0617869. [DOI] [PubMed] [Google Scholar]
- 36.Bojja RS, Cerny RL, Proctor RH, Du L. Determining the biosynthetic sequence in the early steps of the fumonisin pathway by use of three gene-disruption mutants of Fusarium verticillioides. J Agr Food Chem. 2004;52:2855–2860. doi: 10.1021/jf035429z. [DOI] [PubMed] [Google Scholar]
- 37.Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 38.DeZwaan TM, Carroll AM, Valent B, Sweigard JA. Magnaporthe grisea pth11p is a novel plasma membrane protein that mediates appressorium differentiation in response to inductive substrate cues. The Plant Cell Online. 1999;11:2013–2030. doi: 10.1105/tpc.11.10.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Preininger AM, Hamm HE. G protein signaling: insights from new structures. Sci STKE. 2004;2004:re3. doi: 10.1126/stke.2182004re3. [DOI] [PubMed] [Google Scholar]
- 40.Svoboda P, Teisinger J, Novotny J, Bourova L, Drmota T, Hejnova L, Moravcova Z, Lisy V, Rudajev V, Stohr J, Vokurkova A, Svandova I, Durchankova D. Biochemistry of transmembrane signaling mediated by trimeric G proteins. Physiol Res. 2004;53(Suppl 1):S141–S152. [PubMed] [Google Scholar]
- 41.Yu HY, Seo JA, Kim JE, Han KH, Shim WB, Yun SH, Lee YW. Functional analyses of heterotrimeric G protein G alpha and G beta subunits in Gibberella zeae. Microbiology. 2008;154:392–401. doi: 10.1099/mic.0.2007/012260-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen RE, Thorner J. Function and regulation in MAPK signaling pathways: lessons learned from the yeast Saccharomyces cerevisiae. Biochim Biophys Acta. 2007;1773:1311–1340. doi: 10.1016/j.bbamcr.2007.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu JR. Map kinases in fungal pathogens. Fungal Genet Biol. 2000;31:137–152. doi: 10.1006/fgbi.2000.1237. [DOI] [PubMed] [Google Scholar]
- 44.Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 45.Xu J-R, Hamer JE. MAP kinase and cAMP signaling regulate infection structure formation and pathogenic growth in the rice blast fungus Magnaporthe grisea. Genes Dev. 1996;10:2696–2706. doi: 10.1101/gad.10.21.2696. [DOI] [PubMed] [Google Scholar]
- 46.Oide S, Liu J, Yun SH, Wu D, Michev A, Choi MY, Horwitz BA, Turgeon BG. Histidine kinase two-component response regulator proteins regulate reproductive development, virulence, and stress responses of the fungal cereal pathogens Cochliobolus heterostrophus and Gibberella zeae. Eukaryot Cell. 2010;9:1867–1880. doi: 10.1128/EC.00150-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ochiai N, Tokai T, Nishiuchi T, Takahashi-Ando N, Fujimura M, Kimura M. Involvement of the osmosensor histidine kinase and osmotic stress-activated protein kinases in the regulation of secondary metabolism in Fusarium graminearum. Biochem Biophys Res Commun. 2007;363:639–644. doi: 10.1016/j.bbrc.2007.09.027. [DOI] [PubMed] [Google Scholar]
- 48.Torosantucci A, Chiani P, De Bernardis F, Cassone A, Calera JA, Calderone R. Deletion of the two-component histidine kinase gene (CHK1) of Candida albicans contributes to enhanced growth inhibition and killing by human neutrophils in vitro. Infect Immun. 2002;70:985–987. doi: 10.1128/IAI.70.2.985-987.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rep M, van der Does HC, Meijer M, van Wijk R, Houterman PM, Dekker HL, de Koster CG, Cornelissen BJ. A small, cysteine-rich protein secreted by Fusarium oxysporum during colonization of xylem vessels is required for I-3-mediated resistance in tomato. Mol Microbiol. 2004;53:1373–1383. doi: 10.1111/j.1365-2958.2004.04177.x. [DOI] [PubMed] [Google Scholar]
- 50.Lauge R, Goodwin PH, de Wit PJ, Joosten MH. Specific HR-associated recognition of secreted proteins from Cladosporium fulvum occurs in both host and non-host plants. Plant J. 2000;23:735–745. doi: 10.1046/j.1365-313x.2000.00843.x. [DOI] [PubMed] [Google Scholar]
- 51.Westerink N, Brandwagt BF, de Wit PJ, Joosten MH. Cladosporium fulvum circumvents the second functional resistance gene homologue at the Cf-4 locus (Hcr9-4E ) by secretion of a stable avr4E isoform. Mol Microbiol. 2004;54:533–545. doi: 10.1111/j.1365-2958.2004.04288.x. [DOI] [PubMed] [Google Scholar]
- 52.van den Burg HA, Westerink N, Francoijs KJ, Roth R, Woestenenk E, Boeren S, de Wit PJ, Joosten MH, Vervoort J. Natural disulfide bond-disrupted mutants of AVR4 of the tomato pathogen Cladosporium fulvum are sensitive to proteolysis, circumvent Cf-4-mediated resistance, but retain their chitin binding ability. J Biol Chem. 2003;278:27340–27346. doi: 10.1074/jbc.M212196200. [DOI] [PubMed] [Google Scholar]
- 53.van den Burg HA, Spronk CA, Boeren S, Kennedy MA, Vissers JP, Vuister GW, de Wit PJ, Vervoort J. Binding of the AVR4 elicitor of Cladosporium fulvum to chitotriose units is facilitated by positive allosteric protein-protein interactions: the chitin-binding site of AVR4 represents a novel binding site on the folding scaffold shared between the invertebrate and the plant chitin-binding domain. J Biol Chem. 2004;279:16786–16796. doi: 10.1074/jbc.M312594200. [DOI] [PubMed] [Google Scholar]
- 54.Stephenson SA, Hatfield J, Rusu AG, Maclean DJ, Manners JM. CgDN3: an essential pathogenicity gene of colletotrichum gloeosporioides necessary to avert a hypersensitive-like response in the host Stylosanthes guianensis. Mol Plant Microbe Interact. 2000;13:929–941. doi: 10.1094/MPMI.2000.13.9.929. [DOI] [PubMed] [Google Scholar]
- 55.Marx F. Small, basic antifungal proteins secreted from filamentous ascomycetes: a comparative study regarding expression, structure, function and potential application. Appl Microbiol Biotechnol. 2004;65:133–142. doi: 10.1007/s00253-004-1600-z. [DOI] [PubMed] [Google Scholar]
- 56.Mygind PH, Fischer RL, Schnorr KM, Hansen MT, Sönksen CP, Ludvigsen S, Raventós D, Buskov S, Christensen B, De Maria L. Plectasin is a peptide antibiotic with therapeutic potential from a saprophytic fungus. Nature. 2005;437:975–980. doi: 10.1038/nature04051. [DOI] [PubMed] [Google Scholar]
- 57.Marcet-Houben M, Ballester AR, de la Fuente B, Harries E, Marcos JF, Gonzalez-Candelas L, Gabaldon T. Genome sequence of the necrotrophic fungus Penicillium digitatum, the main postharvest pathogen of citrus. BMC Genomics. 2012;13:646. doi: 10.1186/1471-2164-13-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.De Wit PJ, Mehrabi R, Van den Burg HA, Stergiopoulos I. Fungal effector proteins: past, present and future. Mol Plant Pathol. 2009;10:735–747. doi: 10.1111/j.1364-3703.2009.00591.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de Jonge R, Thomma BP. Fungal LysM effectors: extinguishers of host immunity? Trends Microbiol. 2009;17:151–157. doi: 10.1016/j.tim.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 60.Bolton MD, van Esse HP, Vossen JH, de Jonge R, Stergiopoulos I, Stulemeijer IJ, van den Berg GC, Borras-Hidalgo O, Dekker HL, de Koster CG, de Wit PJ, Joosten MH, Thomma BP. The novel Cladosporium fulvum lysin motif effector Ecp6 is a virulence factor with orthologues in other fungal species. Mol Microbiol. 2008;69:119–136. doi: 10.1111/j.1365-2958.2008.06270.x. [DOI] [PubMed] [Google Scholar]
- 61.Muller O, Schreier PH, Uhrig JF. Identification and characterization of secreted and pathogenesis-related proteins in Ustilago maydis. Mol Genet Genomics. 2008;279:27–39. doi: 10.1007/s00438-007-0291-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim S, Ahn IP, Rho HS, Lee YH. MHP1, a Magnaporthe grisea hydrophobin gene, is required for fungal development and plant colonization. Mol Microbiol. 2005;57:1224–1237. doi: 10.1111/j.1365-2958.2005.04750.x. [DOI] [PubMed] [Google Scholar]
- 63.Coppin E, Silar P. Identification of PaPKS1, a polyketide synthase involved in melanin formation and its use as a genetic tool in Podospora anserina. Mycol Res. 2007;111:901–908. doi: 10.1016/j.mycres.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 64.Thompson JE, Fahnestock S, Farrall L, Liao DI, Valent B, Jordan DB. The second naphthol reductase of fungal melanin biosynthesis in Magnaporthe grisea: tetrahydroxynaphthalene reductase. J Biol Chem. 2000;275:34867–34872. doi: 10.1074/jbc.M006659200. [DOI] [PubMed] [Google Scholar]
- 65.Keller NP, Hohn TM. Metabolic pathway gene clusters in filamentous fungi. Fungal Genet Biol. 1997;21:17–29. doi: 10.1006/fgbi.1997.0970. [DOI] [PubMed] [Google Scholar]
- 66.Fox EM, Gardiner DM, Keller NP, Howlett BJ. A Zn(II)2Cys6 DNA binding protein regulates the sirodesmin PL biosynthetic gene cluster in Leptosphaeria maculans. Fungal Genet Biol. 2008;45:671–682. doi: 10.1016/j.fgb.2007.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen H, Lee MH, Daub ME, Chung KR. Molecular analysis of the cercosporin biosynthetic gene cluster in Cercospora nicotianae. Mol Microbiol. 2007;64:755–770. doi: 10.1111/j.1365-2958.2007.05689.x. [DOI] [PubMed] [Google Scholar]
- 68.Schell MA. Control of Virulence and Pathogenicity Genes of Ralstonia Solanacearum by an Elaborate Sensory Network. Annu Rev Phytopathol. 2000;38:263–292. doi: 10.1146/annurev.phyto.38.1.263. [DOI] [PubMed] [Google Scholar]
- 69.Bolker M. Ustilago maydis–a valuable model system for the study of fungal dimorphism and virulence. Microbiology. 2001;147:1395–1401. doi: 10.1099/00221287-147-6-1395. [DOI] [PubMed] [Google Scholar]
- 70.Stergiopoulos I, de Wit PJ. Fungal effector proteins. Annu Rev Phytopathol. 2009;47:233–263. doi: 10.1146/annurev.phyto.112408.132637. [DOI] [PubMed] [Google Scholar]
- 71.Houterman PM, Cornelissen BJ, Rep M. Suppression of plant resistance gene-based immunity by a fungal effector. Plos Pathog. 2008;4:e1000061. doi: 10.1371/journal.ppat.1000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Leonard G, Richards TA. Genome-scale comparative analysis of gene fusions, gene fissions, and the fungal tree of life. Proc Natl Acad Sci U S A. 2012;109:21402–21407. doi: 10.1073/pnas.1210909110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Feschotte C. Transposable elements and the evolution of regulatory networks. Nat Rev Genet. 2008;9:397–405. doi: 10.1038/nrg2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Galagan JE, Calvo SE, Cuomo C, Ma LJ, Wortman JR, Batzoglou S, Lee SI, Basturkmen M, Spevak CC, Clutterbuck J, Kapitonov V, Jurka J, Scazzocchio C, Farman M, Butler J, Purcell S, Harris S, Braus GH, Draht O, Busch S, D'Enfert C, Bouchier C, Goldman GH, Bell-Pedersen D, Griffiths-Jones S, Doonan JH, Yu J, Vienken K, Pain A, Freitag M. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature. 2005;438:1105–1115. doi: 10.1038/nature04341. [DOI] [PubMed] [Google Scholar]
- 75.Kubicek CP, Herrera-Estrella A, Seidl-Seiboth V, Martinez DA, Druzhinina IS, Thon M, Zeilinger S, Casas-Flores S, Horwitz BA, Mukherjee PK. Comparative genome sequence analysis underscores mycoparasitism as the ancestral life style of Trichoderma. Genome Biol. 2011;12:R40. doi: 10.1186/gb-2011-12-4-r40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Clarkson JP, Staveley J, Phelps K, Young CS, Whipps JM. Ascospore release and survival in Sclerotinia sclerotiorum. Mycol Res. 2003;107:213–222. doi: 10.1017/S0953756203007159. [DOI] [PubMed] [Google Scholar]
- 77.Schafer W. Molecular mechanisms of fungal pathogenicity to plants. Annu Rev Phytopathol. 1994;32:461–477. doi: 10.1146/annurev.py.32.090194.002333. [DOI] [Google Scholar]
- 78.Gao JX, Liu T, Chen J. Insertional mutagenesis and cloning of the gene required for the biosynthesis of the non-host-specific toxin in Cochliobolus lunatus that causes maize leaf spot. Phytopathology. 2014;104:332–339. doi: 10.1094/PHYTO-07-13-0190-R. [DOI] [PubMed] [Google Scholar]
- 79.Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25:1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 80.Stanke M, Tzvetkova A, Morgenstern B. AUGUSTUS at EGASP: using EST, protein and genomic alignments for improved gene prediction in the human genome. Genome Biol. 2006;7(Suppl 1):S11. doi: 10.1186/gb-2006-7-s1-s11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Server Rhttp://www.repeatmasker.org/
- 82.Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- 83.Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 84.Chen Y, Yu P, Luo J, Jiang Y. Secreted protein prediction system combining CJ-SPHMM, TMHMM, and PSORT. Mamm Genome. 2003;14:859–865. doi: 10.1007/s00335-003-2296-6. [DOI] [PubMed] [Google Scholar]
- 85.Eisenhaber B, Bork P, Eisenhaber F. Sequence properties of GPI-anchored proteins near the omega-site: constraints for the polypeptide binding site of the putative transamidase. Protein Eng. 1998;11:1155–1161. doi: 10.1093/protein/11.12.1155. [DOI] [PubMed] [Google Scholar]
- 86.Graham MA, Silverstein KA, Cannon SB, VandenBosch KA. Computational identification and characterization of novel genes from legumes. Plant Physiol. 2004;135:1179–1197. doi: 10.1104/pp.104.037531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ostlund G, Schmitt T, Forslund K, Kostler T, Messina DN, Roopra S, Frings O, Sonnhammer EL. InParanoid 7: new algorithms and tools for eukaryotic orthology analysis. Nucleic Acids Res. 2010;38:D196–D203. doi: 10.1093/nar/gkp931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 89.Kumar S, Nei M, Dudley J, Tamura K. MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform. 2008;9:299–306. doi: 10.1093/bib/bbn017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Taylor JW, Berbee ML. Dating divergences in the Fungal Tree of Life: review and new analyses. Mycologia. 2006;98:838–849. doi: 10.3852/mycologia.98.6.838. [DOI] [PubMed] [Google Scholar]
- 91.Lucking R, Huhndorf S, Pfister DH, Plata ER, Lumbsch HT. Fungi evolved right on track. Mycologia. 2009;101:810–822. doi: 10.3852/09-016. [DOI] [PubMed] [Google Scholar]
- 92.Schomburg D, Schomburg I. Enzyme databases. Methods Mol Biol. 2010;609:113–128. doi: 10.1007/978-1-60327-241-4_7. [DOI] [PubMed] [Google Scholar]
- 93.Saier MH, Jr, Tran CV, Barabote RD. TCDB: the Transporter Classification Database for membrane transport protein analyses and information. Nucleic Acids Res. 2006;34:D181–D186. doi: 10.1093/nar/gkj001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Khaldi N, Seifuddin FT, Turner G, Haft D, Nierman WC, Wolfe KH, Fedorova ND. SMURF: Genomic mapping of fungal secondary metabolite clusters. Fungal Genet Biol. 2010;47:736–741. doi: 10.1016/j.fgb.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Anand S, Prasad MV, Yadav G, Kumar N, Shehara J, Ansari MZ, Mohanty D. SBSPKS: structure based sequence analysis of polyketide synthases. Nucleic Acids Res. 2010;38:W487–W496. doi: 10.1093/nar/gkq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Scott-Craig JS, Panaccione DG, Pocard JA, Walton JD. The cyclic peptide synthetase catalyzing HC-toxin production in the filamentous fungus Cochliobolus carbonum is encoded by a 15.7-kilobase open reading frame. J Biol Chem. 1992;267:26044–26049. [PubMed] [Google Scholar]
- 97.Haese A, Schubert M, Herrmann M, Zocher R. Molecular characterization of the enniatin synthetase gene encoding a multifunctional enzyme catalysing N-methyldepsipeptide formation in Fusarium scirpi. Mol Microbiol. 1993;7:905–914. doi: 10.1111/j.1365-2958.1993.tb01181.x. [DOI] [PubMed] [Google Scholar]
- 98.Weber G, Schorgendorfer K, Schneider-Scherzer E, Leitner E. The peptide synthetase catalyzing cyclosporine production in Tolypocladium niveum is encoded by a giant 45.8-kilobase open reading frame. Curr Genet. 1994;26:120–125. doi: 10.1007/BF00313798. [DOI] [PubMed] [Google Scholar]
- 99.Gardiner DM, Howlett BJ. Bioinformatic and expression analysis of the putative gliotoxin biosynthetic gene cluster of Aspergillus fumigatus. FEMS Microbiol Lett. 2005;248:241–248. doi: 10.1016/j.femsle.2005.05.046. [DOI] [PubMed] [Google Scholar]
- 100.Johnson RD, Johnson L, Itoh Y, Kodama M, Otani H, Kohmoto K. Cloning and characterization of a cyclic peptide synthetase gene from Alternaria alternata apple pathotype whose product is involved in AM-toxin synthesis and pathogenicity. Mol Plant Microbe Interact. 2000;13:742–753. doi: 10.1094/MPMI.2000.13.7.742. [DOI] [PubMed] [Google Scholar]
- 101.Benjamini Y, Yekutieli D. The control of the false discovery rate in multiple testing under dependency. Ann Stat. 2001;29:1165–1188. doi: 10.1214/aos/1013699998. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Comparative genomics analysis of C. lunata. The file is composed by 13 tables in separate excel sheets, in which the additional information refer to comparative genomic properties and gene family analysis of C. lunata with other pathogenic fungi. Table S1. The number of genes for selected gene families in C. lunata and other Ascomycetes. Table S2. Major protein families involved in pathogen-host interaction in C. lunata and other Ascomycetes. Table S3. Putative transposase-encoding genes. Table S4. Glycoside hydrolase in C. lunata and other fungi. Table S5. Protease genes in different fungal genomes, classed by MEROPS families. Table S6. Transporters in C. lunata CX-3, C. lunata m118 and B. maydis C5. Table S7. Drug transporters of C. lunata and other fungi. Table S8. – Cytochrome P450 genes in different fungal genomes, classed by CYP families. Table S9. G-protein-coupled receptors in different fungal genomes. Table S10. The number of protein kinases in different fungal genomes. Table S11. Proteins for MAP Kinase pathway in the C. lunata CX-3 genome. Table S12. The pupative small, cysteine-rich peptides encoding genes in C. lunata CX-3 genome. Table S13. Differentially expressed genes in WS18-Pob21-11 compared to WS18. (XLS 146 KB)
Additional file 2: Figure S1: Phylogenetic and domain analyses of C. lunata CX-3 NRPSs compared with known NRPS from other fungi for mycotoxin biosynthesis. (DOC 2 MB)