

Abstract
AIM: To evaluate the possible differences in morphology and immunohistochemical expression of CD3, transforming growth factor β1(TGF-β1), Smad7, α-smooth muscle actin (α-Sma), and collagen types I-VII of small and large intestine in Smad3 null and wild-type mice.
METHODS: Ten null and ten wild-type adult mice were sacrificed at 4 mo of age and the organs (esophagus, small and large bowel, ureters) were collected for histology(hematoxylin and eosin, Masson thrichrome, silver staining), morphometry and immunohistochemistry analysis. TGF-β1 levels of intestinal tissue homogenates were assessed by ELISA.
RESULTS: No macroscopic intestinal lesions were detected both in null and wild-type mice. Histological and morphometric evaluation revealed a significant reduction in muscle layer thickness of small and large intestine in null mice as compared to wild-type mice. Immunohistochemistry evaluation showed a significant increase of CD3+T cell, TGF-β1 and Smad7 staining in the small and large intestine mucosa of Smad3 null mice as compared to wild-type mice. α-Sma and collagen I-VII staining of small and large intestine did not differ between the two groups of mice. TGF-β1 levels of colonic tissue homogenates were significantly higher in null mice than in wild-type mice. In preliminary experiments a significant reduction of TNBS-induced intestinal fibrosis was observed in null mice as compared to wild-type mice.
CONCLUSION: Smad3 null mice are a useful model to investigate the in vivo role of the TGF-β/Smad signalling pathway in intestinal inflammation and fibrosis.
Keywords: Transforming growth factor, TGF-β, Fibrosis, Smad proteins
INTRODUCTION
Smads are a family of eight related proteins which function as signalling intermediates for the transforming growth factor β (TGF-β) superfamily of ligands[1-3]. Upon ligation and activation of TGF-β with its receptors (I, II and III), the phosphorylated Smad2 and 3 form a complex with the common mediator Smad4. The Smad2/3- Smad4 complex can translocate into the nuclei where they enhance specific TGF-β target genes. The inhibitory Smad7 antagonizes TGF-β signalling by interfering with the ligation of Smad2/3 with the activated receptor complex. Experimental evidence from several research groups suggests that disruption of the TGF-β/Smad signalling pathway plays a central role in sustaining both chronic tissue inflammation and fibrosis[4-6].
TGF-β is a multifunctional polypeptide hormone influencing different functions in a variety of cells including regulation of cell proliferation, differentiation and apoptosis, immunoregulation, regulation of the inflammatory response, restitution and healing[5,7,8]. At cellular level, TGF-β affects virtually all stages of the chronic inflammatory and fibrotic disease processes. The effects of TGF-β on the extracellular matrix are more complex than those of any other growth factor and are central to its effects in increasing the maturation and strength of wounds, as well as in the pathological matrix accumulation, characteristic of fibrotic disease[4,9,10]. TGF-β regulates the transcription of a wide spectrum of matrix proteins including collagen, fibronectin, glycosaminoglycans, and matrix-degrading proteases (metalloproteinases) and their inhibitors.
TGF-β/Smad signalling plays an important role in chronic inflammatory diseases, especially in Crohn’s disease (CD)[5,7,11,12]. The transmural infiltrate of CD is responsible for initiating and maintaining a series of connective tissue changes not only involving the mucosa but also the submucosa and muscularis mucosae and propria where a marked increase of type I, III and V collagens and RNA transcripts are observed[13-15]. In CD, there is a marked overexpression of TGF-β1 and TGF-β receptors in the colonic mucosa[16-18]. Fibrosis in CD can therefore be viewed as an aberrant healing response to injury[19]. In addition, TGF-β appears to be involved in intestinal fibrosis present in other enteropathies, such as radiation enteritis, collageneous colitis and intestinal graft-versus-host disease[20-22].
Experimental transgenic animal models are useful tools to study the in vivo function of individual molecules[23-25]. TGF-β knock-out mouse model is characterized by the loss of a critical regulator of immune function which leads to an excessive inflammatory response with massive infiltration of leukocytes in several organs[26]. This condition develops rapidly with onset during the first week of life and results in severe wasting and death by the fourth week of life[27,28]. Unlike the targeted disruptions of Smad2 and 4 which are lethal[29,30], the disruption of Smad3[31] results in the birth of mice which are viable and can survive to adulthood (up to 8 mo of age). Since the Smad3 knock out model provides pivotal information concerning cutaneous wound healing[32-34], it is thought that this model might also be useful to investigate the in vivo role of the TGFβ/Smad signalling pathway in intestinal inflammation and fibrosis.
The present study was to evaluate the small bowel and colonic morphology as well as the immunohistochemical expression of collagens I-VII, α-smooth muscle actin (α-SMA), TGFβ1, Smad7, and CD3 in Smad3 wild-type and null mice.
MATERIALS AND METHODS
Animals
Colonies of Smad3 wild-type, heterozygous and null mice (black Swiss strain) were bred in our laboratory. These animal colonies were developed using pairs of Smad3 heterozygous mice kindly provided by A. Roberts (NCI, Bethesda, MD, USA). Smad3ex8/ex8 mice were generated by targeted disruption of the Smad3 gene by homologous recombination. Targeted embryonic stem-cell clones were injected into a C57BL blastocyst to obtain germline transmission. Mice heterozygous for the targeted disruption were intercrossed to produce homozygous offsprings[31].
All mice were maintained in a specific pathogen-free (SPF) facility and routinely monitored. Mice were kept in microisolator cages and allowed free access to food and water. All mice were examined 4 times a week for signs of colitis including weight loss, diarrhea, rectal bleeding and prolapse[35], as well as signs of systemic inflammation such as piloerection, lethargy and periorbital exudates[36].
DNA extraction and genotype analysis
Mouse tail DNA extraction was performed according to the protocol reported elsewhere[37].
Genotype analysis was carried out by the polymerase chain reaction (PCR) method in which the wild type Smad3 allele was detected using primer 1 (5’-CCAGACTGCCTTGGGAAAAGC-3’) and primer 2 (5’-CCCGAACAGTTGGATTCACACA-3’). Primer 1 is located 5’ to the deletion and primer 2 is located within the deletion. This primer pair amplified a fragment of ~ 400 bp from wild-type and Smad3 ex8/+ mice, but not from Smad3ex8/ex8 mice (Figure 1). DNA was also amplified using primer 1 and primer 3 (5’-CCAGACTGCCCTTGGGATGCCCCTG-3’), which is located in the pLoxpneo, to detect the mutant Smad3 allele. In this case, a 250 bp fragment was detected in mice, heterozygous or homozygous for the mutant Smad3 allele, while no signal was detected in wild-type mice.
Figure 1.
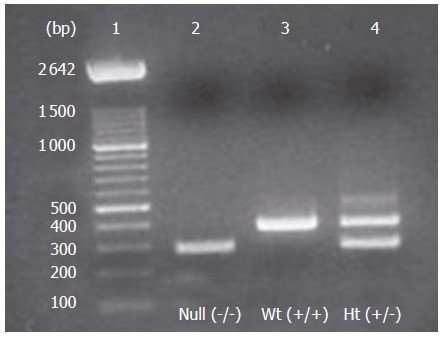
Genotyping of animal offsprings by PCR of cDNA (tail extracts). Lane 1= molecular weight ladder of 100 bp; lane 2= null mice; lane 3= wild-type (Wt) mice; lane 4 = heterozygous(Ht) mice.
Sample recovery and preparation
A total of 20 mice (10 wild-type and 10 null for the Smad3 allele) were sacrificed at 4 mo of age. Laparotomy was performed under ether anesthesia. The esophagus, small bowel, colon and ureters were visualized, rapidly excised and placed in a Petri dish containing sterile saline solution. Tissue samples from the esophagus, small bowel, colon, and ureters were processed for structural and immunohistochemical studies.
Histology and morphometry
Specimens obtained from the esophagus, small bowel, colon and ureters of all animals were washed and immediately immersed in 10% buffered formalin in phosphate buffer saline (PBS) ( pH 7.4) for 3 h at room temperature followed by the standard procedure for paraffin embedding. Serial 3 μm sections were stained with hematoxylin and eosin (HE) to assess the degree of inflammation and with Masson trichrome and silver stain to detect connective tissue. The stained sections were then observed under an Olympus BX51 light microscope (Olympus, Optical Co. Ltd., Tokyo, Japan). The degree of intestinal inflammation was scored as absent, mild, moderate or severe according to the density and extent of both acute and chronic inflammatory infiltrate, loss of goblet cells and bowel wall thickening[35,38]. Intestinal fibrosis was scored as mild, moderate or severe depending on the density and extent of trichrome-positive connective tissue staining and disruption of tissue architecture[38]. A quantitative estimate of mucosa, submucosa and muscular layer thickness of distal esophagus, proximal and distal small bowel, proximal and distal colon, and distal ureters was performed. Morphometric analysis was done in all animals by ten measurements randomly taken in 4 different fields (x 40) in a double blind fashion by two independent pathologists with an agreement always higher than 90%. Light microscopic and IHC microphotographs were taken by Olympus BX-51 Light Microscopy (Olympus, Optical Co. Ltd., Tokyo, Japan) with a videocam (Spot Insight, Diagnostic Instrument, Inc., Sterling Heights, MI, USA) and processed with an image analysis system (IAS-Delta Sistemi, Rome, Italy) software.
Immunohistochemistry
Samples from small bowel and colon obtained as previously described, were also promptly fixed with 10% buffered formalin in PBS (pH 7.4) for 3 h, dehydrated in graded ethanols and embedded in low-temperature-fusion paraffin. Serial 3 μm sections were incubated for 40 min in methanol and 3% hydrogen peroxide solution and then rinsed in PBS. Thereafter, sections were incubated overnight at 4 °C with monoclonal antibodies to CD3, TGFβ-1, Smad7 and α-SMA (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), used at a dilution of 1:100, 1:250, 1:100 and 1:400 respectively in PBS. Additional sections were incubated with a cocktail of monoclonal antibodies to collagen types (I, III, IV, V, VII) (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) in order to demonstrate the morphological and topographic features of collagen distribution in different layers of small bowel and colonic wall.
Samples were then rinsed with PBS for 5 min and incubated with a labeled streptavidin-biotin-peroxidase conjugate kit (Dako LSAB, cod. K0675, Dako-Cytomation, Milan, Italy). After rinsed in PBS for 10 min, the sections were incubated with 3,3-diaminobenzidine-tetrahydrochloride (DAB, Sigma Aldrich, Milan, Italy) for 1-3 min.
To control specificity of the immune reaction, sections were incubated omitting the primary antibody, i.e., incubated with the secondary antibody alone. Finally, the samples were counterstained with Mayer’s hematoxylin and observed under photomicroscope (Olympus BX51 light microscope; Olympus, Optical Co. Ltd, Tokyo, Japan).
Measurement of colonic TGF-β1 protein levels
TGF-β1 protein was determined by ELISA. Briefly, tissue was homogenized in the presence of a mixture of protease inhibitors with a broad specificity for the inhibition of serine, cysteine, aspartic proteases and aminopeptidases (1 mL/20 g). The mixture contained 4, (2-aminoethyl) benzenesulphonyl fluoride (AEBSF), pepstatin A, E-64, bestatin, leupeptin and aprotinin without metal chelators. For determination of TGF-β1 levels, an aliquot of the supernates was treated with 1 mol/L HCl to activate TGF-β1, followed by neutralization with 1 mol/L NaOH using a standard ELISA procedure (Quantikine, R&D Systems, Minneapolis, MN, USA).
Statistical analysis
All statistical analyses were performed in a double-blind fashion and results were computed using an appropriate program (SAS/STAT software). Results were expressed as mean±SD. Statistical significance was performed by the two-tailed Student’s t test for paired data and P < 0.05 was considered statistically significant.
RESULTS
Animal gross phenotype
The phenotype of Smad3 null mice at birth was identical to that in heterozygous and wild-type littermate controls. Nevertheless, the Smad3 null mice at three weeks of age were characterized by the following hallmarks: short body length compared to the tail and a predominant deformity of the anterior paws (Figure 2). About 20% of Smad3 null animals developed a wasting disease (Figure 3) and died between 3-6 mo but none of them developed diarrhea or hematochezia, and only a marked dilatation of the colon was observed in two Smad3 null mice.
Figure 2.

Morphology of Smad3 null and wild-type mice. The majority of Smad3 null mice exhibited a reduced size compared to littermate controls. Severe bending of forepaw joints was present in Smad3 null mice.
Figure 3.
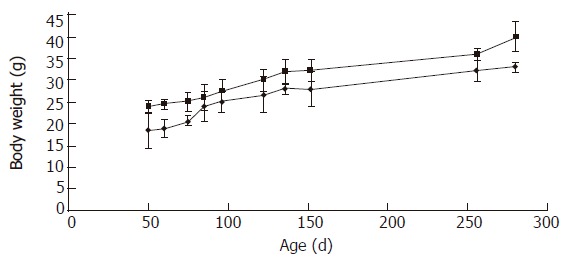
Body Weight changes of wild-type and Smad3 null mice. Each point represents mean weight data pooled from 10 mice. Standard deviations are indicated. Plot of weight (g) vs age (days). Wild-type are indicated as □ (squares), and null as ◊ (diamonds).
About 80 % of null mice survived up to 6 mo of age, while a mortality rate of 6% and 2% was observed at 18 mo of age in heterozygous and wild-type mice, respectively. No serosal or mucosal macroscopic lesions of the small or large intestine were detected either in wild-type mice or in null mice.
Histologic and morphometric evaluation
HE staining of the small and large bowel showed a normal morphology both in wild-type mice and in Smad3 null mice (Figure 4). Masson trichromic staining and silver staining of the colon and small intestine showed a similar collagen distribution in all intestinal layers both in wild-type mice and in Smad3 null mice (Figure 5).
Figure 4.
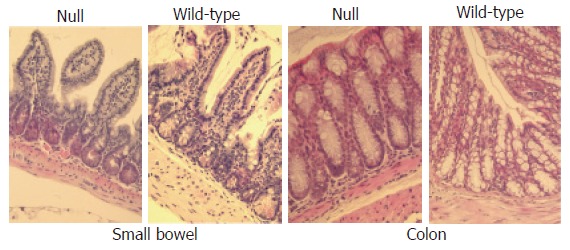
Haematoxylin and eosin-stained sections (x 20) analysis of small and large bowel from wild-type and Smad3 null mice shows normal morphology.
Figure 5.
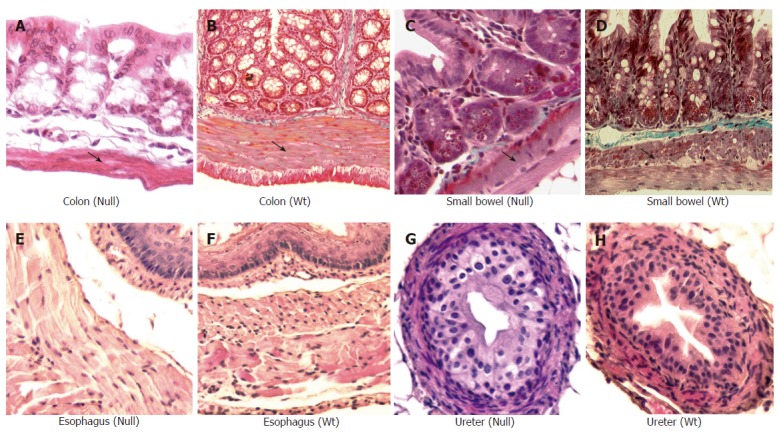
Masson trichrome staining (x 20) of small and large bowel from Smad3 mice. Significant reduction of muscular layer of descending colon of Smad3 null (A) is observed compared to colon from wild-type (WT) mice (B), reduction of muscle layer in cross sections of the proximal small bowel from Smad3 null (C) as compared to wild-type mice (D). Haematoxylin and eosin staining (x 20) of ureter and esophagus of Smad3 mice. Cross section of esophagus from Smad3 null (E) and wild-type mice (F) shows no differences in muscle layer. Cross section of ureter from Smad3 null (G) and wild-type mice (H) shows no differences in muscle layers.
A significant reduction in muscle layer thickness confined to the colon and small intestine was observed in Smad3 null mice compared to wild-type mice, while the mucosa and submucosa layers were similar in the two groups (Table 1, Figure 5). No differences in the thickness of the mucosa, submucosa and muscle layers of the ureter and esophagus were observed either in wild-type mice or in Smad3 null mice (Figure 5).
Table 1.
Muscle layer thickness of gastrointestinal and urinary tract segments from Smad3 wild-type and null mice (mean±SD)
| Wild-type (n) | Null (n) | P value | |
| Colon | 221.79 ± 99.78 (6) | 104.68 ± 50.73 (7) | 0.02 |
| Small bowel | 95.55 ± 38.45 (6) | 46.44 ± 25.96 (6) | 0.03 |
| Esophagus | 189.36 ± 42.60 (5) | 188.49 ± 43.94 (6) | NS |
| Ureters | 32.85 ± 3.74 (5) | 30.31 ± 1.91 (5) | NS |
(n) = Number of mice evaluated. Muscle layer thickness is expressed in micron. NS = not significant.
Immunohistochemistry evaluation
In the colonic and small intestine mucosa of Smad3 null mice, a significant staining of CD3+ T cells, TGF-β1 and Smad7 was observed compared to the intestinal mucosa of wild-type mice (Figure 6). TGF-β1 and Smad7 staining was localized mainly in lymphocytes of the intestinal lamina propria from Smad3 null mice. In small intestine and colon, α-SMA staining was limited to the smooth muscle cells of muscularis mucosa, muscularis propria as well as myocytes of the median vessel layer, with a comparable pattern both in wild-type mice and in null mice (Figure 7). Staining of collagens I-VII of the small intestine and colon was localized mainly in the submucosa and muscularis propria connective tissue and did not differ between the wild-type and null mice (Figure 7).
Figure 6.
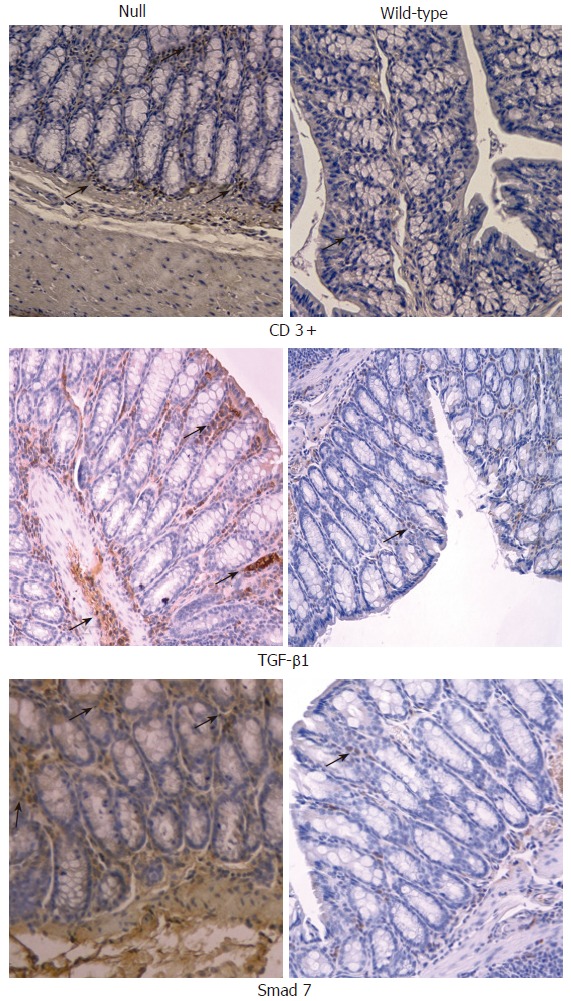
Immunohistochemical analysis (x 20) of CD3+ T cells, TGF- β1 and Smad7 in colon obtained from Smad3 null and wild-type mice. CD3+ T cells were significantly increased within large intestine of Smad3 null mice as compared to the wild-type mice. TGF-β1 and Smad7 were significantly increased within large intestine of Smad3 null mice compared to wild-type mice.
Figure 7.
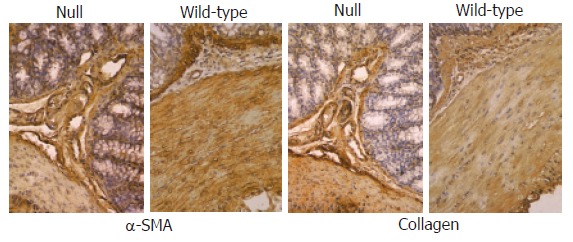
Immunohistochemical analysis (x 20) of α-SMA and collagens I-VII in colon from Smad3 null and wild-type mice. A similar localization of α-SMA antibody was found in miocytes of muscularis mucosae, muscle layer and vessels of both groups of animals. Staining of collagens I-VII in large intestine of Smad3 null and wild-type mice was localized mainly within connective tissue of submucosa and muscularis propria showing identical staining pattern between the two groups of mice.
TGF-β1 levels in colon homogenates
TGF-β1 levels in colonic tissue homogenates were significantly higher in null mice than in wild-type mice (Figure 8).
Figure 8.
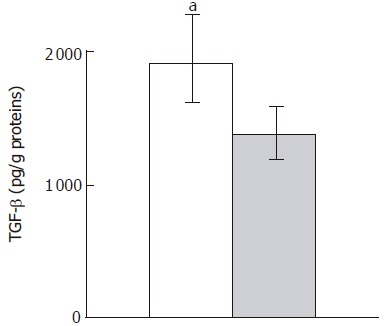
TGF- β1 ELISA of colon homogenates from Smad3 null (solid column) and wild-type mice (dashed column). Data are given as mean±SD. aP < 0.05 vs wild type mice.
DISCUSSION
In this study, we characterized the changes in intestinal structures which may occur in a Smad3 knock out mouse model compared to the littermate wild-type controls. In particular, attention was focused on evaluation of intestinal alterations present in healthy adult animals that could be used as a model to investigate the role of the TGFβ1/Smad3 pathway in the development of chronic intestinal inflammation and fibrosis. The animals studied did not develop any signs of colitis, but Smad3 null mice had a deficit in body weight gain as compared to their controls. Smad3 null mice were smaller than wild-type littermates and about 40% of them showed the presence of medially torqued forepaws associated with kyphosis sometimes. Some aspects of this phenotype are similar to those of mice expressing a transgenic dominant negative type II TGF-β receptor[39]. We did not observe any significant macroscopic intestinal lesions except for colonic dilatation in 20% of Smad3 null mice.
Histopathological analysis of small and large intestine specimens did not reveal any neoplastic lesions, significant intestinal mucosa inflammation (i.e., chronic abscesses or marked neutrophil/monocyte infiltrate) or changes in intestinal connective tissue distribution. On the contrary, mice that died prematurely (1-3 mo of age) often showing signs of systemic inflammation, presented severe histologic lesions of the intestinal mucosa (data not shown) similar to the findings described earlier[31]. These animals were not included in the present study since the aim of the investigation was to evaluate the intestine of healthy adult mice in which an intestinal fibrosis could be experimentally induced.
A significant reduction in smooth muscle layer thickness of small and large intestine was present in Smad3 null mice as compared to wild-type mice. These alterations of colonic smooth muscle layers could account for the colonic dilation observed in Smad3 null mice. In this respect, immunostaining of the colonic mioenteric plexus was also performed which showed a normal appearance (data not shown). Smooth muscle layer thickness from the esophagus and ureters was normal and similar in the two groups of mice. The reason why the intestinal muscle layer thickness was reduced in Smad3 null mice is unknown. Nevertheless, several lines of evidence suggest that TGF-β/Smad3 signalling plays an important role in the development of smooth muscle cells from totipotent or multipotent embryonic stem cells[40,41]. Furthermore, TGF-β/Smad3 signalling is also involved in the differentiation and proliferation of smooth muscle cells [42,43] .
A number of phenotypic changes as observed in our mice with a targeted disruption of Smad3 in exon 8[31], are similarly present also in mice with a disruption of Smad3 in exon 1 or 2[44,45]. In fact, in all these three models a decrease in size and growth rate, and the presence of skeletal abnormalities have been observed. They also show a decreased survival. The deletions of both exon 8 and 1 are associated with different degrees of intestinal inflammation [31,44], while the deletion of exon 2 accompanies the development of metastatic colorectal cancer[45]. The reason for this discrepancy is unknown. This discrepancy could be related to differences in genetic background of the Smad3 null animals used. It is possible that in mice a differential activation of downstream targets exists with a disruption in exons 1, 2 and 8. This hypothesis is supported by in vitro studies indicating that different domains of the Smad3 protein may be involved in activation of diverse downstream pathways[46,47].
Although no evident intestinal mucosa inflammation was found, the immunohistochemical analysis showed an increase in CD3+ T cells within the intestinal mucosa of Smad3 null mice, compared to wild-type mice, which is consistent with previous data[31]. In addition, increased TGF-β1 and Smad7 staining was observed in the intestinal mucosa of Smad3 null mice. The constitutively high TGF-β expression in the intestine could account for the positive counteractive mechanism due to the loss of intracellular transduction signals in Smad3 null mice. TGF-β overexpression could not be attributed to monocytes and macrophages not increased in the intestinal mucosa[31,32], nor to TGF-β autoinduction under the control of Smad3[32,33]. On the other hand, increased TGF-β expression could be attributed to the increased T cells in the intestinal mucosa or to the platelet degranulation not assessed in this study. The mechanism that may upregulate Smad7 expression is not clear. Moreover, Smad7 is strongly and rapidly induced by TGF-β itself[48]. Efficient expression of Smad7 appears to depend upon the cooperation of Smad, Sp1 and AP-1 transcription factors[49]. In Smad3 null mice, the overexpression of TGF-β1 may induce Smad2 phosphorylation which in turn could upregulate Smad7 intracellular expression even in the absence of Smad3. In some cell types, Smad7 expression is induced by other signalling pathways, such as Jak1/Stat1 pathway following stimulation with interferon (INF)-γ and activated nuclear factor (NF)-kB following stimulation with tumor necrosis factor (TNF)-α [50,51]. Whatever the main inducible Smad7 factor is, high Smad7 expression levels interfere with activation of Smad2 and Smad3 or accelerate degradation of TGF-β receptors, inhibiting TGF-β/Smad signalling.
The lack of significant neutrophil/monocyte infiltrate in intestinal mucosa of Smad3 null mice could be related to the impaired chemotactic response toward TGF-β shown by mutant neutrophils and monocytes[32,33]. In contrast, mutant-activated T cells are resistant to the suppressive effect of TGF-β leading to their significant increase in the intestinal mucosa[31]. Furthermore, a reduced number of IgA+ plasma cells has been detected in the intestine of severely affected Smad3 null mice[31]. These data suggest that Smad3 plays a pivotal role in TGF-β-mediated regulation of mucosal immunity and local inflammatory response. Loss of these functions may also be responsible for infection and the high mortality rate of Smad3-null mice.
It has been reported that mice lacking Smad3 show accelerated healing of cutaneous incisional wounds with reduced inflammation and accumulation of matrix[32,33], and decreased cutaneous lesions and fibrosis after exposure to ionizing radiation[34] or subcutaneous injection of bleomycin[52]. Furthermore, loss of Smad3 could attenuate bleomycin-induced lung fibrosis[53], CCl4-induced liver fibrosis[54], and glomerular fibrosis induced by different methods[55] in mice, suggesting that Smad3 plays a pivotal role during tissue injury that leads to skin, lung, liver and kidney fibrosis[56].
Recently, it has also been reported that reduction of Smad3 accelerates re-epithelization in a murine model of colitis[57]. Amelioration of cutaneous radiation-induced fibrosis has also been obtained by halofuginone, which inhibits the formation of phospho-Smad2 and Smad3, increases inhibitory Smad7 expression, and decreases cytosolic and membrane TGF-β type II receptors[58]. There is increasing evidence that Smad3 may take part in recruitment of fibroblasts to the site of injury, differentiation of fibroblasts to myofibroblasts and regulation of collagen synthesis[33,34,59]. Since loss of Smad3 interferes with the effects of TGF-β on chemotaxis and autoinduction in inflammatory cells, and as well as induction of many ECM genes by TGF-β is also Smad3-dependent, this may explain why Smad3 null mice are resistant to cutaneous[33,34,52], pulmonary[53], hepatic[54] and renal fibrosis[55]. Based on these observations, we may hypothesize that mice lacking Smad3 would be also resistant to chronic intestinal inflammation and fibrosis. In fact, by inducing intestinal fibrosis with TNBS according to the protocol of Lawrance et al[38], we observed a significant reduction of intestinal fibrosis in knock out mice as compared to the wild-type mice in preliminary experiments (data not shown).
In conclusion, about 80% of Smad3 knock-out mice survive up to 6 mo of age without developing significant macroscopic and histological lesions of the small and large intestine, with the exception of a reduction in intestinal muscle layer thickness. Preliminary data shown that Smad3 knockout mice are resistant to intestinal fibrosis. This model could be a useful tool to unravel the molecular mechanisms of chronic intestinal inflammation and fibrosis.
Footnotes
S- Editor Wang J and Guo SY L- Editor Wang XL E- Editor Bi L
References
- 1.Shi Y, Massagué J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 2.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 3.Roberts AB, Russo A, Felici A, Flanders KC. Smad3: a key player in pathogenetic mechanisms dependent on TGF-beta. Ann N Y Acad Sci. 2003;995:1–10. doi: 10.1111/j.1749-6632.2003.tb03205.x. [DOI] [PubMed] [Google Scholar]
- 4.Verrecchia F, Mauviel A. Control of connective tissue gene expression by TGF beta: role of Smad proteins in fibrosis. Curr Rheumatol Rep. 2002;4:143–149. doi: 10.1007/s11926-002-0010-4. [DOI] [PubMed] [Google Scholar]
- 5.Beck PL, Podolsky DK. Growth factors in inflammatory bowel disease. Inflamm Bowel Dis. 1999;5:44–60. doi: 10.1097/00054725-199902000-00007. [DOI] [PubMed] [Google Scholar]
- 6.Schuppan D, Koda M, Bauer M, Hahn EG. Fibrosis of liver, pancreas and intestine: common mechanisms and clear targets. Acta Gastroenterol Belg. 2000;63:366–370. [PubMed] [Google Scholar]
- 7.Fiocchi C. TGF-beta/Smad signaling defects in inflammatory bowel disease: mechanisms and possible novel therapies for chronic inflammation. J Clin Invest. 2001;108:523–526. doi: 10.1172/JCI13863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thompson JS, Saxena SK, Sharp JG. Regulation of intestinal regeneration: new insights. Microsc Res Tech. 2000;51:129–137. doi: 10.1002/1097-0029(20001015)51:2<129::AID-JEMT4>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 9.Verrecchia F, Mauviel A. Transforming growth factor-beta signaling through the Smad pathway: role in extracellular matrix gene expression and regulation. J Invest Dermatol. 2002;118:211–215. doi: 10.1046/j.1523-1747.2002.01641.x. [DOI] [PubMed] [Google Scholar]
- 10.Wells RG. Fibrogenesis. V. TGF-beta signaling pathways. Am J Physiol Gastrointest Liver Physiol. 2000;279:G845–G850. doi: 10.1152/ajpgi.2000.279.5.G845. [DOI] [PubMed] [Google Scholar]
- 11.Monteleone G, Kumberova A, Croft NM, McKenzie C, Steer HW, MacDonald TT. Blocking Smad7 restores TGF-beta1 signaling in chronic inflammatory bowel disease. J Clin Invest. 2001;108:601–609. doi: 10.1172/JCI12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hahm KB, Im YH, Parks TW, Park SH, Markowitz S, Jung HY, Green J, Kim SJ. Loss of transforming growth factor beta signalling in the intestine contributes to tissue injury in inflammatory bowel disease. Gut. 2001;49:190–198. doi: 10.1136/gut.49.2.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geboes KP, Cabooter L, Geboes K. Contribution of morphology for the comprehension of mechanisms of fibrosis in inflammatory enterocolitis. Acta Gastroenterol Belg. 2000;63:371–376. [PubMed] [Google Scholar]
- 14.Assche GV. Can we influence fibrosis in Crohn's disease. Acta Gastroenterol Belg. 2001;64:193–196. [PubMed] [Google Scholar]
- 15.Van Assche G, Geboes K, Rutgeerts P. Medical therapy for Crohn's disease strictures. Inflamm Bowel Dis. 2004;10:55–60. doi: 10.1097/00054725-200401000-00009. [DOI] [PubMed] [Google Scholar]
- 16.Babyatsky MW, Rossiter G, Podolsky DK. Expression of transforming growth factors alpha and beta in colonic mucosa in inflammatory bowel disease. Gastroenterology. 1996;110:975–984. doi: 10.1053/gast.1996.v110.pm8613031. [DOI] [PubMed] [Google Scholar]
- 17.Lawrance IC, Maxwell L, Doe W. Inflammation location, but not type, determines the increase in TGF-beta1 and IGF-1 expression and collagen deposition in IBD intestine. Inflamm Bowel Dis. 2001;7:16–26. doi: 10.1097/00054725-200102000-00003. [DOI] [PubMed] [Google Scholar]
- 18.McKaig BC, Hughes K, Tighe PJ, Mahida YR. Differential expression of TGF-beta isoforms by normal and inflammatory bowel disease intestinal myofibroblasts. Am J Physiol Cell Physiol. 2002;282:C172–C182. doi: 10.1152/ajpcell.00048.2001. [DOI] [PubMed] [Google Scholar]
- 19.Pucilowska JB, Williams KL, Lund PK. Fibrogenesis. IV. Fibrosis and inflammatory bowel disease: cellular mediators and animal models. Am J Physiol Gastrointest Liver Physiol. 2000;279:G653–G659. doi: 10.1152/ajpgi.2000.279.4.G653. [DOI] [PubMed] [Google Scholar]
- 20.Wang J, Zheng H, Sung CC, Richter KK, Hauer-Jensen M. Cellular sources of transforming growth factor-beta isoforms in early and chronic radiation enteropathy. Am J Pathol. 1998;153:1531–1540. doi: 10.1016/s0002-9440(10)65741-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ståhle-Bäckdahl M, Maim J, Veress B, Benoni C, Bruce K, Egesten A. Increased presence of eosinophilic granulocytes expressing transforming growth factor-beta1 in collagenous colitis. Scand J Gastroenterol. 2000;35:742–746. doi: 10.1080/003655200750023426. [DOI] [PubMed] [Google Scholar]
- 22.Liem LM, Fibbe WE, van Houwelingen HC, Goulmy E. Serum transforming growth factor-beta1 levels in bone marrow transplant recipients correlate with blood cell counts and chronic graft-versus-host disease. Transplantation. 1999;67:59–65. doi: 10.1097/00007890-199901150-00009. [DOI] [PubMed] [Google Scholar]
- 23.Datto M, Wang XF. The Smads: transcriptional regulation and mouse models. Cytokine Growth Factor Rev. 2000;11:37–48. doi: 10.1016/s1359-6101(99)00027-1. [DOI] [PubMed] [Google Scholar]
- 24.Weinstein M, Yang X, Deng C. Functions of mammalian Smad genes as revealed by targeted gene disruption in mice. Cytokine Growth Factor Rev. 2000;11:49–58. doi: 10.1016/s1359-6101(99)00028-3. [DOI] [PubMed] [Google Scholar]
- 25.Goumans MJ, Mummery C. Functional analysis of the TGFbeta receptor/Smad pathway through gene ablation in mice. Int J Dev Biol. 2000;44:253–265. [PubMed] [Google Scholar]
- 26.Böttinger EP, Letterio JJ, Roberts AB. Biology of TGF-beta in knockout and transgenic mouse models. Kidney Int. 1997;51:1355–1360. doi: 10.1038/ki.1997.185. [DOI] [PubMed] [Google Scholar]
- 27.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nomura M, Li E. Smad2 role in mesoderm formation, left-right patterning and craniofacial development. Nature. 1998;393:786–790. doi: 10.1038/31693. [DOI] [PubMed] [Google Scholar]
- 30.Yang X, Li C, Xu X, Deng C. The tumor suppressor SMAD4/DPC4 is essential for epiblast proliferation and mesoderm induction in mice. Proc Natl Acad Sci U S A. 1998;95:3667–3672. doi: 10.1073/pnas.95.7.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang X, Letterio JJ, Lechleider RJ, Chen L, Hayman R, Gu H, Roberts AB, Deng C. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO J. 1999;18:1280–1291. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ashcroft GS, Yang X, Glick AB, Weinstein M, Letterio JL, Mizel DE, Anzano M, Greenwell-Wild T, Wahl SM, Deng C, et al. Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response. Nat Cell Biol. 1999;1:260–266. doi: 10.1038/12971. [DOI] [PubMed] [Google Scholar]
- 33.Ashcroft GS, Roberts AB. Loss of Smad3 modulates wound healing. Cytokine Growth Factor Rev. 2000;11:125–131. doi: 10.1016/s1359-6101(99)00036-2. [DOI] [PubMed] [Google Scholar]
- 34.Flanders KC, Sullivan CD, Fujii M, Sowers A, Anzano MA, Arabshahi A, Major C, Deng C, Russo A, Mitchell JB, et al. Mice lacking Smad3 are protected against cutaneous injury induced by ionizing radiation. Am J Pathol. 2002;160:1057–1068. doi: 10.1016/S0002-9440(10)64926-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neurath MF, Fuss I, Kelsall BL, Stüber E, Strober W. Antibodies to interleukin 12 abrogate established experimental colitis in mice. J Exp Med. 1995;182:1281–1290. doi: 10.1084/jem.182.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hotchkiss RS, Swanson PE, Cobb JP, Jacobson A, Buchman TG, Karl IE. Apoptosis in lymphoid and parenchymal cells during sepsis: findings in normal and T- and B-cell-deficient mice. Crit Care Med. 1997;25:1298–1307. doi: 10.1097/00003246-199708000-00015. [DOI] [PubMed] [Google Scholar]
- 37.Ren S, Li M, Cai H, Hudgins S, Furth PA. A simplified method to prepare PCR template DNA for screening of transgenic and knockout mice. Contemp Top Lab Anim Sci. 2001;40:27–30. [PubMed] [Google Scholar]
- 38.Lawrance IC, Wu F, Leite AZ, Willis J, West GA, Fiocchi C, Chakravarti S. A murine model of chronic inflammation-induced intestinal fibrosis down-regulated by antisense NF-kappa B. Gastroenterology. 2003;125:1750–1761. doi: 10.1053/j.gastro.2003.08.027. [DOI] [PubMed] [Google Scholar]
- 39.Serra R, Johnson M, Filvaroff EH, LaBorde J, Sheehan DM, Derynck R, Moses HL. Expression of a truncated, kinase-defective TGF-beta type II receptor in mouse skeletal tissue promotes terminal chondrocyte differentiation and osteoarthritis. J Cell Biol. 1997;139:541–552. doi: 10.1083/jcb.139.2.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen S, Lechleider RJ. Transforming growth factor-beta-induced differentiation of smooth muscle from a neural crest stem cell line. Circ Res. 2004;94:1195–1202. doi: 10.1161/01.RES.0000126897.41658.81. [DOI] [PubMed] [Google Scholar]
- 41.Sinha S, Hoofnagle MH, Kingston PA, McCanna ME, Owens GK. Transforming growth factor-beta1 signaling contributes to development of smooth muscle cells from embryonic stem cells. Am J Physiol Cell Physiol. 2004;287:C1560–C1568. doi: 10.1152/ajpcell.00221.2004. [DOI] [PubMed] [Google Scholar]
- 42.Ikedo H, Tamaki K, Ueda S, Kato S, Fujii M, Ten Dijke P, Okuda S. Smad protein and TGF-beta signaling in vascular smooth muscle cells. Int J Mol Med. 2003;11:645–650. [PubMed] [Google Scholar]
- 43.Hu B, Wu Z, Phan SH. Smad3 mediates transforming growth factor-beta-induced alpha-smooth muscle actin expression. Am J Respir Cell Mol Biol. 2003;29:397–404. doi: 10.1165/rcmb.2003-0063OC. [DOI] [PubMed] [Google Scholar]
- 44.Datto MB, Frederick JP, Pan L, Borton AJ, Zhuang Y, Wang XF. Targeted disruption of Smad3 reveals an essential role in transforming growth factor beta-mediated signal transduction. Mol Cell Biol. 1999;19:2495–2504. doi: 10.1128/mcb.19.4.2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu Y, Richardson JA, Parada LF, Graff JM. Smad3 mutant mice develop metastatic colorectal cancer. Cell. 1998;94:703–714. doi: 10.1016/s0092-8674(00)81730-4. [DOI] [PubMed] [Google Scholar]
- 46.Nagarajan RP, Liu J, Chen Y. Smad3 inhibits transforming growth factor-beta and activin signaling by competing with Smad4 for FAST-2 binding. J Biol Chem. 1999;274:31229–31235. doi: 10.1074/jbc.274.44.31229. [DOI] [PubMed] [Google Scholar]
- 47.Hayes SA, Zarnegar M, Sharma M, Yang F, Peehl DM, ten Dijke P, Sun Z. SMAD3 represses androgen receptor-mediated transcription. Cancer Res. 2001;61:2112–2118. [PubMed] [Google Scholar]
- 48.Nakao A, Okumura K, Ogawa H. Smad7: a new key player in TGF-beta-associated disease. Trends Mol Med. 2002;8:361–363. doi: 10.1016/s1471-4914(02)02376-6. [DOI] [PubMed] [Google Scholar]
- 49.Brodin G, Ahgren A, ten Dijke P, Heldin CH, Heuchel R. Efficient TGF-beta induction of the Smad7 gene requires cooperation between AP-1, Sp1, and Smad proteins on the mouse Smad7 promoter. J Biol Chem. 2000;275:29023–29030. doi: 10.1074/jbc.M002815200. [DOI] [PubMed] [Google Scholar]
- 50.Ulloa L, Doody J, Massagué J. Inhibition of transforming growth factor-beta/SMAD signalling by the interferon-gamma/STAT pathway. Nature. 1999;397:710–713. doi: 10.1038/17826. [DOI] [PubMed] [Google Scholar]
- 51.Bitzer M, von Gersdorff G, Liang D, Dominguez-Rosales A, Beg AA, Rojkind M, Böttinger EP. A mechanism of suppression of TGF-beta/SMAD signaling by NF-kappa B/RelA. Genes Dev. 2000;14:187–197. [PMC free article] [PubMed] [Google Scholar]
- 52.Lakos G, Takagawa S, Chen SJ, Ferreira AM, Han G, Masuda K, Wang XJ, DiPietro LA, Varga J. Targeted disruption of TGF-beta/Smad3 signaling modulates skin fibrosis in a mouse model of scleroderma. Am J Pathol. 2004;165:203–217. doi: 10.1016/s0002-9440(10)63289-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao J, Shi W, Wang YL, Chen H, Bringas P Jr, Datto MB, Frederick JP, Wang XF, Warburton D. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am J Physiol Lung Cell Mol Physiol. 2002;282:L585–L593. doi: 10.1152/ajplung.00151.2001. [DOI] [PubMed] [Google Scholar]
- 54.Schnabl B, Kweon YO, Frederick JP, Wang XF, Rippe RA, Brenner DA. The role of Smad3 in mediating mouse hepatic stellate cell activation. Hepatology. 2001;34:89–100. doi: 10.1053/jhep.2001.25349. [DOI] [PubMed] [Google Scholar]
- 55.Wang W, Koka V, Lan HY. Transforming growth factor-beta and Smad signalling in kidney diseases. Nephrology (Carlton) 2005;10:48–56. doi: 10.1111/j.1440-1797.2005.00334.x. [DOI] [PubMed] [Google Scholar]
- 56.Flanders KC. Smad3 as a mediator of the fibrotic response. Int J Exp Pathol. 2004;85:47–64. doi: 10.1111/j.0959-9673.2004.00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tokumasa A, Katsuno T, Tanaga TS, Yokote K, Saito Y, Suzuki Y. Reduction of Smad3 accelerates re-epithelialization in a murine model of colitis. Biochem Biophys Res Commun. 2004;317:377–383. doi: 10.1016/j.bbrc.2004.03.047. [DOI] [PubMed] [Google Scholar]
- 58.Xavier S, Piek E, Fujii M, Javelaud D, Mauviel A, Flanders KC, Samuni AM, Felici A, Reiss M, Yarkoni S, et al. Amelioration of radiation-induced fibrosis: inhibition of transforming growth factor-beta signaling by halofuginone. J Biol Chem. 2004;279:15167–15176. doi: 10.1074/jbc.M309798200. [DOI] [PubMed] [Google Scholar]
- 59.Roberts AB, Piek E, Böttinger EP, Ashcroft G, Mitchell JB, Flanders KC. Is Smad3 a major player in signal transduction pathways leading to fibrogenesis. Chest. 2001;120:43S–47S. doi: 10.1378/chest.120.1_suppl.s43-a. [DOI] [PubMed] [Google Scholar]