

Abstract
Numerous cellular and molecular events have been described in development of gastric cancer. In this article, we overviewed roles of Helicobacter pylori (H pylori) infection on some of the important events in gastric carcinogenesis and discussed whether these cellular and molecular events are reversible after cure of the infection. There are several bacterial components affecting gastric epithelial kinetics and promotion of gastric carcinogenesis. The bacterium also increases risks of genetic instability and mutations due to NO and other reactive oxygen species. Epigenetic silencing of tumor suppressor genes such as RUNX3 may alter the frequency of phenotype change of gastric glands to those with intestinal metaplasia. Host factors such as increased expression of growth factors, cytokines and COX-2 have been also reported in non-cancerous tissue in H pylori-positive subjects. It is noteworthy that most of the above phenomena are reversed after the cure of the infection. However, some of them including overexpression of COX-2 continue to exist and may increase risks for carcinogenesis in metaplastic or dysplastic mucosa even after successful H pylori eradication. Thus, H pylori eradication may not completely abolish the risk for gastric carcinogenesis. Efficiency of the cure of the infection in suppressing gastric cancer depends on the timing and the target population, and warrant further investigation.
Keywords: Helicobacter, Cancer, Gastric acid, p53, In-flammation, Gastric atrophy, Intestinal metaplasia
INTRODUCTION
Gastric cancer is one of the most common neoplasmas worldwide, accounting for over 870 000 new cases and over 650 000 deaths annually[1]. Mortality from gastric cancer is the second most in death from malignancies, following to lung cancer. With exceptions in countries that have developed screening programs for early diagnoses, most patients reach treatment with cancers already in advanced stages[2]. Since surgery can be palliative, and gastric cancers are largely resistant to chemotherapy and radiotherapy, advanced gastric cancer has a poor prognosis. Therefore gastric cancer still remains a major clinical challenge and a great public health concern.
Extensive epidemiologic studies have shown that Helicobacter pylori (H pylori) infection is a major risk factor for developing gastric cancer and its precursor lesions[3]. The bacterium affects more than 50% of the world popultion[4]. The risk of patients with H pylori infection developing gastric cancer is in the order of two- to six-fold according to most retrospective case-control and prospective epidemiologic studies[5]. Furthermore, some of the trials eradica-ting H pylori have shown that cure of the infection reduces development of gastric cancer in high risk populations[6-8]. Thus, a hope is emerging and growing that cure of the H pylori infection may reduce incidence of gastric cancer.
The goal of this review is to clarify whether eradication H pylori results in eradication of gastric cancer. To accomplish this, we will discuss what types of cellular and molecular events occur in the H pylori-infected gastric mucosa; what bacterial factors are involved in the process of gastric carcinogenesis; and what host factors are able or unable to be reversed after the cure of the infection.
CELLULAR BASIS OF H PYLORI-RELATED GASTRIC CARCINOGENESIS
Histopathologic alterations
Chronic infection with H pylori causes active inflammation of gastric mucosa in majority of the population, although it is mostly asymptomatic. The bacterium also alters physiologic and histological behaviors of gastric mucosa, including hypochlorhydria, hyperchlorhydria, and changes in mucosal population of gastric epithelial cells that are specifically differentiated. In the hypochlorhydric population, hypergastrinemia occurs, while parietal cells do not respond to gastrin to produce acid, probably due to corpus inflammation. Apoptotic loss of superficial epithelial cells in the process of differentiation and migration in gastric glands increases[9], while proliferation of neck cells also increases possibly by some sort of compensation and by transactivation of growth stimuli in gastric mucosa[10]. In corpus mucosa, parietal cell population is also diminished in a long term, which is associated with alteration in population of other types of cells in each gland. Together with lowered density of glands partly due to inflammatory and edematous changes in subepithelial tissues, the above changes are known as gastric mucosal atrophy or atrophic gastritis. In addition, epithelial clones with incomplete and complete intestinal phenotypes emerge in the long-term process.
Currently, origins of the epithelial clones for the intestinal metaplasia have not yet been clearly determined. It is likely that epithelial clones for incomplete and complete intestinal metaplasia are developed from gastric epithelial cells by an activation of a series of genes including cdx-1/cdx-2[11-16]. In addition, bone marrow-derived adult somatic stem cells are involved in the regeneration of gastric glands, and may be another source of epithelial population[17]. Although our own study suggest that bone marrow-derived epithelial cells do not harbor permanently in a gastric gland, gastric adenocarcinomas are recently reported to be bone marrow-derived[18]. Stem cells in gastric glands locate neck region, whereas those in intestinal glands reside bottom region, a location completely different from the neck. Transdifferentiation of gastric gland cells to metaplastic cells remains an important question in gastric carcinogenesis.
Bacterial and/or host factors affecting the histologic alterations
Several pathogenic factors of H pylori have been de-monstrated to be involved in the above alterations in gastric mucosa and the following development of gastric diseases.
Ammonia (NH3), produced by H pylori urease, has been shown to cause acute gastric injury[19] in rats in vivo and to accelerate gastric epithelial cell death in vitro[19-21]. Chronic administration of NH3, whose concentration is comparable to that found in H pylori-infected patients, increases epithelial cell replication and epithelial cell turnover, associated with accelerated epithelial cell death, cell exfoliation, preferential loss of parietal cells and gastric mucosal atrophy[22,23]. The damaging effects of NH3 on gastric mucosa are pH-dependent, while sodium hydroxide at the same pH does not cause significant mucosal injury[19]. Ammonia dissolves readily in water where it forms, and is in equilibrium with, ammonium ions (NH4+). With decreases in pH, NH4+ predominates, but increases in pH may materially increase levels of non-ionized NH3[19]. On the other hand, per os administration with ammonium chloride (NH4Cl) results in intragastric NH4+, and does not induce significant mucosal atrophy. Rather, NH4Cl is reported to stimulate antral mucosa to increase gastrin release[24], which possibly induces gastric mucosal hypertrophy[25]. Therefore, not only H pylori but also gastric acid secretion of the host is an important determinant of gastric cell kinetics.
NH3 also increases incidence and size of gastric adenocarcinomas in rats pretreated with N-methyl-N’-nitro-N-nitrosoguanidine (MNNG)[26,27]. Prior administration of NH3 followed by MNNG does not increase incidence of gastric adenocarcinomas in rats, indicating that NH3 may act as a promoter in the chemically-induced gastric carcinogenesis. Immunohistochemical analysis using bromo deoxy-uridine (BrdU) demonstrates that NH3 increases cell replication in gastric tumors as well as non-cancerous tissues surrounding the tumors. Thus, NH3 and the consequent host epithelial responses play important roles not only in increased cell proliferation in untransformed gastric mucosa but also in promotion of gastric cancer.
The other virulence factors including CagA and other cag pathogenicity island (PAI) proteins, VacA and adhesions have been considered to be involved in wide diversity of H pylori-related diseases. For an example, strains containing the cag PAI have been reported to trigger signaling cascades in gastric epithelial cells, resulting in NF-κB activation and other cellular responses. Furthermore, CagA, which can be injected into the host cells, is able to be phosphorylated in the host, and to alter epithelial morphology probably through signaling pathway similar to that of HGF/c-met[28-31]. Roles of phosphorylated CagA protein in gastric epithelium are under extensive investigation and reviewed elsewhere[32]. Since it is related to gastric inflammation, cag PAI may stimulate indirectly excessive production of reactive oxygen species, including nitric oxide, and lead to programmed cell death. Indeed, studies show conflicting results for an association between cag PAI and apoptosis[33,34].
VacA reportedly induces gastric epithelial cell apoptosis[35,36]. It is found that VacA also induces apoptosis of marophages and suppresses T-cell responses[37-40]. Shibayama et al[41] showed that γ-glutamyl transpeptidase induces apoptosis. Furthermore, several apoptotic mediators such as TNF-α, FAS-ligand, TRAILs and their receptors are reported to be upregulated[42-44]. Thus, proapoptotic factors from either the bacterium or the host appear to be involved in altered cell kinetics as well as disturbed immunologic surveillance in gastric mucosa. Once certain clones acquire the resistance from apoptotic or immunologic surveillance, they begin to grow to form clusters of neoplastic phenotypes.
MOLECULAR ALTERATIONS OF H PYLORI-RELATED GASTRIC CARCINOGENESIS
Events promoting gastric carcinogenesis
Gastric cancer is divided into two histologic entities: ‘intestinal-type’ and ‘diffuse-type’. These two types differ in epidemiology and clinical outcome. Molecular profiles are also distinct between these phenotypes[45-47], and actually consist of wide variety of alterations including mutations, loss of heterozygosity (LOH), and epigenetic changes of expression of unmutated genes(Table 1). It is not surprising that numerous reviews have been published regarding this topic[45-52], considering the size of population with gastric cancers or with H pylori infection. In diffuse-type gastric adenocarcinomas, DNA-repair errors, p16 suppression and cyclin E amplification occur frequently in early stages. In early stages of intestinal-type gastric adenocarcinomas, inactivation of APC due to LOH or mutation and non-functioning p53 frequently occur. Events due to changes in tumor microenvironments, i.e., overexpression or transactivation of growth factors such as EGF-family growth factors (TGFα, EGF, HB-EGF, etc), insulin-like growth factors (IGF-1 and IGF-2), transforming growth factor-β, cytokines, and gastrin, also play important roles in phenotypic change in gastric epithelial cells [53,54]. For an example, elevated gastrin may transactivate HB-EGF and its receptors, resulting in upregulation of mitogen-inducible cyclooxygenase (COX-2) and its products (prostaglandin E2, etc)[55,56].
Table 1.
Molecular alteration in the process of gastric carcinogenesis
| Molecules | Major alterations | Comments | Category |
| p53 | Mutation, LOH | Reported in diffuse-type and intestinal-type adenocarcinomas, as well as some precancerous lesions. | Tumor suppressor |
| APC | Mutation, LOH | Reported in diffuse-type and intestinal-type adenocarcinomas, as well as some precancerous lesions. | Tumor suppressor |
| DCC | LOH | Reported in intestinal-type adenocarcinomas. Related to cell adhesion | Tumor suppressor |
| CDH1 | Mutation | Reported in diffuse-type adenocarcinomas. | Tumor suppressor |
| β-catenin | Mutation | Reported in intestinal-type adenocarcinomas. | Tumor suppressor |
| Fhit | LOH or deletion at chr. 3p14.2 | Reported in diffuse-type and, in less frequency, intestinal-type adenocarcinomas, as well as some precancerous lesions. | Tumor suppressor |
| RUNX3 | Hypermethylation | Related to TGF-β/SMAD signaling. | Tumor suppressor |
| K-ras | Mutation | Reported in intestinal-type adenocarcinomas. An element in signal transduction regulating cell proliferation, etc.. | Oncogene |
| bcl-2 | LOH | Reported in intestinal-type adenocarcinomas. Anti-apoptotic factor. | Oncogene |
| c-met | Amplification | Reported in diffuse-type and intestinal-type adenocarcinomas. The HGF receptor / tyrosine-kinase. Upregulation without mutation is also reported after mucosal injury. | Oncogene / Growth stimulus |
| c-erbB2 | Amplification | Reported in intestinal-type adenocarcinomas. One of receptor-tyrosine kinases for EGF-family proteins. | Oncogene / Growth stimulus |
| Cyclin E | Amplification | Reported in diffuse-type and intestinal type adenocarciomas. | Cell cycle regulator |
| K-sam | Amplification | Reported in diffuse-type adenocarcinomas. One of bFGF receptor family proteins, FGFR2. | Oncogene |
| Mismatch repair (MMR) genes | Silencing due to hypermethylation | Reported in diffuse-type and intestinal-type adenocarcinomas, as well as some precancerous lesions. A possible source of mutations of other genes involving gastric carcinogenesis. | Determinants of microsatelite instability (MSI) |
| MMR genes | Mutation | Reported in diffuse-type and intestinal-type adenocarcinomas. There are conflicting data suggesting that mucosa with intestinal metaplasia is prone to and resistant to MSI. | Determinants of MSI |
| EGFR | Overexpression | Reported in diffuse-type and intestinal-type adenocarcinomas. | Growth stimulus |
| EGF | Overexpression | Reported in diffuse-type and intestinal-type adenocarcinomas. | Growth stimulus |
| TGF-α | Overexpression | Reported in diffuse-type and intestinal-type adenocarcinomas, as well as some precancerous lesions. Another EGF-family protein. | Growth stimulus |
| VEGF | Overexpression | Reported in diffuse-type and intestinal-type adenocarcinomas. | Angiogenic factor |
| iNOS | Overexpression | Reported in diffuse-type and intestinal-type adenocarcinomas, as well as some precancerous lesions and mucosa with H. pylori. | Enzyme |
| COX-2 | Overexpression | Reported in diffuse-type and intestinal-type adenocarcinomas, as well as some precancerous lesions and mucosa with H. pylori. Cytokines and growth factors are possible inducer of COX-2. | Enzyme |
| ODC | Overexpression | Reported earlier in gastritis. | Enzyme |
| Telomerase | Activated | Enlongs telomere and prevents cell senescence. | Enzyme |
| CDXs | Overexpression | Reported in diffuse-type and intestinal-type adenocarcinomas, as well as precancerous lesions. Is involved in intestinal metaplasia. | Transcription factor |
| Ets1 | Overexpression | A transcription factor involving angiogenesis. | Transcription factor |
| NF-κB | Overexpression | A transcription factor regulating expression of proinflammatory cytokines, chemokines, iNOS and COX-2. | Transcription factor |
| Sp-1 | Overexpression | Reported in diffuse-type and intestinal-type adenocarcinomas. | Transcription factor |
| SC-1 | Overexpression | Reported in diffuse-type adenocarcinomas. | Apoptosis receptor |
| Fas/CD95 | Overexpression | Reported in diffuse-type adenocarcinomas. | Apoptosis receptor |
| E-cadherin | Mutation | Reported in diffuse-type and intestinal-type adenocarcinomas. | Cell adhesion |
| CD44 | Splicing variant | Reported in diffuse-type and intestinal-type adenocarcinomas. | Cell adhesion |
| Gastrin | Elevation in serum | Elevation of amidated gastrin is reported. Transactivates EGF-family proteins. | Gut hormone |
Recently, COX-2[57-71] attracts attention of many oncologists and gastroenterologists. In fact, some epidemiologic studies have shown that a long-term NSAID-use results in significant reduction of incidence and mortality of digestive cancers including not only colon but also stomach[72,73]. We have shown that the COX-2 overexpression alters cell kinetics, suppresses programmed cell death, induces invasive phenotypes, supports tumor angiogenesis and influences cell adhesion to endothelial cells[54,70,71,74-79]. H pylori infection induces gastric COX-2 upregulation[71,80-86], and cure of the infection reduces the COX-2 expression[70]. However, in mucosa with intestinal metaplasia, COX-2 is overexpressed even after the cure of the infection (Figure 1)[70]. Procarcinogenic effects of COX-2 on stomach could be only partially reversed by successful H pylori eradication. Similar findings were also observed in the case of expression of nitrotyrosine, a product of nitric oxide (NO), in precancerous gastric mucosa. Expression of nitrotyrosine is elevated in gastric mucosa in patients with H pylori gastritis, which is reversible after successful H pylori eradication. However, in gastric mucosa with intestinal metaplasia, nitrotyrosine continue to be overexpressed even after the cure of the H pylori infection, suggesting that NO and other reactive nitrogen species is highly produced in metaplastic lesions[70].
Figure 1.
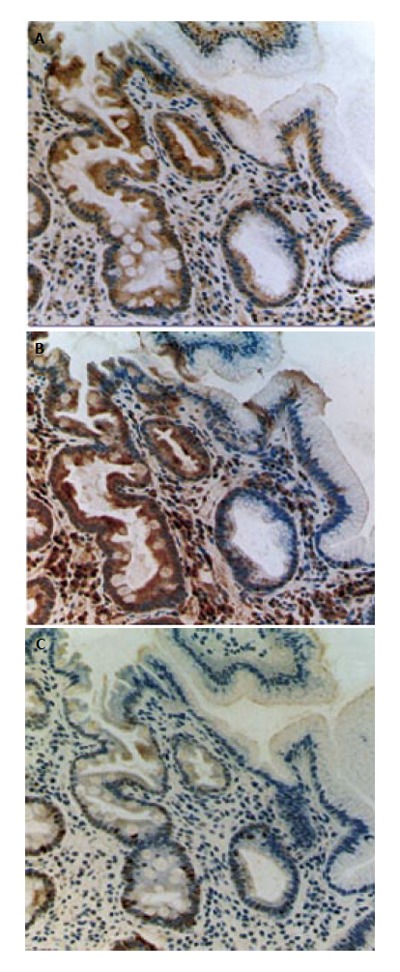
Expression of COX-2, nitrotyrosine and Ki-67 immunoreactivity in human gastric mucosa with intestinal metaplasia after cure of the H pylori infection. A: COX-2 immunostaining; B: nitrotyrosine immunostaining; C: Ki-67 immunostaining. The overexpression of COX-2 and nitrotyrosine, adduct of nitric oxide, are reported in gastric mucosa with H pylori infection[66,68,70,71]. In these photographs, metaplastic gland with goblet cells (in the left side of each photograph) and non-metaplastic gastric glands (in the right side) are shown. COX-2 and nitrotyrosine immunoreactivities continue to exist in gastric mucosa with intestinal metaplasia after the successful H pylori eradication with PPI-triple therapy.
Mismatch repair deficiency
Microsatellite instability (MSI) is defined as the presence of replication errors in simple repetitive microsatellite sequences due to mismatch repair (MMR) deficiency[48]. It is classified as high-frequency (MSI-H), low-frequency (MSI-L) or stable (MSS)[87]. MSI has been recognized as one of the earliest changes in carcinogenesis and results in genomic instability. MSI is detected not only in gastric cancer but also in intestinal metaplasia from subjects both with and without gastric cancer[88], suggesting that MSI can be an early event in gastric carcinogenesis[89-91]. Furthermore, hypermethylation of CpG islands in the promoter region of the hMLH1 gene is associated with decreased hMLH1 protein, and often occurs in gastric cancer cases with MSI-H, indicating that epigenetic inactivation of hMLH1 may underlie MSI[92]. MSI in gastric cancer is associated with antral tumors, intestinal-type differentiation, and a better prognosis. Cancer cases with MSI exhibit mutations in BAX, hMSH3, hMSH6, E2F-4, TGF-β receptor II, and IGF-R II, which have simple tandem repeat sequences within their coding regions[93-99]. H pylori infection and following gastric mucosal alteration are closely related to MSI[100-102]. In particular, Park et al[100] recently reported an immunohistochemical study demonstrating that DNA MMR protein expression (hMLH1 and hMSH2) decreases in patients with H pylori infection. Cure of the infection resulted in significant increases in the percentage of hMLH1 (76.60 ± 20.27, 84.82 ± 12.73, P = 0.01) and hMSH2 (82.36 ± 12.86, 88.11 ± 9.27, P < 0.05) positive epithelial cells[100], suggesting that the effects of H pylori on MSI are reversible at least in a part. On the other hand, MSI results in frame-shift mutations of hMSH3 and hMSH6, and loss of hMSH1 and hMSH2 functions, which may lead gastric epithelial cells to further genetic instability that cannot be reverted by H pylori eradication. Therefore, the precise mechanism for H pylori-induced suppression on MMR protein has not yet been clarified and one of the important topics in H pylori-related gastric carcinogenesis.
Oncogenes
Certain EGF-like growth factors and their receptors are activated by membrane-type proteases called ADAMs (a disintegrin and metalloproteinase) following the sti-mulation including gastrin[56], endothelin and IL-8 that have G-protein coupled receptors[103]. IL-1β is also known to transactivate EGF-receptor via pathways dependent and independent of IL-8[103].
In addition, certain growth factors, their receptors and components of intracellular signaling have mutations or amplifications activating cell growth, inhibiting programmed cell death, and altering cell phenotypes. These oncogenes include HGF receptor (c-met), c-erbB2 (HER-2/neu), c-erbB3, K-sam, ras, c-myc and others, and have been reported to be mutated, amplified, or overexpressed in the process of gastric carcinogenesis[47,52]. Once these oncogenes are mutated, it would be hardly possible for H pylori eradication to suppress oncogenes.
Tumor suppressor genes
Various tumor suppressor genes have been reported to be inactivated and involved in gastric carcinogenesis. For example, inactivation of p53 and p16 has been shown in both diffuse- and intestinal-type gastric cancers[52,104,105]. On the other hand, mutation of adenomatous polyposis coli (APC) gene occurs more often in intestinal-type gastric cancer. Another important tumor suppressor gene in intestinal-type gastric cancer is runt-related gene 3 (RUNX3) coding a subunit of polyomavirus enhancer binding protein 2[106-110], since expression of RUNX3 is greatly reduced in intestinal metaplasias in human stomachs[111] and Runx3-/- mouse gastric epithelial cells have a potential to differentiate into Cdx-2 positive intestinal type cells[112]. The product of the gene appears to interact with smad 2/3, which mediates TGF-β signaling pathway, and induces p21WAF1/Cip1 expression.
Inactivation of these tumor suppressor genes includes, inactivating mutations, LOH, and epigenetic silencing. For example, hot spot mutations on CpG islands in p53 have been reported not only in gastric cancers at early stages but also in non-cancerous tissues with intestinal metaplas-
ia [104,105,113-118]. In stomach, mutated p53 proteins are largely non-functioning and accumulate in the cells. Interestingly, p53 mutation frequently include G:C→A:T transition (Table 2, Figure 1)[119,120], and NO is an important mutagen causing this type of mutation[120-122]. On the other hand, silencing of RUNX3 by promoter hypermethylation is frequently found in gastric cancers and in intestinal metaplasia. Although the silencing of tumor suppressor genes due to mutation may not be reversed, the epigenetic silencing may be reversed in methylation and demethylation processes. At present, there is no evidence indicating H pylori per se increases aberrant hypermethylation of tumor suppressor genes[123]: rather, Epstein-Barr virus-related gastric cancer is associated with a high frequency of DNA hypermethylation, suggesting that viral oncogenesis might involve DNA hypermethylation with inactivation of tumor suppressor genes[124]. However, male gender, intestinal metaplasia and chronic inflammation with monocytic infiltration are strongly associated with increased methylation in non-cancerous gastric mucosa[123], and H pylori infection is one of the major causes of gastric inflammation. Thus, it remains an important question whether cure of the infection reduces the epigenetic alterations in tumor suppressor genes in non-transformed gastric epithelia.
Table 2.
"p53" mutation in gastric cancers of early stages and precancerous gastric lesions. In gastric cancers of early stages and precancerous gastric lesions, LOH and splicing are merely reported. Abbreviations for mutation: Del: deletion; Ins: insertion; F/S: frame shift. Abbreviations for lesion: EGC: early gastric cancer; AD: adenoma, CA/AD: carcinoma in adenoma; D: dysplasia; IM: intestinal metaplasia; N: mucosa without dysplasia, IM or carcinoma. Data are collected from references 104, 105, 113-118. (Modified from Tsuji et al[119,120])
| First author | Year | Case | G:C→A:T | G:C→T:A | A:T→G:C | A:T→C:G | A:T→T:A | G or C | Del | Ins | F/S | Lesions |
| Yokozaki | 1992 | 1 | 2 | 1 | 1 | 1 | EGC | |||||
| Tohdo | 1993 | 5 | 3 | 1 | AD or CA/AD | |||||||
| Uchino | 1993 | 12 | 10 | 2 | 1 | EGC | ||||||
| Correa | 1994 | 8 | 4 | 3 | 1 | D, IM, or N | ||||||
| Hongyo | 1995 | 9 | 10 | 1 | 1 | 2 | Cancer at stage I | |||||
| Sakurai | 1995 | 7 | 4 | AD, CA/AD or EGC | ||||||||
| Tamura | 1995 | 1 | 1 | AD | ||||||||
| Tamura | 1995 | 4 | 3 | 2 | EGC | |||||||
| Ranzani | 1995 | 18 | 13 | 1 | 1 | 1 | 1 | 2 | EGC | |||
| Summary | 45 | 3 | 8 | 3 | 2 | 3 | 4 | 4 | 1 | |||
| (%) | 62 | 4 | 11 | 4 | 3 | 4 | 5 | 5 | 1 |
Telomere and/or telomerase
Activation of telomerase that prevents shortening of telomeres during cell division may play an important role in immortalizing cells[47,125-127]. In brief, telomeres cover the ends of chromosomes and are important in maintaining chromosomal integrity. In intestinal metaplasia, shortening of telomeres[52] as well as telomerase activation[127,128] are observed, suggesting an important role in development of gastric cancer with intestinal type. Interestingly, it has been reported that H pylori infection reactivates telomerase[129,130], and that cure of the infection appears to reduce telomerase activity[130]. Since clinical studies using human subjects may suffer from sampling errors, it remains an open question whether H pylori eradication reverses telomerase activation.
HOST GENETICS OF H PYLORI-RELATED GASTRIC CARCINOGENESIS
Genetic predisposition affecting inflammation and acidity of stomach
Genetic predisposition plays an important role in developing gastric cancer. The most widely reported are IL1B and NAT1 polymorphisms[131-138]. The association of IL1B polymorphism and gastric carcinogenesis was hypothetically explained by El-Omar et al[134] to be a strong acid-inhibiting and proinflammatory capacity of the gene product. Indeed, gastric acid secretion is known to be suppressed by IL-1β, which is mediated by nitric oxide[139]. These genetic factors may have strong association with H pylori infection, since the bacterium induces production of interleukins, inflammation, and elevates intragastric pH, which may result in increase of xenobiotic products. On the other hand, IL-1β and IL-8 were recently reported to transactivate EGF-receptor via ADAM-10 activation[103]. IL-1β is also known to up-regulate COX-2 in gastric epithelium[140]. Therefore, the reason for the association of IL1B polymorphisms and the risk for gastric cancer remains an open question and may require further inves-tigation.
Genetic predisposition possibly independent of acidity of stomach
Another example of the genetic predisposition is families of hereditary nonpolyposis colorectal cancer (HNPCC) kindred of which have an excess of gastric carcinoma; complete intestinal metaplasia and chronic atrophic gastritis restricted to the antrum[141-143]. Interestingly, HNPCC patients frequently have a mutation in one of two DNA mismatch repair genes, hMSH2 or hMLH1, and demonstrate MSI-H. As mentioned earlier, H pylori has an ability to decrease MMR activity. Several genetic predispositions in MSI may share the same mutations to those found in H pylori-induced carcinogenesis. In these cases, the bacterial infection has a potent impact on gastric carcinogenesis, since it could lower the MMR activity more than the hereditary predisposition alone.
Hereditary gastric cancer due to germline mutation of the E-cadherin has been reported[144], which is a risk factor possibly independent of H pylori infection.
DOES H PYLORI ERADICATION ERADI-CATE GASTRIC CANCER
Unlike the typical adenoma-carcinoma sequence of colon, development of gastric cancer appears to be a complex process. Due to the complexity of molecular events of gastric carcinogenesis, factors discussed here do not cover every aspect of gastric carcinogenesis. Rather, we tried to overview some of the possible factors initiating, promoting and supporting the development of gastric cancer. By doing so, we discussed what types of risks exists in H pylori positive subjects and what extent of these risks could be withdrawn after the cure of the infection.
Certain bacterial factors affect gastric epithelial cells directly to support establishment and development of metaplastic or dysplastic clones. Successful H pylori eradi-cation withdraws these bacterial factors and therefore lowers the promotional effects on tumor development. The bacterium also increases genetic instability and ri-sks of mutation. Some host factors such as NO and other reactive oxygen species are induced by H pylori and increase risks of mutation. Although cure of the infection may reduce these risks leading to epithelial mutagenesis, it does not abolish the risk completely. Particularly, in gastric mucosa with intestinal metaplasia and other phenotypically altered tissues, increases in MSI and NO synthesis, as well as COX-2 overexpression are unaltered after the cure of the H pylori infection. Thus, H pylori eradication is an effective strategy in reducing the risk of gastric cancer; however, it is not efficient enough to eradicate gastric cancer. Prevention of the infection, H pylori immunization, H pylori eradication in the youth, selection of the high risk population, and alternative chemopreventive measures may be essential for optimal management of malignancy of the stomach.
Footnotes
S- Editor Xia HHX L- Editor Zhang JZ E- Editor Wu M
References
- 1.Stewart B, Kleihues P, editors . World Cancer Report. Lyon: IARC Press;; 2003. [Google Scholar]
- 2.Hohenberger P, Gretschel S. Gastric cancer. Lancet. 2003;362:305–315. doi: 10.1016/s0140-6736(03)13975-x. [DOI] [PubMed] [Google Scholar]
- 3.Hamilton S, Aaltonen L, editors . Pathology and Genetics. Tumours of the Digestive System. WHO Classification of Tumours. Lyon: IARC Press;; 2000. [Google Scholar]
- 4.Danesh J. Helicobacter pylori infection and gastric cancer: systematic review of the epidemiological studies. Aliment Pharmacol Ther. 1999;13:851–856. doi: 10.1046/j.1365-2036.1999.00546.x. [DOI] [PubMed] [Google Scholar]
- 5.Eslick GD, Lim LL, Byles JE, Xia HH, Talley NJ. Association of Helicobacter pylori infection with gastric carcinoma: a meta-analysis. Am J Gastroenterol. 1999;94:2373–2379. doi: 10.1111/j.1572-0241.1999.01360.x. [DOI] [PubMed] [Google Scholar]
- 6.Uemura N, Mukai T, Okamoto S, Yamaguchi S, Mashiba H, Taniyama K, Sasaki N, Haruma K, Sumii K, Kajiyama G. Effect of Helicobacter pylori eradication on subsequent development of cancer after endoscopic resection of early gastric cancer. Cancer Epidemiol Biomarkers Prev. 1997;6:639–642. [PubMed] [Google Scholar]
- 7.Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, Taniyama K, Sasaki N, Schlemper RJ. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001;345:784–789. doi: 10.1056/NEJMoa001999. [DOI] [PubMed] [Google Scholar]
- 8.Wong BC, Lam SK, Wong WM, Chen JS, Zheng TT, Feng RE, Lai KC, Hu WH, Yuen ST, Leung SY, et al. Helicobacter pylori eradication to prevent gastric cancer in a high-risk region of China: a randomized controlled trial. JAMA. 2004;291:187–194. doi: 10.1001/jama.291.2.187. [DOI] [PubMed] [Google Scholar]
- 9.Xia HH, Talley NJ. Apoptosis in gastric epithelium induced by helicobacter pylori infection: implications in gastric carcinogenesis. Am J Gastroenterol. 2001;96:16–26. doi: 10.1111/j.1572-0241.2001.03447.x. [DOI] [PubMed] [Google Scholar]
- 10.Stoicov C, Saffari R, Cai X, Hasyagar C, Houghton J. Molecular biology of gastric cancer: Helicobacter infection and gastric adenocarcinoma: bacterial and host factors responsible for altered growth signaling. Gene. 2004;341:1–17. doi: 10.1016/j.gene.2004.07.023. [DOI] [PubMed] [Google Scholar]
- 11.Silberg DG, Furth EE, Taylor JK, Schuck T, Chiou T, Traber PG. CDX1 protein expression in normal, metaplastic, and neoplastic human alimentary tract epithelium. Gastroenterology. 1997;113:478–486. doi: 10.1053/gast.1997.v113.pm9247467. [DOI] [PubMed] [Google Scholar]
- 12.Silberg DG, Swain GP, Suh ER, Traber PG. Cdx1 and cdx2 expression during intestinal development. Gastroenterology. 2000;119:961–971. doi: 10.1053/gast.2000.18142. [DOI] [PubMed] [Google Scholar]
- 13.Satoh K, Mutoh H, Eda A, Yanaka I, Osawa H, Honda S, Kawata H, Kihira K, Sugano K. Aberrant expression of CDX2 in the gastric mucosa with and without intestinal metaplasia: effect of eradication of Helicobacter pylori. Helicobacter. 2002;7:192–198. doi: 10.1046/j.1523-5378.2002.00080.x. [DOI] [PubMed] [Google Scholar]
- 14.Mutoh H, Sakurai S, Satoh K, Tamada K, Kita H, Osawa H, Tomiyama T, Sato Y, Yamamoto H, Isoda N, et al. Development of gastric carcinoma from intestinal metaplasia in Cdx2-transgenic mice. Cancer Res. 2004;64:7740–7747. doi: 10.1158/0008-5472.CAN-04-1617. [DOI] [PubMed] [Google Scholar]
- 15.Mutoh H, Sakurai S, Satoh K, Osawa H, Tomiyama T, Kita H, Yoshida T, Tamada K, Yamamoto H, Isoda N, et al. Pericryptal fibroblast sheath in intestinal metaplasia and gastric carcinoma. Gut. 2005;54:33–39. doi: 10.1136/gut.2004.042770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mutoh H, Hakamata Y, Sato K, Eda A, Yanaka I, Honda S, Osawa H, Kaneko Y, Sugano K. Conversion of gastric mucosa to intestinal metaplasia in Cdx2-expressing transgenic mice. Biochem Biophys Res Commun. 2002;294:470–479. doi: 10.1016/S0006-291X(02)00480-1. [DOI] [PubMed] [Google Scholar]
- 17.Komori M, Tsuji S, Tsujii M, Murata H, Iijima H, Yasumaru M, Nishida T, Irie T, Kawano S, Hori M. Efficiency of bone marrow-derived cells in regeneration of the stomach after induction of ethanol-induced ulcers in rats. J Gastroenterol. 2005;40:591–599. doi: 10.1007/s00535-005-1593-0. [DOI] [PubMed] [Google Scholar]
- 18.Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, Cai X, Fox JG, Goldenring JR, Wang TC. Gastric cancer originating from bone marrow-derived cells. Science. 2004;306:1568–1571. doi: 10.1126/science.1099513. [DOI] [PubMed] [Google Scholar]
- 19.Tsujii M, Kawano S, Tsuji S, Fusamoto H, Kamada T, Sato N. Mechanism of gastric mucosal damage induced by ammonia. Gastroenterology. 1992;102:1881–1888. doi: 10.1016/0016-5085(92)90309-m. [DOI] [PubMed] [Google Scholar]
- 20.Nakamura E, Hagen SJ. Role of glutamine and arginase in protection against ammonia-induced cell death in gastric epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2002;283:G1264–G1275. doi: 10.1152/ajpgi.00235.2002. [DOI] [PubMed] [Google Scholar]
- 21.Hagen SJ, Takahashi S, Jansons R. Role of vacuolation in the death of gastric epithelial cells. Am J Physiol. 1997;272:C48–C58. doi: 10.1152/ajpcell.1997.272.1.C48. [DOI] [PubMed] [Google Scholar]
- 22.Tsujii M, Kawano S, Tsuji S, Ito T, Nagano K, Sasaki Y, Hayashi N, Fusamoto H, Kamada T. Cell kinetics of mucosal atrophy in rat stomach induced by long-term administration of ammonia. Gastroenterology. 1993;104:796–801. doi: 10.1016/0016-5085(93)91015-a. [DOI] [PubMed] [Google Scholar]
- 23.Kawano S, Tsujii M, Fusamoto H, Sato N, Kamada T. Chronic effect of intragastric ammonia on gastric mucosal structures in rats. Dig Dis Sci. 1991;36:33–38. doi: 10.1007/BF01300084. [DOI] [PubMed] [Google Scholar]
- 24.Dial EJ, Cooper LC, Lichtenberger LM. Amino acid- and amine-induced gastrin release from isolated rat endocrine granules. Am J Physiol. 1991;260:G175–G181. doi: 10.1152/ajpgi.1991.260.2.G175. [DOI] [PubMed] [Google Scholar]
- 25.Lichtenberger LM, Dial EJ, Romero JJ, Lechago J, Jarboe LA, Wolfe MM. Role of luminal ammonia in the development of gastropathy and hypergastrinemia in the rat. Gastroenterology. 1995;108:320–329. doi: 10.1016/0016-5085(95)90056-x. [DOI] [PubMed] [Google Scholar]
- 26.Tsujii M, Kawano S, Tsuji S, Takei Y, Tamura K, Fusamoto H, Kamada T. Mechanism for ammonia-induced promotion of gastric carcinogenesis in rats. Carcinogenesis. 1995;16:563–566. doi: 10.1093/carcin/16.3.563. [DOI] [PubMed] [Google Scholar]
- 27.Tsujii M, Kawano S, Tsuji S, Nagano K, Ito T, Hayashi N, Fusamoto H, Kamada T, Tamura K. Ammonia: a possible promotor in Helicobacter pylori-related gastric carcinogenesis. Cancer Lett. 1992;65:15–18. doi: 10.1016/0304-3835(92)90207-c. [DOI] [PubMed] [Google Scholar]
- 28.Higashi H, Nakaya A, Tsutsumi R, Yokoyama K, Fujii Y, Ishikawa S, Higuchi M, Takahashi A, Kurashima Y, Teishikata Y, et al. Helicobacter pylori CagA induces Ras-independent morphogenetic response through SHP-2 recruitment and activation. J Biol Chem. 2004;279:17205–17216. doi: 10.1074/jbc.M309964200. [DOI] [PubMed] [Google Scholar]
- 29.Azuma T, Yamazaki S, Yamakawa A, Ohtani M, Muramatsu A, Suto H, Ito Y, Dojo M, Yamazaki Y, Kuriyama M, et al. Association between diversity in the Src homology 2 domain--containing tyrosine phosphatase binding site of Helicobacter pylori CagA protein and gastric atrophy and cancer. J Infect Dis. 2004;189:820–827. doi: 10.1086/381782. [DOI] [PubMed] [Google Scholar]
- 30.Umehara S, Higashi H, Ohnishi N, Asaka M, Hatakeyama M. Effects of Helicobacter pylori CagA protein on the growth and survival of B lymphocytes, the origin of MALT lymphoma. Oncogene. 2003;22:8337–8342. doi: 10.1038/sj.onc.1207028. [DOI] [PubMed] [Google Scholar]
- 31.Azuma T, Yamazaki S, Yamakawa A, Ito Y, Ohtani M, Dojo M, Yamazaki Y, Higashi H, Hatakeyama M. The effects of cure of Helicobacter pylori infection on the signal transduction of gastric epithelial cells. Aliment Pharmacol Ther. 2003;18 Suppl 1:39–44. doi: 10.1046/j.1365-2036.18.s1.2.x. [DOI] [PubMed] [Google Scholar]
- 32.Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer. 2004;4:688–694. doi: 10.1038/nrc1433. [DOI] [PubMed] [Google Scholar]
- 33.Peek RM Jr, Vaezi MF, Falk GW, Goldblum JR, Perez-Perez GI, Richter JE, Blaser MJ. Role of Helicobacter pylori cagA(+) strains and specific host immune responses on the development of premalignant and malignant lesions in the gastric cardia. Int J Cancer. 1999;82:520–524. doi: 10.1002/(sici)1097-0215(19990812)82:4<520::aid-ijc9>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 34.Moss SF, Sordillo EM, Abdalla AM, Makarov V, Hanzely Z, Perez-Perez GI, Blaser MJ, Holt PR. Increased gastric epithelial cell apoptosis associated with colonization with cagA + Helicobacter pylori strains. Cancer Res. 2001;61:1406–1411. [PubMed] [Google Scholar]
- 35.Torres VJ, McClain MS, Cover TL. Interactions between p-33 and p-55 domains of the Helicobacter pylori vacuolating cytotoxin (VacA) J Biol Chem. 2004;279:2324–2331. doi: 10.1074/jbc.M310159200. [DOI] [PubMed] [Google Scholar]
- 36.Cover TL, Krishna US, Israel DA, Peek RM Jr. Induction of gastric epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res. 2003;63:951–957. [PubMed] [Google Scholar]
- 37.Gebert B, Fischer W, Weiss E, Hoffmann R, Haas R. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science. 2003;301:1099–1102. doi: 10.1126/science.1086871. [DOI] [PubMed] [Google Scholar]
- 38.Boncristiano M, Paccani SR, Barone S, Ulivieri C, Patrussi L, Ilver D, Amedei A, D'Elios MM, Telford JL, Baldari CT. The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. J Exp Med. 2003;198:1887–1897. doi: 10.1084/jem.20030621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Menaker RJ, Ceponis PJ, Jones NL. Helicobacter pylori induces apoptosis of macrophages in association with alterations in the mitochondrial pathway. Infect Immun. 2004;72:2889–2898. doi: 10.1128/IAI.72.5.2889-2898.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sundrud MS, Torres VJ, Unutmaz D, Cover TL. Inhibition of primary human T cell proliferation by Helicobacter pylori vacuolating toxin (VacA) is independent of VacA effects on IL-2 secretion. Proc Natl Acad Sci U S A. 2004;101:7727–7732. doi: 10.1073/pnas.0401528101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shibayama K, Kamachi K, Nagata N, Yagi T, Nada T, Doi Y, Shibata N, Yokoyama K, Yamane K, Kato H, et al. A novel apoptosis-inducing protein from Helicobacter pylori. Mol Microbiol. 2003;47:443–451. doi: 10.1046/j.1365-2958.2003.03305.x. [DOI] [PubMed] [Google Scholar]
- 42.Martin JH, Potthoff A, Ledig S, Cornberg M, Jandl O, Manns MP, Kubicka S, Flemming P, Athmann C, Beil W, et al. Effect of H. pylori on the expression of TRAIL, FasL and their receptor subtypes in human gastric epithelial cells and their role in apoptosis. Helicobacter. 2004;9:371–386. doi: 10.1111/j.1083-4389.2004.00269.x. [DOI] [PubMed] [Google Scholar]
- 43.Wu YY, Tsai HF, Lin WC, Chou AH, Chen HT, Yang JC, Hsu PI, Hsu PN. Helicobacter pylori enhances tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in human gastric epithelial cells. World J Gastroenterol. 2004;10:2334–2339. doi: 10.3748/wjg.v10.i16.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koyama S. Flow cytometric measurement of tumor necrosis factor-related apoptosis-inducing ligand and its receptors in gastric epithelium and infiltrating mucosal lymphocytes in Helicobacter pylori-associated gastritis. J Gastroenterol Hepatol. 2003;18:763–770. doi: 10.1046/j.1440-1746.2003.03055.x. [DOI] [PubMed] [Google Scholar]
- 45.Nardone G, Rocco A, Malfertheiner P. Review article: helicobacter pylori and molecular events in precancerous gastric lesions. Aliment Pharmacol Ther. 2004;20:261–270. doi: 10.1111/j.1365-2036.2004.02075.x. [DOI] [PubMed] [Google Scholar]
- 46.Nardone G. Review article: molecular basis of gastric carcinogenesis. Aliment Pharmacol Ther. 2003;17 Suppl 2:75–81. doi: 10.1046/j.1365-2036.17.s2.10.x. [DOI] [PubMed] [Google Scholar]
- 47.Tahara E. Genetic pathways of two types of gastric cancer. IARC Sci Publ. 2004:327–349. [PubMed] [Google Scholar]
- 48.Zheng L, Wang L, Ajani J, Xie K. Molecular basis of gastric cancer development and progression. Gastric Cancer. 2004;7:61–77. doi: 10.1007/s10120-004-0277-4. [DOI] [PubMed] [Google Scholar]
- 49.Juhász M, Herszényi L, Tulassay Z, Malfertheiner P, Ebert MP. Helicobacter pylori and molecular mechanisms of gastric carcinogenesis: targets for prevention and therapy. Expert Rev Anticancer Ther. 2004;4:97–103. doi: 10.1586/14737140.4.1.97. [DOI] [PubMed] [Google Scholar]
- 50.Naumann M, Crabtree JE. Helicobacter pylori-induced epithelial cell signalling in gastric carcinogenesis. Trends Microbiol. 2004;12:29–36. doi: 10.1016/j.tim.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 51.Nardone G, Morgner A. Helicobacter pylori and gastric malignancies. Helicobacter. 2003;8 Suppl 1:44–52. doi: 10.1046/j.1523-5378.2003.00160.x. [DOI] [PubMed] [Google Scholar]
- 52.Tahara E, Kuniyasu H, Yasui W, Yokozaki H. Gene alterations in intestinal metaplasia and gastric cancer. Eur J Gastroenterol Hepatol. 1994;6 Suppl 1:S97–102. [PubMed] [Google Scholar]
- 53.Tsuji S, Kawai N, Tsujii M, Kawano S, Hori M. Review article: inflammation-related promotion of gastrointestinal carcinogenesis--a perigenetic pathway. Aliment Pharmacol Ther. 2003;18 Suppl 1:82–89. doi: 10.1046/j.1365-2036.18.s1.22.x. [DOI] [PubMed] [Google Scholar]
- 54.Tsuji S, Tsujii M, Kawano S, Hori M. Cyclooxygenase-2 upregulation as a perigenetic change in carcinogenesis. J Exp Clin Cancer Res. 2001;20:117–129. [PubMed] [Google Scholar]
- 55.Komori M, Tsuji S, Sun WH, Tsujii M, Kawai N, Yasumaru M, Kakiuchi Y, Kimura A, Sasaki Y, Higashiyama S, et al. Gastrin enhances gastric mucosal integrity through cyclooxygenase-2 upregulation in rats. Am J Physiol Gastrointest Liver Physiol. 2002;283:G1368–G1378. doi: 10.1152/ajpgi.00006.2002. [DOI] [PubMed] [Google Scholar]
- 56.Miyazaki Y, Shinomura Y, Tsutsui S, Zushi S, Higashimoto Y, Kanayama S, Higashiyama S, Taniguchi N, Matsuzawa Y. Gastrin induces heparin-binding epidermal growth factor-like growth factor in rat gastric epithelial cells transfected with gastrin receptor. Gastroenterology. 1999;116:78–89. doi: 10.1016/s0016-5085(99)70231-3. [DOI] [PubMed] [Google Scholar]
- 57.Wu CY, Wang CJ, Tseng CC, Chen HP, Wu MS, Lin JT, Inoue H, Chen GH. Helicobacter pylori promote gastric cancer cells invasion through a NF-kappaB and COX-2-mediated pathway. World J Gastroenterol. 2005;11:3197–3203. doi: 10.3748/wjg.v11.i21.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chang YJ, Wu MS, Lin JT, Sheu BS, Muta T, Inoue H, Chen CC. Induction of cyclooxygenase-2 overexpression in human gastric epithelial cells by Helicobacter pylori involves TLR2/TLR9 and c-Src-dependent nuclear factor-kappaB activation. Mol Pharmacol. 2004;66:1465–1477. doi: 10.1124/mol.104.005199. [DOI] [PubMed] [Google Scholar]
- 59.Jang TJ. Expression of proteins related to prostaglandin E2 biosynthesis is increased in human gastric cancer and during gastric carcinogenesis. Virchows Arch. 2004;445:564–571. doi: 10.1007/s00428-004-1104-3. [DOI] [PubMed] [Google Scholar]
- 60.Wambura C, Aoyama N, Shirasaka D, Kuroda K, Maekawa S, Ebara S, Watanabe Y, Tamura T, Kasuga M. Influence of gastritis on cyclooxygenase-2 expression before and after eradication of Helicobacter pylori infection. Eur J Gastroenterol Hepatol. 2004;16:969–979. doi: 10.1097/00042737-200410000-00004. [DOI] [PubMed] [Google Scholar]
- 61.Sun WH, Yu Q, Shen H, Ou XL, Cao DZ, Yu T, Qian C, Zhu F, Sun YL, Fu XL, et al. Roles of Helicobacter pylori infection and cyclooxygenase-2 expression in gastric carcinogenesis. World J Gastroenterol. 2004;10:2809–2813. doi: 10.3748/wjg.v10.i19.2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nardone G, Rocco A, Vaira D, Staibano S, Budillon A, Tatangelo F, Sciulli MG, Perna F, Salvatore G, Di Benedetto M, et al. Expression of COX-2, mPGE-synthase1, MDR-1 (P-gp), and Bcl-xL: a molecular pathway of H pylori-related gastric carcinogenesis. J Pathol. 2004;202:305–312. doi: 10.1002/path.1512. [DOI] [PubMed] [Google Scholar]
- 63.Kim KM, Oh YL, Ko JS, Choe YH, Seo JK. Histopathology and expression of Ki-67 and cyclooxygenase-2 in childhood Helicobacter pylori gastritis. J Gastroenterol. 2004;39:231–237. doi: 10.1007/s00535-003-1282-9. [DOI] [PubMed] [Google Scholar]
- 64.Thun MJ, Henley SJ, Gansler T. Inflammation and cancer: an epidemiological perspective. Novartis Found Symp. 2004;256:6–21; discussion 22-28, 49-52, 266-269. [PubMed] [Google Scholar]
- 65.Konturek SJ, Bielanski W, Gruchała A, Stachura J, Czesnikiewicz M, Bobrzyński A, Konturek PC, Hahn EG. Severe atrophic gastritis with extreme hypergastrinemia and gene expression of ornithine decarboxylase (ODC) and cyclo-oxygenase-2 (COX-2) expression: comparison with gastric cancer. J Clin Gastroenterol. 2004;38:87–89. doi: 10.1097/00004836-200401000-00022. [DOI] [PubMed] [Google Scholar]
- 66.Sheu BS, Yang HB, Sheu SM, Huang AH, Wu JJ. Higher gastric cycloxygenase-2 expression and precancerous change in Helicobacter pylori-infected relatives of gastric cancer patients. Clin Cancer Res. 2003;9:5245–5251. [PubMed] [Google Scholar]
- 67.Jüttner S, Cramer T, Wessler S, Walduck A, Gao F, Schmitz F, Wunder C, Weber M, Fischer SM, Schmidt WE, et al. Helicobacter pylori stimulates host cyclooxygenase-2 gene transcription: critical importance of MEK/ERK-dependent activation of USF1/-2 and CREB transcription factors. Cell Microbiol. 2003;5:821–834. doi: 10.1046/j.1462-5822.2003.00324.x. [DOI] [PubMed] [Google Scholar]
- 68.Shun CT, Wu MS, Huang SP, Wang HP, Chuang SM, Lin JT. Cyclooxygenase-2 expression correlates with nuclear p53 accumulation in gastric carcinoma. Hepatogastroenterology. 2003;50:988–992. [PubMed] [Google Scholar]
- 69.Caputo R, Tuccillo C, Manzo BA, Zarrilli R, Tortora G, Blanco Cdel V, Ricci V, Ciardiello F, Romano M. Helicobacter pylori VacA toxin up-regulates vascular endothelial growth factor expression in MKN 28 gastric cells through an epidermal growth factor receptor-, cyclooxygenase-2-dependent mechanism. Clin Cancer Res. 2003;9:2015–2021. [PubMed] [Google Scholar]
- 70.Kimura A, Tsuji S, Tsujii M, Sawaoka H, Iijima H, Kawai N, Yasumaru M, Kakiuchi Y, Okuda Y, Ali Z, et al. Expression of cyclooxygenase-2 and nitrotyrosine in human gastric mucosa before and after Helicobacter pylori eradication. Prostaglandins Leukot Essent Fatty Acids. 2000;63:315–322. doi: 10.1054/plef.2000.0220. [DOI] [PubMed] [Google Scholar]
- 71.Sawaoka H, Kawano S, Tsuji S, Tsuji M, Sun W, Gunawan ES, Hori M. Helicobacter pylori infection induces cyclooxygenase-2 expression in human gastric mucosa. Prostaglandins Leukot Essent Fatty Acids. 1998;59:313–316. doi: 10.1016/s0952-3278(98)90079-5. [DOI] [PubMed] [Google Scholar]
- 72.Thun MJ, Namboodiri MM, Calle EE, Flanders WD, Heath CW Jr. Aspirin use and risk of fatal cancer. Cancer Res. 1993;53:1322–1327. [PubMed] [Google Scholar]
- 73.Wang WH, Huang JQ, Zheng GF, Lam SK, Karlberg J, Wong BC. Non-steroidal anti-inflammatory drug use and the risk of gastric cancer: a systematic review and meta-analysis. J Natl Cancer Inst. 2003;95:1784–1791. doi: 10.1093/jnci/djg106. [DOI] [PubMed] [Google Scholar]
- 74.Kakiuchi Y, Tsuji S, Tsujii M, Murata H, Kawai N, Yasumaru M, Kimura A, Komori M, Irie T, Miyoshi E, et al. Cyclooxygenase-2 activity altered the cell-surface carbohydrate antigens on colon cancer cells and enhanced liver metastasis. Cancer Res. 2002;62:1567–1572. [PubMed] [Google Scholar]
- 75.Sawaoka H, Tsuji S, Tsujii M, Gunawan ES, Sasaki Y, Kawano S, Hori M. Cyclooxygenase inhibitors suppress angiogenesis and reduce tumor growth in vivo. Lab Invest. 1999;79:1469–1477. [PubMed] [Google Scholar]
- 76.Murata H, Kawano S, Tsuji S, Tsuji M, Sawaoka H, Kimura Y, Shiozaki H, Hori M. Cyclooxygenase-2 overexpression enhances lymphatic invasion and metastasis in human gastric carcinoma. Am J Gastroenterol. 1999;94:451–455. doi: 10.1111/j.1572-0241.1999.876_e.x. [DOI] [PubMed] [Google Scholar]
- 77.Sawaoka H, Kawano S, Tsuji S, Tsujii M, Gunawan ES, Takei Y, Nagano K, Hori M. Cyclooxygenase-2 inhibitors suppress the growth of gastric cancer xenografts via induction of apoptosis in nude mice. Am J Physiol. 1998;274:G1061–G1067. doi: 10.1152/ajpgi.1998.274.6.G1061. [DOI] [PubMed] [Google Scholar]
- 78.Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M, DuBois RN. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell. 1998;93:705–716. doi: 10.1016/s0092-8674(00)81433-6. [DOI] [PubMed] [Google Scholar]
- 79.Tsuji S, Kawano S, Sawaoka H, Takei Y, Kobayashi I, Nagano K, Fusamoto H, Kamada T. Evidences for involvement of cyclooxygenase-2 in proliferation of two gastrointestinal cancer cell lines. Prostaglandins Leukot Essent Fatty Acids. 1996;55:179–183. doi: 10.1016/s0952-3278(96)90095-2. [DOI] [PubMed] [Google Scholar]
- 80.Franco L, Talamini G, Carra G, Doria D. Expression of COX-1, COX-2, and inducible nitric oxide synthase protein in human gastric antrum with Helicobacter pylori infection. Prostaglandins Other Lipid Mediat. 1999;58:9–17. doi: 10.1016/s0090-6980(99)00020-9. [DOI] [PubMed] [Google Scholar]
- 81.Fu S, Ramanujam KS, Wong A, Fantry GT, Drachenberg CB, James SP, Meltzer SJ, Wilson KT. Increased expression and cellular localization of inducible nitric oxide synthase and cyclooxygenase 2 in Helicobacter pylori gastritis. Gastroenterology. 1999;116:1319–1329. doi: 10.1016/s0016-5085(99)70496-8. [DOI] [PubMed] [Google Scholar]
- 82.McCarthy CJ, Crofford LJ, Greenson J, Scheiman JM. Cyclooxygenase-2 expression in gastric antral mucosa before and after eradication of Helicobacter pylori infection. Am J Gastroenterol. 1999;94:1218–1223. doi: 10.1111/j.1572-0241.1999.01070.x. [DOI] [PubMed] [Google Scholar]
- 83.Kim JM, Kim JS, Jung HC, Song IS, Kim CY. Upregulated cyclooxygenase-2 inhibits apoptosis of human gastric epithelial cells infected with Helicobacter pylori. Dig Dis Sci. 2000;45:2436–2443. doi: 10.1023/a:1005611613542. [DOI] [PubMed] [Google Scholar]
- 84.Takahashi S, Fujita T, Yamamoto A. Role of cyclooxygenase-2 in Helicobacter pylori- induced gastritis in Mongolian gerbils. Am J Physiol Gastrointest Liver Physiol. 2000;279:G791–G798. doi: 10.1152/ajpgi.2000.279.4.G791. [DOI] [PubMed] [Google Scholar]
- 85.Sung JJ, Leung WK, Go MY, To KF, Cheng AS, Ng EK, Chan FK. Cyclooxygenase-2 expression in Helicobacter pylori-associated premalignant and malignant gastric lesions. Am J Pathol. 2000;157:729–735. doi: 10.1016/S0002-9440(10)64586-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tatsuguchi A, Sakamoto C, Wada K, Akamatsu T, Tsukui T, Miyake K, Futagami S, Kishida T, Fukuda Y, Yamanaka N, et al. Localisation of cyclooxygenase 1 and cyclooxygenase 2 in Helicobacter pylori related gastritis and gastric ulcer tissues in humans. Gut. 2000;46:782–789. doi: 10.1136/gut.46.6.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Werner M, Becker KF, Keller G, Höfler H. Gastric adenocarcinoma: pathomorphology and molecular pathology. J Cancer Res Clin Oncol. 2001;127:207–216. doi: 10.1007/s004320000195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hayden JD, Martin IG, Cawkwell L, Quirke P. The role of microsatellite instability in gastric carcinoma. Gut. 1998;42:300–303. doi: 10.1136/gut.42.2.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hamamoto T, Yokozaki H, Semba S, Yasui W, Yunotani S, Miyazaki K, Tahara E. Altered microsatellites in incomplete-type intestinal metaplasia adjacent to primary gastric cancers. J Clin Pathol. 1997;50:841–846. doi: 10.1136/jcp.50.10.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ottini L, Palli D, Falchetti M, D'Amico C, Amorosi A, Saieva C, Calzolari A, Cimoli F, Tatarelli C, De Marchis L, et al. Microsatellite instability in gastric cancer is associated with tumor location and family history in a high-risk population from Tuscany. Cancer Res. 1997;57:4523–4529. [PubMed] [Google Scholar]
- 91.Leung WK, Kim JJ, Kim JG, Graham DY, Sepulveda AR. Microsatellite instability in gastric intestinal metaplasia in patients with and without gastric cancer. Am J Pathol. 2000;156:537–543. doi: 10.1016/S0002-9440(10)64758-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Leung SY, Yuen ST, Chung LP, Chu KM, Chan AS, Ho JC. hMLH1 promoter methylation and lack of hMLH1 expression in sporadic gastric carcinomas with high-frequency microsatellite instability. Cancer Res. 1999;59:159–164. [PubMed] [Google Scholar]
- 93.Lee HS, Choi SI, Lee HK, Kim HS, Yang HK, Kang GH, Kim YI, Lee BL, Kim WH. Distinct clinical features and outcomes of gastric cancers with microsatellite instability. Mod Pathol. 2002;15:632–640. doi: 10.1038/modpathol.3880578. [DOI] [PubMed] [Google Scholar]
- 94.Menoyo A, Alazzouzi H, Espín E, Armengol M, Yamamoto H, Schwartz S Jr. Somatic mutations in the DNA damage-response genes ATR and CHK1 in sporadic stomach tumors with microsatellite instability. Cancer Res. 2001;61:7727–7730. [PubMed] [Google Scholar]
- 95.Ohmiya N, Matsumoto S, Yamamoto H, Baranovskaya S, Malkhosyan SR, Perucho M. Germline and somatic mutations in hMSH6 and hMSH3 in gastrointestinal cancers of the microsatellite mutator phenotype. Gene. 2001;272:301–313. doi: 10.1016/s0378-1119(01)00517-0. [DOI] [PubMed] [Google Scholar]
- 96.Ogata S, Tamura G, Endoh Y, Sakata K, Ohmura K, Motoyama T. Microsatellite alterations and target gene mutations in the early stages of multiple gastric cancer. J Pathol. 2001;194:334–340. doi: 10.1002/path.895. [DOI] [PubMed] [Google Scholar]
- 97.Kobayashi K, Okamoto T, Takayama S, Akiyama M, Ohno T, Yamada H. Genetic instability in intestinal metaplasia is a frequent event leading to well-differentiated early adenocarcinoma of the stomach. Eur J Cancer. 2000;36:1113–1119. doi: 10.1016/s0959-8049(00)00066-6. [DOI] [PubMed] [Google Scholar]
- 98.Yamamoto H, Perez-Piteira J, Yoshida T, Terada M, Itoh F, Imai K, Perucho M. Gastric cancers of the microsatellite mutator phenotype display characteristic genetic and clinical features. Gastroenterology. 1999;116:1348–1357. doi: 10.1016/s0016-5085(99)70499-3. [DOI] [PubMed] [Google Scholar]
- 99.Chung YJ, Park SW, Song JM, Lee KY, Seo EJ, Choi SW, Rhyu MG. Evidence of genetic progression in human gastric carcinomas with microsatellite instability. Oncogene. 1997;15:1719–1726. doi: 10.1038/sj.onc.1201343. [DOI] [PubMed] [Google Scholar]
- 100.Park DI, Park SH, Kim SH, Kim JW, Cho YK, Kim HJ, Sohn CI, Jeon WK, Kim BI, Cho EY, et al. Effect of Helicobacter pylori infection on the expression of DNA mismatch repair protein. Helicobacter. 2005;10:179–184. doi: 10.1111/j.1523-5378.2005.00309.x. [DOI] [PubMed] [Google Scholar]
- 101.Kim JJ, Tao H, Carloni E, Leung WK, Graham DY, Sepulveda AR. Helicobacter pylori impairs DNA mismatch repair in gastric epithelial cells. Gastroenterology. 2002;123:542–553. doi: 10.1053/gast.2002.34751. [DOI] [PubMed] [Google Scholar]
- 102.Yao Y, Tao H, Kim JJ, Burkhead B, Carloni E, Gasbarrini A, Sepulveda AR. Alterations of DNA mismatch repair proteins and microsatellite instability levels in gastric cancer cell lines. Lab Invest. 2004;84:915–922. doi: 10.1038/labinvest.3700117. [DOI] [PubMed] [Google Scholar]
- 103.Tanida S, Joh T, Itoh K, Kataoka H, Sasaki M, Ohara H, Nakazawa T, Nomura T, Kinugasa Y, Ohmoto H, et al. The mechanism of cleavage of EGFR ligands induced by inflammatory cytokines in gastric cancer cells. Gastroenterology. 2004;127:559–569. doi: 10.1053/j.gastro.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 104.Yokozaki H, Kuniyasu H, Kitadai Y, Nishimura K, Todo H, Ayhan A, Yasui W, Ito H, Tahara E. p53 point mutations in primary human gastric carcinomas. J Cancer Res Clin Oncol. 1992;119:67–70. doi: 10.1007/BF01209657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tohdo H, Yokozaki H, Haruma K, Kajiyama G, Tahara E. p53 gene mutations in gastric adenomas. Virchows Arch B Cell Pathol Incl Mol Pathol. 1993;63:191–195. doi: 10.1007/BF02899260. [DOI] [PubMed] [Google Scholar]
- 106.Li QL, Ito K, Sakakura C, Fukamachi H, Inoue K, Chi XZ, Lee KY, Nomura S, Lee CW, Han SB, et al. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell. 2002;109:113–124. doi: 10.1016/s0092-8674(02)00690-6. [DOI] [PubMed] [Google Scholar]
- 107.Guo WH, Weng LQ, Ito K, Chen LF, Nakanishi H, Tatematsu M, Ito Y. Inhibition of growth of mouse gastric cancer cells by Runx3, a novel tumor suppressor. Oncogene. 2002;21:8351–8355. doi: 10.1038/sj.onc.1206037. [DOI] [PubMed] [Google Scholar]
- 108.Ito Y. Oncogenic potential of the RUNX gene family: 'overview'. Oncogene. 2004;23:4198–4208. doi: 10.1038/sj.onc.1207755. [DOI] [PubMed] [Google Scholar]
- 109.Sakakura C, Hagiwara A, Miyagawa K, Nakashima S, Yoshikawa T, Kin S, Nakase Y, Ito K, Yamagishi H, Yazumi S, et al. Frequent downregulation of the runt domain transcription factors RUNX1, RUNX3 and their cofactor CBFB in gastric cancer. Int J Cancer. 2005;113:221–228. doi: 10.1002/ijc.20551. [DOI] [PubMed] [Google Scholar]
- 110.Chi XZ, Yang JO, Lee KY, Ito K, Sakakura C, Li QL, Kim HR, Cha EJ, Lee YH, Kaneda A, et al. RUNX3 suppresses gastric epithelial cell growth by inducing p21(WAF1/Cip1) expression in cooperation with transforming growth factor {beta}-activated SMAD. Mol Cell Biol. 2005;25:8097–8107. doi: 10.1128/MCB.25.18.8097-8107.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Oshimo Y, Oue N, Mitani Y, Nakayama H, Kitadai Y, Yoshida K, Ito Y, Chayama K, Yasui W. Frequent loss of RUNX3 expression by promoter hypermethylation in gastric carcinoma. Pathobiology. 2004;71:137–143. doi: 10.1159/000076468. [DOI] [PubMed] [Google Scholar]
- 112.Fukamachi H, Ito K, Ito Y. Runx3-/- gastric epithelial cells differentiate into intestinal type cells. Biochem Biophys Res Commun. 2004;321:58–64. doi: 10.1016/j.bbrc.2004.06.099. [DOI] [PubMed] [Google Scholar]
- 113.Hongyo T, Buzard GS, Palli D, Weghorst CM, Amorosi A, Galli M, Caporaso NE, Fraumeni JF Jr, Rice JM. Mutations of the K-ras and p53 genes in gastric adenocarcinomas from a high-incidence region around Florence, Italy. Cancer Res. 1995;55:2665–2672. [PubMed] [Google Scholar]
- 114.Ranzani GN, Luinetti O, Padovan LS, Calistri D, Renault B, Burrel M, Amadori D, Fiocca R, Solcia E. p53 gene mutations and protein nuclear accumulation are early events in intestinal type gastric cancer but late events in diffuse type. Cancer Epidemiol Biomarkers Prev. 1995;4:223–231. [PubMed] [Google Scholar]
- 115.Sakurai S, Sano T, Nakajima T. Clinicopathological and molecular biological studies of gastric adenomas with special reference to p53 abnormality. Pathol Int. 1995;45:51–57. doi: 10.1111/j.1440-1827.1995.tb03379.x. [DOI] [PubMed] [Google Scholar]
- 116.Tamura G, Sakata K, Maesawa C, Suzuki Y, Terashima M, Satoh K, Sekiyama S, Suzuki A, Eda Y, Satodate R. Microsatellite alterations in adenoma and differentiated adenocarcinoma of the stomach. Cancer Res. 1995;55:1933–1936. [PubMed] [Google Scholar]
- 117.Correa P, Shiao YH. Phenotypic and genotypic events in gastric carcinogenesis. Cancer Res. 1994;54:1941s–1943s. [PubMed] [Google Scholar]
- 118.Uchino S, Noguchi M, Ochiai A, Saito T, Kobayashi M, Hirohashi S. p53 mutation in gastric cancer: a genetic model for carcinogenesis is common to gastric and colorectal cancer. Int J Cancer. 1993;54:759–764. doi: 10.1002/ijc.2910540509. [DOI] [PubMed] [Google Scholar]
- 119.Tsuji S, Kawano S, Tsujii M, Sun W, Gunawan ES, Murata H, Hori M. H pylori and gastric carcinogenesis. Meta-analysis of p53 mutations Gut. 1997;41(Suppl.) doi: 10.1097/00004836-199700001-00030. European Helicobacter pylori Study Group): A51. [DOI] [PubMed] [Google Scholar]
- 120.Tsuji S, Tsujii M, Sun WH, Gunawan ES, Murata H, Kawano S, Hori M. Helicobacter pylori and gastric carcinogenesis. J Clin Gastroenterol. 1997;25 Suppl 1:S186–S197. doi: 10.1097/00004836-199700001-00030. [DOI] [PubMed] [Google Scholar]
- 121.Nguyen T, Brunson D, Crespi CL, Penman BW, Wishnok JS, Tannenbaum SR. DNA damage and mutation in human cells exposed to nitric oxide in vitro. Proc Natl Acad Sci U S A. 1992;89:3030–3034. doi: 10.1073/pnas.89.7.3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wink DA, Kasprzak KS, Maragos CM, Elespuru RK, Misra M, Dunams TM, Cebula TA, Koch WH, Andrews AW, Allen JS. DNA deaminating ability and genotoxicity of nitric oxide and its progenitors. Science. 1991;254:1001–1003. doi: 10.1126/science.1948068. [DOI] [PubMed] [Google Scholar]
- 123.Kang GH, Lee HJ, Hwang KS, Lee S, Kim JH, Kim JS. Aberrant CpG island hypermethylation of chronic gastritis, in relation to aging, gender, intestinal metaplasia, and chronic inflammation. Am J Pathol. 2003;163:1551–1556. doi: 10.1016/S0002-9440(10)63511-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kim TY, Jong HS, Jung Y, Kim TY, Kang GH, Bang YJ. DNA hypermethylation in gastric cancer. Aliment Pharmacol Ther. 2004;20 Suppl 1:131–142. doi: 10.1111/j.1365-2036.2004.01984.x. [DOI] [PubMed] [Google Scholar]
- 125.Miyachi K, Fujita M, Tanaka N, Sasaki K, Sunagawa M. Correlation between telomerase activity and telomeric-repeat binding factors in gastric cancer. J Exp Clin Cancer Res. 2002;21:269–275. [PubMed] [Google Scholar]
- 126.Yang SM, Fang DC, Luo YH, Lu R, Battle PD, Liu WW. Alterations of telomerase activity and terminal restriction fragment in gastric cancer and its premalignant lesions. J Gastroenterol Hepatol. 2001;16:876–882. doi: 10.1046/j.1440-1746.2001.02540.x. [DOI] [PubMed] [Google Scholar]
- 127.Jong HS, Park YI, Kim S, Sohn JH, Kang SH, Song SH, Bang YJ, Kim NK. Up-regulation of human telomerase catalytic subunit during gastric carcinogenesis. Cancer. 1999;86:559–565. [PubMed] [Google Scholar]
- 128.Kameshima H, Yagihashi A, Yajima T, Kobayashi D, Denno R, Hirata K, Watanabe N. Helicobacter pylori infection: augmentation of telomerase activity in cancer and noncancerous tissues. World J Surg. 2000;24:1243–1249. doi: 10.1007/s002680010246. [DOI] [PubMed] [Google Scholar]
- 129.Lan J, Xiong YY, Lin YX, Wang BC, Gong LL, Xu HS, Guo GS. Helicobacter pylori infection generated gastric cancer through p53-Rb tumor-suppressor system mutation and telomerase reactivation. World J Gastroenterol. 2003;9:54–58. doi: 10.3748/wjg.v9.i1.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chung IK, Hwang KY, Kim IH, Kim HS, Park SH, Lee MH, Kim CJ, Kim SJ. Helicobacter pylori and telomerase activity in intestinal metaplasia of the stomach. Korean J Intern Med. 2002;17:227–233. doi: 10.3904/kjim.2002.17.4.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Troost E, Hold GL, Smith MG, Chow WH, Rabkin CS, McColl KE, El-Omar EM. The role of interleukin-1beta and other potential genetic markers as indicators of gastric cancer risk. Can J Gastroenterol. 2003;17 Suppl B:8B–12B. doi: 10.1155/2003/397060. [DOI] [PubMed] [Google Scholar]
- 132.El-Omar EM, Rabkin CS, Gammon MD, Vaughan TL, Risch HA, Schoenberg JB, Stanford JL, Mayne ST, Goedert J, Blot WJ, et al. Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology. 2003;124:1193–1201. doi: 10.1016/s0016-5085(03)00157-4. [DOI] [PubMed] [Google Scholar]
- 133.El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, et al. The role of interleukin-1 polymorphisms in the pathogenesis of gastric cancer. Nature. 2001;412:99. doi: 10.1038/35083631. [DOI] [PubMed] [Google Scholar]
- 134.El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature. 2000;404:398–402. doi: 10.1038/35006081. [DOI] [PubMed] [Google Scholar]
- 135.Figueiredo C, Machado JC, Pharoah P, Seruca R, Sousa S, Carvalho R, Capelinha AF, Quint W, Caldas C, van Doorn LJ, et al. Helicobacter pylori and interleukin 1 genotyping: an opportunity to identify high-risk individuals for gastric carcinoma. J Natl Cancer Inst. 2002;94:1680–1687. doi: 10.1093/jnci/94.22.1680. [DOI] [PubMed] [Google Scholar]
- 136.Machado JC, Pharoah P, Sousa S, Carvalho R, Oliveira C, Figueiredo C, Amorim A, Seruca R, Caldas C, Carneiro F, et al. Interleukin 1B and interleukin 1RN polymorphisms are associated with increased risk of gastric carcinoma. Gastroenterology. 2001;121:823–829. doi: 10.1053/gast.2001.28000. [DOI] [PubMed] [Google Scholar]
- 137.Boissy RJ, Watson MA, Umbach DM, Deakin M, Elder J, Strange RC, Bell DA. A pilot study investigating the role of NAT1 and NAT2 polymorphisms in gastric adenocarcinoma. Int J Cancer. 2000;87:507–511. [PubMed] [Google Scholar]
- 138.Katoh T, Boissy R, Nagata N, Kitagawa K, Kuroda Y, Itoh H, Kawamoto T, Bell DA. Inherited polymorphism in the N-acetyltransferase 1 (NAT1) and 2 (NAT2) genes and susceptibility to gastric and colorectal adenocarcinoma. Int J Cancer. 2000;85:46–49. doi: 10.1002/(sici)1097-0215(20000101)85:1<46::aid-ijc8>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 139.Esplugues JV, Barrachina MD, Calatayud S, Pique JM, Whittle BJ. Nitric oxide mediates the inhibition by interleukin-1 beta of pentagastrin-stimulated rat gastric acid secretion. Br J Pharmacol. 1993;108:9–10. doi: 10.1111/j.1476-5381.1993.tb13431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fan XM, Wong BC, Lin MC, Cho CH, Wang WP, Kung HF, Lam SK. Interleukin-1beta induces cyclo-oxygenase-2 expression in gastric cancer cells by the p38 and p44/42 mitogen-activated protein kinase signaling pathways. J Gastroenterol Hepatol. 2001;16:1098–1104. doi: 10.1046/j.1440-1746.2001.02593.x. [DOI] [PubMed] [Google Scholar]
- 141.Cristofaro G, Lynch HT, Caruso ML, Attolini A, DiMatteo G, Giorgio P, Senatore S, Argentieri A, Sbano E, Guanti G. New phenotypic aspects in a family with Lynch syndrome II. Cancer. 1987;60:51–58. doi: 10.1002/1097-0142(19870701)60:1<51::aid-cncr2820600110>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 142.Lynch HT, Smyrk TC, Lanspa SJ, Jenkins JX, Lynch PM, Cavalieri J, Lynch JF. Upper gastrointestinal manifestations in families with hereditary flat adenoma syndrome. Cancer. 1993;71:2709–2714. doi: 10.1002/1097-0142(19930501)71:9<2709::aid-cncr2820710904>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 143.Frei JV. Hereditary nonpolyposis colorectal cancer (Lynch syndrome II). Diploid malignancies with prolonged survival. Cancer. 1992;69:1108–1111. doi: 10.1002/cncr.2820690507. [DOI] [PubMed] [Google Scholar]
- 144.Lynch HT, Grady W, Lynch JF, Tsuchiya KD, Wiesner G, Markowitz SD. E-cadherin mutation-based genetic counseling and hereditary diffuse gastric carcinoma. Cancer Genet Cytogenet. 2000;122:1–6. doi: 10.1016/s0165-4608(00)00273-9. [DOI] [PubMed] [Google Scholar]