

Abstract
AIM: To explore the mechanism by which H pylori causes activation of gastric epithelial cells.
METHODS: A VacA (+) and CagA (+) standard H pylori line NCTC 11637 and a human gastric adenocarcinoma derived gastric epithelial cell line BGC-823 were applied in the study. MTT assay and 3H-TdR incorporation test were used to detect the proliferation of BGC-823 cells and Western blotting was used to detect the activity and existence of related proteins.
RESULTS: Incubation with H pylori extract increased the proliferation of gastric epithelial cells, reflected by both live cell number and DNA synthesis rate. The activity of extracellular signal-regulated protein kinase (ERK) signal transduction cascade increased within 20 min after incubation with H pylori extract and appeared to be a sustained event. MAPK/ERK kinase (MEK) inhibitor PD98059 abolished the action of H pylori extract on both ERK activity and cell proliferation. Incubation with H pylori extract increased c-Fos expression and SRE-dependent gene expression. H pylori extract caused phosphorylation of several proteins including a protein with molecular size of 97.4 kDa and tyrosine kinase inhibitor genistein inhibited the activation of ERK and the proliferation of cells caused by H pylori extract.
CONCLUSION: Biologically active elements in H pylori extract cause proliferation of gastric epithelial cells through activating tyrosine kinase and ERK signal transduction cascade.
Keywords: H pylori, Gastric cancer cells, Proliferation, Mitogen-activated protein kinase
INTRODUCTION
H pylori is an important pathogen associated with gastritis and peptic ulcers[1]. It has also been defined as a carcinogen[2]. The mechanisms of pathogenic and carcinogenic effects of H pylori infection are under intensive investigation. Research data suggest that H pylori might stimulate the proliferation of gastric epithelial cells both in vitro and in vivo[3-5]. This effect of H pylori has an important pathogenic significance because increased cell proliferation may elevate the gastric mutation rate and is a predisposing factor for neoplasia. The mechanism, especially the intracellular signal transduction pathway associated with the stimulating effect of H pylori on the proliferation of gastric epithelial cells, is under extensive study.
Protein kinases, which regulate the protein phospho-rylation, are considered to play the most important role in regulating protein function and cell activity. Mitogen-activated protein kinase (also known as MAP Kinase) is a crucial member of the protein kinase family. So far, four MAP Kinase cascades have been characterized in mammalian cells. Among them, extracellular signal-regulated protein kinase (ERK) cascade is most well characterized. The activation of signal transduction pathways by growth factors, hormones and neurotransmitters is mediated through ERK cascade[6,7]. It is the basic signal transduction pathway regulating cell proliferation and differentiation. Recent research data indicate that activation of ERK cascade is involved in
H pylori- induced proliferation of gastric epithelial cells[8-10]. However, more details of H pylori-induced activation of ERK cascade are needed to be explored.
MATERIALS AND METHODS
Materials
Human gastric epithelial cell line BGC-823 (ICLC HTL98007) was provided by Institute of Tumor Research of Beijing. VacA (+) and CagA (+) international standard H pylori strain NCTC11637 was provided by Institute of Microbiology and Epidemiology of China, Beijing. DMEM culture medium was from GiBco (Grand Island, NY). Brucella broth was from Becton Dickinson (Franklin Lakes, NJ). New born calf serum (NBCS) was from No.2 Factory of Beijing Milk Company (Beijing, China). Antibody against phosphorylated ERK was from Sigma (St. Louis, MO). All other primary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). HRP-conjugated secondary antibody was from Jackson ImmunoResearch Laboratories (West Grove, PA). ECL chemoluminescence reagents were from Amersham (Buckinghamshire, England). Tyrosine kinase inhibitors genistein and luciferin were from Sigma (St. Louis, MO). MAPK/ERK inhibitor PD98059 was from Calbiochem (San Diego, CA). Plasmid DNA constructs encoding SRE-luciferase and CRE-luciferase were kind gifts from Dr. Renate Pilz in University of California, San Diego.
Cell culture
Human gastric cancer cell line BGC-823 was kept in DMEM containing 100 mL/L serum and incubated in 5% CO2 at 37°C. The medium was changed every second day and the cells were sub-cultured at 80%-90%
confluence.
Preparation of H pylori extract
Healthy H pylori was scraped down from horse serum agar plate and transferred into Brucella broth containing 100 mL/L serum. The bacterial suspension was incubated in micro-oxygen environment (80% N2, 15% CO2, 5% O2) at 37°C for 24 h with continuous shaking. The growth of H pylori was confirmed by Gram staining before harvest. H pylori cells were precipitated by centrifugation at 5000 r/min for 15 min at 4°C and washed with PBS. The bacteria were re-suspended in serum free cell culture medium (DMEM) and sonicated (100 W, 15 s × 6, at 30 s interval). The breakdown of the bacteria was confirmed by microscopy. The suspension was centrifuged at 10 000 r/min for 30 min at 4°C. The supernatant (H pylori extract) was adjusted to protein concentration of 1 g/L and kept at -20°C until use.
Preparation of protein samples
The harvested cells (1 × 107) were washed three times with PBS and re-suspended in 40 μL suspension buffer containing 100 mmol/L NaCl, 10 mmol/L Tris-HCl (pH7.6), 1 mmol/L EDTA (pH8.0), 1 mg/L aprotinin, 100 mg/L PMSF. The same volume of boiled 2 × SDS protein loading buffer containing 100 mmol/L Tris-HCl (pH6.8), 200 mmol/L DTT, 4% SDS, 0.2% bromophenol blue, 20% glycerol was added into the cell suspension. The sample was boiled for 10 min to lyse the cells and sonicated for 1 min to break down the DNA. After centrifugation at 12 000 r/min for 10 min, the supernatant was kept at -20°C until use. For preparation of membrane protein samples, the cells were harvested, washed with PBS, and lysed with extract buffer containing 20 mmol/L HEPES (pH7.4), 10 mL/L Triton X-100, 5 mmol/L EDTA, 50 mmol/L NaCl, 30 μmol/L β-glycerophosphate, 50 mmol/L sodium fluoride, 50 mg/L aprotinin, 10 mg/L leupeptin. The lysate was centrifuged at 100 000 × g for 1 h at 4°C and the supernatant was kept at -20°C until use.
Western blotting
The protein sample was loaded with 30 μg protein per lane on SDS-PAGE gel. After electrophoresis, the proteins on the gel were transferred onto nitrocellulose (NC) membrane as described previously. The NC membrane was blocked with 50 g/L milk in TBS-T for 1 h at room temperature (RT), incubated with first antibody for 2 h and with secondary antibody for 50 min at RT and finally washed three times after each incubation. ECL chemoluminescence reagent was used to show the positive bands on the membrane.
MTT assay
A total of 1 × 104 trypsin-dispersed BGC-823 cells in 0.1 mL culture medium were seeded into each well of the 96-well plates and cultured for 24 h. Then, the cells were incubated with medium alone or with medium plus H pylori extract at different concentrations. After 24 h of further incubation, 10 μL of MTT (6 g/L, Sigma) was added to each well and the incubation was continued for 4 h at 37°C. Finally, the culture medium was removed and 200 μL of dimethylsulphoxide (DMSO) was added to each well. The absorbance was determined with an ELISA reader at 570 nm. The “change percentage” of the absorbency (A) value of the cells treated with H pylori extract compared with A value of the cells without treatment was calculated as:
Change (%) = A value (H pylori group-control)/ A value of control group×100%
3H-TdR incorporation
The cells were cultured in the same way as described in MTT assay. When the culture medium was changed and H pylori extract was added, 0.2 μCi of 3H-TdR was also added into each well. After further incubation for 24 h, the medium was discarded and the cells were trypsin-dispersed and collected onto a membrane. The membrane was heated dry at 65°C and put into a plastic vial containing 6 mL of scintillation liquid. The count per minute (CPM) was calculated with a Beckman scintillation counter. The “change percentage” of CPM of the cells treated with H pylori extract compared with CPM of the cells not treated with H pylori extract was calculated as:
Change (%) = CPM (H pylori group-control)/ CPM of control group×100%
Reporter gene assay
The cells were transfected with plasmid DNA encoding SRE-luciferase or CRE-luciferase, and β-galactosidase. After incubation with H pylori extract for 8 h, the cells were harvested and the activity of luciferase and β-galactosidase was measured by luminescence-based assays. The luciferase activity was calculated as the fold of increase after normalized by the β-galactosidase activity.
Statistical analysis
The results of MTT assay, 3H-TdR incorporation test and reporter gene assay data were expressed as means ± SD. The significance of the difference between control group and experimental group was evaluated by Student’s t-test.
RESULTS
H pylori extract stimulated proliferation of BGC-823 cells
The MTT assay showed that H pylori extract could stimulate the proliferation of BGC-823 cells. The A value reflecting the number of viable cells, increased significantly in the cells incubated with 50 mg/L and 100 mg/L H pylori extract for 24 h (Figure 1A). Similar result was also obtained in 3H-TdR-incorporation test. The 3H incorporation increased dose-dependently when the cells were incubated with 20 mg/L, 50 mg/L, and 100 mg/L H pylori extract respectively for 24 h (Figure 1B).
Figure 1.
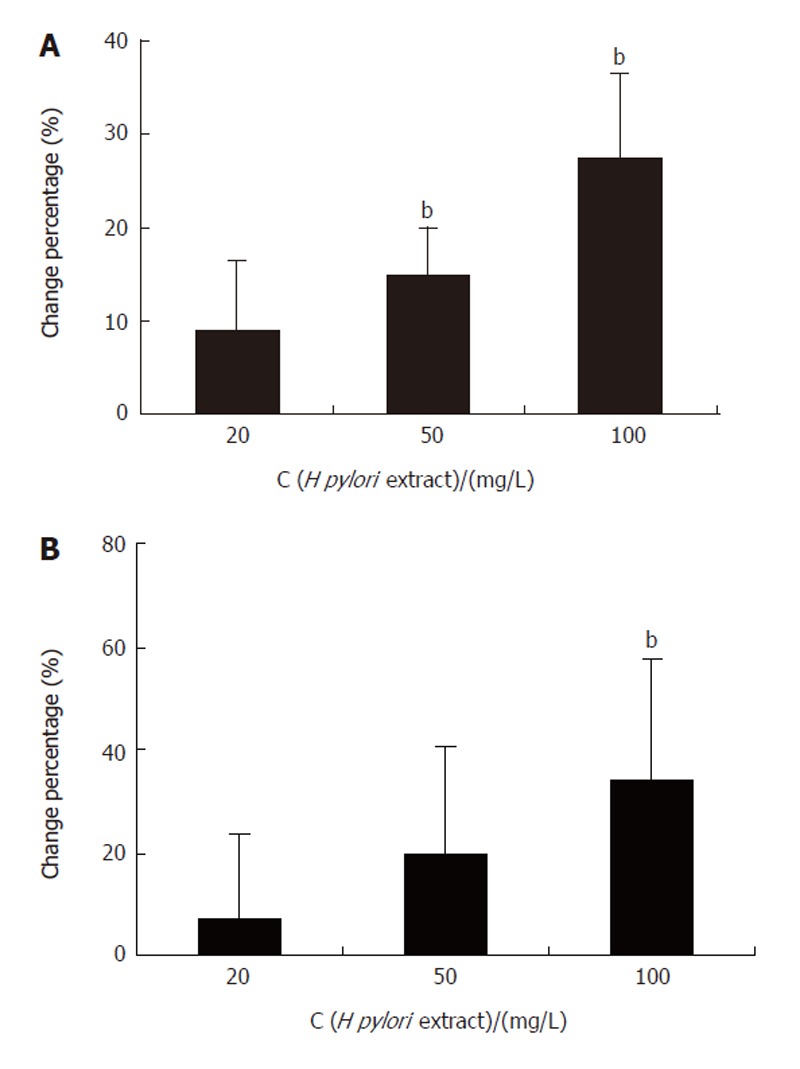
MTT assay (A) and 3H-TdR-incorporation test (B) showing H pylori extract- stimulated proliferation of BGC-823 cells. bP < 0.01 vs control.
H pylori extract activated extracellular signal-regulated kinase in BGC-823 cells
The separated protein samples from cells incubated with 50 mg/L H pylori extract for different periods of time were first probed with antibody against phosphorylated ERK to analyze the activity of ERK and re-probed with anti-ERK2 antibody to show the protein content of this kinase. The results showed that in serum-starved cells, the activity level of ERK was very low. During incubation with H pylori extract, the activity of ERK increased obviously within 20 min and stayed at a high level (about 5 times of control) for more than 8 h ( Figure 2).
Figure 2.
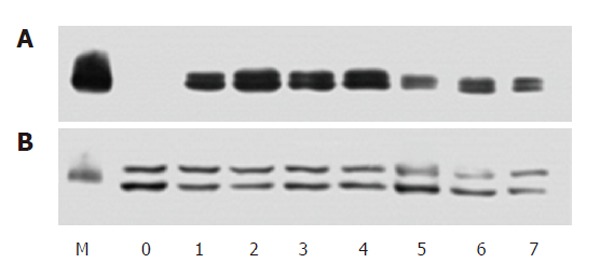
Western blotting showing phosphorylated ERK (A) and total ERK (B) in serum-starved BGC-823 cells incubated with H pylori extract. M: Protein molecular marker; 0: Control; 1-7: Incubation with 50 mg/L H pylori extract for 20, 40, 60 min and 3, 6, 12, 24 h.
PD98059 blocked the stimulating effect of H pylori extract on ERK activity and proliferation of BGC-823 cells
PD98059, an inhibitor of MAPK/ERK kinase (MEK) activating ERK directly, was used to confirm the specificity of ERK activation induced by H pylori extract. Western blotting showed that PD98059 abolished the stimulating effect of H pylori extract on ERK activity (Figure 3). MTT assay showed that the inhibitor also blocked the stimulating effect of H pylori extract on proliferation of the cells
Figure 3.
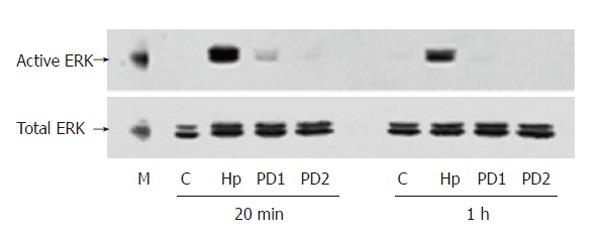
Western blotting showing PD98059-blocked stimulating effect of H pylori extract on ERK activation in BGC-823 cells. M: Molecular marker; C: Control; Hp: 50 mg/L H pylori extract; PD1: 25 μmol/L PD98059 + 50 mg/L H pylori extract; PD2: 50 μmol/L PD98059 + 50 mg/L H pylori extract.
(Figure 4).
Figure 4.
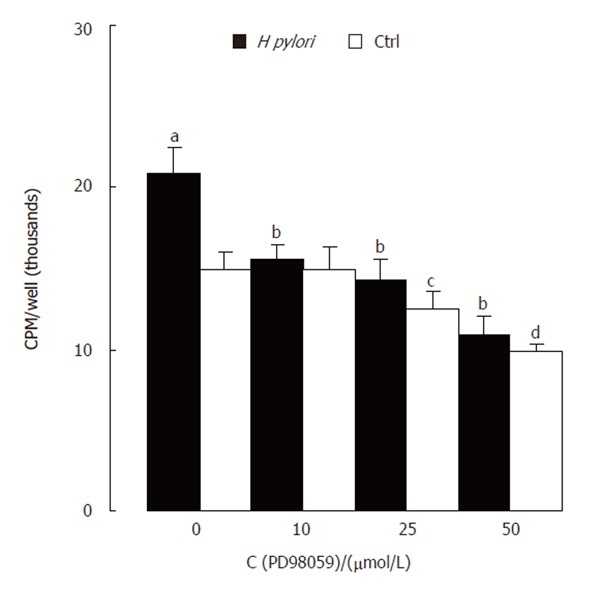
MTT assay showing PD98059-blocked proliferation-stimulating effect of H pylori extract. aP < 0.05, cP < 0.05, dP < 0.01 vs control only; bP < 0.01 vs Hp only.
Genistein inhibited ERK activation induced by H pylori extract
Genistein is an inhibitor of tyrosine kinase. Western blotting with antibody against phosphorylated ERK showed that treating the cells with genistein inhibited the ERK activation induced by H pylori extract (Figure 5).
Figure 5.
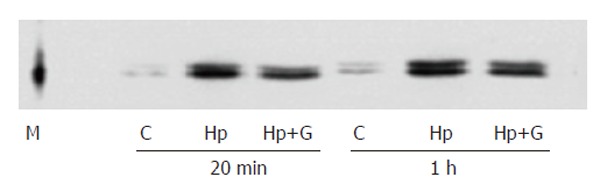
Western blotting showing genistein-prevented ERK activation by H pylori extract. M: Molecular marker; C: Control; Hp: H pylori extract; Hp + G: Genistein and H pylori extract.
H pylori extract caused tyrosine phosphorylation of membrane protein
An antibody against pan-tyrosine phosphorylation of proteins was used to detect the tyrosine phosphorylation caused by H pylori extract. Western blotting showed that during incubation with H pylori extract, several proteins were phosphorylated on tyrosine within 20 min of incubation (Figure 6). A tyrosine-phosphorylated protein with molecular size around 97.4 kDa was especially arrested because it also existed in membrane protein sample extracted from the cells (Data not shown).
Figure 6.

Western blotting showing H pylori extract-caused tyrosine pho-sphorylation of cell lysate of BGC-823. M: Molecular marker; 1: control; 2-7: H pylori extract incubated for 20, 40 min and 1, 3, 6, 12 h.
H pylori extract increased expression of c-fos
Western blotting showed that when the cells were incubated with 50 mg/L H pylori extract, the expression of c-Fos protein increased obviously. During incubation, the expression of c-Fos started to increase around 40 min after incubation, reached its peak around 1 h and lasted for 6 h (Figure 7).
Figure 7.

Western blotting showing H pylori extract- increased expression of c-Fos. 1: Control; 2-8: H pylori extract incubated for 20, 40 min and 1, 3, 6, 12 and 24 h.
H pylori extract stimulated SRE-dependent reporter gene expression
Reporter gene assay showed that H pylori extract specifically stimulated SRE-dependent expression of luciferase while it had no effect on CRE-driven luciferase expression (Figure 8), indicating that H pylori extract could increase c-Fos expression through SRE cis-element of the promoter.
Figure 8.
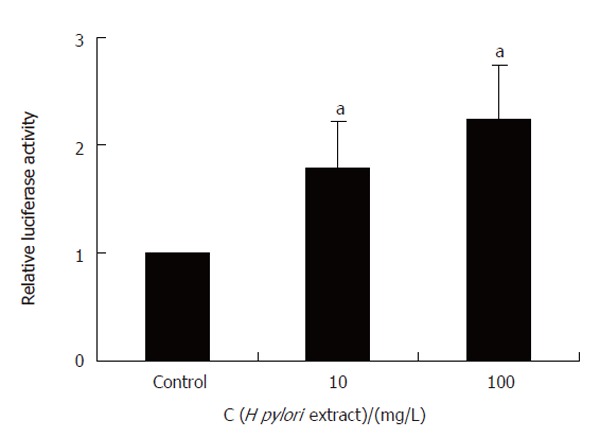
H pylori extract-stimulated SRE-dependent reporter gene expression. aP < 0.05 vs control.
DISCUSSION
Gastric H pylori infection is the most common infection in humans. It is responsible for virtually all cases of gastritis and most cases of peptic ulcers[1]. Moreover,
H pylori is defined as a group 1 carcinogen in humans by International Agency for Research on Cancer, a working party of WHO[2]. The conclusion is, however, mainly based on the epidemiological data and clinical investigations. Even though laboratory data on carcinogenesis of H pylori are accumulating during recent years[11-13], more efforts are still needed to elucidate the mechanism of H pylori-induced malignant diseases. Gastric carcinoma develops through a sequence of events from normal mucosa to gastric carcinoma[14]. H pylori might be closely associated with this process. Several mechanisms have been proposed by which H pylori infection might lead to predisposition for gastric cancer[15]. One explanation is that H pylori- associated chronic inflammation may increase the rate of cell turnover in gastric mucosa. Rapidly replicating DNA is more susceptible to damage. Alongside increased reactive oxygen species and decreased ascorbic acid content during inflammatory reactions, H pylori -induced proliferation may be the major factor for gastric dysplasia and gastric cancer[16,17]. In H pylori hosts with gastritis, increased proliferation of gastric epithelial cells has been confirmed[3,4]. Laboratory data have also shown that H pylori might directly stimulate proliferation of gastric epithelial cells[5,10,11]. The former reveals an increased cell turnover rate in H pylori host and the latter suggests that increased proliferation of gastric epithelial cells might be a direct effect of H pylori.
Probing separated proteins with anti-phosphorylated ERK antibody and re-probing them with anti-ERK2 antibody provide a sensitive way to detect ERK activity[18]. With this method, our study showed that during incubation with H pylori extract, the activity of ERK increased obviously in serum-starved cells and the activation of ERK was a sustained event. This is significant because sustained activation of ERK appears to be required for many cells to pass the gastrointestinal restriction point to enter S phase, in which cellular DNA is replicated[19,20]. MTT assay and 3H-TdR-incorporation test confirmed the stimulating effect of H pylori extract on cell proliferation in our study. Since ERK-mediated cascade is the basic signal transduction pathway regulating cell proliferation, this pathway can mediate proliferation-stimulating effects of
H pylori.
ERK is in the middle of the signal transduction pathway. Currently, it is believed that ERK may be activated through several pathways[21]. One is the receptor tyrosine kinase-Ras-MEK pathway. Growth factor ligand binds to membrane receptor and causes tyrosine-phosphorylation of the receptor. Consequential action of Ras and then Raf, which are components of the cascade, causes the activation of MEK (also known as MAP kinase), a direct upstream regulator of ERK[22]. PD98059 is a specific inhibitor of MEK[23]. Our results showed that PD98059 could block both ERK activity-stimulating and proliferation-stimulating effects of H pylori extract. Ras activity was not investigated in this experiment. However, we used tyrosine kinase inhibitor genistein to explore the possible involvement of tyrosine kinase in H pylori extract-induced signaling. The results showed that genistein inhibited H pylori extract-induced activation of ERK. Then, we used antibody against pan-tyrosine phosphorylation to detect tyrosine phosphorylation in both whole cell lysate and membrane protein from the cells. The results showed that in samples from the cells treated with H pylori extract, there was obvious tyrosine phosphorylation of a membrane protein with molecular size around 97.4 kDa, suggesting that it is very likely that H pylori extract activates ERK through receptor tyrosine kinase-Ras-MEK pathway.
As to the down-stream events of ERK activation, it was reported that some ERK activated by MEK can translocate to the nuclei and phosphorylate at least two transcription factors, c-Myc and Elk-1[24,25]. The phosphorylated factors may lead to increased production of c-Fos mRNA and finally lead to cell activation[26]. Mitsuno et al[27] reported that co-culturing human gastric cancer cells with Cag-positive H pylori strains can activate c-fos gene through SRE. Our results showed that H pylori extract stimulated SRE-dependent gene transcription and increased the expression of c-Fos protein, indicating that the signaling process behind activation of ERK by H pylori can derive biological elements.
Several methods, including co-culture of live H pylori with target cells, use of water extract and sonication extract to stimulate target cells, have been used to study the effect of biologically active components of H pylori on the activity of target cells. For example, during co-culture, H pylori may secrete active factors or act on the target cells through type IV secretion system[28], water extracting can get the surface component of the bacteria[29], and sonication extracting can efficiently release active components from the body of bacterial cells[30,31]. In this experiment, sonication of H pylori extract showed that soluble component from the bacteria activated the MAPK cascade of gastric cells. Further work is needed to identify the molecular features of the component in our laboratory.
In summary, H pylori extract has direct proliferation-stimulating effects on gastric epithelial cells and the activation of gastric epithelial cells can be induced by H pylori through MAPK-mediated signal transduction pathway, suggesting that biologically active elements in
H pylori contribute to the tumorigenesis effect of the bacteria.
ACKNOWLEDGMENTS
We sincerely thank Professor Renate B Pilz in University of California, USA, for her kind gift of DNA constructs.
Footnotes
S- Editors Pan BR L- Editor Wang XL E- Editor Ma WH
References
- 1.Lee A. The nature of Helicobacter pylori. Scand J Gastroenterol Suppl. 1996;214:5–8; discussion 9-12. [PubMed] [Google Scholar]
- 2.Schistosomes liver flukes and Helicobacter pylori IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7-14 June 1994. IARC Monogr Eval Carcinog Risks Hum. 1994;61:1–241. [PMC free article] [PubMed] [Google Scholar]
- 3.Brenes F, Ruiz B, Correa P, Hunter F, Rhamakrishnan T, Fontham E, Shi TY. Helicobacter pylori causes hyperproliferation of the gastric epithelium: pre- and post-eradication indices of proliferating cell nuclear antigen. Am J Gastroenterol. 1993;88:1870–1875. [PubMed] [Google Scholar]
- 4.Bechi P, Balzi M, Becciolini A, Maugeri A, Raggi CC, Amorosi A, Dei R. Helicobacter pylori and cell proliferation of the gastric mucosa: possible implications for gastric carcinogenesis. Am J Gastroenterol. 1996;91:271–276. [PubMed] [Google Scholar]
- 5.Fan XG, Kelleher D, Fan XJ, Xia HX, Keeling PW. Helicobacter pylori increases proliferation of gastric epithelial cells. Gut. 1996;38:19–22. doi: 10.1136/gut.38.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Force T, Bonventre JV. Growth factors and mitogen-activated protein kinases. Hypertension. 1998;31:152–161. doi: 10.1161/01.hyp.31.1.152. [DOI] [PubMed] [Google Scholar]
- 7.Rubinfeld H, Seger R. The ERK cascade: a prototype of MAPK signaling. Mol Biotechnol. 2005;31:151–174. doi: 10.1385/MB:31:2:151. [DOI] [PubMed] [Google Scholar]
- 8.Strowski MZ, Cramer T, Schäfer G, Jüttner S, Walduck A, Schipani E, Kemmner W, Wessler S, Wunder C, Weber M, et al. Helicobacter pylori stimulates host vascular endothelial growth factor-A (vegf-A) gene expression via MEK/ERK-dependent activation of Sp1 and Sp3. FASEB J. 2004;18:218–220. doi: 10.1096/fj.03-0055fje. [DOI] [PubMed] [Google Scholar]
- 9.Sebkova L, Pellicanò A, Monteleone G, Grazioli B, Guarnieri G, Imeneo M, Pallone F, Luzza F. Extracellular signal-regulated protein kinase mediates interleukin 17 (IL-17)-induced IL-8 secretion in Helicobacter pylori-infected human gastric epithelial cells. Infect Immun. 2004;72:5019–5026. doi: 10.1128/IAI.72.9.5019-5026.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foryst-Ludwig A, Neumann M, Schneider-Brachert W, Naumann M. Curcumin blocks NF-kappaB and the motogenic response in Helicobacter pylori-infected epithelial cells. Biochem Biophys Res Commun. 2004;316:1065–1072. doi: 10.1016/j.bbrc.2004.02.158. [DOI] [PubMed] [Google Scholar]
- 11.Keates S, Keates AC, Warny M, Peek RM, Murray PG, Kelly CP. Differential activation of mitogen-activated protein kinases in AGS gastric epithelial cells by cag+ and cag- Helicobacter pylori. J Immunol. 1999;163:5552–5559. [PubMed] [Google Scholar]
- 12.Penta R, De Falco M, Iaquinto G, De Luca A. Helicobacter pylori and gastric epithelial cells: from gastritis to cancer. J Exp Clin Cancer Res. 2005;24:337–345. [PubMed] [Google Scholar]
- 13.Zhu Y, Zhong X, Zheng S, Du Q, Xu W. Transformed immortalized gastric epithelial cells by virulence factor CagA of Helicobacter pylori through Erk mitogen-activated protein kinase pathway. Oncogene. 2005;24:3886–3895. doi: 10.1038/sj.onc.1208551. [DOI] [PubMed] [Google Scholar]
- 14.Correa P. Human gastric carcinogenesis: a multistep and multifactorial process--First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention. Cancer Res. 1992;52:6735–6740. [PubMed] [Google Scholar]
- 15.De Koster E, Buset M, Fernandes E, Deltenre M. Helicobacter pylori: the link with gastric cancer. Eur J Cancer Prev. 1994;3:247–257. doi: 10.1097/00008469-199403030-00003. [DOI] [PubMed] [Google Scholar]
- 16.McColl KE, el-Omar E. Helicobacter pylori and disturbance of gastric function associated with duodenal ulcer disease and gastric cancer. Scand J Gastroenterol Suppl. 1996;215:32–37. doi: 10.3109/00365529609094531. [DOI] [PubMed] [Google Scholar]
- 17.Konturek PC. Physiological, immunohistochemical and molecular aspects of gastric adaptation to stress, aspirin and to H. pylori-derived gastrotoxins. J Physiol Pharmacol. 1997;48:3–42. [PubMed] [Google Scholar]
- 18.Sun QY, Luria A, Rubinstein S, Breitbart H. Protein kinase inhibitors induce the interphase transition by inactivating mitogen-activated protein kinase in mouse eggs. Zygote. 1998;6:277–284. doi: 10.1017/s0967199498000227. [DOI] [PubMed] [Google Scholar]
- 19.Pages G, Lenormand P, L’Allemain G, Chambard JC, Meloche S, Pouyssegur J. Mitogen-activated protein kinases p42mapk and p44mapk are required for fibroblast proliferation. Proc Natl Acad Sci USA. 1993;90:8319–8323. doi: 10.1073/pnas.90.18.8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 21.Post GR, Brown JH. G protein-coupled receptors and signaling pathways regulating growth responses. FASEB J. 1996;10:741–749. doi: 10.1096/fasebj.10.7.8635691. [DOI] [PubMed] [Google Scholar]
- 22.Denhardt DT. Signal-transducing protein phosphorylation cascades mediated by Ras/Rho proteins in the mammalian cell: the potential for multiplex signalling. Biochem J. 1996;318(Pt 3):729–747. doi: 10.1042/bj3180729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alessi DR, Cuenda A, Cohen P, Dudley DT, Saltiel AR. PD 098059 is a specific inhibitor of the activation of mitogen-activated protein kinase kinase in vitro and in vivo. J Biol Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- 24.Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- 25.Treisman R. Journey to the surface of the cell: Fos regulation and the SRE. EMBO J. 1995;14:4905–4913. doi: 10.1002/j.1460-2075.1995.tb00173.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Treisman R. Regulation of transcription by MAP kinase cascades. Curr Opin Cell Biol. 1996;8:205–215. doi: 10.1016/s0955-0674(96)80067-6. [DOI] [PubMed] [Google Scholar]
- 27.Mitsuno Y, Maeda S, Yoshida H, Hirata Y, Ogura K, Akanuma M, Kawabe T, Shiratori Y, Omata M. Helicobacter pylori activates the proto-oncogene c-fos through SRE transactivation. Biochem Biophys Res Commun. 2002;291:868–874. doi: 10.1006/bbrc.2002.6530. [DOI] [PubMed] [Google Scholar]
- 28.Bourzac KM, Guillemin K. Helicobacter pylori-host cell interactions mediated by type IV secretion. Cell Microbiol. 2005;7:911–919. doi: 10.1111/j.1462-5822.2005.00541.x. [DOI] [PubMed] [Google Scholar]
- 29.Mai UE, Perez-Perez GI, Wahl LM, Wahl SM, Blaser MJ, Smith PD. Soluble surface proteins from Helicobacter pylori activate monocytes/macrophages by lipopolysaccharide-independent mechanism. J Clin Invest. 1991;87:894–900. doi: 10.1172/JCI115095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liljemark WF, Bloomquist CG. Isolation of a protein-containing cell surface component from Streptococcus sanguis which affects its adherence to saliva-coated hydroxyapatite. Infect Immun. 1981;34:428–434. doi: 10.1128/iai.34.2.428-434.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Singh R, Banerjee A, Kaul P, Barse B, Banerjee UC. Release of an enantioselective nitrilase from Alcaligenes faecalis MTCC 126: a comparative study. Bioprocess Biosyst Eng. 2005;27:415–424. doi: 10.1007/s00449-005-0013-4. [DOI] [PubMed] [Google Scholar]