

Abstract
To study the clinical correlates of the H63D mu-tation we have analysed the phenotype of H63D homo-zygotes identified through mutation analysis in a referral laboratory. A total of 366 blood samples referred for HFE analysis were screened for C282Y and H63D mutations. Four H63D homozygotes were identified. All had raised serum ferritin but normal transferrin
saturation. They were negative for hepatitis B and C and only one patient consumed excess alcohol. In all 4 cases ultrasonography revealed fatty liver. In two patients a liver biopsy was done and showed mild siderosis with an unusual distribution and macrovesicular steatosis. These data confirm the association between fatty liver, hyperferritinemia and increased hepatic iron, but do not clarify whether siderosis was related to steatosis rather than homozygosity for the H63D mutation. Patients with fatty liver may complicate the interpretation of data in population studies of the expression of H63D homozygosity.
Keywords: Hyperferritinemia, HFE gene, H63D homozygosity, Fatty liver
INTRODUCTION
Hereditary haemochromatosis (HH) is a common autosomal recessive disorder of iron metabolism with an incidence of up to 1 in 200 and an estimated frequency of carriers of 1 in 10 among people of Northern European descent[1-3]. The disease is characterized by enhanced gastrointestinal absorption of iron to excessive accumulation in tissue, which may result in damage to liver and other target organs[4]. C282Y missense mutation in the HFE gene was found to be strongly related to the occurrence of HH[2]. In a UK study, homozygosity for the C282Y mutation was found to account for 91 % of HH[5]. A second missense mutation in the HFE gene, H63D, is found in around 4% of patients with HH, but its role in iron overload is still debated[2,6-8]. The H63D mutation is variably distributed worldwide. It is more prevalent than the C282Y mutation so that approximately one in five of the European population are H63D heterozygotes[3,9]. Individuals who are compound heterozygous for C282Y and H63D can have iron overload in the range diagnostic of haemochromatosis, although the penetrance of the genotype is low[2,10,11]. Similarly, homozygosity for H63D has been associated with iron overload, ranging from asymptomatic subjects to patients with typical haemochromatosis. As with compound heterozygosity for C282Y/H63D, the penetrance is low and the phenotypic presentation of this genotype varied considerably[11,12]. The aim of this study was to analyse the phenotypic expression of H63D homozygotes identified through the genetic screening of patients referred to our Centre for HFE mutation analysis.
366 consecutive blood samples, referred to the Centre for Hepatology at the Royal Free and University College Medical School (UCL), were analysed for HFE mutations. Mutation analysis was requested on the basis of biochemical or clinical suspicion of HH, family screening or known diagnosis of haemochromatosis. Samples were obtained after informed written consent, where appropriate. C282Y and H63D mutations were detected by polymerase chain reaction (PCR) amplification of total genomic DNA followed by restriction digestion with RsaI and MboI enzymes respectively, as previously described[5].
CASE REPORTS
Four males were found to be homozygous for the H63D mutation. Their main features are summarised in Table 1.
Table 1.
Clinical, biochemical and serological features of the four H63D homozygous patients
| Case 1 | Case 2 | Case 3 | Case 4 | |
| Age (yr) | 36 | 35 | 44 | 66 |
| BMI (kg/m2) | 30 | 24 | 32.5 | 31 |
| Blood pressure (mmHg) | 140/100 | 130/80 | 160/100 | 150/95 |
| Alcohol intake (g/wk) | 8 | 0 | 60 | 0 |
| Blood sugar (mmol/L) | 4.2 | 4.6 | 5.6 | 9.5 |
| Total cholesterol (mmol/L) | 4.1 | 4.3 | 5.5 | 6 |
| Triglicerides (mmol/L) | 1.6 | 1.5 | 3.6 | 2.9 |
| AST (U/L) | 46 | 69 | 44 | 15 |
| ALT (U/L) | 117 | 175 | 112 | 18 |
| γGT (U/L) | 50 | 45 | 70 | 30 |
| Ferritin (µg/L) | 454 | 350 | 568 | 423 |
| HCV/HBV serology | neg | neg | neg | neg |
Legend: BMI=body mass index; wk=week; neg=negative. Normal ranges: BMI 18 - 25 kg/m2; blood pressure max < 120mmHg, min<80mmHg (see the seventh report of the Joint National Committee on high blood pressure, NIH publication no 03 - 5233, December 2003); blood sugar < 6mmol/L.
Case 1
English male patient aged 36 years with suspected iron overload indicated by a serum ferritin concentration of 454 µg/L (reference range: 39-340). Serum iron, transferrin saturation and total iron binding capacity (TIBC) were normal. He was referred because of abnormal levels of liver enzymes and elevated serum ferritin found during investigation for dyspepsia. He consumed 8 g of alcohol per week. He had a positive family history for obesity and maturity onset diabetes mellitus. There was no family history of haemochromatosis. On examination he was well, overweight and mildly hypertensive (140/100). Abdominal examination was normal. Liver function tests showed: ALT 117 U/L, AST 46 U/L and γGT 50 U/L. Viral markers for hepatitis B and C were negative. Ultrasonography revealed a large liver, with diffuse hyperechogenicity, characteristic of fatty change. Since the liver enzymes and the ferritin remained persistently elevated, a liver biopsy was performed. It showed moderate macrovesicular steatosis (grade 2 on a scale of 0 to 3) with grade 1 siderosis (on a scale of 0 to 4)[13] in periportal hepatocytes and Kupffer cells, with evidence of pericellular fibrosis of zone 3 (stage 1 of steatohepatitis according to Brunt et al[14]). The sinusoidal cells had unusual granular siderosis (Figures 1A, 1B, 1C). The hepatic iron concentration was increased being 185 µg/100g dry weight (reference range: 35-136). The patient was seen every three months for 2 years to monitor transaminases and ferritin. On the basis of the result of the liver biopsy, venesection therapy was started, together with dietary restrictions. Over a period of two years, he was treated with seven phlebotomies and approximately 1.3 g of iron were removed. The serum ferritin concentration returned to normal at 95 µg/L as did liver function tests except for minimal elevation of ALT at 45 U/L.
Figure 1.
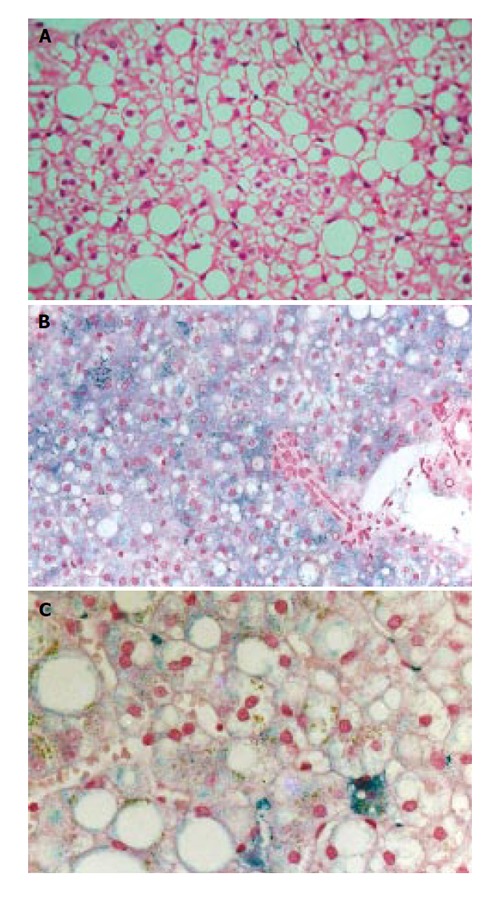
Case 1 liver histology. A: Hematoxylin and eosin staining shows grade 2 hepatic steatosis; B: Perls’ staining shows grade 1 hepatocyte siderosis with predominant periportal distribution; C: Higher power of Perls’ staining shows clustered Kupffer cell siderosis (right lower field) and also irregular large granular deposits in sinusoidal cells.
Case 2
Male patient aged 35 years from Lebanon with mild abnormality of iron indices. In 1996 he had a routine check-up and was found to have abnormal ALT (175 U/L) and AST (69 U/L), together with a minimally elevated serum ferritin (350 µg/L). Serum iron, TIBC and transferrin saturation were normal, as were the other liver function tests. The family history was negative for haemochromatosis. The patient did not drink alcohol. Viral markers for hepatitis B and C were negative. Between 1989 and 1995 he had been a blood donor giving approximately 2 units (approximately 450 mL each) of blood every year. On examination he had a normal build with gynaecomastia. Blood pressure was normal. Abdominal examination showed a palpable liver on deep inspiration (2 cm) but no other abnormalities. The alpha-1-antitrypsin level was 1.0 g/L (normal range 1.2-2.6 g/L). Analysis showed him to have phenotype MZ. Ultrasound examination revealed a hyperechogenic liver compatible with hepatic steatosis. A liver biopsy was performed and showed moderate macrovesicular steatosis (grade 2) without fibrosis (stage 0 of steatohepatitis[14]). There was grade 2 siderosis in periportal hepatocytes, and focal Kupffer cell iron. The sinusoidal cell iron had a granular pattern similar to that seen for case 1 (Figures 2A, 2B, 2C). No cholestasis or alpha-1-antitrypsin staining was detected.
Figure 2.
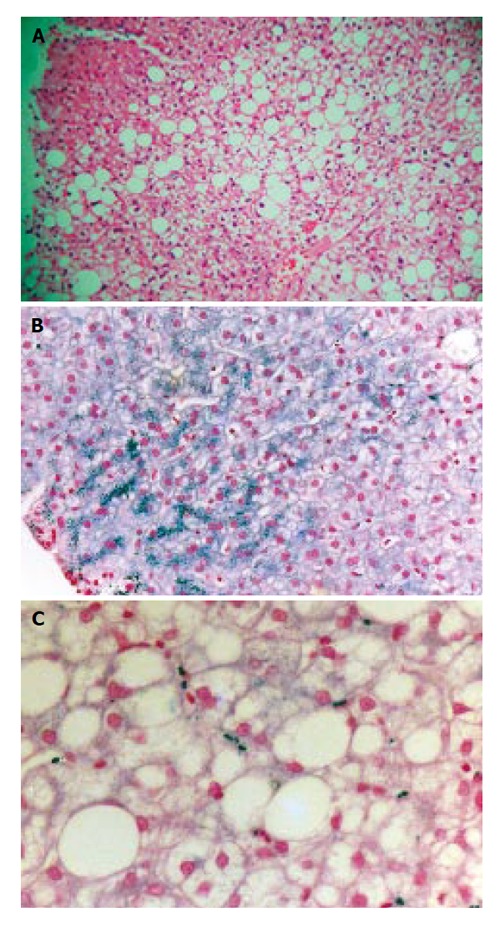
Case 2 liver histology. A: Hematoxylin and eosin staining shows grade 2 hepatic steatosis; B: Perls’ staining shows grade 2 hepatocyte siderosis with predominant periportal distribution; C: Higher power shows focal Kupffer cell siderosis and more granular irregular sinusoidal siderosis.
The patient was seen every three months for 4 years to monitor transaminases and ferritin, which remained persistently elevated.
Case 3
Irish male patient aged 44 years referred with possible iron overload indicated by a ferritin concentration of 568 µg/L. Serum iron, transferrin saturation and TIBC were normal. He was referred to our clinic in November 1998 to investigate raised levels of liver transaminases: ALT was 112 U/L, AST 44 U/L and γGT 70 U/L. The family history was positive for obesity and maturity onset diabetes mellitus and was negative for haemochromatosis. Physical examination showed a middle aged obese man (103 kg/178 cm; Body Mass Index (BMI) =32.5 kg/m2). Blood pressure was elevated in the supine position (160/100) and the hypertension was treated with Tenif one per day (atenolol 50 mg, nifedipine 20 mg) and Valsartan (80 mg/day). The patient had also a history of moderate alcohol abuse (alcohol intake: 60 g/week) and elevated triglyceride levels (3.61 mmol/L). Viral markers for hepatitis B and C were negative. Ultrasonography of the abdomen revealed a liver of normal size, but with echoreflectivity consistent with fatty liver. No liver biopsy was done. The patient was seen every three months for 2 years to monitor weight, transaminases and ferritin. He abstained from alcohol. Dietary therapy was started and the patient lost 6 kg in weight. No venesection was performed. During follow-up, liver function tests remained mildly abnormal with an ALT of 84 U/L. The other enzymes fell to normal levels and triglycerides fell to 1.73 mmol/L. Despite abstinence and losing weight, his ferritin remained elevated (482 µg/L). The possibility of iron overload was raised and the patient was genotyped for mutations in the HFE gene and found to be homozygous for H63D.
Case 4
English male patient aged 66 years with an elevated serum ferritin concentration (423 µg/L) on a background of type 2 diabetes and peripheral neuropathy. Serum iron, transferrin saturation and TIBC were normal. He was diagnosed as hypertensive in February 1988 and he is currently treated with Enalapril. He also had atrial fibrillation for which he has been treated with warfarin. He was treated for diabetes mellitus with gliclazide, metformin and acarbose. Physical examination showed no features of chronic liver disease and no hepatomegaly. Liver function tests were normal. Viral markers for hepatitis B and C were negative. Abdominal ultrasonography revealed an enlarged and fatty liver. Since his ferritin levels remained in the region of 400 µg/L, the patient was tested for mutations in the HFE gene and found to be homozygous for H63D mutation.
DISCUSSION
Four H63D homozygotes were identified by screening 366 blood samples referred for genetic analysis in the HFE gene. All the four patients had high serum ferritin. It is noteworthy that in all the cases an abdominal ultrasonography showed fatty liver. In the two cases where liver biopsy was done and histology showed not only mild siderosis of hepatocytes, typical of iron overload of HFE-related haemochromatosis, but also siderosis of sinusoidal cells, with a granular pattern that could be related to nonalcoholic steatohepatitis (NASH). Both patients had significant macrovesicular steatosis. One patient was treated with dietary restriction and venesection, after which liver function tests and serum ferritin concentration returned to normal. These findings suggest that hepatic steatosis, together with a biological effect of the H63D mutation, could be responsible for the hepatic siderosis in these patients. Recent evidence suggests that H63D homozygosity could lead to iron overload with variable penetrance and phenotype[12]. However, an association with fatty liver has not been previously reported. Another study of more than 10,000 blood donors suggested an effect of H63D homozygosity on iron metabolism[15], although the mean serum iron indices for this genotype were within the normal range, possibly reflecting the fact that blood donors are in general healthy and young (mean age was 38 years for men and 36 years for women). These findings have been reinforced experimentally by the demonstration that transgenic mice homozygous for the H63D mutation have elevated transferrin saturation and hepatic iron concentration compared to wild type mice[16].
H63D homozygosity could thus contribute to iron overload but the phenotypic expression may be influenced by cofactors. Sex, age, diet and modifier genes are likely to influence penetrance of the genotype. In this series all 4 cases were male and this reflects the relative protection of women from iron overload by menstruation and pregnancy. Furthermore, three of the cases had one or more metabolic disorders which are part of the insulin resistance syndrome (IRS)[17]. Case 1 was overweight (BMI>25 kg/m2) and mildly hypertensive, Case 3 was obese (BMI>30 kg/m2) and had hypertriglyceridemia and hypertension requiring antihypertensive treatment. Case 4 was obese, he had type 2 diabetes mellitus requiring therapy, hypertrigliceridemia and hypertension under therapy.
Although hyperferritinemia may be associated with hepatic steatosis per se[18,19], in two patients presented here hepatic siderosis with an unusual histological pattern was present. Homozygosity for H63D may have played a role, but the unusual cellular pattern raises the possibility that in some cases of hepatic steatosis there are other changes in cellular iron handling which result in iron accumulation. Alterations in cytokines and hence hepcidin may result in accumulation of iron in sinusoidal cells[20,21]. A role for the mutations in the HFE gene in other diseases, however, has been suggested by their over-representation in subjects with NASH and with the dysmetabolic iron overload syndrome (DIOS) characterized by an association between iron overload and insulin resistance[22-24]. The molecular mechanism explaining these associations is not clear. Hepatic steatosis has been recognised as the first of two “hits” in the pathogenesis of NASH, since the presence of oxidisable fat within the liver is enough to trigger lipid peroxidation[25]. However, many patients with fatty liver do not progress to necroinflammation[26]. It has been suggested that the second hit for the development of NASH may be oxidative stress, leading to necroinflammation[27]. Several potential second hits have been suggested. Iron even in relatively low concentrations could synergize with lipid overload and induction of ethanol-inducible cytochrome P450 2E1 (CYP2E1) to increase oxidative stress in hepatocytes[28].
If iron leads to oxidative stress and to progression of non-alcoholic fatty liver to NASH, venesection therapy is theoretically beneficial, and this may have contributed to the normalisation of serum ferritin concentration and improvement of liver function tests in Case 1. In conclusion, our findings confirm a link between fatty liver and mild iron accumulation. Whether homozygosity for H63D contributed to the association is uncertain. Further studies of iron regulatory proteins are needed in hepatic steatosis.
Footnotes
Supported by the European Commission Fifth Framework Pro-gramme Grant No. QLK6-CT-1999-02237. GS was supported by a Clinical Fellowship from the European Commission (Leonardo da Vinci Grant I/99/2/09209/PL/II.1.2.a/FPI)
S- Editor Wang J L- Editor Zhang JZ E- Editor Wu M
References
- 1.Edwards CQ, Griffen LM, Goldgar D, Drummond C, Skolnick MH, Kushner JP. Prevalence of hemochromatosis among 11,065 presumably healthy blood donors. N Engl J Med. 1988;318:1355–1362. doi: 10.1056/NEJM198805263182103. [DOI] [PubMed] [Google Scholar]
- 2.Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, Domingo R Jr, Ellis MC, Fullan A, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13:399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- 3.Merryweather-Clarke AT, Pointon JJ, Shearman JD, Robson KJ. Global prevalence of putative haemochromatosis mutations. J Med Genet. 1997;34:275–278. doi: 10.1136/jmg.34.4.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bacon BR, Powell LW, Adams PC, Kresina TF, Hoofnagle JH. Molecular medicine and hemochromatosis: at the crossroads. Gastroenterology. 1999;116:193–207. doi: 10.1016/S0016-5085(99)70244-1. [DOI] [PubMed] [Google Scholar]
- 5.A simple genetic test identifies 90% of UK patients with ha-emochromatosis. The UK Haemochromatosis Consortium. Gut. 1997;41:841–844. doi: 10.1136/gut.41.6.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jazwinska EC, Cullen LM, Busfield F, Pyper WR, Webb SI, Powell LW, Morris CP, Walsh TP. Haemochromatosis and HLA-H. Nat Genet. 1996;14:249–251. doi: 10.1038/ng1196-249. [DOI] [PubMed] [Google Scholar]
- 7.Gochee PA, Powell LW, Cullen DJ, Du Sart D, Rossi E, Olynyk JK. A population-based study of the biochemical and clinical expression of the H63D hemochromatosis mutation. Gastroenterology. 2002;122:646–651. doi: 10.1016/S0016-5085(02)80116-0. [DOI] [PubMed] [Google Scholar]
- 8.Fairbanks VF, Brandhagen DJ, Thibodeau SN, Snow K, Wollan PC. H63D is an haemochromatosis associated allele. Gut. 1998;43:441–442. doi: 10.1136/gut.43.3.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rochette J, Pointon JJ, Fisher CA, Perera G, Arambepola M, Arichchi DS, De Silva S, Vandwalle JL, Monti JP, Old JM, et al. Multicentric origin of hemochromatosis gene (HFE) mutations. Am J Hum Genet. 1999;64:1056–1062. doi: 10.1086/302318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aguilar Martinez P, Biron C, Blanc F, Masmejean C, Jeanjean P, Michel H, Schved JF. Compound heterozygotes for hemochromatosis gene mutations: may they help to understand the pathophysiology of the disease. Blood Cells Mol Dis. 1997;23:269–276. doi: 10.1006/bcmd.1997.0143. [DOI] [PubMed] [Google Scholar]
- 11.De Gobbi M, D'Antico S, Castagno F, Testa D, Merlini R, Bondi A, Camaschella C. Screening selected blood donors with biochemical iron overload for hemochromatosis: a regional experience. Haematologica. 2004;89:1161–1167. [PubMed] [Google Scholar]
- 12.Aguilar-Martinez P, Bismuth M, Picot MC, Thelcide C, Pageaux GP, Blanc F, Blanc P, Schved JF, Larrey D. Variable phenotypic presentation of iron overload in H63D homozygotes: are genetic modifiers the cause. Gut. 2001;48:836–842. doi: 10.1136/gut.48.6.836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scheuer PJ, Lefkowitch JH. Liver biopsy interpretation, 6th ed. London: Saunders. 2000:p. 274. [Google Scholar]
- 14.Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol. 1999;94:2467–2474. doi: 10.1111/j.1572-0241.1999.01377.x. [DOI] [PubMed] [Google Scholar]
- 15.Jackson HA, Carter K, Darke C, Guttridge MG, Ravine D, Hutton RD, Napier JA, Worwood M. HFE mutations, iron deficiency and overload in 10,500 blood donors. Br J Haematol. 2001;114:474–484. doi: 10.1046/j.1365-2141.2001.02949.x. [DOI] [PubMed] [Google Scholar]
- 16.Tomatsu S, Orii KO, Fleming RE, Holden CC, Waheed A, Britton RS, Gutierrez MA, Velez-Castrillon S, Bacon BR, Sly WS. Contribution of the H63D mutation in HFE to murine hereditary hemochromatosis. Proc Natl Acad Sci U S A. 2003;100:15788–15793. doi: 10.1073/pnas.2237037100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607. doi: 10.2337/diabetes.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 18.Bacon BR, Farahvash MJ, Janney CG, Neuschwander-Tetri BA. Nonalcoholic steatohepatitis: an expanded clinical entity. Gastroenterology. 1994;107:1103–1109. doi: 10.1016/0016-5085(94)90235-6. [DOI] [PubMed] [Google Scholar]
- 19.Loguercio C, De Simone T, D'Auria MV, de Sio I, Federico A, Tuccillo C, Abbatecola AM, Del Vecchio Blanco C. Non-alcoholic fatty liver disease: a multicentre clinical study by the Italian Association for the Study of the Liver. Dig Liver Dis. 2004;36:398–405. doi: 10.1016/j.dld.2004.01.022. [DOI] [PubMed] [Google Scholar]
- 20.Walker AP, Partridge J, Srai SK, Dooley JS. Hepcidin: what every gastroenterologist should know. Gut. 2004;53:624–627. doi: 10.1136/gut.2003.030304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 22.Bonkovsky HL, Jawaid Q, Tortorelli K, LeClair P, Cobb J, Lambrecht RW, Banner BF. Non-alcoholic steatohepatitis and iron: increased prevalence of mutations of the HFE gene in non-alcoholic steatohepatitis. J Hepatol. 1999;31:421–429. doi: 10.1016/S0168-8278(99)80032-4. [DOI] [PubMed] [Google Scholar]
- 23.Moirand R, Mortaji AM, Loreal O, Paillard F, Brissot P, Deugnier Y. A new syndrome of liver iron overload with normal transferrin saturation. Lancet. 1997;349:95–97. doi: 10.1016/S0140-6736(96)06034-5. [DOI] [PubMed] [Google Scholar]
- 24.Mendler MH, Turlin B, Moirand R, Jouanolle AM, Sapey T, Guyader D, Le Gall JY, Brissot P, David V, Deugnier Y. Insulin resistance-associated hepatic iron overload. Gastroenterology. 1999;117:1155–1163. doi: 10.1016/S0016-5085(99)70401-4. [DOI] [PubMed] [Google Scholar]
- 25.Day CP, James OF. Steatohepatitis: a tale of two”hits”. Gastroenterology. 1998;114:842–845. doi: 10.1016/S0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 26.Teli MR, James OF, Burt AD, Bennett MK, Day CP. The natural history of nonalcoholic fatty liver: a follow-up study. Hepatology. 1995;22:1714–1719. doi: 10.1002/hep.1840220616. [DOI] [PubMed] [Google Scholar]
- 27.Berson A, De Beco V, Letteron P, Robin MA, Moreau C, El Kahwaji J, Verthier N, Feldmann G, Fromenty B, Pessayre D. Steatohepatitis-inducing drugs cause mitochondrial dysfunction and lipid peroxidation in rat hepatocytes. Gastroenterology. 1998;114:764–774. doi: 10.1016/S0016-5085(98)70590-6. [DOI] [PubMed] [Google Scholar]
- 28.Tsukamoto H, Horne W, Kamimura S, Niemelä O, Parkkila S, Ylä-Herttuala S, Brittenham GM. Experimental liver cirrhosis induced by alcohol and iron. J Clin Invest. 1995;96:620–630. doi: 10.1172/JCI118077. [DOI] [PMC free article] [PubMed] [Google Scholar]