

Abstract
Background
Gastrointestinal nematodes are one of the most serious causes of disease in domestic ruminants worldwide. There is considerable variation in resistance to gastrointestinal nematodes within and between sheep breeds, which appears to be due to underlying genetic diversity. Through selection of resistant animals, rapid genetic progress has been demonstrated in both research and commercial flocks. Recent advances in genome sequencing and genomic technologies provide new opportunities to understand the ovine host response to gastrointestinal nematodes at the molecular level, and to identify polymorphisms conferring nematode resistance.
Results
Divergent lines of Romney and Perendale sheep, selectively bred for high and low faecal nematode egg count, were genotyped using the Illumina® Ovine SNP50 BeadChip. The resulting genome-wide SNP data were analysed for selective sweeps on loci associated with resistance or susceptibility to gastrointestinal nematode infection. Population differentiation using FST and Peddrift revealed sixteen regions, which included candidate genes involved in chitinase activity and the cytokine response. Two of the sixteen regions identified were contained within previously identified QTLs associated with nematode resistance.
Conclusions
In this study we identified fourteen novel regions associated with resistance or susceptibility to gastrointestinal nematodes. Results from this study support the hypothesis that host resistance to internal nematode parasites is likely to be controlled by a number of loci of moderate to small effects.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-15-637) contains supplementary material, which is available to authorized users.
Background
Gastrointestinal nematodes are one of the most serious causes of disease in domestic ruminants worldwide [1, 2]. Production losses due to parasitism are two-fold; the direct cost of anthelmintic treatment and production losses due to ill-thrift and in extreme cases death [3]. In the face of the increasing incidence of anthelmintic resistance and the need to minimise drench residues in animal products, new strategies for control are required [4].
Breeding for host resistance has been shown to be a viable method of nematode control [5]. Host resistance is heritable, with wide variability among individuals, and rapid genetic progress has been demonstrated in both research and commercial flocks [6, 7]. Moreover, computer simulation models have shown that selection for host resistance, using the phenotype low faecal worm egg count, should be stable over a short time frame such as 20 years [8]. This is supported by field data, where it was shown that when gastrointestinal nematodes were exposed to genetically resistant or susceptible sheep over a sustained period of time they showed no evidence of adaptation to their host [9]. These findings support the hypothesis that resistance is determined by many genes each with a relatively small effect [10] and that selection for parasite resistance based on faecal egg count (FEC) is sustainable in the medium to long-term.
With sheep it is possible to manipulate breeding lines to produce strong phenotypic differences, in well-defined pedigrees, in a relatively short space of time. Reduction of variation in genomic regions surrounding a beneficial mutation due to strong and recent selection is known as a “selective sweep”; identification of regions that have undergone selective sweeps can help to reveal genes underlying phenotypic differences. Different statistics pick up different patterns of variation left by selection of a beneficial mutation. Wright’s fixation index (FST) is a single marker test that detects highly differentiated alleles, where positive selection in one area causes larger frequency differences between populations as compared to neutrally evolving alleles. Peddrift [11] is a program that also uses single markers to calculate exact probabilities of allele frequency differences, by using the recorded pedigree structure to take into account minor allele frequencies, genetic drift, founder and sampling effects. Evidence of selection is shown by divergence from the expected distribution (given by a P-value). Unlike FST and Peddrift, tests based on linkage disequilibrium, such as the extended haplotype homozygosity (EHH) statistic and its derivatives, are dependent on SNP spacing and frequency, as they are multi-marker tests. The integrated haplotype score (iHS) [12] and cross population EHH (XP-EHH) [13] tests are both based on extended haplotype. While iHS detect partial selective sweeps a moderate frequency (~50-80%), XP-EHH detects alleles that have risen to near fixation in one population (>80% frequency), but remain polymorphic in the population as a whole. Studies that search for signatures of selective sweeps tend to use multiple tests as they are largely complementary; iHS and XP-EHH have been used to search for recent positive selection in humans [13, 14], as well as other species such as cattle [15, 16].
Recent advances in genomic technologies have provided new opportunities to detect regions in the sheep genome that have undergone selection. The advent of the SNP50 BeadChip provided 54,241 evenly spaced Single Nucleotide Polymorphisms (SNP) across the sheep genome for association analysis. The chip has already been utilised to map causal mutations for traits controlled by a single locus [17–21] and detect signatures of selection among sheep breeds [22, 23]. The identification of genes or linked markers that have a significant association to parasite resistance could accelerate the genetic improvement of resistance to internal nematodes through marker-assisted selection [24]. Additionally, identification of genes under selection in animals selected for resistance or susceptibility to gastrointestinal parasites will help in our understanding of the biological processes underlying this trait.
The SNP50 BeadChip provides a rapid way to detect regions under selection, which can be further fine-mapped using Sequenom® or other technologies. To this end, lines of sheep that have been selected for resistance, resilience, or susceptibility coupled with high-density genetic maps are a key resource that would enable future marker assisted selection of animals without the need for parasite challenge. Here we utilise data from Romney and Perendale parasite selection lines to conduct whole genomic screens for selection, in the hope of identifying loci, within and between the two breeds, that affect individual host resistance or susceptibility to nematode parasites.
Results
Quality control
After quality control (see methods) the final data set consisted of 46,736 SNP for the Romney data set and 48,436 SNP for the Perendale data set. In total 177 Romney (82 high FEC and 95 low FEC) and 146 Perendale (72 high FEC and 74 low FEC) animals passed the quality control. The average MAF of the remaining SNP over all samples was 0.24 (SD = 0.16) in the Romney data set and 0.26 (SD = 0.15) in the Perendale data set.
Genome-wide analysis
Two analytical methods, FST [25] and Peddrift [11], were used to detect differentiation between resistant and susceptible animals based on SNP allele frequencies. While FST is a generic population differentiation statistic, Peddrift is specific to this example in that it was designed to account for structure within the population surveyed.
As individual SNP may not show a strong signal, a 5-SNP moving average (WIN5) was used to identify regions with strong signatures of selection over multiple SNP, which also reduces noise [26]. The average WIN5 FST value in the Romney selection lines (Figure 1A) was 0.0567 (SD = 0.0386), while differentiation was lower in the Perendale selection lines (Figure 1B) with an average WIN5 FST of 0.0299 (SD = 0.0388). A total of 16 genomic regions contained the top 0.1% of markers (Table 1) ranked using WIN5 –log10(combined Peddrift P-values) (Figure 1C), with four regions containing genes that have previously been implicated or are candidates for resistance or susceptibility to gastrointestinal nematodes. The first region, on chromosome 1 (region 2), contains the leukocyte surface antigen CD53, as well as DENND2D and three genes from the chitinase family, acidic mammalian chitinase (CHIA), chitinase 3-like 2 (CHI3L2) and oviduct-specific glycoprotein (OVGP1). Selection was also observed on chromosome 4 (region 5), chromosome 16 (region 14) and chromosome 19 (region 15), containing genes previously implicated in resistance to gastrointestinal nematodes.
Figure 1.
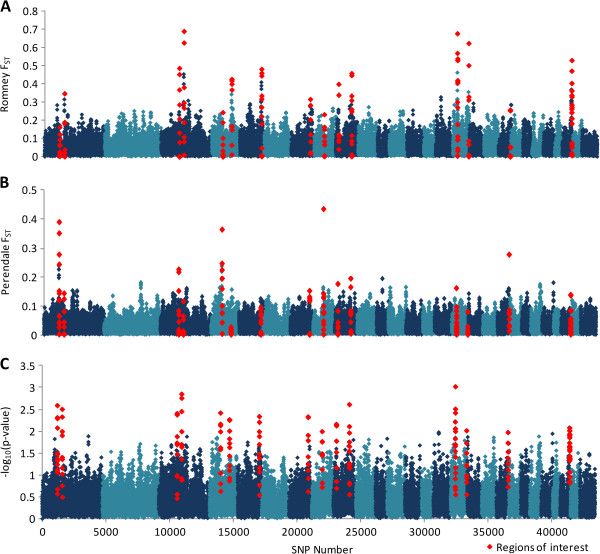
Genome wide signatures of selection. A moving window of 5 FST values between the resistant and susceptible Romney (A) and Perendale (B) lines. (C) A moving average (of 5 SNP) showing the one-tailed probability of the chi-squared distribution of the combined Romney and Perendale Peddrift P-values. Results are expressed as -logl0 (significance probability). Regions of interest as defined by WIN5 –log10 (combined Peddrift P-values) (Table 1) are shown in red.
Table 1.
Genomic regions containing the top 0.1% of SNP, ranked using a moving (5 SNP) average (WIN5) of –log 10 (combined Peddrift P-values)
| Region | Chr | Position (Mb) | SNP50 BeadChip SNP | Top SNP | Peak SNP | Peak SNP rank | F ST Peak | PEDDRIFT WIN5 -log 10 (p-value) | Genes | Candidate gene(s) in region (OARv3.1) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Romney | Perendale | Romney | Perendale | Combined | |||||||||
| 1 | 1 | 66.74-67.43 | 14 | 4 | s43910 | 5 | 0.1632 | 0.3901 | 0.6799 | 3.0134 | 2.5988 | 1 | |
| 2 | 1 | 87.38-88.13 | 10 | 3 | OAR1_93166689 | 7 | 0.3475 | 0.1424 | 2.1441 | 1.3027 | 2.5124 | 18 | CD53, CHI3L2, CHIA, DENND2D |
| 3 | 3 | 78.72-79.32 | 12 | 3 | s08153 | 11 | 0.4868 | 0.2270 | 1.7146 | 1.6205 | 2.4161 | 3 | |
| 4 | 3 | 98.12-98.51 | 9 | 4 | OAR3_104545117_X | 2 | 0.6897 | 0.1148 | 3.1276 | 0.7050 | 2.8569 | 0 | |
| 5 | 4 | 44.99-45.39 | 10 | 4 | OAR4_47833230 | 10 | 0.2439 | 0.3645 | 0.6647 | 3.0036 | 2.4283 | 2 | RELN |
| 6 | 4 | 83.23-83.69 | 12 | 4 | OAR4_88693058_X | 20 | 0.4264 | 0.0256 | 2.9894 | 0.3238 | 2.2755 | 0 | |
| 7 | 5 | 95.21-95.69 | 11 | 3 | OAR5_103935962 | 14 | 0.4816 | 0.0927 | 2.7849 | 0.6253 | 2.3464 | 2 | |
| 8 | 7 | 81.54-82.04 | 9 | 2 | OAR7_89131104 | 15 | 0.3167 | 0.1509 | 1.8827 | 1.6553 | 2.3387 | 10 | |
| 9 | 8 | 41.02-41.48 | 11 | 1 | OAR8_44326031_X | 39 | 0.2327 | 0.4345 | 0.9548 | 2.1834 | 2.0028 | 1 | |
| 10 | 9 | 11.31-11.69 | 8 | 3 | OAR9_11436829 | 26 | 0.3990 | 0.1750 | 1.8185 | 1.2966 | 2.1687 | 0 | |
| 11 | 9 | 66.27-67.09 | 13 | 2 | OAR9_70612779 | 4 | 0.4586 | 0.1935 | 1.9607 | 1.6057 | 2.6214 | 0 | |
| 12 | 16 | 16.67-16.88 | 7 | 2 | s59518 | 23 | 0.6775 | 0.1604 | 2.1737 | 0.9608 | 2.2246 | 2 | |
| 13 | 16 | 17.33-17.7 | 11 | 5 | s61002 | 1 | 0.5391 | 0.0953 | 3.2562 | 0.7822 | 3.0273 | 0 | |
| 14 | 16 | 66.35-66.7 | 9 | 1 | s54054 | 35 | 0.6238 | 0.0791 | 2.4545 | 0.4334 | 2.0232 | 4 | NSUN2 |
| 15 | 19 | 54.68-55.19 | 9 | 1 | OAR19_58095077 | 44 | 0.2582 | 0.2780 | 0.9065 | 2.1367 | 1.9812 | 3 | HRH1 |
| 16 | 25 | 39.57-40.32 | 19 | 2 | OAR25_41988307 | 31 | 0.5301 | 0.1371 | 2.5086 | 0.9984 | 2.0855 | 1 | |
The top 5% of SNP were used to define the boundaries of each region (see Materials & Methods). Position is taken from build 3.1 of the sheep genome. The number of SNP50 BeadChip SNP and the number of top SNP (0.1%) are given in each region. Peak SNP is defined as the SNP with the lowest WIN5 –log10(combined Peddrift P-value) in the region, and the rank of that SNP is also shown. Maximum FST and –log10 (Peddrift P-value) are shown for each breed. The number of genes in each region, along with candidates (defined as those with a previously reported role in immunity) was taken from Ensembl release 74.
Investigation of selection sweep on OAR1
In total, 41 extra SNPs were genotyped in region 2; after quality control using the same criteria applied to the SNP50 BeadChip data, 15 of these SNP were used for further analyses. As a consequence of genotyping extra SNP, the peak FST value in the region increased slightly from 0.3475 (Table 1) to 0.3895.
The LD correlation coefficient r2 in region 2 was calculated for each of the selection lines separately (Additional files 1, 2, 3 and 4). All four analyses showed a haplotype block between 12 SNP (Table 2) in region 2. The Romney selection lines showed high linkage disequilibrium (r2 > 0.8) within the haplotype block, consistent with selection being imposed on the locus [27].
Table 2.
The 11 SNP core haplotype shown by Sweep (v1.1) to be in LD in region 2 (Table 1 ) on chromosome 1
| SNP ID | Chr | Position | Platform | SNP | Haplotype | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (Derived/ancestral) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||||
| OAR1_93117979 | 1 | 87702641 | Sequenom® | T/C | C | C | C | C | C | C | C | T |
| OAR1_93118073 | 1 | 87702735 | Sequenom® | C/T | T | T | T | T | T | T | T | C |
| OAR1_93124341.1 | 1 | 87709264 | SNP50 BeadChip | T/C | C | C | C | C | C | T | T | T |
| OAR1_93154178 | 1 | 87740396 | Sequenom® | A/G | A | G | G | G | G | A | G | A |
| s50853 | 1 | 87751371 | Sequenom® | G/C | G | C | C | G | G | C | C | C |
| OAR1_93166689.1 | 1 | 87753155 | SNP50 BeadChip | G/T | T | T | T | T | T | T | T | G |
| OAR1_93219257.1 | 1 | 87791568 | SNP50 BeadChip | C/T | C | C | C | C | C | C | C | T |
| OAR1_93226648 | 1 | 87801889 | Sequenom® | A/G | G | G | G | G | G | G | G | A |
| OAR1_93231706 | 1 | 87807624 | Sequenom® | G/T | T | T | T | G | T | T | T | T |
| OAR1_93231813 | 1 | 87807731 | Sequenom® | T/C | T | T | T | C | T | C | T | T |
| s57213.1 | 1 | 87826285 | SNP50 BeadChip | C/T | C | C | T | C | T | C | C | T |
| OAR1_93251748 | 1 | 87828912 | Sequenom® | A/G | - | - | - | - | - | - | - | - |
| Selection line | Haplotype frequency | |||||||||||
| Romney resistant | - | - | - | 0.3211 | - | - | - | 0.6789 | ||||
| Romney susceptible | - | - | - | 0.8841 | - | - | - | 0.0976 | ||||
| Perendale resistant | 0.0203 | - | 0.0203 | 0.6554 | 0.0338 | 0.0203 | 0.0405 | - | ||||
| Perendale susceptible | - | 0.0278 | - | 0.3264 | 0.0764 | - | - | 0.5417 | ||||
Haplotypes 1–8 were present in either the Romney or Perendale animals, or both. Haplotype frequencies are given for each selection line.
Analysis using Sweep (v1.1) showed that 11 of the 12 above SNP created a haplotype block. In the Romney lines two contrasting haplotypes were observed, denoted 4 and 8 (Table 2). In the Romney lines haplotype 4 was present in 88.4% of the susceptible population, and 32.1% of the resistant population. In the Perendale animals the frequency of haplotype 4 was higher in the resistant animals (65.5%) compared to the susceptible animals (32.5%). There were six additional haplotypes observed in the Perendale selection lines, although they were less frequent (2-8% of the population).
The integrated haplotype score (iHS; Figure 2B) [12] and cross-population extended haplotype homozygosity test (XP-EHH; Figure 2C) [13] are designed to uncover selected alleles with higher frequency than expected to their haplotype length. The most significant results were in the Romney susceptible animals where iHS values approached significance (P = 0.0518).
Figure 2.
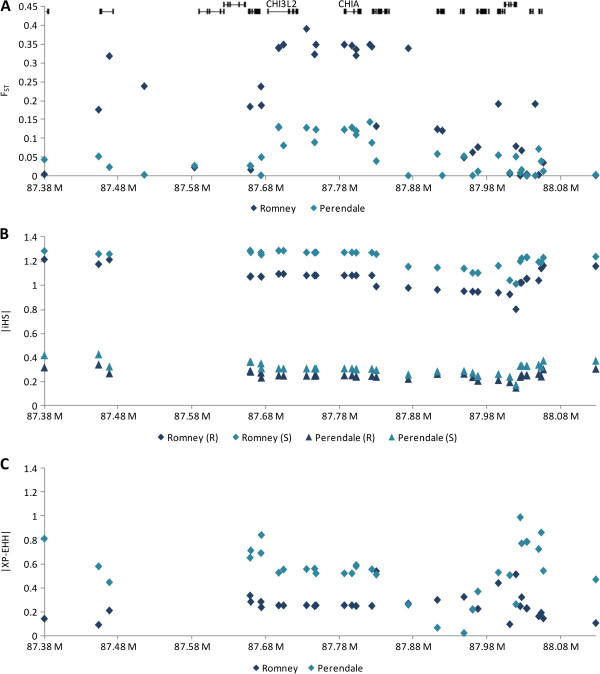
Signatures of selection observed in region 2 (Table 1 ) in Romney and Perendale selection lines. (A) FST between resistant and susceptible lines, -log P-values from standardised |iHS| (B) and |XP-EHH| (C) analyses.
Sequencing
After examination of the signals of selection in region 2 (Figure 2), the candidate gene CHIA (chitinase, acidic) was chosen for exon sequencing. CHIA has previously been associated with the control of helminth infection [28]. Other genes in the region include CD53, CHI3L2, CHIA, and DENND2D. Sequencing the CHIA exons of animals homozygous for both haplotypes showed the presence of several SNP (Additional file 5), however there were no SNP that distinguished the animals of different haplotype or selection line. One SNP at base 1174 of the ovine CHIA mRNA could potentially differentiate animals homozygous for haplotype 4 or 8. This, however, would require genotyping in further animals to validate.
Discussion
Using single-marker tests for differentiation between selection lines, multiple areas were discovered where allele frequency differed between resistant and susceptible lines (Figure 1). This was expected, as variation in complex traits such as resistance to parasites are understood to be controlled by many polymorphisms, each of a small effect [10]. The classic model of a selective sweep involves a beneficial allele being rapidly driven to fixation (‘hard sweep’). However, with complex traits selection may occur through polygenic adaptation, where adaptation occurs by simultaneous selection on variants at many loci. Selection under a polygenic adaptation model would result in modest allele frequency changes across the genome, which may be undetectable using current methods for detecting selection [29]. Despite this, the ‘hard sweep’ and polygenic models are not mutually exclusive, and the alleles with largest effect sizes may sweep to fixation [30]. Polygenic traits will therefore show increased FST across the genomes, with alleles of a large effect showing increased FST in that particular region.
Divergent lines of Romney [6, 31] and Perendale [7] sheep were selectively bred for high and low FEC by AgResearch Ltd from 1978 and 1986, respectively (Table 3). One of the aims of this study was to discover if the Romney and Perendale selection lines have utilised the same genetic strategy in developing resistance or susceptibility to internal parasites. Combined Peddrift values were used to define the regions to examine for candidate genes as the test was designed to account for structure within each of the populations surveyed. While peaks were observed in both lines, these were better defined when smoothing, via a 5-SNP window, was applied (Figure 1C).
Table 3.
| Trait | Romney | Perendale | ||
|---|---|---|---|---|
| High | Low | High | Low | |
| Average yearly flock size (rams/ewes) | 6/100 | 4.6/50 | ||
| Divergence of log-transformed FEC between lines | 2.73 | 0.85 | ||
| Divergence in average BVs between lines | 2.83 | 1.77 | ||
| Fold-difference in FEC mean between lines | 11.3 | 4.9 | ||
| Heritability | 0.28 ± 0.02 | 0.18 ± 0.03 | ||
| Back-transformed FEC means (eggs/g) | 3823 | 339 | 556 | 114 |
It must be noted that the strongest signals of selection were observed in the Romney selection lines, and the strength of the selection would have affected the combined data. As expected, the most extreme values for all statistics in the Romney selection lines were larger than those observed in the Perendale selection lines.
The Perendale lines have not been selected for as long (23 versus 31 years) and the genetic divergence in the selected trait is only half as large (Table 3). The effective population size of the foundation animals is also likely to have had a strong bearing on the differences between the breeds, although this is difficult to determine due to the combination of two separate Romney selection lines. The effective population size (Ne) for NZ industry Romneys 5 generations ago was estimated as 226 and for Perendale 109, derived from extensive analysis of more than 10,000 New Zealand animals genotyped with the SNP50 BeadChip [32]. As an interbreed of Cheviot and Romney [33] the Perendale animals are also likely to have a higher (Ne) [34, 35] than a pure breed, which may contribute to the observed pattern in the data. However, since its establishment, the Perendale breed has had a smaller population, which may contribute to low LD between closely spaced markers distances but greater LD between distant markers.
In the regions showing signatures of selection, candidate genes were defined as those with a previously reported role in immunity. We recognise that by examining in detail only those genes with obvious functional links to immunity we have eliminated some genes that could have novel and unexpected effects on the trait concerned, or may contain as yet unidentified features that have an effect separate from the gene itself. However, we believe our approach is a tractable solution, with the data and annotation currently available, and there will be potential to extend this analysis in the future. For example, several regions that appeared to be under selection from our analyses appear to contain no underlying genes (Table 1; Additional file 6). The current annotation of the sheep genome is not as comprehensive as that in humans or even cattle, and these areas cannot be completely dismissed as containing no genes or regulatory elements. This can only be improved following the recent annotation of version 3.1 of the sheep genome by Ensembl (Ensembl release 74). It has also been observed that while some proposed candidates for selection have strong support in the form of a functional mutation with an identified phenotypic effect, often the functional target is unknown [36].
The discovery that the same core haplotype (haplotype 4) in region 2 (Table 2) is observed in both susceptible Romney and resistant Perendale animals does not have an obvious explanation, but could be due to epistatic effects or a recent novel mutation. There was no correlation between haplotype and average estimated FEC breeding value. Following this, there are several possible reasons for the observed differences. It appears that selection in region 2 is primarily occurring in the Romney susceptible line. This is supported by the greater number of haplotypes that were observed in the Perendale selection lines in the Sweep (v1.1) analysis. Sequencing the CHIA exons of animals homozygous for both haplotypes showed the presence of several SNP; however none were responsible for the observed haplotype. The observed effect could also be driven by a regulatory element, such as a transcription factor, that could be interacting with a locus or loci in other parts of the genome [37]. In addition, while perhaps not the most likely scenario, a causal mutation in the region could have occurred separately in Perendale and Romneys, on the opposite haplotype block, which would explain the differences observed. Unravelling this, however, is complicated by the fact that the Perendale breed was formed in 1956 by crossing a Cheviot over a Romney, thus half of the Perendale genome is in effect of Romney origin.
Comparison with other studies showed that only two of the regions identified using Peddrift values (Table 1) were contained within a previously identified QTL (Sheep QTLdb [38]). Region 8 overlaps a QTL located on chromosome 7 (CSAP35E–MCM149; OAR7:44,018,971-81,694,614) for resistance to Haemonchus contortus infestation in merino sheep [39]. The QTL was not considered by the authors a good candidate for fine-mapping because evidence for the QTL decreased with confirmatory mapping. Region 16 is contained within two suggestive QTL detected on chromosome 25 in a genome scan for for resistance to Haemonchus contortus resistance in Romane x Martinik Black Belly backcross lambs [40]. The suggestive QTL, for sex ratio in the adult worm population (0.4-40.7 Mb; OARv2.0) and packed cell volume after second challenge (6.6-44 Mb; OARv2.0), were discovered using linkage analysis with SNP data.
Previously many studies have focussed specifically on chromosomes 3 and 20, which contain interferon gamma (IFNG) and the major histocompatibility complex (MHC) region respectively. The SNP50 BeadChip contains four SNP within the IFNG locus (OAR3:151,528,059-151,532,204); the maximum WIN5 –log10 (combined Peddrift P-values) in the region was 0.62, which was only slightly higher than the genome-wide average of 0.42. The Romney and Perendale FST peaks were 0.0505 and 0.0377 respectively, which when compared to the genome wide distributions (Figure 2A & B) is fairly low. Both Romney and Perendale selection lines showed no obvious signals of selection on the other common candidate region, the MHC region on chromosome 20, with a chromosome-wide WIN5 –log10 (combined Peddrift P-values) peak of 1.56. While this value is reasonably high when compared to the genome-wide distribution (Figure 1), the highest ranked SNP in the region, going by WIN5 –log10 (combined Peddrift P-values), was 167th (OAR20_1876702).
Four regions (Table 1) contained genes that have previously been implicated or are candidates for resistance or susceptibility to gastrointestinal nematodes; OAR 1 (CD53, CHI3L2, CHIA and DENND2D), OAR 4 (RELN), OAR 16 (NSUN2) and OAR 19 (HRH1).
The leukocyte surface antigen CD53 contributes to the transduction of CD2-generated signals in T cells and natural killer (NK) cells [41]. NK cells have been shown to produce significant amounts of IL-5, which contributes to eosinophil recruitment in an in vivo mouse model of allergic inflammation [42], and may also be involved in T-cell-independent eosinophil recruitment after helminth infections [43]. The CD53 protein forms functional interactions with prominent leukocyte receptors including MHC molecules and the surface of B cells [44], and has been shown to be down-regulated upon stimulation of human neutrophils with TNF-α [45]. In humans CD53 deficiency has been associated with recurrent infectious diseases caused by bacteria, fungi and viruses [46], and polymorphisms in the gene have been associated with regulation of TNF-α levels [47]; up-regulation of TNF-α has been observed in the abomasal lymph node of sheep challenged with T. circumcincta 5 days after infection [48], and abomasal mucosa of resistant DRB1*1101 carrier lambs at 3 days post infection [49].
Chitinases are a group of digestive enzymes that break down glycosidic bonds in chitin, which is present in fungi and the exoskeletal elements of some animals, including nematodes and arthropods [50]. Mammalian chitinases and chitinase-like proteins are known to be up-regulated and secreted in TH2 induced inflammatory responses, including nematode infection [51] suggesting these genes are plausible candidate genes for mediating resistance status.
CHIA [52] has previously been associated with the development of the immune response in mammals and control of helminth infection [28]. Induction of CHIA is via a TH2 specific, IL-13 mediated pathway, and has been implicated in TH2 dominated disorders such as asthma [53]. In mice it has been shown that chitin is a recognition element for tissue infiltration by innate immune cells implicated in allergic and helminth immunity (including eosinophils and basophils) and this process can be negatively regulated by a vertebrate chitinase [54]. Despite this, there is no evidence in the literature that CHIA has previously been implicated in increased resistance or susceptibility to gastrointestinal parasites in ungulates.
Chitinase-like proteins can bind chitin, however, due to mutations in their active domains they do not have chitinolytic enzyme activity [28]. The chitinase-like molecule, CHI3L1, has been shown to be up-regulated in the abomasum of sheep in response to T. circumcincta challenge of previously infected animals [55]. CHIA expression levels were also examined in the same study, and while expression was observed up-regulation of transcripts was not significant. Expression of CHI3L2 (UGID: 1230481; http://www.ncbi.nlm.nih.gov/UniGene) has been observed in the abomasum of 18 and 21 week old steers exposed to Ostertagia ostertagi [56]. Expression also been observed in the abomasal lymph node of resistant and susceptible Blackface lambs infected with T. circumcincta in comparison to sham-infected controls [57]. In human macrophages CHI3L2 has been found to be upregulated by IL-4 and TGF-β [58].
While the TH1/TH2 dichotomy has not been proven in sheep, the components involved in response to gastrointestinal parasite infection are typical of a TH2 pathway; immunity is associated with the production of TH2-associated cytokines, increased numbers of mast cells, peripheral and tissue eosinophilia, and elevated production of multiple antibody isotypes [59–62]. HRH1 is predominantly expressed on TH1 cells, in an IL3-upregulatable manner [63]. Mice lacking HRH1 had lower percentages of Interferon-γ (IFNG)-producing cells, and produced higher levels of antigen-specific IgG1 and IgE. Mice lacking either HRH1 or HRH2 tended to have a higher frequency of IL4-producing cells. Jutel et al. [63] concluded that histamine secreted from mast cells and basophils potently influences TH1 and TH2 responses, as well as antibody isotypes, as a regulatory loop in inflammatory reactions. In Blackface lambs challenged over a period of three months with T. circumcincta, significantly increased levels of HRH1 expression in the abomasal lymph node were observed [57].
While the genes DENND2D, RELN and NSUN2 do not have obvious roles in immunity, they have previously been reported as being upregulated in susceptible animals. The DENND2D protein was found to be more abundant in genetically susceptible sheep following infection by H. contortus [64]. RELN was upregulated in Suffolk (susceptible) compared to Texel (resistant) animals three days post infection with T. circumcincta [65]. Finally, in a study comparing gene expression in the duodenum following natural infection of lambs from the Perendale selection lines used in this study, NSUN2 was found to be more highly expressed in susceptible animals [66].
For complex traits, where many loci of small to moderate effect are likely to influence phenotype, the 50,000 SNP available for ovine analysis, which are also biased to high MAF SNP, may not provide enough information. In sheep, single markers were estimated to explain a maximum of 0.48% or 0.08% of the phenotypic variance in FEC following challenge with either T. colubriformis or H. contortus respectively [10]. It has been suggested in cattle, based on the FST difference between adjacent loci, that 150,000 evenly spaced SNP would be required to study selective signatures across the bovine genome [67].
In humans, the search for selective sweeps is aided by a large number of densely spaced SNP, with over 3.1 million SNP available from Phase II of the HapMap project (approximately one marker per 1 kb) [68]. Densely spaced SNP give greater power when using statistical tests that rely on linkage disequilibrium (LD), as signals of selection are less likely to be lost. The SNP50 BeadChip, while providing uniform genome-wide coverage, is estimated to have only one marker every 46 kb. Fine-mapping, where more SNP are genotyped in an area of interest using, for example, Sequenom® technology, allows further analysis of LD in areas that appear to be under selection. With the information obtained from more SNP, definition of LD in the area increases, improving the ability to be able to localise causal variants using numerous statistical methods, such as iHS and XP-EHH, that have been developed to identify signatures left in the genome by selection.
As previously discussed the SNP50 BeadChip has already been used to map causal mutations for traits controlled by a single locus, and furthermore used to validate and detect selection sweeps in sheep [22, 23]. While it is perhaps surprising that only two of the regions under selection correlated with a previously identified QTL, this lends support to the widely held theory that parasite resistance is under the control of many loci with a moderate effect. New genomic approaches, including the SNP50 BeadChip, and sequencing of whole genomes [69] and transcriptomes [70], provide an opportunity to rapidly look for and find genome-wide signals of selection [71, 72].
Conclusions
Genome wide analysis of selection signatures revealed 16 regions, which included genes involved in chitinase activity and the cytokine response. Many of the signals of selection found in this study are novel observations; further knowledge of the genes involved in gastrointestinal parasite resistance or susceptibility can only increase our understanding of the mechanisms involved.
Methods
Ethics statement
This study was carried out in strict accordance of the guidelines of the 1999 New Zealand Animal Welfare Act and was approved by the AgResearch’s Invermay Animal Ethics committees (Permit Numbers include: 497; 551; 593; 636; 10441; 10820).
Selection lines
Divergent lines of Romney [6, 31] and Perendale [7] sheep were selectively bred for high and low FEC by AgResearch Ltd from 1978 and 1986, respectively (Table 3). The Perendale selection flocks were established from an initial group of 111 rams, ranked for FEC, with the high and low FEC animals mated with 148 foundation dams. The number of foundation animals for the Romney selection lines is more difficult to define, due to divergent lines from two separate locations being merged to make the final selection lines in 1993 [6]. Selection lines have now been discontinued. Animals were selected as lambs solely on the basis of FEC following a natural mixed species nematode challenge. The predominant parasites were of the Trichostrongylus and Teladorsagia genera, however Cooperia, Haemonchus, and Nematodirus species were also present, with other genera being less abundant [6, 7, 31]. In the 1984–89 lamb crops, of the Perendale selection lines, the natural challenge was augmented further by an artificial challenge with H. contortus larvae.
Genotyping data
Animals were genotyped using the SNP50 BeadChip (Additional file 7), using high concentration DNA obtained from heparinised blood [73]. In total 180 Romney (83 high FEC animals and 97 low FEC animals) and 149 Perendale (74 high FEC animals and 75 low FEC animals) animals were genotyped. Using pedigree information, animals were chosen to be as unrelated as possible, however 66 sires and dams were also included (17 sires and 10 dams from the Romney lines, and 3 sires and 36 dams from the Perendale lines).
SNP locations for version 3.1 of the sheep genome were obtained from CSIRO (http://www.livestockgenomics.csiro.au/sheep/oar3.1.php; OARv3.1). Minor allele frequency (MAF) and call rate was calculated for each SNP. Quality control checks [74] excluded those SNP that had a call rate less than 99% and a MAF (over all animals of a breed) of less than 2%. Individual animals were removed from the analysis if there were more than 1% genotyping failure. Additionally, SNP not in Hardy-Weinberg equilibrium (HWE; p < 10-6) within selection line were also excluded. The Bonferroni correction was used to address the problem of multiple comparisons [75]. An experiment-wise significance level (α = 0.05) was chosen, and the number of tests was taken to be the number of SNP (n = 50,000), giving β = α /n = 1 × 10-6 as the test-wise significance level for HWE. This is conservative as the Bonferroni correction factor is based on independent tests. While departure from HWE may be caused by selection, it is more likely that extreme violations indicate a poorly performing SNP [76].
Genome-wide analysis
Two single-marker tests for differentiation, FST and Peddrift, were used to distinguish signals of selection between selection lines from whole-genome data. FST, which describes the proportion of variance within a species that is due to population subdivision, was calculated using Fisher’s [25] method for each breed:
Where the variance of p is computed across sub-populations, and p(1-p) is the expected frequency of heterozygotyes. The value of FST can theoretically range from 0 (no differentiation) to 1 (complete differentiation, in which subpopulations are fixed for different alleles).
Allele frequency differences at each SNP were also compared using Peddrift [11]. Peddrift calculates exact probabilities of allele frequency differences, taking into account genetic drift, founder and sampling effects. The method simulates genotypes through the actual pedigree data. Evidence of selection is shown by divergence from the expected Chi-squared (X2) distribution. Peddrift was run for both Romney and Perendale lines together using known pedigrees (with 2,000,000 simulations) to estimate the distribution of X2 under the null hypothesis of no selection. Results are expressed for each breed as a P-value for each marker.
To explore regions under selection across both breeds, the Peddrift P-values for each SNP were combined; if they have the same overall hypothesis, results from two independent tests can be combined using Fisher’s method [25], using the formula:
where pi is the P-value for the ith hypothesis test. The combined P-value was found by comparing X2 to a χ22k distribution. To reduce noise a 5 SNP moving average (WIN5) of –log10 of the combined P-values was used; signatures of selection are shown by SNP in a region showing low P-values. The concordance between Peddrift p-values for each SNP in Romney and Perendale was investigated by setting a p-value upper threshold of 0.01. There were 21 SNP under this threshold in both breeds, more than would be expected if there was no association in the two breeds by chance (14), suggesting that some regions had been selected in common which supports using the combined approach.
SNP were ranked using WIN5 –log10 (combined Peddrift P-values), and the top 0.1% of markers (n = 44) were used to determine regions under selection. The method of Kijas et al. [23] was used to define the boundaries of the regions; neighbouring markers were included until two consecutive markers ranked outside of the top 5%. The second marker that ranked outside of the top 5% was excluded and the position of the region determined using sheep genome assembly 3.1.
Candidate regions were annotated using Ensembl release 74 (as of 1/2014), and gene function determined using Online Mendelian Inheritance in Man (OMIM) and a literature search. Candidate genes were defined as those with a known role in the immune response. Sheep QTL were obtained from the Sheep QTLdb [38].
Detecting signatures of selection
Fine-mapping allows further analysis of LD in areas that appear to be under selection; with the information obtained from more SNP, definition of LD in the area increases, improving the ability to be able to localise causal variants. One region on chromosome 1 (region 2) was chosen for fine-mapping with a denser set of SNP (Additional file 8), using iPLEX™ genotyping assay for the Sequenom® MassARRAY® platform. This region was chosen for fine-mapping as it contained multiple candidate genes. Selection sweep statistics were subsequently used to clarify the observed signals of selection.
All known SNP in region 2 were examined for suitability for sequencing on the Sequenom® MassARRAY® platform; these included SNP discovered on both the Solexa and 454 platforms (http://www.sheephapmap.org/genseq.php). In total 41 extra SNP were genotyped.
Linkage disequilibrium (LD) between two loci was visualised using the correlation coefficient r2 within each selection line using Haploview [77], with areas of strong LD indicating areas under selection.
Haplotype phase estimation was performed using fastPHASE [78]. Haplotypes were subsequently used to calculate the selection statistics EHH, XP-EHH and iHS. The EHH statistic was computed using Sweep v1.1 [79], while the iHS and XP-EHH statistics were calculated using scripts obtained from the Pritchard lab (http://hgdp.uchicago.edu/Software/). Standardized iHS (|iHS|) was calculated using the genome-wide empirical distributions, following the method of Voight et al. [12]. Ancestral alleles for the SNP50 BeadChip SNP were obtained from Dr Clare Gill of Texas A&M University (2009, pers. comm.), and were determined by running 11 outgroup bovid species on the SNP50 BeadChip. A cross-species megaBLAST of Sequenom® primers against Bos taurus, Sus scrofa, Canis familiaris, Equus caballus and Homo sapiens was used to discover ancestral alleles for the remaining SNP.
Sequencing
Four animals were chosen for sequencing using standard amplicon sequencing (Additional file 9) with BigDye technology on an AB3730XL (Applied Biosystems). Animals consisted of one resistant and susceptible animal from each breed. Animals were selected based on homozygosity of an 11 SNP core haplotype shown by Sweep (v1.1) to be in LD (Table 2). Forward and reverse sequences were combined into contigs using Vector NTI® (Invitrogen), and consensus sequences BLASTed back against Ovis aries CHIA mRNA (XM_004002314.1) to search for SNP.
Data availability
The data sets supporting the results of this article are included within the article (and its additional files).
Electronic supplementary material
{kind=link}
Additional file 1: Linkage disequilibrium (LD), as shown by r 2 , for region 2 (OAR1:87384757–88132568) SNP in the Romney resistant selection line animals. (PNG 162 KB)
{kind=link}
Additional file 2: Linkage disequilibrium (LD), as shown by r 2 , for region 2 (OAR1:87384757–88132568) SNP in the Romney susceptible selection line animals. (PNG 152 KB)
{kind=link}
Additional file 3: Linkage disequilibrium (LD), as shown by r 2 , for region 2 (OAR1:87384757–88132568) SNP in the Perendale resistant selection line animals. (PNG 159 KB)
{kind=link}
Additional file 4: Linkage disequilibrium (LD), as shown by r 2 , for region 2 (OAR1:87384757–88132568) SNP in the Perendale susceptible selection line animals. (PNG 164 KB)
Additional file 5: SNP in CHIA discovered using sequencing. Haplotype: Sweep (v1.1) haplotype, avBV: Average FEC breeding value, Positions refer to base in CHIA (XM_004002314.1). (CSV 307 bytes)
Additional file 6: All known genes in regions of interest as of 01/2014. (CSV 4 KB)
Additional file 7: Pedigree information and Illumina® Ovine SNP50 BeadChip data for Romney and Perendale selection line animals. (ZIP 6 MB)
Additional file 8: SNP used on iPLEX™ genotyping assay for the Sequenom® MassARRAY® platform. Multiplex number; SNP ID; Amplification primer sequence; Extension primer sequence; Ancestral allele (AA). (CSV 4 KB)
Additional file 9: PCR primers used for direct sequencing of CHIA exons. (CSV 911 bytes)
Acknowledgements
Funding is gratefully acknowledged from the Ovita Consortium and AgResearch New Zealand. The authors would also like to thank Dr. Rudiger Brauning, Dr. Natalie Pickering, Tim Manley, Rayna Anderson & Joanne Gillum who helped and supported this research. We also thank the anonymous reviewers for their useful suggestions.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
KMM planned, performed the analyses and drafted the manuscript. JCM provided the dataset and supervised the analysis. KGD provided statistical and analysis support and NJG supervised the analysis. All authors have contributed to the editing of the article, and approved the final manuscript.
Contributor Information
Kathryn M McRae, Email: kathryn.mcrae@teagasc.ie.
John C McEwan, Email: john.mcewan@agresearch.co.nz.
Ken G Dodds, Email: ken.dodds@agresearch.co.nz.
Neil J Gemmell, Email: neil.gemmell@otago.ac.nz.
References
- 1.Familton AS, McAnulty RW. Life cycles and development of nematode parasites of ruminants. In: Barrell GK, editor. Sustainable control of internal parasites in ruminants. Lincoln University, New Zealand: Animal Industries Workshop; 1997. pp. 67–80. [Google Scholar]
- 2.Perry BD, Randolph TF. Improving the assessment of the economic impact of parasitic diseases and of their control in production animals. Vet Parasitol. 1999;84(3–4):145–168. doi: 10.1016/S0304-4017(99)00040-0. [DOI] [PubMed] [Google Scholar]
- 3.Miller CM, Waghorn TS, Leathwick DM, Candy PM, Oliver AMB, Watson TG. The production cost of anthelmintic resistance in lambs. Vet Parasitol. 2012;186(3–4):376–381. doi: 10.1016/j.vetpar.2011.11.063. [DOI] [PubMed] [Google Scholar]
- 4.Sargison ND. Pharmaceutical treatments of gastrointestinal nematode infections of sheep—Future of anthelmintic drugs. Vet Parasitol. 2012;189(1):79–84. doi: 10.1016/j.vetpar.2012.03.035. [DOI] [PubMed] [Google Scholar]
- 5.Morris C, Watson T, Bisset S, Vlassoff A, Douch P. Breeding sheep in New Zealand for resistance or resilience to nematode parasites. In: Gray G, Woolaston R, Eaton B, editors. Breeding for Resistance to Infectious Diseases of Small Ruminants. Canberra: Australian Centre for International Agricultural Research; 1995. pp. 77–98. [Google Scholar]
- 6.Morris CA, Vlassoff A, Bisset S, Baker R, Watson TG, West CJ, Wheeler M. Continued selection of Romney sheep for resistance or susceptibility to nematode infection: estimates of direct and correlated responses. Anim Sci. 2000;70:17–27. [Google Scholar]
- 7.Morris CA, Wheeler M, Watson TG, Hosking BC, Leathwick DM. Direct and correlated responses to selection for high or low faecal nematode egg count in Perendale sheep. N Z J Agric Res. 2005;48:1–10. doi: 10.1080/00288233.2005.9513625. [DOI] [Google Scholar]
- 8.Kemper K, Goddard M, Bishop S. Adaptation of gastrointestinal nematode parasites to host genotype: single locus simulation models. Genet Sel Evol. 2013;45(1):14. doi: 10.1186/1297-9686-45-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kemper KE, Elwin RL, Bishop SC, Goddard ME, Woolaston RR. Haemonchus contortus and Trichostrongylus colubriformis did not adapt to long-term exposure to sheep that were genetically resistant or susceptible to nematode infections. Int J Parasitol. 2009;39(5):607–614. doi: 10.1016/j.ijpara.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 10.Kemper KE, Emery DL, Bishop SC, Oddy H, Hayes BJ, Dominik S, Henshall JM, Goddard ME. The distribution of SNP marker effects for faecal worm egg count in sheep, and the feasibility of using these markers to predict genetic merit for resistance to worm infections. Genet Res. 2011;93(3):203–219. doi: 10.1017/S0016672311000097. [DOI] [PubMed] [Google Scholar]
- 11.Dodds KG, McEwan JC. Proceedings of the 12th Conference of the Association for the Advancement of Animal Breeding and Genetics: 6th-10th April 1997. Dubbo, Australia : University of New England, Armidale, New South Wales: AAABG; 1997. Calculating exact probabilities of allele frequency differences in divergent selection lines; pp. 556–560. [Google Scholar]
- 12.Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4(3):e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, Xie X, Byrne EH, McCarroll SA, Gaudet R, Schaffner SF, Lander ES, Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, Pasternak S, Wheeler DA, Willis TD, Yu F, Yang H, Zeng C, Gao Y, Hu H, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449(7164):913–918. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pickrell JK, Coop G, Novembre J, Kudaravalli S, Li JZ, Absher D, Srinivasan BS, Barsh GS, Myers RM, Feldman MW, Pritchard JK. Signals of recent positive selection in a worldwide sample of human populations. Genome Res. 2009;19(5):826–837. doi: 10.1101/gr.087577.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gautier M, Naves M: Footprints of selection in the ancestral admixture of a New World Creole cattle breed.Mol Ecol2011:3128–3143. [DOI] [PubMed]
- 16.Hayes BJ, Chamberlain AJ, Maceachern S, Savin K, McPartlan H, MacLeod I, Sethuraman L, Goddard ME. A genome map of divergent artificial selection between Bos taurus dairy cattle and Bos taurus beef cattle. Anim Genet. 2008;40(2):176–184. doi: 10.1111/j.1365-2052.2008.01815.x. [DOI] [PubMed] [Google Scholar]
- 17.Becker D, Tetens J, Brunner A, Bürstel D, Ganter M, Kijas J, Drögemüller C, International Sheep Genomics Consortium for the Microphthalmia in Texel sheep is associated with a missense mutation in the paired-like homeodomain 3 (PITX3) gene. PLoS One. 2010;5(1):e8689. doi: 10.1371/journal.pone.0008689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shariflou MR, Wade CM, Kijas J, McCulloch R, Windsor PA, Tammen I, Nicholas FW: Brachygnathia, cardiomegaly and renal hypoplasia syndrome (BCRHS) in Merino sheep maps to a 1.1-megabase region on ovine chromosome OAR2.Anim Genet2012:231–233. [DOI] [PubMed]
- 19.Mömke S, Kerkmann A, Wöhlke A, Ostmeier M, Hewicker-Trautwein M, Ganter M, Kijas J, Distl O, for the International Sheep C A frameshift mutation within LAMC2 is responsible for Herlitz type junctional epidermolysis bullosa (HJEB) in black headed mutton sheep. PLoS One. 2011;6(5):e18943. doi: 10.1371/journal.pone.0018943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao X, Dittmer KE, Blair HT, Thompson KG, Rothschild MF, Garrick DJ. A novel nonsense mutation in the DMP1 gene identified by a genome-wide association study is responsible for inherited rickets in Corriedale sheep. PLoS One. 2011;6(7):e21739. doi: 10.1371/journal.pone.0021739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnston SE, McEwan JC, Pickering NK, Kijas JW, Beraldi D, Pilkington JG, Pemberton JM, Slate J. Genome-wide association mapping identifies the genetic basis of discrete and quantitative variation in sexual weaponry in a wild sheep population. Mol Ecol. 2011;20(12):2555–2566. doi: 10.1111/j.1365-294X.2011.05076.x. [DOI] [PubMed] [Google Scholar]
- 22.Moradi MH, Nejati-Javaremi A, Moradi-Shahrbabak M, Dodds K, McEwan J. Genomic scan of selective sweeps in thin and fat tail sheep breeds for identifying of candidate regions associated with fat deposition. BMC Genet. 2012;13(1):10. doi: 10.1186/1471-2156-13-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kijas JW, Lenstra JA, Hayes B, Boitard S, Porto Neto LR, San Cristobal M, Servin B, McCulloch R, Whan V, Gietzen K, Paiva S, Barendse W, Ciani E, Raadsma H, McEwan J, Dalrymple B, other members of the International Sheep Genomics C Genome wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012;10(2):e1001258. doi: 10.1371/journal.pbio.1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kemper KE. The implications for the host-parasite relationship when sheep are bred for enhanced resistance to gastrointestinal nematodes. PhD Thesis. Melbourne: The University of Melbourne; 2010. [Google Scholar]
- 25.Fisher RA. Statistical methods for research workers. Edinburgh: Oliver and Boyd; 1925. [Google Scholar]
- 26.Weir BS, Cardon LR, Anderson AD, Nielsen DM, Hill WG. Measures of human population structure show heterogeneity among genomic regions. Genome Res. 2005;15(11):1468–1476. doi: 10.1101/gr.4398405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McVean G. The structure of linkage disequilibrium around a selective sweep. Genetics. 2007;175(3):1395–1406. doi: 10.1534/genetics.106.062828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee CG, Da Silva CA, Dela Cruz CS, Ahangari F, Ma B, Kang M-J, He C-H, Takyar S, Elias JA. Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu Rev Physiol. 2011;73(1):479–501. doi: 10.1146/annurev-physiol-012110-142250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pritchard JK, Pickrell JK, Coop G. The genetics of human adaptation: hard sweeps, soft sweeps, and polygenic adaptation. Curr Biol. 2010;20(4):R208–R215. doi: 10.1016/j.cub.2009.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pritchard JK, Di Rienzo A. Adaptation – not by sweeps alone. Nat Rev Genet. 2010;11(10):665–667. doi: 10.1038/nrg2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morris CA, Vlassoff A, Bisset SA, Baker RL, West CJ, Hurford AP. Responses of Romney sheep to selection for resistance or susceptibility to nematode infection. Anim Sci. 1997;64(2):319–329. doi: 10.1017/S1357729800015897. [DOI] [Google Scholar]
- 32.Prieur V. The use of a 50K SNP array to assess linkage disequilibrium and effective population size in New Zealand sheep. MSc Thesis. Agroparistech: Paris; 2013. [Google Scholar]
- 33.Perendale Sheep Society of New Zealand [http://www.perendalenz.com/]
- 34.Wright S. Evolution in Mendelian populations. Genetics. 1931;16(2):97–159. doi: 10.1093/genetics/16.2.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wright S. Size of population and breeding structure in relation to evolution. Science. 1938;87:430–431. [Google Scholar]
- 36.Sabeti PC, Schaffner SF, Fry B, Lohmueller J, Varilly P, Shamovsky O, Palma A, Mikkelsen TS, Altshuler D, Lander ES. Positive natural selection in the human lineage. Science. 2006;312(5780):1614–1620. doi: 10.1126/science.1124309. [DOI] [PubMed] [Google Scholar]
- 37.Schork AJ, Thompson WK, Pham P, Torkamani A, Roddey JC, Sullivan PF, Kelsoe JR, O'Donovan MC, Furberg H, Schork NJ, Andreassen OA, Dale AM, The Tobacco Genetics Consortium, The Bipolar Disorder Psychiatric Genomics Consortium, The Schizophrenia Psychiatric Genomics Consortium All SNPs are not created equal: genome-wide association studies reveal a consistent pattern of enrichment among functionally annotated SNPs. PLoS Genet. 2013;9(4):e1003449. doi: 10.1371/journal.pgen.1003449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu Z-L, Park CA, Wu X-L, Reecy JM. Animal QTLdb: an improved database tool for livestock animal QTL/association data dissemination in the post-genome era. Nucleic Acids Res. 2013;41(D1):D871–D879. doi: 10.1093/nar/gks1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marshall K, Maddox JF, Lee SH, Zhang Y, Kahn L, Graser HU, Gondro C, Walkden-Brown SW, van der Werf JH. Genetic mapping of quantitative trait loci for resistance to Haemonchus contortus in sheep. Anim Genet. 2009;40(3):262–272. doi: 10.1111/j.1365-2052.2008.01836.x. [DOI] [PubMed] [Google Scholar]
- 40.Sallé G, Jacquiet P, Gruner L, Cortet J, Sauvé C, Prevot F, Grisez C, Bergeaud JP, Schibler L, Tircazes A, Francois D, Pery C, Bouvier F, Thouly JC, Brunel JC, Legarra A, Elsen JM, Bouix J, Rupp R, Moreno CR. A genome scan for QTL affecting resistance to Haemonchus contortus in sheep. J Anim Sci. 2012;90(13):4690–4705. doi: 10.2527/jas.2012-5121. [DOI] [PubMed] [Google Scholar]
- 41.Bell GM, Seaman WE, Niemi EC, Imboden JB. The OX-44 molecule couples to signaling pathways and is associated with CD2 on rat T lymphocytes and a natural killer cell line. J Exp Med. 1992;175(2):527–536. doi: 10.1084/jem.175.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walker C, Checkel J, Cammisuli S, Leibson PJ, Gleich GJ. IL-5 Production by NK Cells Contributes to Eosinophil Infiltration in a Mouse Model of Allergic Inflammation. J Immunol. 1998;161(4):1962–1969. [PubMed] [Google Scholar]
- 43.Meeusen ENT, Balic A. Do Eosinophils have a Role in the Killing of Helminth Parasites? Parasitol Today. 2000;16(3):95–101. doi: 10.1016/S0169-4758(99)01607-5. [DOI] [PubMed] [Google Scholar]
- 44.Szöllósi J, Horejsí V, Bene L, Angelisová P, Damjanovich S. Supramolecular complexes of MHC class I, MHC class II, CD20, and tetraspan molecules (CD53, CD81, and CD82) at the surface of a B cell line JY. J Immunol. 1996;157(7):2939–2946. [PubMed] [Google Scholar]
- 45.Mollinedo F, Martín-Martín B, Gajate C, Lazo PA. Physiological activation of human neutrophils down-regulates CD53 cell surface antigen. J Leukoc Biol. 1998;63(6):699–706. doi: 10.1002/jlb.63.6.699. [DOI] [PubMed] [Google Scholar]
- 46.Mollinedo F, Fontán G, Barasoain I, Lazo PA. Recurrent infectious diseases in human CD53 deficiency. Clin Diagn Lab Immunol. 1997;4(2):229–231. doi: 10.1128/cdli.4.2.229-231.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bos SD, Lakenberg N, van der Breggen R, Houwing-Duistermaat JJ, Kloppenburg M, de Craen AJ, Beekman M, Meulenbelt I, Slagboom PE. A genome-wide linkage scan reveals CD53 as an important regulator of innate TNF-alpha levels. Eur J Hum Genet. 2010;18(8):953–959. doi: 10.1038/ejhg.2010.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Craig NM, Miller HRP, Smith WD, Knight PA. Cytokine expression in naïve and previously infected lambs after challenge with Teladorsagia circumcincta. Vet Immunol Immunopathol. 2007;120(1–2):47–54. doi: 10.1016/j.vetimm.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 49.Hassan M, Hanrahan JP, Good B, Mulcahy G, Sweeney T. A differential interplay between the expression of TH1/TH2/Treg related cytokine genes in Teladorsagia circumcincta infected DRB1*1101 carrier lambs. Vet Res. 2011;42(1):45. doi: 10.1186/1297-9716-42-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fuhrman JA, Piessens WF. Chitin synthesis and sheath morphogenesis in Brugia malayi microfilariae. Mol Biochem Parasitol. 1985;17(1):93–104. doi: 10.1016/0166-6851(85)90130-6. [DOI] [PubMed] [Google Scholar]
- 51.Nair MG, Gallagher IJ, Taylor MD, Loke P, Coulson PS, Wilson RA, Maizels RM, Allen JE. Chitinase and Fizz family members are a generalized feature of nematode infection with selective upregulation of Ym1 and Fizz1 by antigen-presenting cells. Infect Immun. 2005;73(1):385–394. doi: 10.1128/IAI.73.1.385-394.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Boot RG, Blommaart EF, Swart E, Ghauharali-van der Vlugt K, Bijl N, Moe C, Place A, Aerts JM. Identification of a novel acidic mammalian chitinase distinct from chitotriosidase. J Biol Chem. 2001;276(9):6770–6778. doi: 10.1074/jbc.M009886200. [DOI] [PubMed] [Google Scholar]
- 53.Zhu Z, Zheng T, Homer RJ, Kim Y-K, Chen NY, Cohn L, Hamid Q, Elias JA. Acidic mammalian chitinase in asthmatic TH2 inflammation and IL-13 pathway activation. Science. 2004;304(5677):1678–1682. doi: 10.1126/science.1095336. [DOI] [PubMed] [Google Scholar]
- 54.Reese TA, Liang HE, Tager AM, Luster AD, Van Rooijen N, Voehringer D, Locksley RM. Chitin induces accumulation in tissue of innate immune cells associated with allergy. Nature. 2007;447(7140):92–96. doi: 10.1038/nature05746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Knight PA, Pate J, Smith WD, Miller HR. An ovine chitinase-like molecule, chitinase-3 like-1 (YKL-40), is upregulated in the abomasum in response to challenge with the gastrointestinal nematode, Teladorsagia circumcincta. Vet Immunol Immunopathol. 2007;120(1–2):55–60. doi: 10.1016/j.vetimm.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 56.Sonstegard TS, Van Tassell CP, Matukumalli LK, Harhay GP, Bosak S, Rubenfield M, Gasbarre LC. Germplasm Release. 2004. Production of EST from cDNA libraries derived from immunologically activated bovine gut. [Google Scholar]
- 57.Gossner A, Wilkie H, Joshi A, Hopkins J. Exploring the abomasal lymph node transcriptome for genes associated with resistance to the sheep nematode Teladorsagia circumcincta. Vet Res. 2013;44(1):68. doi: 10.1186/1297-9716-44-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gratchev A, Schmuttermaier C, Mamidi S, Gooi L, Goerdt S, Kzhyshkowska J. Expression of osteoarthritis marker YKL-39 is stimulated by transforming growth factor Beta (TGF-beta) and IL-4 in differentiating macrophages. Biomark Insights. 2008;3:39–44. [PMC free article] [PubMed] [Google Scholar]
- 59.Gill HS, Altmann K, Cross ML, Husband AJ. Induction of T helper 1- and T helper 2-type immune responses during Haemonchus contortus infection in sheep. Immunology. 2000;99(3):458–463. doi: 10.1046/j.1365-2567.2000.00974.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lacroux C, Nguyen THC, Andreoletti O, Prevot F, Grisez C, Bergeaud J-P, Gruner L, Brunel J-C, Francois D, Dorchies P, Jacquiet P. Haemonchus contortus (Nematoda: Trichostrongylidae) infection in lambs elicits an unequivocal TH2 immune response. Vet Res. 2006;37(4):607–622. doi: 10.1051/vetres:2006022. [DOI] [PubMed] [Google Scholar]
- 61.Jankovic D, Liu Z, Gause WC. TH1- and TH2-cell commitment during infectious disease: asymmetry in divergent pathways. Trends Immunol. 2001;22(8):450–457. doi: 10.1016/S1471-4906(01)01975-5. [DOI] [PubMed] [Google Scholar]
- 62.Maizels RM, Yazdanbakhsh M. Immune Regulation by helminth parasites: cellular and molecular mechanisms. Nat Rev Immunol. 2003;3(9):733–744. doi: 10.1038/nri1183. [DOI] [PubMed] [Google Scholar]
- 63.Jutel M, Watanabe T, Klunker S, Akdis M, Thomet OAR, Malolepszy J, Zak-Nejmark T, Koga R, Kobayashi T, Blaser K, Akdis CA. Histamine regulates T-cell and antibody responses by differential expression of H1 and H2 receptors. Nature. 2001;413(6854):420–425. doi: 10.1038/35096564. [DOI] [PubMed] [Google Scholar]
- 64.Nagaraj SH, Harsha HC, Reverter A, Colgrave ML, Sharma R, Andronicos N, Hunt P, Menzies M, Lees MS, Sekhar NR, Pandey A, Ingham A. Proteomic analysis of the abomasal mucosal response following infection by the nematode, Haemonchus contortus, in genetically resistant and susceptible sheep. J Proteomics. 2012;75(7):2141–2152. doi: 10.1016/j.jprot.2012.01.016. [DOI] [PubMed] [Google Scholar]
- 65.Ahmed AM. Immunological and transcriptomic analysis of genetically resistant and susceptible sheep to gastrointestinal nematodes. PhD Thesis. Dublin: University College Dublin; 2013. [Google Scholar]
- 66.Keane OM, Dodds KG, Crawford AM, McEwan JC. Transcriptional profiling of Ovis aries identifies Ovar-DQA1 allele frequency differences between nematode-resistant and susceptible selection lines. Physiol Genomics. 2007;30(3):253–261. doi: 10.1152/physiolgenomics.00273.2006. [DOI] [PubMed] [Google Scholar]
- 67.Barendse W, Harrison BE, Bunch RJ, Thomas MB, Turner LB. Genome wide signatures of positive selection: the comparison of independent samples and the identification of regions associated to traits. BMC Genomics. 2009;10:178. doi: 10.1186/1471-2164-10-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, Pasternak S, Wheeler DA, Willis TD, Yu F, Yang H, Zeng C, Gao Y, Hu H, Hu W, Li C, Lin W, Liu S, Pan H, Tang X, Wang J, Wang W, Yu J, Zhang B, Zhang Q, The International HapMap Consortium et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449(7164):851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li R, Fan W, Tian G, Zhu H, He L, Cai J, Huang Q, Cai Q, Li B, Bai Y, Zhang Z, Zhang Y, Wang W, Li J, Wei F, Li H, Jian M, Li J, Zhang Z, Nielsen R, Li D, Gu W, Yang Z, Xuan Z, Ryder OA, Leung FC-C, Zhou Y, Cao J, Sun X, Fu Y. The sequence and de novo assembly of the giant panda genome. Nature. 2010;463(7279):311–317. doi: 10.1038/nature08696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grossman SR, Shylakhter I, Karlsson EK, Byrne EH, Morales S, Frieden G, Hostetter E, Angelino E, Garber M, Zuk O, Lander ES, Schaffner SF, Sabeti PC. A composite of multiple signals distinguishes causal variants in regions of positive selection. Science. 2010;327(5967):883–886. doi: 10.1126/science.1183863. [DOI] [PubMed] [Google Scholar]
- 72.Karlsson EK, Baranowska I, Wade CM, Salmon Hillbertz NHC, Zody MC, Anderson N, Biagi TM, Patterson N, Pielberg GR, Kulbokas EJ, Comstock KE, Keller ET, Mesirov JP, von Euler H, Kampe O, Hedhammar A, Lander ES, Andersson G, Andersson L, Lindblad-Toh K. Efficient mapping of mendelian traits in dogs through genome-wide association. Nat Genet. 2007;39(11):1321–1328. doi: 10.1038/ng.2007.10. [DOI] [PubMed] [Google Scholar]
- 73.Montgomery G, Sise J. Extraction of DNA from sheep white blood cells. N Z J Agric Res. 1990;33(3):437–441. doi: 10.1080/00288233.1990.10428440. [DOI] [Google Scholar]
- 74.The Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447(7145):661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Abdi H. Encyclopedia of Measurement and Statistics. Thousand Oaks (CA): Sage; 2007. [Google Scholar]
- 76.Teo YY, Fry AE, Clark TG, Tai ES, Seielstad M. On the usage of HWE for identifying genotyping errors. Ann Hum Genet. 2007;71(5):701–703. doi: 10.1111/j.1469-1809.2007.00356.x. [DOI] [PubMed] [Google Scholar]
- 77.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 78.Scheet P, Stephens M. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am J Hum Genet. 2006;78(4):629–644. doi: 10.1086/502802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sabeti PC, Reich DE, Higgins JM, Levine HZ, Richter DJ, Schaffner SF, Gabriel SB, Platko JV, Patterson NJ, McDonald GJ, Ackerman HC, Campbell SJ, Altshuler D, Cooper R, Kwiatkowski D, Ward R, Lander ES. Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419(6909):832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Linkage disequilibrium (LD), as shown by r 2 , for region 2 (OAR1:87384757–88132568) SNP in the Romney resistant selection line animals. (PNG 162 KB)
Additional file 2: Linkage disequilibrium (LD), as shown by r 2 , for region 2 (OAR1:87384757–88132568) SNP in the Romney susceptible selection line animals. (PNG 152 KB)
Additional file 3: Linkage disequilibrium (LD), as shown by r 2 , for region 2 (OAR1:87384757–88132568) SNP in the Perendale resistant selection line animals. (PNG 159 KB)
Additional file 4: Linkage disequilibrium (LD), as shown by r 2 , for region 2 (OAR1:87384757–88132568) SNP in the Perendale susceptible selection line animals. (PNG 164 KB)
Additional file 5: SNP in CHIA discovered using sequencing. Haplotype: Sweep (v1.1) haplotype, avBV: Average FEC breeding value, Positions refer to base in CHIA (XM_004002314.1). (CSV 307 bytes)
Additional file 6: All known genes in regions of interest as of 01/2014. (CSV 4 KB)
Additional file 7: Pedigree information and Illumina® Ovine SNP50 BeadChip data for Romney and Perendale selection line animals. (ZIP 6 MB)
Additional file 8: SNP used on iPLEX™ genotyping assay for the Sequenom® MassARRAY® platform. Multiplex number; SNP ID; Amplification primer sequence; Extension primer sequence; Ancestral allele (AA). (CSV 4 KB)
Additional file 9: PCR primers used for direct sequencing of CHIA exons. (CSV 911 bytes)
Data Availability Statement
The data sets supporting the results of this article are included within the article (and its additional files).