

Abstract
The thrombophilia which can be either congenital or acquired in adult life has major implications in the abdominal vessels. The resulting portal vein thrombosis, Budd-Chiari syndrome and mesenteric vein thrombosis have a variety of consequences ranging from acute abdomen to chronic hepatomegaly and even totally asymptomatic patient in whom the only finding is pancytopenia. The complications like esophageal varices, portal gastropathy, ascites, severe hypersplenism, liver failure requiring liver transplantation are well known. Interesting features of collateral venous circulation showing itself as pseudocholangiocarcinoma sign and its possible clinical reflection as cholestasis are also known from a long time. The management strategies for these complications of intraabdominal vessel thrombosis are not different from their counterpart which is cirrhotic portal hypertension, but the prognosis is unquestionably better in former cases. In this review we presented and discussed the abdominal venous thrombosis, etiology and the resulting clinical pictures. There are controversial issues both in nomenclature, and management including anticoagulation problems and follow up strategies. In light of the current knowledge, we discussed some controversial issues in literature and presented our experience and our proposals about this group of patients.
Keywords: Portal vein thrombosis, Pseudocholangiocarcinoma sign, Thrombophilia, Budd-Chiari syndrome, Mesenteric vein thrombosis
INTRODUCTION
The thrombosis in the major vessels of abdomen causes a wide spectrum of clinical pictures ranging from a totally asymptomatic patient to a patient with acute abdominal pain and even impending liver failure in patients with underlying chronic liver disease. As liver is the main organ of synthesis of many essential proteins in the body, it bears the burden of thrombosis of major vessels in the abdomen whether this may be a partial or total thrombosis with resulting liver disease, portal hypertension and cirrhosis requiring different management strategies including liver transplantation.
In this clinical setting, the thrombosis of the abdominal vessels has a special place in medicine since the clinicopathological pictures and courses of these processes are usually heterogenous, protean and fluctuant in nature. In general practice physicians pay extra attention to the thrombosis of coronary, pulmonary, mesenteric or cerebral circulation but not to abdominal venous circulation. We believe that the thrombotic occlusion of all major vessels of abdominal cavity has severe clinical consequences and chronic complications.
In this review we will describe thrombophilic conditions and discuss the potential consequences of thrombosis in the major abdominal vessels with potential clinical implications.
THROMBOPHILIA
Normal coagulation hemostasis involves the interaction of an initial vascular reaction (vasoconstriction), thrombocytic activation (white clot formation) and formation of thrombin via activation of coagulation cascade. The balance between the forces favoring formation of a clot and forces against it is the normal state which is controlled with very delicate systems. Although very complicated and several mechanisms play a role in the vascular and thrombocytic steps of clot formation, these steps are beyond the scope of this review and will not be discussed further.
Thrombophilia is a term which is proposed as an opposite term against hemophilia. Thrombophilia can be defined as a disturbed state of the coagulation-fibrinolysis balance in favor of thrombosis formation (congenital or acquired in adult life) in which thrombosis (in arterial and/or venous vasculature) is observed more frequently than normal population. For the gastroenterologists and surgeons, the congenital and acquired causes of the thrombophilia are important not only due to their potential risks to patients’ lives but also their preventability with the advent of genetic tests and surveillance and their treatability with new invasive techniques and new anticoagulant drugs.
Thrombophilia can be grouped under two major headlines: inherited and acquired.
CAUSES OF INHERITED THROMBOPHILIA
Factor V Leiden mutation (FVLM)
The most common cause of inherited thrombophilia is FVLM. This mutation was firstly defined by Bertina et al[1] in 1994, after studies of Dahlback and colleagues[2] who showed a resistance factor against activated protein C (APC). The prevalence of this mutation differs between populations throughout the world. The prevalence of this mutation is very heterogenous that some native populations of Africa, America and Asia show no mutation whereas in some districts of Scandinavian peninsula the prevalence rises to 15%. In Turkey, the prevalence is about 8-9% and this prevalence rate decreases going west from Asia Minor towards Europe to 2-4%[3].
Factor V (FV) is synthesized in liver and megakaryocytes which must be activated into form of FVA to play a crucial role in prothrombinase complex along with activated factor X to produce thrombin. The mutation involves a point at the 1691st nucleotide of the exon 10 in first chromosome making G-C change. This results in a change in aminoacid sequences which is depicted as A506G (Arginine at 506 changed by Glutamine). The mutation containing FVA is resistant to degradation by protein C (which is a natural anticoagulant protein) and undegraded factor rises over time increasing the risk of uncontrolled thrombin and therefore thrombus formation. The diagnosis of FVLM depends partially on the activated protein C resistance (APCR) as a screening tool but direct PCR test to detect mutation can also be applied. APCR is a standard test which is widely available in most laboratories but modified APCR has a very high level of sensitivity and specificity and should be applied to patients who are on warfarin treatment, anti-phospholipid syndrome patients, pregnant women, protein S (PS) deficiency and patients with high levels of factor VIII[4].
When compared with normal population, the impact of FVLM on thrombosis formation is a risk about 3-7 times higher in heterozygotes and 50-100 times higher in homozygotes.
Prothrombin mutation (PM)
PM is firstly defined in 1996 by Poort et al[5]. The mutation involves a base pair change in the 20210th position (guanosine vs. adenosin) resulting in excessive generation of prothrombin which forms excessive procoagulant accumulation[6]. This mutation has a general population prevalence of about 2% compared to the prevalence increased to 6% in patients who had their first attack of venous thrombosis. Like FVLM, the genetic basis is also important for the prevalence. In African and Asian populations the PM is rare when compared to Caucasians[7].
Protein C (PC) and protein S (PS) deficiency
PC is synthesized in liver by a vitamin K dependent mechanism. During coagulation cascade, this protein is activated by thrombin into its active form, namely activated PC (APC). PC is encoded in chromosome 2 and up to date about 160 mutations have been defined[4,8-10]. The PC activity is usually determined by functional tests. The heterozygote PC deficiency reveals itself by PC activity less than 50%, whereas in homozygote deficiency PC activity is below 5%. The prevalence of PC deficiency is about 0.2% in general population and in the population who had first venous thrombosis attack the prevalence is found to be about 3%[11]. An important factor to point out is that the acquired PC deficiency occurs in patients with liver disease (due to decreased synthetic capacity), oral anticoagulation treatment and acute thrombosis cases (due to utilization).
PS is naturally a cofactor of activated PC during inactivation processes of activated factors V and VIII. This protein is synthesized from liver, endothelial cells, megakaryocytes and in testis by vitamin K dependent reactions. There are many mutations responsible for PS deficiency so that genotyping and analysis for a suspected PS deficiency is not practical. The screening for PS deficiency is functional activity testing. The protein S deficiency is accepted as a weak risk factor for thrombosis formation which is about 2 times more than normal population. The importance of liver disease in assessing these natural anti-coagulant deficiencies is discussed later.
Antithrombin (AT) deficiency
AT is a natural serine protease inhibitor with a very potential controlling functions over coagulation cascade. AT inhibits the steps of coagulation depending on mainly thrombin and less importantly activated factors IX, X, XI, and XII. In the presence of heparin or heparin like molecules the inhibitor function is 1000 times more potent[12]. The prevalence of deficiency of AT in the general population is about 0.02-0.2%, while it accounts for 1% of venous thrombosis etiologies[11,13].
CAUSES OF ACQUIRED THROMBOPHILIA
In young adults, the annual risk of venous thrombosis is 1/10000 for event/person but this figure increases to 1/100 for event/person in patients over 70 years of age[14]. Age is considered as an independent risk factor for venous thrombosis mainly due to inactivity (related with venous congestion), comorbid illnesses related with age and degenerative changes in the vasculature that occur with aging[15].
Malignant disease is also an important cause of thrombosis. Cancer patients have a crude risk of 10-20% to have venous thrombosis in the rest of their lives. The thrombosis related deaths row in the second place in all causes of deaths in this patient population[16-18]. Myeloproliferative disorders (MPD) merit a special discussion in this section. MPD forms a group of special blood disorders which may be termed as half-malignant due to their natural course and this group of patients frequently suffer from venous thrombosis. Thrombocytosis and the increased hematocrit which are natural characteristics of this disorders which also cause thrombosis in the venous systems[19].
Other well known acquired thrombophilic conditions are surgery (orthopedic and neurosurgery), drugs (i.e., oral contraceptives, hormone replacement treatment, tamoxifen, L-Asparaginase), antiphospholipid syndrome, pregnancy and Behcet’s disease[20].
The relative risks of common thrombophilic conditions[21,22] are shown in Table 1.
Table 1.
| Thrombophilic condition | Relative risk of thrombosis |
| Inherited | |
| Heterozygote deficiency of natural anticoagulants (PC, PS, AT) | 10-fold |
| Heterozygote FVLM | 5-8 fold |
| Homozygote FVLM | 50-80 fold |
| Heterozygote PM | 2-4 fold |
| Homozygote PM | 10-fold |
| Acquired | |
| Oral contraceptive use | 4-fold |
| Surgery | 6-fold |
| Immobilization | 11-fold |
| Anti-phospholipid syndrome | 10-fold |
| Combined | |
| Oral contraceptive use + Heterozygote FVLM | 35-fold |
PORTAL VENOUS THROMBOSIS: ETIOLOGY, CLINICAL CONSEQUENCES
Portal vein thrombosis (PVT) refers to the total or near-total obstruction of blood flow secondary to thrombus formation. This thrombus may extend towards liver involving intrahepatic portal veins or may extend distally to involve splenic veins or mesenteric veins. On some occasions extensive involvement of all of these vessels may occur with an increased risk of intestinal ischemia. Therefore, the involved segment of portal venous system and the degree of compensatory mechanisms determine the clinical presentation. Although PVT has been observed most commonly in the setting of cirrhosis, discussion presented in the text from this point specifies chronic, non-cirrhotic PVT unless otherwise specified.
PVT has considerable consequences for the liver. Upon cessation of blood flow liver loses about two thirds of its blood supply. Interestingly, while the acute arterial blockage usually results in severe hepatic failure or death, this condition is tolerated well and the patients are almost asymptomatic due to compensatory mechanisms. First compensatory mechanism is the well known “arterial vasodilation” of the hepatic artery (which is a vascular reflex seen in every dual-vessel supplied organ) also observed during portal vein clamping in liver surgery[23]. This “arterial rescue” stabilizes the liver functions at a normal level in the very acute stages of PVT. The second rescue mechanism is the “venous rescue” involving rapid development of collateral vessels to bypass the obstructed segment. These vessels begin to form in a few days after the obstruction and organize into a cavernomatous transformation (Figures 1 and 2) (see later section for detailed discussion) in 3-5 wk[24,25]. We believe that when symptomatic, acute PVT rather be called “acute mesenteric thrombosis” due to extensive co-involvement of the superior mesenteric vein and branches. This condition has very acutely developing deleterious effects over intestinal circulation compromising the patient’s life before development of portal hypertension and its consequences.
Figure 1.
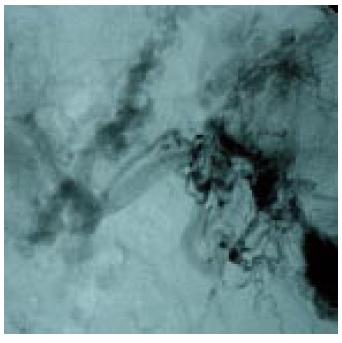
Conventional splenoportography showing a case of portal cavernomatous transformation with portosystemic collaterals, and extensive esophageal-gastric varicose veins.
Figure 2.
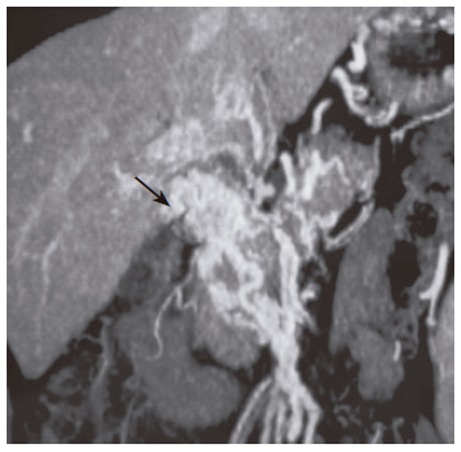
CT-angiography of portal system. Arrow shows the portal cavernomatous transformation with portosystemic collaterals.
Besides these compensatory mechanisms, liver bears the burden of decreased blood to some extent. The decreased portal blood flow stimulates apoptosis in hepatocytes of rats when portal vein is obliterated in a gradual fashion[26] and increases mitotic activity of hepatocytes in the unaffected lobe. The latter effect is well known from selective pre-surgery portal vein obliteration performed in an intention to stimulate the hypertrophy of the opposite lobe of the liver used in cancer surgery. Gradual loss of hepatic mass may be responsible for the occurrence of mild to moderate degree of hepatic synthetic dysfunction observed in advanced stages.
Etiology
PVT has various causes. The design of the studies and the parameters evaluating the cause of PVT have changed in parallel to the evolution in medical and genetic technology. In previous studies between 1979-1997, PVT were mostly attributed to trauma (5-17%), intra-abdominal sepsis (5-36%), umbilical sepsis (5-12%)[27] (important cause of PVT in children), pancreatitis (4-5%) and prothrombotic disorders (2-28 %), but in nearly 50% of the patients etiology remained unidentified[28-32]. But with the advent of better medical care, potent antibiotic treatment, advanced medical technology, discovery of various causes of genetic thrombophilia and with better understanding of the coagulation system, this profile has also changed.
Identification of etiology usually starts by exclusion of local factors such as cirrhosis, primary or metastatic cancer of liver, pylephlebitis, liver cysts, vascular abnormalities (like webs or aneurysms) and pancreatitis. Besides routine investigations such as liver biopsies or Doppler-USG studies, magnetic resonance or CT based (3 dimensional angiographic images also available) techniques are very helpful at this stage. If no local lesion is found, then the studies are directed for a possible thrombophilic condition. After all, if no factor can be found, then the condition should be named as “idiopathic PVT”.
In a study by Denninger et al[33], the underlying prothrombotic condition was identified in 72 % of PVT patients. There was one or more prothrombotic condition in 26 out of 36 patients studied and primary myeloproliferative disorders were the leading cause with a prevalence of 30 % in this study. Other studies by Valla et al[34-36] and De Stefano et al[37] report the similar figure that as a cause of PVT, myeloproliferative disorders (overt or occult) rank first in the thrombophilic conditions. Valla et al also proposed that overt or latent myeloproliferative disorders may be responsible for thrombophilic milieu for PVT in 48% of patients.
The importance and impact of genetic thrombophilic factors in PVT are investigated in various studies. Table 2 shows the recent and most comprehensive studies about this topic published between 1999-2005. One of the most interesting findings about the results of these studies is the differences between them. Although all of the studies investigated the same patient population, the results are totally different from each other. The explanations for this situation may be the patient selection bias which is the problem of the tertiary care facilities, small number of patients, non-standard evaluation of the same parameters and genetic differences of the patient population.
Table 2.
Role of genetic thrombophilic factors in development of PVT
| Thrombophilic factor | Chamourad et al[41] | Egesel et al[40] | Janssen et al[39] | Denninger et al[33] | Primignani et al[38] |
| n | 10 | 32 | 92 | 36 | 65 |
| FVLM | 10 | 30 | 8 | 2,8 | 3 |
| PM | 40 | NS1 | 3 | 14 | 22 |
| PC deficiency | NS | 26 | 7 | 0 | 0 |
| PS deficiency | NS | 43 | 2 | 30 | 2 |
| AT deficiency | NS | 26 | 1 | 4,5 | 3 |
All values except number of patients (n) indicate percentage of patients. 1NS, not studied.
The investigation of PC, PS and AT deficiencies in patients with PVT is a challenge for the clinician to intepret when the results are found to be lower than normal. There are studies investigating whether the low values are result of liver dysfunction or indicate a frank deficiency. One of the most important studies is performed in children. Mack et al[42] studied the coagulation parameters (including factors synthesized exclusively in liver) in 11 children with PVT who underwent a surgical correction (mesenterico-left portal vein bypass: Rex Shunt) of the portal venous system before and after surgery. The investigators found a significant correction of both coagulation factors (factors II, V and VII) indicated also by improvement in prothrombin time and of PC and PS levels after surgery. The same parameters studied in other 2 children receiving distal splenorenal shunt in this study and 7 children in an-other study receiving H-type mesocaval shunt[43] did not improve as the Rex shunting supporting the importance of lower portal vein flow in the development of secondary PC and PS deficiencies. Fisher et al[44] studied the same topic by combining the family investigation of PC and PS deficiencies in patients with PVT. They found out that there were only 3 familial cases of natural anticoagulant deficiencies out of 18 patients indicating a secondary finding. Another support to this finding and the previous results is that they have found significant reductions in AT, PC and PS concentrations after distal splenorenal shunt surgery (3 patients). All of the mentioned studies confirm that the lower portal blood flow has a great impact over the synthesis of natural anticoagulant and coagulant proteins. The shunt procedures directing the blood from portal system towards systemic circulation may worsen this as evidenced from comparison of results of splenorenal shunt vs. Rex shunt. In our opinion this phenomenon of secondary loss of coagulant and anti-coagulant proteins must be considered carefully in clinical practice when anticoagulation with warfarin is considered.
Secondary deficiencies of genetic thrombophilia factors like AT, PC and PS is still a matter of diagnotic challenge. To overcome this problem, there seems to be a very narrow range of solutions such as family study (first degree relatives) of deficient proteins in an attempt to detect familial aggregation of mutant genes and primary genetic studies of the patient to detect the mutant gene(s). These studies are important but not practical and not applicable in every facility. A practical screening method for detection of natural anticoagulant deficiencies in patients with liver disease was proposed by Pabinger et al[45] and used firstly by Janssen et al[39] in a clinical study which utilizes the ratio of natural anti-coagulant factor to coagulation factors (synthesized exclusively in liver). The formula is the ratio of PS or PC or AT (whichever tested) to [(factor II + factor X) /2]. If the result of this ratio is lower than 70 %, then this may indicate a deficiency of the tested natural anti-coagulant protein significantly disproportionate to decreased synthesis of proteins from liver. This formula is easy to use in clinics and is practical to determine a possible genetic deficiency.
The concept of PVT can not be solely attributed to one factor. It is now widely accepted that the PVT occurs from both a primary thrombophilic milieu and a factor that triggers the formation of the pathological thrombus in portal circulation. The aforementioned factors are currently considered as thrombophilic background rather than the primary etiologic factor according to the concept of multifactorial theory of thrombogenesis. The other co-factors required to develop a thrombus can be grouped as local and systemic factors most of which are potentially curable or preventable factors.
Clinical consequences
The consequences of the PVT vary from one patient to another and depend on some important factors. The condition may present itself as one of the complications but due to lack of convincing evidence in the literature, the most common type of initial presentation of PVT (not related with cirrhosis) is still not known. In our personal experience, most common type of presentation is variceal bleeding followed by pancytopenia due to hypersplenism. Undefined cholestasis related with pseudocholangiocarcinoma sign (without icterus) is also one of the rare presentations.
With the advent and widely distribution of USG and Doppler-USG, the condition is becoming to be diagnosed very earlier and a patient presenting with ascites (which is a very late finding in course of PVT) is almost not seen. Webb and Sherlock[27] reported in 1979 that out of 97 patients with PVT 13 presented with ascites which seems to be a very high rate (13.5%). There are still some PVT cases presenting as ascites in underdeveloped parts of the world related with absence of early diagnosis with similar rates around 13%[46].
Varices Esophageal and gastric varices related bleeding contribute to the most important cause of morbidity and hospitalization in this group of patients. When the characteristics of variceal bleeding are considered, major differences between cirrhosis vs PVT related bleeding must be pointed out. Firstly, the risk of variceal bleeding in cirrhosis is approximately 80-120 times more than PVT (non-cirrhotic) related variceal bleeding[47-49] and esophageal varices (irrespective of size) are observed in about 90% of patients. Gastric varices are mostly seen concomitantly with esophageal varices in about 40% of the patients in PVT while portal gastropathy is also a rare feature of this condition[49,50]. Compared to portosystemic varices observed in cirrhotic patients, the varices in PVT patients have some special charateristics: (1) “varice-on-varice” finding is less commonly observed; (2) the sizes of the varices are smaller; (3) associated portal gastropathy is less commonly present[50]. The retrospective study (investigated the determinants of survival in a heterogenous group of patients including 124 non-cirrhotic vs 48 cirrhotic PVT cases) of Janssen et al[47] showed that either the presence of varices or bleeding episodes of varices had no impact on survival rates and the most common cause of death was malignancy related causes. Twenty four percent of the study population had malignancies like hepatocellular cancer, pancreatic cancer, or other malignancy elsewhere metastatic to the liver which presented itself in liver as PVT indicating end stage malignancies.
Although the risk of variceal bleeding is low in non-cirrhotic patients, the management of bleeding varices is not different from cirrhotic patients. Unfortunately there is not any study in the literature comparing endoscopic procedures vs surgical procedures in acutely bleeding varices and about the topic of primary prophylaxis. We believe that primary prophylaxis with endoscopic procedures in an effort to clear varices should be applied with careful monitoring and secondary prophylaxis with combination of endoscopic procedures and medical treatment despite beta-blockers may work although there is no objective and sufficient evidence.
In contrast, beta-blockers may result in more sluggish portal flow as is the same issue for nitrates and terlipressin increasing the risk of progression of thrombosis and even worsening of portal hypertension. This decision must be adjusted with the patient’s condition rather than a standard medical care.
Pseudocholangiocarcinoma sign One of the interesting and misleading clinical conditions that may occur during the course of the PVT is “pseudocholangiocarcinoma sign” (PSCS). After the obstruction in the portal system, the “venous rescue” begins to occur immediately which takes about 5 wk[24] forming new vessels around intrahepatic, extrahepatic biliary tracts and around gallbladder (majorly, vascular plexus of Saint and Petren enlarge and dilate to become large serpentine vessels) named as “portal cavernomatous transformation”. Sometimes these vessels can be very small in caliber to visualize depending on the extent of portal flow and capacity of new vessel formation, but if a careful ERCP is performed (Figure 3) the direct effects of these vessels on biliary ducts or main bile duct as strictures, displacements, thumprinting effects or irregularity can be seen in at least 80% of the patients[49]. This condition mimics a cholangiocellular cancer in ERCP and therefore called PSCS[51,52].
Figure 3.
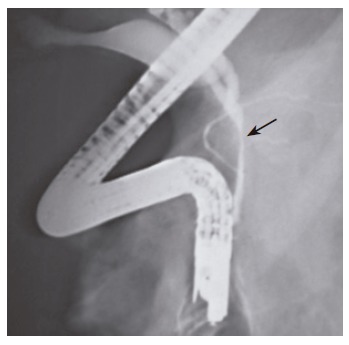
ERCP of a patient with portal cavernomatous transformation. Arrow shows the site of depression in the main bile duct.
The clinical impact of these changes results in a wide spectrum of findings ranging from totally normal biochemical and physical findings to overt cholestasis with elevated cholestatic enzymes and liver injury (very rarely observed). Although a term called “portal biliopathy” and a classification system are proposed[53], we believe that this term and classification system are impractical to use because: (1) new collaterals form depending on the site and level of portal vein obstruction, (2) wide range of anatomical variations between human beings in this anatomical site resulting in different types of cavernous transformations, (3) the nature of new vessel formation is unpredictable and not standard in all portal vein thrombosis patients, (4) the classification system does not have impact over treatment or follow up strategy. We propose that only defining the presence or absence of PSCS is a satisfactory practice not to complicate the picture further because the clinically evident cholestasis and/jaundice is observed in only about 5%[54]. Therefore this physiological compensatory response is not a “-pathy” but just an adaptive change with a low degree of clinical importance.
The clinically evident cholestasis either presenting itself as repeating biliary stones or cholangitis or liver injury can be treated with conventional methods including stenting, papillotomy or even surgery.
3Hypersplenism and issue of anticoagulation treatment Hypersplenism is a finding of latent or chronic PVT and this complication is important when anticoagulation treatment is considered. Hypersplenism is almost always present except in the rare patient with a partial thrombus in one branch of the portal vein with the other branch remaining intact presenting with a mild degree of portal hypertension. The levels of all blood elements begin to decrease as the condition worsens and severe hypersplenism is now considered one of the most important indications in this group of patients to undergo a shunt surgery combined with or without a splenectomy. In the clinical setting of hypersplenism with low platelet counts combined with esophageal varices it raises concerns about the safety of anticoagulation. Unfortunately, the literature lacks information about the safety and long-term results of anticoagulation in well designed prospective controlled studies. A study to clarify this controversial topic was a retrospective analysis. In this study Condat et al[48] included 136 PVT patients in whom 84 received anticoagulant treatment. The study has some limitations like heterogenous groups (pre-treatment endoscopies of all patients and standardization of a homogenous varice distribution between two groups were not accomplished) and lack of information about comorbid conditions of the participating patients, but the study has a low statistical degree of evidence to favor anticoagulation treatment. Anticoagulation was not found to be a risk factor for bleeding in this study while non-anticoagulation resulted in more thrombotic recurrences as expected. In our personal experience it may be a good practice to select patients according to their co-morbid conditions, degree of hypersplenism (low platelet counts may be a relative contraindication for anticoagulation) and the condition of varices (eradication combined with or without medical prophylaxis before start of treatment may be considered to decrease bleeding related risks).
BUDD-CHIARI SYNDROME: ETIOLOGY, CLINICAL CONSEQUENCES
Budd-Chiari syndrome (BCS) is a disorder which (besides its rarity and heterogenous clinical presentations) can potentially result in mortality and severe morbidities including liver failure requiring transplantation and even hepatocellular cancer. Veno-occlusive liver disease (also defined as sinusoidal obstruction syndrome) is a different pathology and therefore not included in this discussion.
There are various classifications of BCS in the literature. Classification systems include; 1) differrences of etiology (primary: related with thrombophilia and secondary: due to tumor or a mass occupying lesion), 2) differences of anatomical involvement (site of obstruction: small and/or large hepatic veins, isolated inferior vena cava involvement, or combination of all), 3) progression of disease (acute/fulminant and chronic/indolent). The forthcoming discussion about BCS involves primary BCS unless otherwise specified.
Etiology
Major underlying mechanism resulting in BCS is thrombosis. The etiology of thrombosis is very similar to PVT as discussed earlier, myeloproliferative disorders are the leading cause in 20-50% of all causes which may be overt or latent in presentation[33,39]. The latent myeloproliferative disorders (for instance; endogenous erythroid colonies) are not easily detectable and requires an expert laboratory and sensitive tests (bone marrow is cultured in vitro to observe whether there is spontaneous erythroid colonies formed in the absence of erythropoietin stimulation). The mirror image of relation between myeloproliferative diseases and BCS is also interesting. In an autopsy study by Wanless et al[55], the hepatic vein thrombosis was found in 6% of all subjects having a history of myeloproliferative disease. Paroxysmal nocturnal hemoglobinuria is also an acquired thrombophilic disorder characterized by occasional hemoglobinuria, venous thrombosis and anemia. It poses a high risk for development of BCS, about up to 10 % of all paroxysmal nocturnal hemoglobinuria patients have thrombosed hepatic veins[56].
Behcet’s disease is a subtype of large vessel vasculitis observed mostly in Turkish population which is related with hepatic vein thrombosis very frequently. Behcet’s disease is associated with recurrent oral and genital ulcers with skin findings (erythema nodosum, pathergy reaction), neurologic involvement and eye inflammation. BCS occuring in Behcet’s disease must be treated with an appropriate combination of colchicine, thalidomide, penicillins and anti-coagulating agents[20,57,58].
Pregnancy and use of oral contraceptives are also related with BCS but studies in the literature have conflicting results due to study designs and the number of subjects included. The studies will not be discussed in detail but the exact relation of both conditions must be studied with adequate number of patients, defining other concurrent acquired and inherited thrombophilic conditions, estrogen content of the oral contraceptive studied. Nevertheless, both of these conditions have a theoretical risk for BCS.
Inherited causes of thrombophilia like FVLM, AT, PC and PS deficiencies account for a considerable percent age in the list of etiology of BCS. FVLM is found to be associated with 25-30% of all cases[59,60], whereas PC deficiency (accounts for 9-20%) leads in all natural anticoagulant deficiencies[33,39].
Other acquired causes of BCS include oral contraceptive use, pregnancy, Behcet’s disease and inflammatory bowel disease.
Clinical consequences
The obstructed hepatic veins present to clinic in variable forms. The most common findings are ascites, hepatomegaly, pain in the abdomen and less frequently jaundice. Although the clinical findings are more florid with drastic consequences in fulminant forms, indolent BCS is an insidious process in which development of findings mimic chronic liver disease of any other kind. The latter course results in development of portal hypertension and eventually cirrhosis combined with classical complications like variceal bleeding, intractable ascites and hypersplenism. Indolent course of BCS is characterized by pathological changes involving a variety of histological patterns like veno-portal fibrosis, veno-centric cirrhosis and nodular regenerative hyperplasia.
The studies describing the pathological changes unique to liver in chronic BCS have found that 1) the liver has an evolving change in terms of vascular dynamics to eventually result in changes in portal system (either intrahepatic or extrahepatic portal system becomes affected in time which shows itself as micro- or macro-thrombosis), 2) the liver is heterogenous, and there is great variability in random sampling (indicating that the liver biopsy must be multiple in number to clearly define the pathology and histological stage in BCS), 3) the regenerative hyperplasia with nodule formation of changing sizes are common which represents the areas with increased arterial supply with a patent venous drainage[61,62].
Portal venous system must always be kept in mind when clinical presentation and management is considered. Portal system is almost always affected in patients with BCS. This results from various reasons: 1) perfusion of the portal system has a profound decrease since the sinusoidal pressure abruptly increases after initiation of BCS, 2) the enlarged and hypertrophied caudate lobe in response to BCS has a mass effect over main portal branches and intrahepatic portal venules resulting in stasis and vulnerability to thrombosis and further depleting liver perfusion. In patients with chronic BCS, the portal system is found to be involved in nearly 50% of patients when adequately studied either confined to the liver or as PVT.
The diagnosis of BCS can be made by utilizing Doppler-USG with a sensitivity of more than 85% in most cases[63]. We believe that when combined with clinical presentation, Doppler-USG is readily the diagnostic tool of option when compared with CT or MRI techniques. One option that can be applied is directly having a non-invasive venography with CT or MRI (Figure 4) to clear out lesions in inferior vena cava, hepatic veins and portal veins. But this is expensive and not widely available. The portal vein involvement must always be investigated during initial evaluation for further follow-up and management decisions since portal vein thrombosis has been shown to be a poor prognostic finding[64]. The liver biopsy must also be a part of routine evaluation of BCS since it may be related with concomitant liver diseases, but there is no consensus in literature for the definition of an adequate biopsy in BCS. We believe there must be multiple biopsies from both lobes to clearly define, diagnose and estimate prognosis of BCS due to heterogenous nature of liver involvement.
Figure 4.
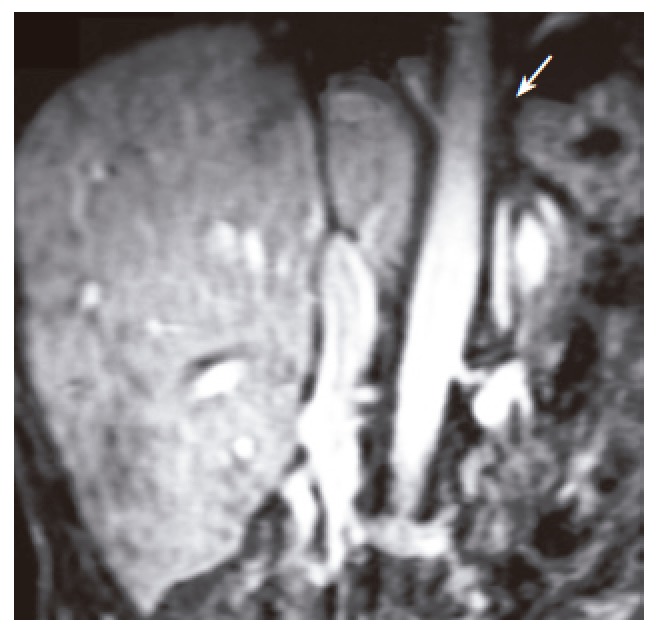
An MRI venography of hepatic veins and inferior vena cava. Arrow shows the site of obstruction in vena cava and unvisible hepatic veins.
Chronic liver disease eventually develops in nearly all BCS patients depending on the severity of liver involvement, potential of collateral development, co-morbid conditions (especially renal involvement, congestive or ischemic heart disease), thrombosis of other organs, progression of thrombosis in liver. Independent factors predicting survival include Child-Pugh score, age of the patient (younger age predicts better prognosis), renal functions and presence of portal vein involvement.
Management of the patients is directed towards prevention of recurrence of thrombosis and activate the natural thrombolytic potential (anticoagulation and antiplatelet treatment), treatment of complications of liver dysfunction (ascites, infections, renal failure) and relief of hepatic venous obstruction. In this sense, medical treatment has been proved efficacious in patients with an indolent, non-progressive (both histologically and biochemically) course[65,66]. The repeated biopsies (in every 4-5 years) combined with biochemical follow-up usually suffices for this purpose. Doppler USG also must be a part of controls for both hepatic and portal venous system evaluations. The risk of anticoagulation and potential bleeding elsewhere including esophageal varices will not be discussed in detail. But we recommend that anticoagulation must be tailored for every patient according to the presence and stage of varices, history of previous bleeding, presence of thrombophilic condition and presence of portal vein thrombosis.
A failing liver with signs of decompensation indicated by refractory ascites, deterioration of Child-Pugh scores and renal failure preventing adequate medical therapy is definite and urgent indications for the need of relief of hepatic venous obstruction by means of vascular interventions including angioplasty, transjugular intrahepatic portosystemic shunt (TIPS) or surgery. TIPS and hepatic vein angioplasty are cost-effective techniques with a potential risk of recurrence of thrombosis[67,68]. The effectiveness of vascular interventions has decreased the need for a shunt surgery which is rarely performed now. In selected patients with refractory, progressive disease despite adequate medical therapy and vascular interventions liver transplantation should be an option for absolute treatment. Sometimes presence of portal venous thrombosis itself precludes an angiographic intervention and results in need for a liver transplantation.
Liver tansplantation performed for BCS has a five-year survival rate as high as 95%[69]. Complications of transplantation are recurrence of thrombosis and development of secondary malignancies due to long-term immune-suppression. The presence of a myeloproliferative disorder underlying the BCS is not accepted as a contraindication for transplantation[70].
MESENTERIC VEIN THROMBOSIS: ETIOLOGY, CLINICAL CONSEQUENCES
Mesenteric vein thrombosis (MVT) is an acute thrombotic disorder of vessels before formation of main portal vein. Some authors proposed to use the time-dependent definition for MVT, namely acute, subacute and chronic. We believe this classification should be abandoned and the term “MVT” be used for only acute venous obstruction of superior and/or inferior mesenteric veins, whereas chronic MVT itself is already PVT. Subacute thrombosis is an intermediary form of thrombosis in which the patient has abdominal pain and other intestinal symptoms. Therefore, the term MVT is used for acute mesenteric venous thrombosis in the rest of the discussion.
MVT presents itself mostly as a severe abdominal pain located mostly at periumbilical area with a blunt and colicky nature. It may be associated with nausea, vomiting, increased bowel movements and sometimes bloody diarrhea. With progression in time, the clinical picture converts itself into an acute abdomen with findings of rebound tenderness, fever, septicemia and finally a full blown peritonitis which is an inevitable consequence of erroneous diagnosis and treatment. Therefore, early diagnosis and rapid initiation of treatment is mandatory for a good clinical outcome.
Etiology
With the improving diagnostic tests in thrombophilia research, the number of idiopathic cases has decreased to 20 %. We believe this percentage will also decrease in time with advent of new techniques and development of new concepts of research. Currently the most important risk factors are considered as presence of a large spleen, cirrhosis, surgery, abdominal inflammation (pancreatitis, abcess, inflammatory bowel disease, diverticulitis), intraabdominal cancer, thrombophilic conditions (acquired or inherited). The importance of inherited thrombophilia is difficult to evaluate (sparing mutations like FVLM and PM) since the measurements of PC, PS and AT in the acute thrombosis stage and in the anti-coagulation treatment stage of the disease is not useful. The literature is lacking convincing evidence about the importance and prevalence of these disorders in the setting of MVT. One good example for this topic is the study by Kumar et al[71]. In this study, the risk factors for MVT (comparing isolated MVT vs combined MVT with portosplenic venous thrombosis) are studied and they reported the inherited thrombophilic conditions (PC, PS and AT deficiencies, FVLM) in 27% and 5% of isolated MVT and combined MVT with portosplenic thrombosis, respectively. Interestingly, they found out that the risk of inherited thrombophilia was lower in patients with more extensive vessel involvement. Although the method and criteria of diagnosis of deficiencies of natural anticoagulants are not explained in detail due to retrospective design, this study is important to point the etiology and clinical characteristics of MVT in detail.
Although myeloproliferative disorders play a great role both in PVT and BCS, it does not seem to play the same role in case of MVT. This difference in presentation between MVT and PVT has important clinical implications for further investigations.
Consequences
The mortality rate of MVT is about 20-50%, increasing in parallel to the older age and presence of comorbid clinical conditions (cirrhosis, cardiac failure, etc.). We believe the most important factor to determine the morbidity and mortality is the prompt diagnosis and rapid initiation of treatment. Clinical suspicion has utmost importance in early diagnosis due to the obscure nature of the initial clinical presentation. History of venous thrombosis, older age, use of oral contraceptives, presence of other thrombophilic conditions are indications to rule out a possible MVT. Although the gold standard diagnostic modality is a conventional angiography, CT establishes diagnosis in nearly 90 % of patients[72,73].
After diagnosis the treatment must be initiated with anti-coagulation unless the patient has peritoneal irritation findings in which surgery is indicated. The most extensive analysis including 45 studies (a total of 3692 patients) about survival in mesenteric ischemia was performed by Schoots et al[74]. They found that MVT has a better survival (including study populations requiring surgical resection of bowel and conservative treatment groups) compared to arterial or non-thrombotic ischemia of the bowel. This finding seems to be related with a limited involvement of bowel in MVT.
CONCLUSION
Thrombophilia has a wide range of clinical presentations but abdominal organs like liver and intestines bear the most important and interesting sequelae. The consequences of thrombophilia like PVT, BCS and MVT are potentially treatable if only diagnosed early and prompt treatments are initiated. With the advent of new technologies in research of thrombophilia, clinicians will be able to diagnose the idiopathic thrombosis group of patients which is a topic of controversy. Although anticoagulant treatment is mandatory, some limitations and contraindications may prevent their use. Lastly, these group of patients must be followed with a consulting surgeon who has experience in this field to decide the best timing for a shunt surgery and for possible acute operation indications.
Footnotes
Supported by Hacettepe University Office of Scientific Research Center
S- Editor Wang J L- Editor Zhang JZ E- Editor Liu WF
References
- 1.Bertina RM, Koeleman BP, Koster T, Rosendaal FR, Dirven RJ, de Ronde H, van der Velden PA, Reitsma PH. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature. 1994;369:64–67. doi: 10.1038/369064a0. [DOI] [PubMed] [Google Scholar]
- 2.Dahlbäck B, Carlsson M, Svensson PJ. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: prediction of a cofactor to activated protein C. Proc Natl Acad Sci U S A. 1993;90:1004–1008. doi: 10.1073/pnas.90.3.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lucotte G, Mercier G. Population genetics of factor V Leiden in Europe. Blood Cells Mol Dis. 2001;27:362–367. doi: 10.1006/bcmd.2001.0388. [DOI] [PubMed] [Google Scholar]
- 4.Greer JP, Foerster J, Lukens JN, Rodgers GM, Paraskevas F, Glader BE. Wintrobe's Clinical Hematology. 10th edition. Williams and Wilkins Company. 1998:1781–1818. [Google Scholar]
- 5.Poort SR, Rosendaal FR, Reitsma PH, Bertina RM. A common genetic variation in the 3'-untranslated region of the prothrombin gene is associated with elevated plasma prothrombin levels and an increase in venous thrombosis. Blood. 1996;88:3698–3703. [PubMed] [Google Scholar]
- 6.Franco RF, Reitsma PH. Genetic risk factors of venous thrombosis. Hum Genet. 2001;109:369–384. doi: 10.1007/s004390100593. [DOI] [PubMed] [Google Scholar]
- 7.Rosendaal FR, Doggen CJ, Zivelin A, Arruda VR, Aiach M, Siscovick DS, Hillarp A, Watzke HH, Bernardi F, Cumming AM, et al. Geographic distribution of the 20210 G to A prothrombin variant. Thromb Haemost. 1998;79:706–708. [PubMed] [Google Scholar]
- 8.De Stefano V, Finazzi G, Mannucci PM. Inherited thrombophilia: pathogenesis, clinical syndromes, and management. Blood. 1996;87:3531–3544. [PubMed] [Google Scholar]
- 9.Makris M, Rosendaal FR, Preston FE. Familial thrombophilia: genetic risk factors and management. J Intern Med Suppl. 1997;740:9–15. [PubMed] [Google Scholar]
- 10.Kottke-Marchant K, Comp P. Laboratory issues in diagnosing abnormalities of protein C, thrombomodulin, and endothelial cell protein C receptor. Arch Pathol Lab Med. 2002;126:1337–1348. doi: 10.5858/2002-126-1337-LIIDAO. [DOI] [PubMed] [Google Scholar]
- 11.Colman RW, Hirsh J, Marder VJ, Clowes AW, George JN. Hemostasis and Thrombosis. 4th Edition. Lippincott: Williams & Wilkins; 2001. pp. 1497–1516. [Google Scholar]
- 12.Colman RW, Hirsh J, Marder VJ, Clowes AW, George JN. Hemostasis and Thrombosis. 4th Edition. Lippincott: Williams & Wilkins; 2001. pp. 321–335. [Google Scholar]
- 13.Crowther MA, Kelton JG. Congenital thrombophilic states associated with venous thrombosis: a qualitative overview and proposed classification system. Ann Intern Med. 2003;138:128–134. doi: 10.7326/0003-4819-138-2-200301210-00014. [DOI] [PubMed] [Google Scholar]
- 14.Nordström M, Lindblad B, Bergqvist D, Kjellström T. A prospective study of the incidence of deep-vein thrombosis within a defined urban population. J Intern Med. 1992;232:155–160. doi: 10.1111/j.1365-2796.1992.tb00565.x. [DOI] [PubMed] [Google Scholar]
- 15.Rosendaal FR. Risk factors for venous thrombotic disease. Thromb Haemost. 1999;82:610–619. [PubMed] [Google Scholar]
- 16.Green KB, Silverstein RL. Hypercoagulability in cancer. Hematol Oncol Clin North Am. 1996;10:499–530. doi: 10.1016/s0889-8588(05)70349-x. [DOI] [PubMed] [Google Scholar]
- 17.Donati MB. Cancer and thrombosis. Haemostasis. 1994;24:128–131. doi: 10.1159/000217092. [DOI] [PubMed] [Google Scholar]
- 18.Harrington KJ, Bateman AR, Syrigos KN, Rintoul R, Bhidayasiri R, McCormack M, Thomas H. Cancer-related thromboembolic disease in patients with solid tumours: a retrospective analysis. Ann Oncol. 1997;8:669–673. doi: 10.1023/a:1008230706660. [DOI] [PubMed] [Google Scholar]
- 19.Matei D, Brenner B, Marder VJ. Acquired thrombophilic syndromes. Blood Rev. 2001;15:31–48. doi: 10.1054/blre.2001.0148. [DOI] [PubMed] [Google Scholar]
- 20.Bayraktar Y, Ozaslan E, Van Thiel DH. Gastrointestinal manifestations of Behcet's disease. J Clin Gastroenterol. 2000;30:144–154. doi: 10.1097/00004836-200003000-00006. [DOI] [PubMed] [Google Scholar]
- 21.Mannucci PM. Laboratory detection of inherited thrombophilia: a historical perspective. Semin Thromb Hemost. 2005;31:5–10. doi: 10.1055/s-2005-863799. [DOI] [PubMed] [Google Scholar]
- 22.Vandenbroucke JP, Koster T, Briët E, Reitsma PH, Bertina RM, Rosendaal FR. Increased risk of venous thrombosis in oral-contraceptive users who are carriers of factor V Leiden mutation. Lancet. 1994;344:1453–1457. doi: 10.1016/s0140-6736(94)90286-0. [DOI] [PubMed] [Google Scholar]
- 23.Henderson JM, Gilmore GT, Mackay GJ, Galloway JR, Dodson TF, Kutner MH. Hemodynamics during liver transplantation: the interactions between cardiac output and portal venous and hepatic arterial flows. Hepatology. 1992;16:715–718. doi: 10.1002/hep.1840160316. [DOI] [PubMed] [Google Scholar]
- 24.Ohnishi K, Okuda K, Ohtsuki T, Nakayama T, Hiyama Y, Iwama S, Goto N, Nakajima Y, Musha N, Nakashima T. Formation of hilar collaterals or cavernous transformation after portal vein obstruction by hepatocellular carcinoma. Observations in ten patients. Gastroenterology. 1984;87:1150–1153. [PubMed] [Google Scholar]
- 25.De Gaetano AM, Lafortune M, Patriquin H, De Franco A, Aubin B, Paradis K. Cavernous transformation of the portal vein: patterns of intrahepatic and splanchnic collateral circulation detected with Doppler sonography. AJR Am J Roentgenol. 1995;165:1151–1155. doi: 10.2214/ajr.165.5.7572494. [DOI] [PubMed] [Google Scholar]
- 26.Bilodeau M, Aubry MC, Houle R, Burnes PN, Ethier C. Evaluation of hepatocyte injury following partial ligation of the left portal vein. J Hepatol. 1999;30:29–37. doi: 10.1016/s0168-8278(99)80005-1. [DOI] [PubMed] [Google Scholar]
- 27.Webb LJ, Sherlock S. The aetiology, presentation and natural history of extra-hepatic portal venous obstruction. Q J Med. 1979;48:627–639. [PubMed] [Google Scholar]
- 28.Cardin F, Graffeo M, McCormick PA, McIntyre N, Burroughs A. Adult "idiopathic" extrahepatic venous thrombosis. Importance of putative "latent" myeloproliferative disorders and comparison with cases with known etiology. Dig Dis Sci. 1992;37:335–339. doi: 10.1007/BF01307724. [DOI] [PubMed] [Google Scholar]
- 29.Stringer MD, Heaton ND, Karani J, Olliff S, Howard ER. Patterns of portal vein occlusion and their aetiological significance. Br J Surg. 1994;81:1328–1331. doi: 10.1002/bjs.1800810923. [DOI] [PubMed] [Google Scholar]
- 30.Orozco H, Takahashi T, Mercado MA, Prado E, Chan C. Surgical management of extrahepatic portal hypertension and variceal bleeding. World J Surg. 1994;18:246–250. doi: 10.1007/BF00294409. [DOI] [PubMed] [Google Scholar]
- 31.Orloff MJ, Orloff MS, Rambotti M. Treatment of bleeding esophagogastric varices due to extrahepatic portal hypertension: results of portal-systemic shunts during 35 years. J Pediatr Surg. 1994;29:142–51; discussion 151-4. doi: 10.1016/0022-3468(94)90309-3. [DOI] [PubMed] [Google Scholar]
- 32.Vleggaar FP, van Buuren HR, Schalm SW. Endoscopic sclerotherapy for bleeding oesophagogastric varices secondary to extrahepatic portal vein obstruction in an adult Caucasian population. Eur J Gastroenterol Hepatol. 1998;10:81–85. doi: 10.1097/00042737-199801000-00015. [DOI] [PubMed] [Google Scholar]
- 33.Denninger MH, Chaït Y, Casadevall N, Hillaire S, Guillin MC, Bezeaud A, Erlinger S, Briere J, Valla D. Cause of portal or hepatic venous thrombosis in adults: the role of multiple concurrent factors. Hepatology. 2000;31:587–591. doi: 10.1002/hep.510310307. [DOI] [PubMed] [Google Scholar]
- 34.Valla D, Casadevall N, Huisse MG, Tulliez M, Grange JD, Muller O, Binda T, Varet B, Rueff B, Benhamou JP. Etiology of portal vein thrombosis in adults. A prospective evaluation of primary myeloproliferative disorders. Gastroenterology. 1988;94:1063–1069. doi: 10.1016/0016-5085(88)90567-7. [DOI] [PubMed] [Google Scholar]
- 35.Valla D, Casadevall N, Lacombe C, Varet B, Goldwasser E, Franco D, Maillard JN, Pariente EA, Leporrier M, Rueff B. Primary myeloproliferative disorder and hepatic vein thrombosis. A prospective study of erythroid colony formation in vitro in 20 patients with Budd-Chiari syndrome. Ann Intern Med. 1985;103:329–334. doi: 10.7326/0003-4819-103-3-329. [DOI] [PubMed] [Google Scholar]
- 36.Valla DC, Condat B. Portal vein thrombosis in adults: pathophysiology, pathogenesis and management. J Hepatol. 2000;32:865–871. doi: 10.1016/s0168-8278(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 37.De Stefano V, Teofili L, Leone G, Michiels JJ. Spontaneous erythroid colony formation as the clue to an underlying myeloproliferative disorder in patients with Budd-Chiari syndrome or portal vein thrombosis. Semin Thromb Hemost. 1997;23:411–418. doi: 10.1055/s-2007-996117. [DOI] [PubMed] [Google Scholar]
- 38.Primignani M, Martinelli I, Bucciarelli P, Battaglioli T, Reati R, Fabris F, Dell'era A, Pappalardo E, Mannucci PM. Risk factors for thrombophilia in extrahepatic portal vein obstruction. Hepatology. 2005;41:603–608. doi: 10.1002/hep.20591. [DOI] [PubMed] [Google Scholar]
- 39.Janssen HL, Meinardi JR, Vleggaar FP, van Uum SH, Haagsma EB, van Der Meer FJ, van Hattum J, Chamuleau RA, Adang RP, Vandenbroucke JP, et al. Factor V Leiden mutation, prothrombin gene mutation, and deficiencies in coagulation inhibitors associated with Budd-Chiari syndrome and portal vein thrombosis: results of a case-control study. Blood. 2000;96:2364–2368. [PubMed] [Google Scholar]
- 40.Egesel T, Büyükasik Y, Dündar SV, Gürgey A, Kirazli S, Bayraktar Y. The role of natural anticoagulant deficiencies and factor V Leiden in the development of idiopathic portal vein thrombosis. J Clin Gastroenterol. 2000;30:66–71. doi: 10.1097/00004836-200001000-00013. [DOI] [PubMed] [Google Scholar]
- 41.Chamouard P, Pencreach E, Maloisel F, Grunebaum L, Ardizzone JF, Meyer A, Gaub MP, Goetz J, Baumann R, Uring-Lambert B, et al. Frequent factor II G20210A mutation in idiopathic portal vein thrombosis. Gastroenterology. 1999;116:144–148. doi: 10.1016/s0016-5085(99)70238-6. [DOI] [PubMed] [Google Scholar]
- 42.Mack CL, Superina RA, Whitington PF. Surgical restoration of portal flow corrects procoagulant and anticoagulant deficiencies associated with extrahepatic portal vein thrombosis. J Pediatr. 2003;142:197–199. doi: 10.1067/mpd.2003.93. [DOI] [PubMed] [Google Scholar]
- 43.Dubuisson C, Boyer-Neumann C, Wolf M, Meyer D, Bernard O. Protein C, protein S and antithrombin III in children with portal vein obstruction. J Hepatol. 1997;27:132–135. doi: 10.1016/s0168-8278(97)80292-9. [DOI] [PubMed] [Google Scholar]
- 44.Fisher NC, Wilde JT, Roper J, Elias E. Deficiency of natural anticoagulant proteins C, S, and antithrombin in portal vein thrombosis: a secondary phenomenon. Gut. 2000;46:534–539. doi: 10.1136/gut.46.4.534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pabinger I, Allaart CF, Hermans J, Briët E, Bertina RM. Hereditary protein C-deficiency: laboratory values in transmitters and guidelines for the diagnostic procedure. Report on a study of the SSC Subcommittee on Protein C and Protein S. Protein C Transmitter Study Group. Thromb Haemost. 1992;68:470–474. [PubMed] [Google Scholar]
- 46.Sarin SK, Aggarwal SR. Idiopathic portal hypertension. Digestion. 1998;59:420–423. doi: 10.1159/000007502. [DOI] [PubMed] [Google Scholar]
- 47.Janssen HL, Wijnhoud A, Haagsma EB, van Uum SH, van Nieuwkerk CM, Adang RP, Chamuleau RA, van Hattum J, Vleggaar FP, Hansen BE, et al. Extrahepatic portal vein thrombosis: aetiology and determinants of survival. Gut. 2001;49:720–724. doi: 10.1136/gut.49.5.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Condat B, Pessione F, Hillaire S, Denninger MH, Guillin MC, Poliquin M, Hadengue A, Erlinger S, Valla D. Current outcome of portal vein thrombosis in adults: risk and benefit of anticoagulant therapy. Gastroenterology. 2001;120:490–497. doi: 10.1053/gast.2001.21209. [DOI] [PubMed] [Google Scholar]
- 49.Sarin SK, Agarwal SR. Extrahepatic portal vein obstruction. Semin Liver Dis. 2002;22:43–58. doi: 10.1055/s-2002-23206. [DOI] [PubMed] [Google Scholar]
- 50.Bayraktar Y, Balkanci F, Uzunalimoglu B, Gokoz A, Koseoglu T, Batman F, Gurakar A, Van Thiel DH, Kayhan B. Is portal hypertension due to liver cirrhosis a major factor in the development of portal hypertensive gastropathy. Am J Gastroenterol. 1996;91:554–558. [PubMed] [Google Scholar]
- 51.Bayraktar Y, Balkanci F, Kayhan B, Ozenç A, Arslan S, Telatar H. Bile duct varices or "pseudo-cholangiocarcinoma sign" in portal hypertension due to cavernous transformation of the portal vein. Am J Gastroenterol. 1992;87:1801–1806. [PubMed] [Google Scholar]
- 52.Bayraktar Y, Balkanci F, Ozenc A, Arslan S, Koseoglu T, Ozdemir A, Uzunalimoglu B, Telatar H, Gurakar A, Van Thiel DH. The "pseudo-cholangiocarcinoma sign" in patients with cavernous transformation of the portal vein and its effect on the serum alkaline phosphatase and bilirubin levels. Am J Gastroenterol. 1995;90:2015–2019. [PubMed] [Google Scholar]
- 53.Chandra R, Kapoor D, Tharakan A, Chaudhary A, Sarin SK. Portal biliopathy. J Gastroenterol Hepatol. 2001;16:1086–1092. doi: 10.1046/j.1440-1746.2001.02562.x. [DOI] [PubMed] [Google Scholar]
- 54.Dilawari JB, Chawla YK. Pseudosclerosing cholangitis in extrahepatic portal venous obstruction. Gut. 1992;33:272–276. doi: 10.1136/gut.33.2.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wanless IR, Peterson P, Das A, Boitnott JK, Moore GW, Bernier V. Hepatic vascular disease and portal hypertension in polycythemia vera and agnogenic myeloid metaplasia: a clinicopathological study of 145 patients examined at autopsy. Hepatology. 1990;12:1166–1174. doi: 10.1002/hep.1840120515. [DOI] [PubMed] [Google Scholar]
- 56.Socié G, Mary JY, de Gramont A, Rio B, Leporrier M, Rose C, Heudier P, Rochant H, Cahn JY, Gluckman E. Paroxysmal nocturnal haemoglobinuria: long-term follow-up and prognostic factors. French Society of Haematology. Lancet. 1996;348:573–577. doi: 10.1016/s0140-6736(95)12360-1. [DOI] [PubMed] [Google Scholar]
- 57.Bayraktar Y, Balkanci F, Bayraktar M, Calguneri M. Budd-Chiari syndrome: a common complication of Behçet's disease. Am J Gastroenterol. 1997;92:858–862. [PubMed] [Google Scholar]
- 58.Bismuth E, Hadengue A, Hammel P, Benhamou JP. Hepatic vein thrombosis in Behçet's disease. Hepatology. 1990;11:969–974. doi: 10.1002/hep.1840110610. [DOI] [PubMed] [Google Scholar]
- 59.Deltenre P, Denninger MH, Hillaire S, Guillin MC, Casadevall N, Brière J, Erlinger S, Valla DC. Factor V Leiden related Budd-Chiari syndrome. Gut. 2001;48:264–268. doi: 10.1136/gut.48.2.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mahmoud AE, Elias E, Beauchamp N, Wilde JT. Prevalence of the factor V Leiden mutation in hepatic and portal vein thrombosis. Gut. 1997;40:798–800. doi: 10.1136/gut.40.6.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tanaka M, Wanless IR. Pathology of the liver in Budd-Chiari syndrome: portal vein thrombosis and the histogenesis of veno-centric cirrhosis, veno-portal cirrhosis, and large regenerative nodules. Hepatology. 1998;27:488–496. doi: 10.1002/hep.510270224. [DOI] [PubMed] [Google Scholar]
- 62.Cazals-Hatem D, Vilgrain V, Genin P, Denninger MH, Durand F, Belghiti J, Valla D, Degott C. Arterial and portal circulation and parenchymal changes in Budd-Chiari syndrome: a study in 17 explanted livers. Hepatology. 2003;37:510–519. doi: 10.1053/jhep.2003.50076. [DOI] [PubMed] [Google Scholar]
- 63.Bolondi L, Gaiani S, Li Bassi S, Zironi G, Bonino F, Brunetto M, Barbara L. Diagnosis of Budd-Chiari syndrome by pulsed Doppler ultrasound. Gastroenterology. 1991;100:1324–1331. [PubMed] [Google Scholar]
- 64.Mahmoud AE, Helmy AS, Billingham L, Elias E. Poor prognosis and limited therapeutic options in patients with Budd-Chiari syndrome and portal venous system thrombosis. Eur J Gastroenterol Hepatol. 1997;9:485–489. doi: 10.1097/00042737-199705000-00014. [DOI] [PubMed] [Google Scholar]
- 65.Min AD, Atillasoy EO, Schwartz ME, Thiim M, Miller CM, Bodenheimer HC. Reassessing the role of medical therapy in the management of hepatic vein thrombosis. Liver Transpl Surg. 1997;3:423–429. doi: 10.1002/lt.500030410. [DOI] [PubMed] [Google Scholar]
- 66.Zeitoun G, Escolano S, Hadengue A, Azar N, El Younsi M, Mallet A, Boudet MJ, Hay JM, Erlinger S, Benhamou JP, et al. Outcome of Budd-Chiari syndrome: a multivariate analysis of factors related to survival including surgical portosystemic shunting. Hepatology. 1999;30:84–89. doi: 10.1002/hep.510300125. [DOI] [PubMed] [Google Scholar]
- 67.Fisher NC, McCafferty I, Dolapci M, Wali M, Buckels JA, Olliff SP, Elias E. Managing Budd-Chiari syndrome: a retrospective review of percutaneous hepatic vein angioplasty and surgical shunting. Gut. 1999;44:568–574. doi: 10.1136/gut.44.4.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Blum U, Rössle M, Haag K, Ochs A, Blum HE, Hauenstein KH, Astinet F, Langer M. Budd-Chiari syndrome: technical, hemodynamic, and clinical results of treatment with transjugular intrahepatic portosystemic shunt. Radiology. 1995;197:805–811. doi: 10.1148/radiology.197.3.7480760. [DOI] [PubMed] [Google Scholar]
- 69.Srinivasan P, Rela M, Prachalias A, Muiesan P, Portmann B, Mufti GJ, Pagliuca A, O'Grady J, Heaton N. Liver transplantation for Budd-Chiari syndrome. Transplantation. 2002;73:973–977. doi: 10.1097/00007890-200203270-00026. [DOI] [PubMed] [Google Scholar]
- 70.Melear JM, Goldstein RM, Levy MF, Molmenti EP, Cooper B, Netto GJ, Klintmalm GB, Stone MJ. Hematologic aspects of liver transplantation for Budd-Chiari syndrome with special reference to myeloproliferative disorders. Transplantation. 2002;74:1090–1095. doi: 10.1097/00007890-200210270-00006. [DOI] [PubMed] [Google Scholar]
- 71.Kumar S, Kamath PS. Acute superior mesenteric venous thrombosis: one disease or two. Am J Gastroenterol. 2003;98:1299–1304. doi: 10.1111/j.1572-0241.2003.07338.x. [DOI] [PubMed] [Google Scholar]
- 72.Vogelzang RL, Gore RM, Anschuetz SL, Blei AT. Thrombosis of the splanchnic veins: CT diagnosis. AJR Am J Roentgenol. 1988;150:93–96. doi: 10.2214/ajr.150.1.93. [DOI] [PubMed] [Google Scholar]
- 73.Harward TR, Green D, Bergan JJ, Rizzo RJ, Yao JS. Mesenteric venous thrombosis. J Vasc Surg. 1989;9:328–333. [PubMed] [Google Scholar]
- 74.Schoots IG, Koffeman GI, Legemate DA, Levi M, van Gulik TM. Systematic review of survival after acute mesenteric ischaemia according to disease aetiology. Br J Surg. 2004;91:17–27. doi: 10.1002/bjs.4459. [DOI] [PubMed] [Google Scholar]