

Abstract
AIM: To assess the combinative role of aflatoxin B1 (AFB1), cyanobacterial toxins (cyanotoxins), and hepatitis B virus (HBV) x gene in hepatotumorigenicity.
METHODS: One-week-old animals carrying HBV x gene and their wild-type littermates were intraperitoneally (ip) injected with either single-dose AFB1 [6 mg/kg body weight (bw)], repeated-dose cyanotoxins (microcystin-LR or nodularin, 10 μg/kg bw once a week for 15 wk), DMSO (vehicle control) alone, or AFB1 followed by cyanotoxins a week later, and were sacrificed at 24 and 52 wk post-treatment.
RESULTS: AFB1 induced liver tumors in 13 of 29 (44.8%) transgenic mice at 52 wk post-treatment, significantly more frequent than in wild-type mice (13.3%). This significant difference was not shown in the 24-wk study. Compared with AFB1 exposure alone, MC-LR and nodularin yielded approximately 3-fold and 6-fold increases in the incidence of AFB1-induced liver tumors in wild-type animals at 24 wk, respectively. HBV x gene did not further elevate the risk associated with co-exposure to AFB1 and cyanotoxins. With the exception of an MC-LR-dosed wild-type mouse, no liver tumor was observed in mice treated with cyanotoxins alone at 24 wk. Neither DMSO-treated transgenic mice nor their wild-type littermates had pathologic alterations relevant to hepatotumorigenesis in even up to 52 wk.
CONCLUSION: HBV x gene and nodularin promote the development of AFB1-induced liver tumors. Co-exposure to AFB1 and MC-LR tends to elevate the risk of liver tumors at 24 wk relative to exposure to one of them. The combinative effect of AFB1, cyanotoxins and HBVx on hepatotumorigenesis is weak at 24 wk.
Keywords: Aflatoxins, Cyanobacteria, Hepatitis B virus, Liver neoplasms, Transgenic mice
INTRODUCTION
Hepatocellular carcinoma (HCC) is the sixth most common cancer and the third leading cause of cancer mortality worldwide[1]. In the People’s Republic of China, HCC is the second leading cause of cancer mortality with 28 463 deaths in 1999[2]. Epidemiological studies revealed that multiple environmental factors were associated with HCC in some high-risk areas. Chronic hepatitis B virus (HBV) infection and dietary aflatoxin exposure are two major etiological risk factors for HCC, which has been studied in the past two decades[3]. For individuals chronically infected with HBV, concurrent exposure to dietary aflatoxins increases the risk of HCC by at least three-fold[4]. Continuous hepatocyte death and regeneration caused by host immune response to viral antigens contributes to the pathogenesis of chronic HBV infection[5]. The HBV x (HBVx) gene, one of the four genes in the HBV genome, encodes a polypeptide of 154 amino acids and is highly conserved among all mammalian hepadnaviruses. Interestingly, in the absence of HBV x gene, duck hepatitis B virus (DHBV) infection did not yield HCC, but the frequent occurrence of HCC was caused by infections of woodchuck hepatitis virus (WHV) and ground squirrel hepatitis virus (GSHV) with the presence of HBV x gene[6]. Compared to other regions of the genome, the viral DNA sequences encoding HBV X and/or truncated envelope PreS2/S proteins were more frequently detected in the primary tumors and tumor-derived cell lines. Although HBVx does not bind to DNA directly, it is capable of trans-activating cellular genes and is considered to be a tumor promoter during liver carcinogenesis[7]. Some HBV x gene transgenic animals showed an increased susceptibility to chemical hepatocarcinogens[8,9]. Studies using transgenic mouse lines lend further support for the epidemiological finding that chronic HBV infection acts synergistically with aflatoxins in hepatocelluar carcinogenesis[10,11].
However, other risk factors, such as chronic exposure to cyanobacterial toxins (cyanotoxins) through drinking water, were relatively less studied or even overlooked. The mortality rate of HCC was closely associated with the source of drinking water in southern China at high risk of HCC[12]. Wide contamination of cyanotoxins in drinking water sources in these endemic areas suggests that the high incidence of HCC may be associated, in part, with chronic exposure to cyanotoxins via drinking water[13]. Cyanobacteria produce a wide range of secondary metabolites that are hazardous to humans, livestock, and wildlife[14]. Several bloom-forming cyanobacterial genera, including Microcystis, Anabaena, Planktothrix and Nostoc, are capable of producing the cyclic heptapeptide toxins, microcystins and Nodularia, the latter further metabolized to the cyclic pentapeptide nodularin. These toxins have a number of common structural features, in particular, the unique β-C20 amino acid 3-amino-9-methoxy-2,6,8-trimethyl-10-phenyldeca-4,6-dienoic acid. The most widely studied microcystins is microcystin-LR (MC-LR) with an LD50 between 32.5 μg/kg body weight (bw) and 10.9 mg/kg
bw in rodents by intraperitoneal (ip) injection, oral, and aerosol inhalation routes[15-17]. The direct cause of death in toxin-treated rodents was hemorrhagic shock and liver necrosis. MC-LR is a potent tumor promoter as well[18]. A two-stage carcinogenesis test showed that MC-LR modulated aflatoxin B1-induced hepatocarcinogenicity. In rats given AFB1 as an initiator, the subsequent treatment with MC-LR resulted in a synergistic increase in the development of glutathione S-transferase placental form (GST-P)-positive liver cell foci[19]. The hepatotoxicity of MC-LR is, at least in part, explained by its ability to inhibit protein phosphatases 1 and 2A and consequently induces hyperphosphorylation of various proteins, including tumor suppressor gene products, retinoblastoma (Rb) and p53[20]. Nodularin injures mammalian and fish livers in the same manner as microcystins; it also inhibits protein phosphatases and acts as a tumor promoter[21]. The result of a two-stage carcinogenesis experiment indicated that nodularin has the potential to stimulate growth of the initiated liver cells more strongly than does microcystin-LR and, additionally, an initiating capacity similar to diethylnitrosamine (DEN)[22].
Although epidemiological studies conducted in high-risk areas in China have implicated the synergistic effect of chronic HBV infection, dietary AFB1 exposure and chronic exposure to cyanotoxin-contaminated drinking water on the development of HCC, no experimental design has been made so far to study comprehensively the combinative effect of these three environmental risk factors on the development of liver tumors in animals. In this study, we attempted to use the established HBV x gene transgenic mouse model to assess the combinative role of chemical and viral infection in hepatotumorigenicity.
MATERIALS AND METHODS
Reagents
Aflatoxin B1 (AFB1) was purchased from Sigma Chemical Co. (St. Louis, MO). MC-LR (> 90% purity in HPLC), generously provided by Dr. Li-Rong Song, Department of Phycology, Institute of Hydrobiology, Chinese Academy of Sciences, was dissolved in saline to a concentration of 0.5 mg/mL. Nodularin (> 95.0% purity in HPLC), kindly provided by Dr. Wayne W. Carmichael, Wright State University, was dissolved in dimethylsulfoxide (DMSO, Burdick & Jackson, Muskegon, MI) to a concentration of 5 mg/mL. All stock solutions were kept at -20 °C until use.
Hepatitis B virus x gene transgenic mice
The development of the HBV x gene transgenic mice used in this study has been reported elsewhere[23]. Briefly, a recombinant retroviral expression vector plasmid pSHDX42 containing a 0.59 kilobase (kb) HBV subtype adr DNA fragment which spans nucleotide position 1 248 to 1 841 in the viral genome was constructed. A retroviral enhancer LTR, SV40 polyadenylation sequence, and a universal terminator from the expression vector pLXSHD were also inserted into pSHDX42 to initiate and terminate the transcription of the x gene. The DNA segment in the vector pSHDX42 contained the entire coding region of the x gene (map positions 1376 to 1840) and was released from pSHDX42 by BamH П/Sph І digestion. The purified DNA was microinjected into the pronuclei of fertilized eggs of C57BL/6J mice purchased from the Shanghai Laboratory Animal Center of Chinese Academy of Sciences. The DNA-injected eggs were transferred into the oviduct of pseudopregnant female ICR mice purchased from the Shanghai Laboratory Animal Center of Chinese Academy of Sciences. Genomic DNA and total RNA were extracted from F1 generation tail and liver tissues. Transgenic mice were identified by the methods of nested-primer PCR and Southern blotting of tail genomic DNA as well as RT-PCR of liver mRNA and immunohistochemical staining of HBV X protein in the liver. Six identified transgenic males were mated with wild-type C57BL/6J females to create a male transgenic colony for the following carcinogen treatments. Age- and litter-matched male offsprings were divided into six groups of transgenic and six groups of wild-type mice. All individual mice were identified according to the digit code and were arbitrarily assigned to treatment groups. All mice were maintained and cared for in accordance with the guidelines established by the National Institute of Health.
Nested-primer PCR
In a previous study, the nested-primer PCR of HBV x gene fragment from mouse tail genomic DNA was demonstrated to be compatible with RT-PCR of mRNA from liver samples in identifying transgenic animals[23]. In this study, only nested-primer PCR was used to screen the transgenic mice from the colony. The PCR primers were designed on human HBV subtype adr x gene sequence. The pair of outer primers included the forward, GTACCGACGATCCCACACGA, and the reverse, ATTAGGCAGACGTGAAAAAG, complementary to position 1375 to 1394 and 1841 to 1822 of the x gene sequence, respectively. The inner primer set consisted of the forward, GAAACAAATGCAGGGCAGCCGCGACTTAG, and the reverse, CAGTCTTTGAAG TATGCCTCAAGGTCGGT, which were complementary to the nucleotide positions 1421 to 1449 and 1719 to 1691 of the sequence, respectively. They generated a DNA product of 439 bp.
The reaction mixture of the first PCR consisted of 2 μL of DNA extract in a total volume of 20 μL, with final concentrations of 10 mmol/L Tris-HCl (pH 8.3), 50 mmol/L KCl, 1.5 mmol/L MgCl2 (10 × PCR buffer, Promega, Madison, WI), a 10 pmol/L concentration of each outer primer, 0.8 U of AmpliTaq DNA polymerase (Sangon, Shanghai, China), and a 10 mmol/L concentration of each dNTP (Promega, Madison, WI). The reaction mixture of the second PCR was identical with the exception of 3 μL of the first reaction products, 10 pmol/L of inner primers and 2.5 U of AmpliTaq DNA polymerase. Reactions with the outer primer set were thermal cycled once at 95 °C for 5 min, 30 times at 94 °Cfor 50 s, 55 °C for 1 min, and then once at 72 °C
for 1 min. For the nested PCR product, reactions were thermal cycled once at 94 °C for 5 min, 30 times at 94 °C for 50 s, 60 °C for 50 s, and 72 °C for 1 min, and then once at 72 °C for 10 min. The PCR products were analyzed by electrophoresis on 20 g/L agarose gels, stained with ethidium bromide, and visualized on an UV transilluminator.
Administration of toxins
Both one-week-old male transgenic mice and their wild-type littermates were used for the studies. All dosing was administered by ip injection based on body weight (bw) at the time of treatment. Animals treated with solvent vehicle (DMSO) were used as the controls. In the tumorigenic study with AFB1, animals were treated with a single dose of AFB1 at 6 mg/kg bw and were then euthanized either at 24 wk or 52 wk post-treatment. In the tumorigenic studies with cyanotoxins, either nodularin or MC-LR was ip administered to animals at 10 μg/kg bw once a week for 15 wk, and all animals were sacrificed at 24 wk post-treatment. In the co-tumorigenic studies with AFB1 and cyanotoxins, the animals were initially given a single dose of AFB1 at 6 mg/kg bw and then, a week later, were treated with 10 μg/kg bw of MC-LR/nodularin once a week for 15 wk. This experiment was terminated at 24 wk after the treatment with AFB1.
Histopathology
Animals were sacrificed under ethyl ether anesthesia at 24 wk or 52 wk after the initial treatment. The skin, musculature, and internal organs were examined for visible abnormalities, and tissues with tumors and lesions were preserved in 100 mL/L neutral-buffered formalin and embedded in paraffin. Sections were stained with hematoxylin-eosin for histopathological alterations and with periodic acid Schiff’s reagent for glycogen accumulation in the hepatocyte. All sections were evaluated by two senior pathologists who were blind to the dosing regimen, and focal hepatic lesions were histologically classified into three morphologic entities: hepatocellular foci, hepatocelluar adenomas or hepatocellular carcinomas.
Statistical analysis
Data were expressed as the incidence of hepatic tumors including carcinoma and adenoma. For animals with both liver adenoma and carcinoma, the statistical analysis of liver tumors was based on the worst level of pathology, carcinoma. The incidences of liver tumors induced by different treatments were statistically evaluated by χ2 test of a 2 × 2 contingency table or Fisher exact test. One-sided P values less than 0.05 were considered statistically significant.
RESULTS
Synergistic hepatotumorigenesis of AFB1 and HBVx
During the observation period (24 wk or 52 wk), there was no difference with respect to general fitness between DMSO-treated transgenic and wild-type mice. Macroscopic examination at autopsy revealed no visible tumors in these two control groups of animals. In some AFB1-treated animals, however, liver tumors were usually multifocal and formed large masses (Figure 1-A1). Upon microscopic inspection, neither neoplastic nor dysplastic alterations were recognized in the livers of DMSO-treated transgenic mice at both 24 and 52 wk post-treatment, but a few inflammatory cells were observed at 52 wk. Some animals treated with AFB1 developed foci of altered hepatocytes, concentrated mainly in centrilobular areas in which foci consisted of relatively large hepatocytes with vacuolations poorly stained with hematoxylin-eosin staining in the cytoplasm (Figure 1-A2). These vacuolations contained glycogen, as shown in periodic acid Schiff’s staining (Figure 1-A3). Clusters of small basophilic cells were observed in some AFB1-treated animals as well. These phenotypic changes were found in both AFB1-treated transgenic and wild-type animals. Histologically, tumors displayed extensive pleomorphism and a significantly higher mitotic rate than the adjacent normal liver tissue (Figure 1-A4). They generally were composed of enlarged cells with lipid or glycogen in the cytoplasm.
Figure 1.
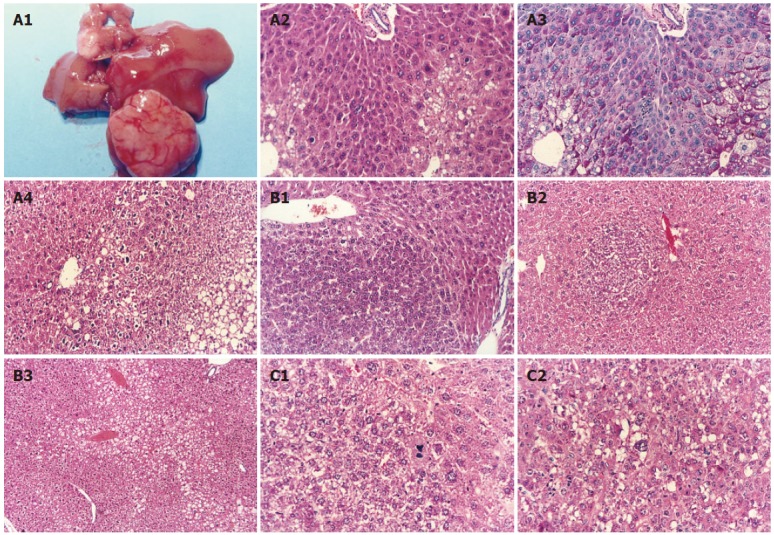
Histopathological alterations identified in liver tissues of mice treated with either AFB1 (A) and cyanobacterial toxins alone (B) or in combination (C). A1: Grossly identified hepatocelluar carcinoma (HCC) in an HBVx gene transgenic mouse at 52 wk post-treatment; A2: altered hepatocyte foci in an HBVx gene transgenic mouse (HE × 100); A3: periodic acid Schiff’s stain for glycogen (× 100) in the continuous liver tissue section of A2; A4: hepatocellular carcinoma in a wild-type mouse at 52 wk post-treatment (HE × 40). The border appears to be disrupted by neoplastic cells penetrating into the adjacent parenchyma; B1: basophilic adenoma in a microcystin-LR-treated wild-type mouse (HE × 40); B2: basophilic hepatocyte foci in microcystin-LR-treated transgenic mice (HE × 10); B3: clear hepatocyte foci in nodularin-treated wild-type mice (HE × 10); C1: HCC in a wild-type mouse treated with both AFB1 and microcystin-LR (HE × 100). Tumor cells had a great variability in cell and nuclear size, and large cells with large hyperchromatic nuclei were present; C2: HCC in a wild-type mouse treated with both AFB1 and nodularin (HE × 100).
During the 52-wk observation period, 6 of 29 (20.7%) AFB1-treated transgenic animals developed HCC, whereas only 1 of 15 (6.7%) AFB1-treated wild-type mice was found to have HCC. In addition, small neoplastic nodules (adenoma) were identified in 7 of 29 (24.1%) AFB1-treated transgenic animals and 1 of 15 (6.7%) AFB1-treated wild-type animals at the 52-wk interval. Overall, the incidence of liver tumors reached 44.8% (13/29) in AFB1-treated transgenic animals during the observation period of 52 wk, which was significantly higher than that in AFB1-treated wild-type littermates (13.3%) at the same interval (χ2 = 4.36, P < 0.05). In the short-term tumorigenic study (Table 1), no HCC was recognized in any of AFB1-treated animals at 24 wk post-treatment; basophilic adenoma was identified in 1 of 11 (9.1%) AFB1-treated transgenic mice and 1 of 20 (5.0%) AFB1-treated wild-type mice. No statistically significant difference in the incidence of liver tumors was found between these two groups at 24 wk post-treatment. Compared with transgenic animals treated with AFB1 and sacrificed at 24 wk post-treatment, AFB1-treated transgenic mice were more likely to develop liver tumors 52 wk after the treatment (P < 0.05). In wild-type animals, the AFB1 treatment did not significantly elevate the risk of developing liver tumors over time.
Table 1.
Incidences of liver tumors caused by exposure to aflatoxin B1 (AFB1) and cyanobacterial toxins (CT) alone or in combination in HBV x (HBVx) gene transgenic mice and their wild-type littermates at 24 wk post-treatment
| Group |
Treatments |
n | Adenoma (%) | Carcinoma (%) | Total (%) | ||
| HBVx gene | AFB1 | CT | |||||
| 1 | - | - | - | 40 | 0 (0) | 0 (0) | 0 (0) |
| 2 | + | - | - | 10 | 0 (0) | 0 (0) | 0 (0) |
| 3 | - | + | - | 20 | 1 (5.0) | 0 (0) | 1 (5.0) |
| 4 | + | + | - | 11 | 1 (9.1) | 0 (0) | 1 (9.1) |
| HBVx gene | AFB1 | MC-LR | |||||
| 5 | - | - | + | 26 | 1 (3.9) | 0 (0) | 1 (3.9) |
| 6 | + | - | + | 11 | 0 (0) | 0 (0) | 0 (0) |
| 7 | - | + | + | 21 | 2 (9.5) | 2 (9.5) | 4 (19.0)a |
| 8 | + | + | + | 15 | 2 (13.3) | 1 (6.7) | 3 (20.0)a |
| HBVx gene | AFB1 | NOD | |||||
| 9 | - | - | + | 26 | 0 (0) | 0 (0) | 0 (0) |
| 10 | + | - | + | 11 | 0 (0) | 0 (0) | 0 (0) |
| 11 | - | + | + | 9 | 2 (22.2) | 1 (11.1) | 3 (33.3)b,c |
| 12 | + | + | + | 11 | 4 (36.4) | 0 (0) | 4 (36.4)d,e,f |
MC-LR: Microcystin-LR; NOD: nodularin. aP < 0.05 vs group 1, bP < 0.01 vs group 1, cP < 0.05 vs group 9, dP < 0.01 vs group 1, eP < 0.05 vs group 3, fP < 0.01 vs group 9.
Contribution of cyanotoxins to the occurrence of AFB1-induced liver tumors during the short period
To determine if AFB1 interacts with cyanotoxins to promote the development of liver tumors, wild-type mice were treated with a single injection of AFB1, followed by repeated injections of either MC-LR or nodularin a week later. Of 21 wild-type mice treated with AFB1, followed by MC-LR, liver tumors were observed in 4 (19.0%) animals at 24 wk post-treatment (Table 1). Mitotic figures were observed in some tumors. The hepatocytic nuclei were variable in size and chromatin content (dense nuclear chromatin pattern) (Figure 1-C1). By contrast, the incidence of liver tumors in wild-type mice treated with AFB1 or MC-LR alone was 5.0% (1/20) and 3.9% (1/26) at the same interval, respectively (Table 1). Although the difference in short-term risk of liver tumors was not statistically significant, co-exposure to AFB1 and MC-LR tended to elevate the risk during the 24-wk period. Alternatively, the incidence of liver tumors in wild-type animals treated with both AFB1 and nodularin was 33.3% (3/9), which was significantly higher than those in wild-type mice treated with nodularin alone (0%, P < 0.05) and slightly higher than those in wild-type mice treated with AFB1 alone (5.0%, P = 0.07) (Table 1). The tumor cells were enlarged and pleomorphic in shape. The nuclei were hyperchromic and exhibited irregular nuclear contour with prominent nucleoli and increased mitotic figures. Small vacuoles found in the dysplastic cells were also observed in the cytoplasm of the neoplastic hepatocytes (Figure 1-C2). This finding suggests a synergistic interaction of AFB1 and nodularin in the pathogenesis of liver tumors.
In addition to liver tumors, other pathological changes included small eosinophilic, basophilic and clear hepatocellular foci in these groups of animals.
Contribution of cyanotoxins and HBV x gene to the development of liver tumors during the short period
Although a wild-type mouse treated with MC-LR was found to have a basophilic adenoma (Figure 1-B1), no hepatic tumors were observed in all transgenic and other wild-type animals treated with either MC-LR or nodularin. None of DMSO-treated transgenic animals was found to have pathological alterations relevant to tumorigenesis during the 24-wk period. A few small clusters of basophilic and clear hepatocytes were identified in cyanotoxin-treated animals (Figure 1-B2, B3). This finding reveals that cyanotoxins and HBV x gene may play a critical role in the pathogenesis of liver tumors as a tumor promoter rather than an initiator.
Contribution of co-exposure to AFB1 and cyanotoxins to the pathogenesis of liver tumors in HBV x gene transgenic mice during the short period
As shown in Table 1, transgenic mice treated with AFB1, followed by MC-LR have a similar risk of liver tumors to their wild-type littermates with the same treatment at 24 wk (20.0% versus 19.0%). Consistently, the HBV x gene did not significantly enhance the risk of liver tumors induced by both AFB1 and nodularin compared to wild-type mice with the same dosing regimen (36.4% versus 33.3%). These findings suggest no appreciably combinative hepatotumorgenecity of AFB1, cyanotoxins and HBV x gene during the 24-wk observation.
DISCUSSION
The nature of coexistence of many types of biotoxins in complex environmental samples, such as food and water, has been reported worldwide. Clear evidence has shown that human populations, particularly HBV carriers in high-risk areas of HCC, are exposed to multiple potent biotoxins, such as aflatoxins and cyanotoxins, through their daily diet and drinking water[24]. To test the epidemiological finding of a combinative risk of HCC associated with co-exposures to AFB1, cyanotoxins and HBV, we performed a series of tumorigenic studies in HBV x gene transgenic mice treated with AFB1 and/or cyanotoxins. Although a little inflammation was observed at 52 wk, there was no obvious cell death or regeneration process in DMSO-treated transgenic mice. Even no tumors developed in them during the observation up to 52 wk. Expression of HBV x gene in the liver of AFB1-treated mice was associated with a greater than 2-fold increase in the incidence of liver tumors at 52 wk post-treatment as compared with AFB1-treated wild-type littermates. This finding indicates that the HBV X protein functions as a cofactor during AFB1-mediated hepatocarcinogenesis. The cofactor role of HBV x antigen in the development of hepatocellular carcinoma was also found in the studies of mice carrying HBV x gene exposed to hepatocarcinogen DEN[8,9]. Alternatively, studies of co-tumorgenicity of HBV and AFB1 using other HBV transgenic mouse models have established that the fragment of HBV genome encoding the entire HBV surface antigen confers a 4- to 20-fold increase in the incidence of liver tumors[11,25].
The underlying mechanism by which the HBV X protein predisposes hepatocytes to the carcinogenic effect of chemical agents has not been fully understood. One possible explanation is the HBVx-induced modulation of specific phase I and II metabolizing enzymes that are involved in bioactivation and elimination of AFB1. Over-expression of HBV surface antigen and x gene was associated with up-regulated expression of specific cytochrome P450 (CYP) isoforms[10] and responsible for bioactivation of AFB1 to the ultimate carcinogenic form, exo-AFB1-8,9-oxide[26]. Additionally, hepatocellular HepG2 cells transfected with HBV demonstrated significant decreases in total GST activity and the level of GST α class[27]. This class of GST is well known to catalyze the reaction in which the AFB1-8,9-epoxide is detoxified in the form of glutathionyl-AFB1 conjugate. Expression of HBV x gene alone, however, was insufficient to induce transactivation of CYP and GST genes or to alter the antioxidant system[28]. Therefore, this possible explanation of the synergistic carcinogenic effect of AFB1 and HBV x gene remains controversial. A second possible mechanism is the ability of the x gene to inhibit DNA repair, leading to accumulation of DNA mutations induced by chemical carcinogens[29]. HBV X protein has been shown to interact with protein or protein complexes that are directly involved in DNA repair. HBV X protein interferes with the nucleotide excision repair pathway through altering activities of helicases[30,31] or interacting with X-associated protein1 to affect the recognition of bulky lesions in DNA[32]. Another possible mechanism is HBVx transactivation of a wide variety of cellular and viral genes as well as the induction of signaling pathways[7,33]. HBV X protein can induce cell proliferation and deregulate cell-cycle checkpoints, which promotes the survival and proliferation of altered hepatocytes after exposure to a mutagenic agent. HBV X protein transactivates the proto-oncogenes like c jun/fos, c myc and cytokine-encoding genes such as tumor necrosis factor α and transforming growth factor β. On the other hand, HBV X protein can inactivate the cellular tumor suppressor gene product, p53[34]. These findings indicate HBV X protein acts as a tumor promoter in hepatocarcinogenesis. For the virus itself, HBV X protein was reported to enhance the virus replication by activating viral gene expression[35]. Although HBV X protein alone was reported to induce liver tumors in HBVx transgenic mice[36-38], our study showed that HBV X protein alone was insufficient to induce pathologic changes relevant to carcinogenesis, which was consistent with the studies by Lee et al[39] and Reifenberg et al[40]. The discrepancy may be a result of different genetic backgrounds of transgenic models. In studies showing liver tumors in HBVx transgenic mice, a mouse strain with a very high incidence of spontaneous liver tumors was used[36]. Since our transgenic mice were derived from C57BL/6J mice, the expected incidence of spontaneous liver tumors was less than 4%[38]. In addition, the factor responsible for the difference may include the DNA fragment of HBV genome which was subcloned to the vector. The segment of DNA used in our study contained only the entire coding region of the HBV x gene; by contrast, Yu et al[38] used the DNA fragment containing the sequences of HBV x gene, the pre C, and the transcriptional enhancer. Another possibility is the integration site of target gene to the host genome and the copy number of transgene. Our result indicated that the expression of x protein alone predisposes hepatocytes to AFB1-induced carcinogenesis, although x protein itself does not induce liver tumors.
In the short-term study of the contribution of cyanotoxins alone and in combination with two other factors to the risk of liver tumors, a few altered hepatocellular foci were identified in transgenic and wild-type mice exposed to cyanotoxins alone. With the exception of an MC-LR-treated wild-type mouse with basophilic adenoma, however, no hepatic tumor was found in animals treated with cyanotoxins alone. HBV x gene was not shown to promote the hepatotumorigenicity of cyanotoxins. An important finding in this study, however, was derived from wild-type animals receiving both AFB1 and cyanotoxins (MC-LR or nodularin). For wild-type animals, the treatment with both AFB1 and MC-LR was more likely to induce liver tumors than the treatment with one of them during the short-term observation, although the increase in risk was non-significant. Additionally, wild-type mice with the treatment of both AFB1 and nodularin have a higher risk of liver tumors 24 wk post-exposure than wild-type animals treated with one of them. The difference in risk reached the statistically significant or marginally significant level. These findings suggest that MC-LR and nodularin possess an ability to modulate the development of liver tumors in the AFB1-initiated animal model. Also, these data proved the previous finding that cyanotoxins are poor initiators and are strong tumor promoters[18,19,22]. Moreover, nodularin was indicated to be a more potent tumor promoter than MC-LR, which is consistent with two-stage carcinogenesis study of modulations of DEN-initiated GST-P-positive foci by cyanotoxins[18,22]. As opposed to what we expected, HBV x gene was not found to further enhance the tumor incidence caused by both AFB1 and cyanotoxins. These results indicate that tumor promotion of cyanobacterial toxins is stronger than HBV X protein alone during 24-wk period. The tumor promoting activity of cyanotoxins may be attributable to inhibition of protein phosphotase 1 and 2A and subsequent hyperphosphorylation of proteins[41]. The possibility of combinative hepatocarcinogenesis of AFB1, cyanotoxins and HBV x gene could not been ruled out based on our finding. In this study, the tumorigenic effect of these factors was observed during the short period. Generally, the in vivo study of carcinogenesis was carried out in a period of more than a year. Actually, our preliminary study found that two out of four HBV x gene transgenic animals developed liver tumors 52 wk after the treatment with AFB1, followed by cyanotoxins. The incidences of AFB1-induced liver tumors were 5.0% and 9.1% in wild-type and transgenic animals, respectively, at 24 wk after the treatment. The same treatment, however, caused a greater than two-fold increase in the incidence in wild-type mice and approximately five-fold increase in transgenic mice at 52 wk post-treatment. Therefore, observation duration is a critical factor in studies of tumorigenicity of potential carcinogens. If the observation period is prolonged, the difference in tumor risk, which was not found significant in the short-term study, may be discernible.
Another limitation of this study was the route of administration of toxins. Repeated injections of cyanotoxins may result in tumor occurrence, although no tumor was observed in almost all animals treated with cyanotoxins. It would be ideal to conduct future studies where the animals are fed AFB1-contaminated food and cyanotoxin-contaminated drinking water to best fit a model relevant to human exposure.
In conclusion, our study shows that AFB1 is a strong tumor initiator, and that HBV x gene and nodularin significantly promote the development of AFB1-induced liver tumors. Although the significance of the contribution of MC-LR to AFB1-initiated hepatotumorigenesis is weak, co-exposure to AFB1 and MC-LR tended to elevate the risk of liver tumors at 24 wk relative to exposure to one of them. HBV x gene could not further increase the contribution of cyanotoxins to AFB1-initiated liver tumors during the short period. Accordingly, a long-term study of interactive hepatocarcinogenesis of these three factors is needed to confirm the present finding.
ACKNOWLEDGEMENTS
The authors sincerely thank Yan Jin, Shuang-Yuan Kuang and Yi-Qian Wu in Shanghai Cancer Institute for their assistance with screening of HBV x gene transgenic mice, and also thank Professors Shi-Ya Zhu and Yuan-Ding Xu in Fudan University for their work in pathological examinations of all sections.
Footnotes
Supported by the Key Project of National Natural Science Foundation of China, No. 39730380
S- Editor Wang J L- Editor Kumar M E- Editor Ma WH
References
- 1.Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA Cancer J Clin. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- 2.Yang L, Parkin DM, Li L, Chen Y. Time trends in cancer mortality in China: 1987-1999. Int J Cancer. 2003;106:771–783. doi: 10.1002/ijc.11300. [DOI] [PubMed] [Google Scholar]
- 3.Wogan GN. Impacts of chemicals on liver cancer risk. Semin Cancer Biol. 2000;10:201–210. doi: 10.1006/scbi.2000.0320. [DOI] [PubMed] [Google Scholar]
- 4.Sun Z, Lu P, Gail MH, Pee D, Zhang Q, Ming L, Wang J, Wu Y, Liu G, Wu Y, et al. Increased risk of hepatocellular carcinoma in male hepatitis B surface antigen carriers with chronic hepatitis who have detectable urinary aflatoxin metabolite M1. Hepatology. 1999;30:379–383. doi: 10.1002/hep.510300204. [DOI] [PubMed] [Google Scholar]
- 5.Szabó E, Páska C, Kaposi Novák P, Schaff Z, Kiss A. Similarities and differences in hepatitis B and C virus induced hepatocarcinogenesis. Pathol Oncol Res. 2004;10:5–11. doi: 10.1007/BF02893401. [DOI] [PubMed] [Google Scholar]
- 6.Koike K. Hepatitis B virus HBx gene and hepatocarcinogenesis. Intervirology. 1995;38:134–142. doi: 10.1159/000150424. [DOI] [PubMed] [Google Scholar]
- 7.Bréchot C, Gozuacik D, Murakami Y, Paterlini-Bréchot P. Molecular bases for the development of hepatitis B virus (HBV)-related hepatocellular carcinoma (HCC) Semin Cancer Biol. 2000;10:211–231. doi: 10.1006/scbi.2000.0321. [DOI] [PubMed] [Google Scholar]
- 8.Madden CR, Finegold MJ, Slagle BL. Hepatitis B virus X protein acts as a tumor promoter in development of diethylnitrosamine-induced preneoplastic lesions. J Virol. 2001;75:3851–3858. doi: 10.1128/JVI.75.8.3851-3858.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Slagle BL, Lee TH, Medina D, Finegold MJ, Butel JS. Increased sensitivity to the hepatocarcinogen diethylnitrosamine in transgenic mice carrying the hepatitis B virus X gene. Mol Carcinog. 1996;15:261–269. doi: 10.1002/(SICI)1098-2744(199604)15:4<261::AID-MC3>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 10.Kirby GM, Chemin I, Montesano R, Chisari FV, Lang MA, Wild CP. Induction of specific cytochrome P450s involved in aflatoxin B1 metabolism in hepatitis B virus transgenic mice. Mol Carcinog. 1994;11:74–80. doi: 10.1002/mc.2940110204. [DOI] [PubMed] [Google Scholar]
- 11.Sell S, Hunt JM, Dunsford HA, Chisari FV. Synergy between hepatitis B virus expression and chemical hepatocarcinogens in transgenic mice. Cancer Res. 1991;51:1278–1285. [PubMed] [Google Scholar]
- 12.Yu SZ. Primary prevention of hepatocellular carcinoma. J Gastroenterol Hepatol. 1995;10:674–682. doi: 10.1111/j.1440-1746.1995.tb01370.x. [DOI] [PubMed] [Google Scholar]
- 13.Ueno Y, Nagata S, Tsutsumi T, Hasegawa A, Watanabe MF, Park HD, Chen GC, Chen G, Yu SZ. Detection of microcystins, a blue-green algal hepatotoxin, in drinking water sampled in Haimen and Fusui, endemic areas of primary liver cancer in China, by highly sensitive immunoassay. Carcinogenesis. 1996;17:1317–1321. doi: 10.1093/carcin/17.6.1317. [DOI] [PubMed] [Google Scholar]
- 14.Carmichael WW. Cyanobacteria secondary metabolites--the cyanotoxins. J Appl Bacteriol. 1992;72:445–459. doi: 10.1111/j.1365-2672.1992.tb01858.x. [DOI] [PubMed] [Google Scholar]
- 15.Burrows WD, Renner SE. Biological warfare agents as threats to potable water. Environ Health Perspect. 1999;107:975–984. doi: 10.1289/ehp.99107975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lovell RA, Schaeffer DJ, Hooser SB, Haschek WM, Dahlem AM, Carmichael WW, Beasley VR. Toxicity of intraperitoneal doses of microcystin-LR in two strains of male mice. J Environ Pathol Toxicol Oncol. 1989;9:221–237. [PubMed] [Google Scholar]
- 17.Yoshida T, Makita Y, Nagata S, Tsutsumi T, Yoshida F, Sekijima M, Tamura S, Ueno Y. Acute oral toxicity of microcystin-LR, a cyanobacterial hepatotoxin, in mice. Nat Toxins. 1997;5:91–95. doi: 10.1002/nt.1. [DOI] [PubMed] [Google Scholar]
- 18.Nishiwaki-Matsushima R, Ohta T, Nishiwaki S, Suganuma M, Kohyama K, Ishikawa T, Carmichael WW, Fujiki H. Liver tumor promotion by the cyanobacterial cyclic peptide toxin microcystin-LR. J Cancer Res Clin Oncol. 1992;118:420–424. doi: 10.1007/BF01629424. [DOI] [PubMed] [Google Scholar]
- 19.Sekijima M, Tsutsumi T, Yoshida T, Harada T, Tashiro F, Chen G, Yu SZ, Ueno Y. Enhancement of glutathione S-transferase placental-form positive liver cell foci development by microcystin-LR in aflatoxin B1-initiated rats. Carcinogenesis. 1999;20:161–165. doi: 10.1093/carcin/20.1.161. [DOI] [PubMed] [Google Scholar]
- 20.Yatsunami J, Komori A, Ohta T, Suganuma M, Fujiki H. Hyperphosphorylation of retinoblastoma protein and p53 by okadaic acid, a tumor promoter. Cancer Res. 1993;53:239–241. [PubMed] [Google Scholar]
- 21.Yoshizawa S, Matsushima R, Watanabe MF, Harada K, Ichihara A, Carmichael WW, Fujiki H. Inhibition of protein phosphatases by microcystins and nodularin associated with hepatotoxicity. J Cancer Res Clin Oncol. 1990;116:609–614. doi: 10.1007/BF01637082. [DOI] [PubMed] [Google Scholar]
- 22.Ohta T, Sueoka E, Iida N, Komori A, Suganuma M, Nishiwaki R, Tatematsu M, Kim SJ, Carmichael WW, Fujiki H. Nodularin, a potent inhibitor of protein phosphatases 1 and 2A, is a new environmental carcinogen in male F344 rat liver. Cancer Res. 1994;54:6402–6406. [PubMed] [Google Scholar]
- 23.Zhu HZ, Cheng GX, Chen JQ, Kuang SY, Cheng Y, Zhang XL, Li HD, Xu SF, Shi JQ, Qian GS, et al. Preliminary study on the production of transgenic mice harboring hepatitis B virus X gene. World J Gastroenterol. 1998;4:536–539. doi: 10.3748/wjg.v4.i6.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wogan GN, Hecht SS, Felton JS, Conney AH, Loeb LA. Environmental and chemical carcinogenesis. Semin Cancer Biol. 2004;14:473–486. doi: 10.1016/j.semcancer.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 25.Ghebranious N, Sell S. Hepatitis B injury, male gender, aflatoxin, and p53 expression each contribute to hepatocarcinogenesis in transgenic mice. Hepatology. 1998;27:383–391. doi: 10.1002/hep.510270211. [DOI] [PubMed] [Google Scholar]
- 26.Mykkänen H, Zhu H, Salminen E, Juvonen RO, Ling W, Ma J, Polychronaki N, Kemiläinen H, Mykkänen O, Salminen S, et al. Fecal and urinary excretion of aflatoxin B1 metabolites (AFQ1, AFM1 and AFB-N7-guanine) in young Chinese males. Int J Cancer. 2005;115:879–884. doi: 10.1002/ijc.20951. [DOI] [PubMed] [Google Scholar]
- 27.Jaitovitch-Groisman I, Fotouhi-Ardakani N, Schecter RL, Woo A, Alaoui-Jamali MA, Batist G. Modulation of glutathione S-transferase alpha by hepatitis B virus and the chemopreventive drug oltipraz. J Biol Chem. 2000;275:33395–33403. doi: 10.1074/jbc.M003754200. [DOI] [PubMed] [Google Scholar]
- 28.Chomarat P, Rice JM, Slagle BL, Wild CP. Hepatitis B virus-induced liver injury and altered expression of carcinogen metabolising enzymes: the role of the HBx protein. Toxicol Lett. 1998;102-103:595–601. doi: 10.1016/s0378-4274(98)00254-9. [DOI] [PubMed] [Google Scholar]
- 29.Kew MC. Synergistic interaction between aflatoxin B1 and hepatitis B virus in hepatocarcinogenesis. Liver Int. 2003;23:405–409. doi: 10.1111/j.1478-3231.2003.00869.x. [DOI] [PubMed] [Google Scholar]
- 30.Jaitovich-Groisman I, Benlimame N, Slagle BL, Perez MH, Alpert L, Song DJ, Fotouhi-Ardakani N, Galipeau J, Alaoui-Jamali MA. Transcriptional regulation of the TFIIH transcription repair components XPB and XPD by the hepatitis B virus x protein in liver cells and transgenic liver tissue. J Biol Chem. 2001;276:14124–14132. doi: 10.1074/jbc.M010852200. [DOI] [PubMed] [Google Scholar]
- 31.Jia L, Wang XW, Harris CC. Hepatitis B virus X protein inhibits nucleotide excision repair. Int J Cancer. 1999;80:875–879. doi: 10.1002/(sici)1097-0215(19990315)80:6<875::aid-ijc13>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 32.Becker SA, Lee TH, Butel JS, Slagle BL. Hepatitis B virus X protein interferes with cellular DNA repair. J Virol. 1998;72:266–272. doi: 10.1128/jvi.72.1.266-272.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Block TM, Mehta AS, Fimmel CJ, Jordan R. Molecular viral oncology of hepatocellular carcinoma. Oncogene. 2003;22:5093–5107. doi: 10.1038/sj.onc.1206557. [DOI] [PubMed] [Google Scholar]
- 34.Staib F, Hussain SP, Hofseth LJ, Wang XW, Harris CC. TP53 and liver carcinogenesis. Hum Mutat. 2003;21:201–216. doi: 10.1002/humu.10176. [DOI] [PubMed] [Google Scholar]
- 35.Xu Z, Yen TS, Wu L, Madden CR, Tan W, Slagle BL, Ou JH. Enhancement of hepatitis B virus replication by its X protein in transgenic mice. J Virol. 2002;76:2579–2584. doi: 10.1128/jvi.76.5.2579-2584.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koike K, Moriya K, Iino S, Yotsuyanagi H, Endo Y, Miyamura T, Kurokawa K. High-level expression of hepatitis B virus HBx gene and hepatocarcinogenesis in transgenic mice. Hepatology. 1994;19:810–819. [PubMed] [Google Scholar]
- 37.Lakhtakia R, Kumar V, Reddi H, Mathur M, Dattagupta S, Panda SK. Hepatocellular carcinoma in a hepatitis B 'x' transgenic mouse model: A sequential pathological evaluation. J Gastroenterol Hepatol. 2003;18:80–91. doi: 10.1046/j.1440-1746.2003.02902.x. [DOI] [PubMed] [Google Scholar]
- 38.Yu DY, Moon HB, Son JK, Jeong S, Yu SL, Yoon H, Han YM, Lee CS, Park JS, Lee CH, et al. Incidence of hepatocellular carcinoma in transgenic mice expressing the hepatitis B virus X-protein. J Hepatol. 1999;31:123–132. doi: 10.1016/s0168-8278(99)80172-x. [DOI] [PubMed] [Google Scholar]
- 39.Lee TH, Finegold MJ, Shen RF, DeMayo JL, Woo SL, Butel JS. Hepatitis B virus transactivator X protein is not tumorigenic in transgenic mice. J Virol. 1990;64:5939–5947. doi: 10.1128/jvi.64.12.5939-5947.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Reifenberg K, Löhler J, Pudollek HP, Schmitteckert E, Spindler G, Köck J, Schlicht HJ. Long-term expression of the hepatitis B virus core-e- and X-proteins does not cause pathologic changes in transgenic mice. J Hepatol. 1997;26:119–130. doi: 10.1016/s0168-8278(97)80018-9. [DOI] [PubMed] [Google Scholar]
- 41.Fujiki H, Sueoka E, Suganuma M. Carcinogenesis of microcystins. In: Watanabe MF, Carmichael WW, Fujiki H, editors. Toxic Microcystis. New York: CRC Press; 1996. pp. 203–232. [Google Scholar]