

Abstract
Background
C2-8 is a small molecule inhibitor of polyglutamine aggregation and can reduce photoreceptor neurodegeneration in a Drosophila model of Huntington's disease (HD). Further preclinical studies have shown that oral administration of C2-8 in R6/2 HD transgenic mice can penetrate into the brain, reduce mHTT-exon1 aggregation, improve motor performance and diminish striatal neuron atrophy.
Objective
In this independent preclinical study, we aimed to evaluate the pharmacokinetic properties and therapeutic efficacy of C2-8 intraperitoneal (IP) delivery in the R6/2 HD mouse.
Methods
R6/2 mice were IP injected with low dose C2-8 (10 mg/kg), high dose C2-8 (20 mg/kg), or vehicle twice daily from 3 weeks to 3 months old. Longitudinal behavioral tests (accelerating Rotarod and wire-hang) were performed to evaluate the motor deficits, and neuropathology was measured by unbiased stereology.
Results
We confirmed that the compound has good blood-brain-barrier penetration after acute or sub-chronic intraperitoneal delivery. Chronic treatment with C2-8 in R6/2 mice results in a significant reduction of nuclear mHTT aggregate volume in the brains, replicating a key finding of C2-8 as a polyglutamine aggregation inhibitor in vivo. However, by comparing HD mice with C2-8 treatment to those with vehicle treatment, we were unable to demonstrate significant amelioration of motor deficits using Rotarod and wire-hang tests. Moreover, we did not observe improvement in the striatal neurodegenerative pathology, as measured by brain weight, striatal volume, and striatal neuron volume in the C2-8 treated R6/2 mice.
Conclusions
Our study supports the practice of independent preclinical studies for novel molecules in HD therapeutic development and suggests that the use of alternative delivery strategies and full-length HD mouse models are likely needed to further assess whether the aggregate-inhibiting properties of C2-8 can be consistently translated into a preclinical benefit in HD mice.
Keywords: Huntington's disease, huntingtin, preclinical, aggregate, R6/2, C2-8
Introduction
C2-8 is a small molecule that was developed based on a yeast high-throughput screen for inhibitors of aggregation formed by a polyglutamine (polyQ)-expanded mutant Huntingtin (mHTT)-exon1 fragment fused with enhanced green fluorescent protein (EGFP, Clontech) [1]. C2-8 is a structural analog of the original C2 compound from the screen, which can not only inhibit polyQ aggregation, but is also relatively non-toxic in mammalian cells compared to other small molecule hits. This compound was shown to inhibit mHTT polyQ fragment aggregation in vitro, in transiently transfected mammalian cells and in brain slice cultures from R6/2 transgenic mice [1, 2]. The original study also suggests a neuroprotective effect of C2-8 in a Drosophila transgenic model expressing mHTT-exon1, since C2-8 can dose-dependently reduce photoreceptor degeneration in this model [1]. Chopra and colleagues further evaluated the preclinical potential of C2-8 as a therapeutic compound by examining its drug-like properties and disease modification efficacy in the R6/2 transgenic mouse model of HD, which expresses mHTT-exon1 fragment under the human HTT promoter [2, 3]. They found that C2-8 was non-toxic, orally available, had favorable brain pharmacokinetics, and had no adverse pharmacologic interactions. Furthermore, oral administration of C2-8 in R6/2 mice reduced mHTT-exon1 aggregate size in vivo and was neuroprotective, as measured by significant improvement of motor deficits (i.e. Rotarod) and some aspects of neurodegenerative pathology (i.e. improved striatal neuronal atrophy but not brain weight loss or striatal volume loss, measured by unbiased stereology). Thus, this original study showed that C2-8 has drug-like properties, can reduce mHTT aggregation in multiple model organisms, and has evidence of ameliorating mHTT-fragment induced toxicities in an invertebrate and mouse model of HD.
The main objective of current study is to provide an independent evaluation of the pharmacokinetic properties and preclinical efficacy of C2-8 compounds in the R6/2 mouse model of HD. Our study adhered in many aspects to the experimental protocols of the original study [1], particularly the phenotypic readouts used to evaluate the preclinical efficacy. However, our study also has certain differences from the original study [2], due to methodological adjustments and the distinct sources of animal and compound, which should be taken into consideration when interpreting the findings. Our replication study data include pharmacokinetic, behavioral and neuropathological studies, and the findings should be informative on the potential of C2-8 as a mHTT-targeted small molecule therapeutic for HD.
Materials and Methods
Randomization, allocation concealment and blinding
Our C2-8 preclinical study design followed the basic guidelines recently recommended by the National Institute of Neurological Disorders and Stroke (NINDS) panel [4]. Both adequate generation and allocation concealment in randomization schemes have been strictly ensured. Among three individuals involved in this project, one randomization coordinator was in charge of allocation, preparation and accounting for the trial injections. Another researcher, who administered the compounds to mice, and the third researcher, who performed the behavioral studies, were not aware of the treatment allocation of any mice. For all outcome assessments (behavioral and pathology), mice were re-coded and kept blinded to the researchers. The researchers were kept blind to the treatment assignment and genotypes until the final analyses were completed.
Data handling
Based on the proposed exclusion criteria, data from 3 mice that died during the study were excluded. There were no outliers defined and no other data were removed from analysis.
C2-8 compounds
C2-8 was synthesized by Dr. Mohammad Khanfar in Dr. Richard B. Silverman's laboratory at Northwestern University. We have obtained at total of 8.02 grams of C2-8 from the Silverman lab.
Animals and drug treatments
Ovary-transplanted female R6/2 breeders (B6CBATg(HDexon1)62Gbp/1J, JAX Stock #002810) were obtained from The Jackson Laboratory (JAX) and crossed to wild-type B6/CBA-F1 males. The female offspring from the F1 generation were genotyped following the JAX standard protocol, using primers oIMR1594 (5′CCGCTCAGGTTCTGCTTTTA3′) and oIMR1596 (5′TGGAAGGACTTGAGGGACTC3′). Using these primers, we confirmed that all mice used in this trial had approximately 160 ± 10 CAG repeats. Female transgenic mice with similar repeat lengths were randomized by a computer generated list and assigned to three groups: vehicle control, low dose C2-8 (10 mg/kg) and high dose of C2-8 (20 mg/kg). The drug solution was made fresh daily prior to injection as following: C2-8 compound was weighed in a 15 ml tube, 100% ethanol (5% of final volume) was added and the mixture was vortexed for 5 minutes; cremorphor (10% of final volume) was then added and the mixture was vortexed for another 10 minutes; the drug solution was brought up to the desired volume with sterilized PBS and sonicated in glass vials for 30 minutes. The same solution without C2-8 compound is used for injection as vehicle control. Drug administration, by intraperitoneal (IP) injection twice daily (at 9 A.M. and 9 P.M.), started at 3 weeks of age and continued until 90 days of age. All mice were maintained and bred under standard conditions consistent with National Institutes of Health guidelines and approved by the University of California, Los Angeles Institutional Animal Care and Use Committees.
Pharmacokinetic (PK) studies
In the first PK study, a single bolus dose of 500 mg/kg C2-8 was administered by IP injection to wild-type mice (CBA × C57BL6 F1, N =3 per time point), and the plasma and brain levels of C2-8 were determined at different post-dosing intervals (0, 3, 6, 12 and 24 hours) using LC/MS/MS at Apredica (Water-town, MA), the company used in the original PK study.
In another PK study, we performed chronic IP injections to assess steady state blood and brain C2-8 concentrations. Wild-type mice (N = 3 per group, 2-months old) were IP administered 20 mg/kg C2-8 twice per day (b.i.d.) for 5 days. Blood and brains were harvested at 1 hour, 4 hours and 8 hours after the last dose for measuring C2-8 levels in both tissues. Three mice without C2-8 injections were used as negative controls.
Behavioral tests
Motor performance was assessed by accelerating Rotarod at 5, 8, and 11 weeks of age with established protocols [5]. Briefly, animals were trained on a Ugo Basile 7650 Rotarod (Stoelting, Ugo Basile, Biological Research apparatus; Varese, Italy) for three trials per day for 2 days at 5 weeks of age using an accelerating paradigm, and then tested for three trials per day for 3 consecutive days at 5, 8, and 11 weeks of age. For each mouse, at least 10 minutes rest was guaranteed between trials. 5 minutes was the maximal time for one trial. If a mouse dropped off in less than 20 seconds, it was re-tested once. Any latency to fall equal to or greater than 20 seconds was recorded as it was. The average of the three trials in the last day during each three consecutive days was used for statistical analysis.
Wire-hang testing was performed at 9.5 and 12.5 weeks of age, following a published protocol [6]. Briefly, mice were placed on a wire cage top that was then gently inverted. The time that each mouse remained on the cage top was recorded. Three trials were performed for each mouse for three consecutive days, and the average of the last day was used for statistical analysis.
Rotarod and wire-hang data were analyzed using repeated measure two-way ANOVA followed by post-hoc analysis with SPSS (Version 17.0). Results were considered statistically significant at p < 0.05.
Neuropathology
At 90 days of age, mice were euthanized and perfused with 4% paraformaldehyde in 0.01 M PBS (pH 7.4). The brain weight was obtained using our established protocols [5]. Serial coronal brain sections (50μm thickness) were prepared for unbiased design stereology of mHTT aggregates, striatal neuronal volume and striatal volumes using the standardized setup and protocol established in our lab [5]. Briefly, Nissl (thionin) staining was performed on every eighth coronal section and striatal volumes were then estimated using the Cavalieri method, and the same sections were used to measure the striatal neuronal volume. Another set of coronal brain sections were stained with mAb EM48 (Millipore, 1:150) followed by biotinylated secondary antibody and Vectastain ABC kit (Vector Laboratories, Burlingame, CA). Striatal neuronal volume and intranuclear mHTT aggregate volume were then determined using the four-point vertical nucleator method as the original study [2]. All analyses were performed blind using StereoInvestigator software (MicroBrightField, Williston, VT), and a Leica DMLB microscope with a motorized stage. For stereological studies, coefficient of sampling error (Gundersen's CE, m= 1) was used to determine the precision of the estimates or sampling variation within each brain. Within the current study, the CE was controlled to be <10%. Data were analyzed by one-way ANOVA followed by Tukey post-hoc analysis using SPSS (version 17.0).
Results
We performed pharmacokinetic (PK) studies to verify the brain availability of C2-8 via IP injection. The rationale for choosing the IP delivery route, instead of the twice-daily oral gavage procedure, included potential stress associated with chronic oral gavage [7], the susceptibility of a subset of R6/2 mice to stress-induced seizures [3], and the simplicity and lower variability of IP drug delivery compared to the oral route [7].
To test whether IP delivery of C2-8 can achieve PK parameters comparable to the original study that utilized oral gavage, we first used a bolus dose of 500 mg/kg C2-8 IP injected in adult wild-type mice to try to mimic the dosing regimen (200 mg/kg, oral gavage, b.i.d.) used in the original study. C2-8 showed good bio-availability after a single bolus injection, with both plasma and brain maintaining high C2-8 levels at all time points examined (Fig. 1A). In another chronic PK study, adult wild-type mice (n = 3) were administered 20 mg/kg C2-8 b.i.d. for 5 days (Fig. 1B). At 1, 4 and 8 hours after the last dose, the average C2-8 levels in the brains were 2052, 772 and 134 pmol/g brain, respectively, which is equivalent to 41.0, 15.4 and 2.7 fold of the original IC50 value (i.e. 50 nM) for C2-8 to inhibit polyQ aggregation in the PC12 cells [1]. Based on our PK findings, particularly the sub-chronic PK result, we conclude that our IP delivery regiment can lead to sufficient levels of C2-8 in the mouse brain to enable the evaluation of its in vivo effects on mHTT aggregation and modification of behavioral and other pathological deficits in R6/2 model of HD.
Fig. 1.
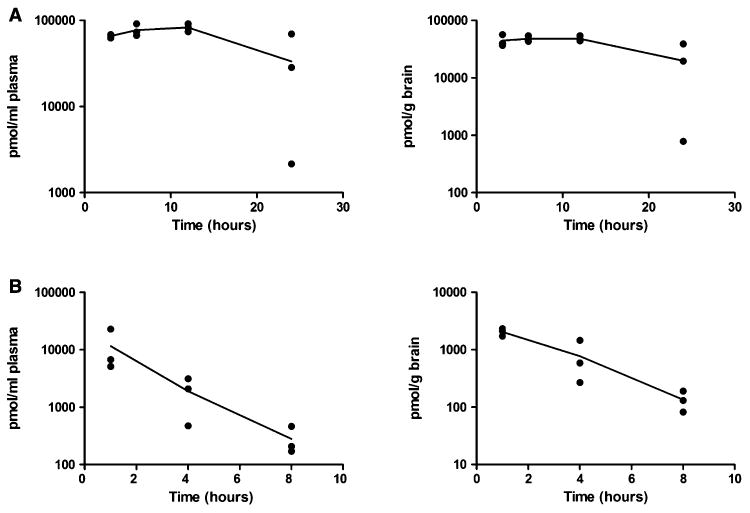
C2-8 has good brain bioavailability. (A) Plasma and brain levels of C2-8 in wild-type mice after single bolus IP injection administration of 500 mg/kg C2-8 were plotted. Levels were determined 3, 6, 12 and 24 h after dosing. N = 3 per time point. (B) Plasma and brain levels of C2-8 in wild-type mice with 5-day I.P. injection of 20 mg/kg C2-8 b.i.d. at 1, 4 and 8 hours after the last dose. N = 3 per time point. The lines connect the means values for each time point.
We then proceed to evaluate the preclinical therapeutic efficacy of C2-8 in the R6/2 mouse model of HD [3]. Mice were randomly assigned to three groups and administered vehicle or C2-8 (at doses of 10 or 20 mg/kg) by IP injection twice daily (with 12 hour intervals). The R6/2 mice received treatment starting at 22 days of age and continued until either spontaneous death or euthanasia at 90 days of age. Two HD mice in the low dose C2-8 group and one mouse in the high dose C2-8 group died during the study and had their data excluded. The remaining mice (high dose: N = 13, low dose: N = 14, vehicle: N = 11) survived until the terminal point and finished the behavioral tests. Additionally, we included 8 wild-type female mice from the same breeding cohort as vehicle administration controls. Body weights were measured twice a week during the time and significant weight loss was observed in R6/2 mice compared to the WT mice at the late stages (after 9–10 weeks of age), but within the R6/2 mice, vehicle or drug treatment groups showed no significant difference in body weights (Fig. S1).
We performed Rotarod testing to assess motor deficits at 5, 8 and 11 weeks of age. The three groups of R6/2 transgenic mice showed similar latency to fall at all three time points. At 5 weeks of age, all three R6/2 mouse groups performed at similar levels on Rotarod compared to wild-type controls (Fig. S2) and were all significantly worse than the wild-type mice at 8 and 11 weeks of age (P < 0.0001, repeated measure two-way ANOVA followed by Tukey post-test). However, C2-8 treatment at two different doses did not significantly affect Rotarod performance compared to vehicle treatment in R6/2 mice (Fig. 2A; Fig. S2; Drug effect: F(2,70) = 0.29, P = 0.7483, repeated measure two-way ANOVA). We also performed wire-hang testing in R6/2 mice at 9.5 and 12.5 weeks of age. At both ages mutant mice in vehicle, low dose and high dose drug treated groups had similar wire-hang endurance compared to the vehicle groups (Fig. 2B; F(2,35) = 0.14, P = 0.8738, two-way ANOVA repeated measure). Thus, our study did not show any significant effects of C2-8 in modifying two behavioral deficits in R6/2 mice.
Fig. 2.
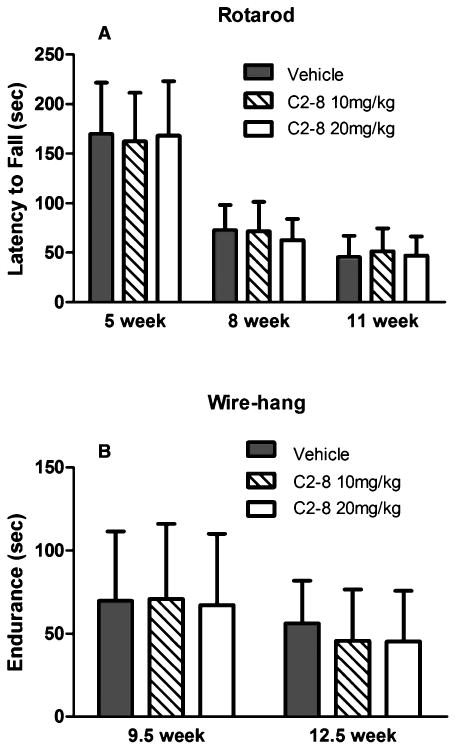
C2-8 treatment does not show a significant rescue effect on behavioral deficits in R6/2 mice. (A) Rotarod performance of C2-8-treated and vehicle-treated R6/2 mice at 5, 8, and 11 weeks of age. C2-8-treated mice performed similar to vehicle controls on the Rotarod at all three time points (drug effect: F(2,70) = 0.29, P value = 0.7483). (B) Wire-hang endurance of R6/2 mice showed no improvement at 9.5 and 12.5 weeks of age with C2-8 treatment (F(2,35) = 0.14, P value = 0.8738). Values represent mean ± S.E.M. Vehicle: n = 11; C2-8 10 m/kg: n = 14; C2-8 20 mg/kg: n = 13. Data were analyzed with repeated measure two-way ANOVA.
We next performed neuropathological studies in our cohort of R6/2 and wild-type mice to test whether C2-8 improves brain atrophy and mHTT aggregation phenotypes in vivo. To assess brain atrophy, we measured brain weight and used unbiased stereology to quantify striatal volume in C2-8 and vehicle treated R6/2 mice at 90 days of age. The three groups of R6/2 mice had comparable forebrain and cerebellum weights (Fig. 3; forebrain: F(2,35) = 0.86, P = 0.4308; cerebellum: F(2,35) = 1.37, P = 0.2660; one-way ANOVA followed by Tukey post-test). Moreover, unbiased stereology revealed that the vehicle, low and high dose C2-8-treated R6/2 mice had similar striatal volumes (F(2,27) = 1.559, P = 0.2287). Importantly, our stereological study did confirm that all three groups of R6/2 mice have significantly smaller striatal volumes compared those from wild-type littermates (Fig. 4; P < 0.001, one-way ANOVA followed by Tukey post-test). In addition to brain volume, we also performed unbiased stereological study of striatal neuronal volumes (Fig. 5) and did not detect any significant difference among R6/2 mice either treated with vehicles or with C2-8 (Fig. 5; F(2,27) = 0.4569, P value = 0.6381). Together, our neuropathological studies do not support efficacy of C2-8 administration in improving striatal atrophy phenotype in our cohort of R6/2 HD mice.
Fig. 3.
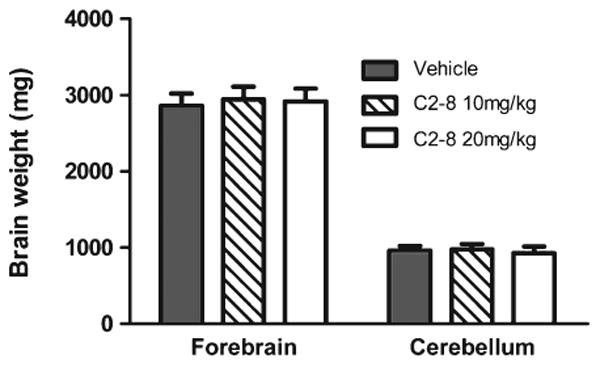
C2-8 treatment has no effect on forebrain or cerebellum weight in R6/2 mice. Forebrain: F(2,35) = 0.86, P value = 0.4308; cerebellum: F(2,35) = 1.37, P value = 0.2660. Values represent mean ± S.E.M. Vehicle: n = 11; C2-8 10m/kg: n = 14; C2-8 20 mg/kg: n = 13. Data were analyzed with one-way ANOVA.
Fig. 4.
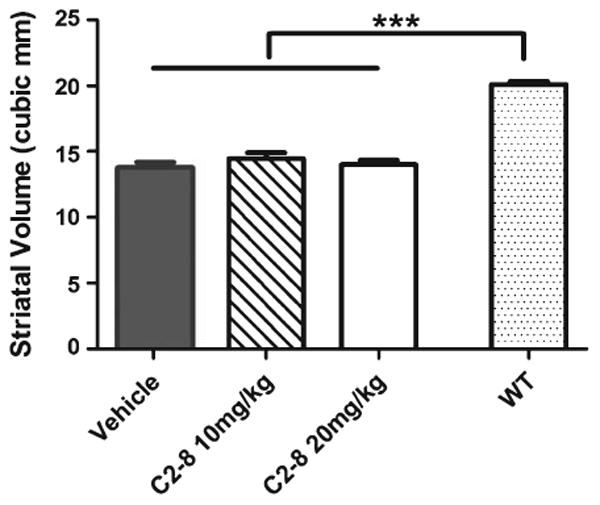
C2-8 treatment does not affect R6/2 mouse striatal volume. All groups of R6/2 mice have similar striatal volumes (F(2,27) = 1.559, P value = 0.2287), which were all significantly lower than wild-type mice (***P < 0.001). Values represent mean ± S.E.M, n =10 per group. Data were analyzed with one-way ANOVA followed by Tukey post-test.
Fig. 5.
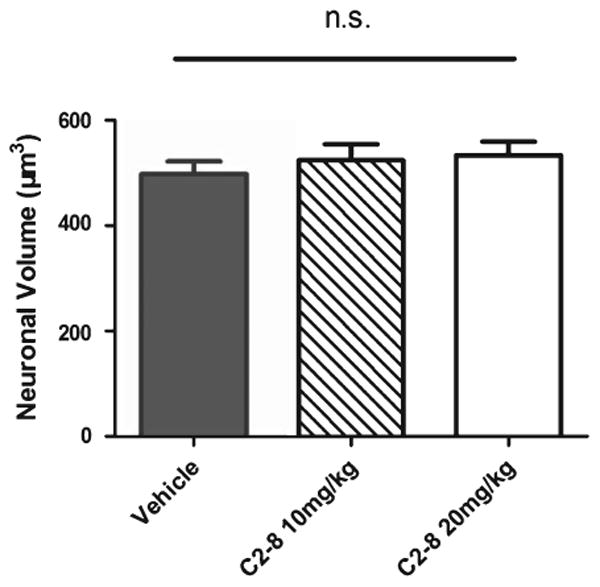
C2-8 treatment does not affect R6/2 mouse striatal neuron volume. All groups of R6/2 mice have similar striatal neuronal volume (F(2,27) = 0.4569, P value = 0.6381). Values represent mean ± S.E.M, n = 10 per group. Data were analyzed with one-way ANOVA followed by Tukey post-test.
Since C2-8 was originally identified as a small molecule that can consistently suppress mHTT aggregation in cellular models and animal models of HD [1, 2], an important goal of our study is to assess whether our IP route of administration could still reduce mHTT aggregate size, as demonstrated in the original study which used oral administration [2]. We used the nucleator method to quantify striatal intranuclear mHTT aggregate volumes in vehicle or C2-8 treated R6/2 mice at 90 days of age (Fig. 6A). We were able to detect a significant effect with the low-dose C2-8 (10 mg/kg) treated group compared to the vehicle treated group, and a further reduction of mHTT aggregate volume in the high-dose (20 mg/kg) C2-8 treated group (Fig. 6B; F(2,27) = 9.339, P = 0.0008, one-way ANOVA followed by Tukey post-test). This finding supports the original observation that C2-8 treated R6/2 mice have smaller mHTT aggregates in the striatum [2] and demonstrates that IP administration of C2-8 can also achieve the primary pharmacodynamic effects of this compound in vivo.
Fig. 6.
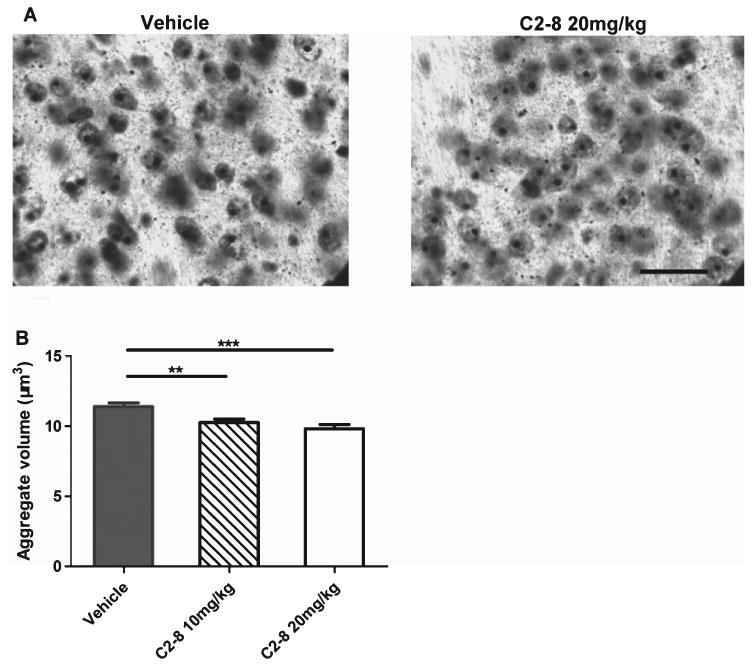
C2-8 treatment leads to significant reduction of mutant huntingtin aggregate volume in R6/2 mouse striatum. (A) Representative immunodetection of intraneuronal mutant huntingtin aggregates using EM48 antibody in the neostriatum of 13-week-old R6/2 mice with vehicle or 20 mg/kg C2-8 treatment (scale bar = 25 μm). (B) C2-8 treatment significantly reduced the striatal intranuclear aggregate volume in R6/2 mice (F(2,27) = 9.339, = 0.0008). Values represent mean ± S.E.M., n = 10; **P < 0.01, ***P < 0.001, one-way ANOVA followed by Tukey post-test.
Discussion
We have performed an independent preclinical study to evaluate the PK properties and therapeutic efficacy of C2-8 in the R6/2 mouse model of HD. Our study showed that both a single bolus IP injection and chronic administration of an experimental dose of C2-8 can lead to good brain availability, which is comparable to the previous study [2]. With regard to preclinical efficacy, we were unable to demonstrate any significant reduction of behavioral deficits in Rotarod and wire-hang tests with low or high dose C2-8 administration compared to vehicle treatment in R6/2 mice. Neuropathologically, we were able to reproduce the original finding [2] by demonstrating the lack of C2-8 effects on R6/2 brain weight or striatal volume phenotypes, and a significant reduction of nuclear mHTT aggregate volume with C2-8 administration. However, unlike the previous study, we did not reveal any improvement of striatal neuronal cell volume with C2-8 treatment in R6/2 mice.
Our study has been carefully designed to provide independent verification of the preclinical efficacy of C2-8 in R6/2 mouse model of HD [2], with specific methodological adjustments that were implemented upon consultation of independent groups contributing to the current study design (i.e. NINDS and the original C2-8 preclinical study group). One such methodological change, aimed to reduce the stress that may be elicited by the twice-daily oral gavage procedure, was to administer C2-8 via IP injection. We chose the IP dose based on communication with the original group of investigators who performed the C2-8 preclinical studies [1] and our independent PK study demonstrates that adequate levels of C2-8 can be achieved in the brain upon chronic IP delivery of C2-8, at least judged by the IC50 level of C2-8 in inhibiting of polyQ aggregation in PC12 cells (Fig. 1).
Our preclinical study design and reporting followed the rigorous standards recently suggested by NINDS [4]. We used a stringent randomization, blinding, and allocation concealment scheme. We also paid special attention to the sample size estimation, based on the power and effect size analysis of the original published study with the R6/2 model, to ensure the numbers of animals per group are enough to exclude the possibility of false negative conclusions. Furthermore, we have enacted high standards of data handling throughout our study, including predetermined stopping points, prevention of ad hoc exclusion of data, and the use of comprehensive statistics software (e.g. SPSS) to analyze data. Thus, our C2-8 replication study has been rigorously designed, interpreted and reported.
A key finding of the current study is our independent verification that chronic administration of C2-8 can lead to a significant reduction of mHTT aggregate volume in the striatum of R6/2 mice. The C2-8 compound was developed based on an initial high-throughput screening against mHTT-exon1 aggregation in a yeast polyQ model [1]. Subsequent validation and optimization of such aggregate-suppressing compounds in a PC12 cell model overexpressing mHTT-exon1 lead to the development of C2-8, which has an IC50 value of 50 nM in the PC12 assay. Importantly, the aggregate suppression activity of C2-8 has been consistently demonstrated in additional models, including an R6/2 hippocampal slice model [1] and R6/2 mice [2]. Our current study provides independent validation that chronic administration of C2-8 can lead to significant reduction of nuclear inclusion volumes in R6/2 mice. Furthermore, our finding also suggests that chronic IP administration of C2-8 can achieve the primary pharmacodynamic effect of this compound in vivo.
Prior study suggests that C2-8 administration in a Drosophila model or R6/2 transgenic mouse model of HD also ameliorates mHTT-exon1-induced toxicities, as measured by motor behavior improvement, reduction of striatal neuron atrophy in HD mice, or dose-dependent rescue of eye degeneration in the HD fly model [1, 2]. Our current study, despite showing a significant reduction of intranuclear mHTT aggregate volume, failed to demonstrate any significant improvement of motor behavior deficits or striatal neuron atrophy or volume loss in the R6/2 model. There are multiple reasons that may account for the differences in preclinical efficacy of C2-8 in our current study compared to the one by Chopra et al. [2]. These include the source of R6/2 mice (ours are bred from ovary transplanted females directly from Jackson Laboratory and the prior study is using a R6/2 colony maintained by the original investigators), the CAG repeat length of R6/2 mice (ours are 160 ± 10, while repeat lengths were not reported in the original study), potential differences in housing conditions at the different institutions, the different sources of C2-8 compound (ours was synthesized by Dr. Silverman's lab, while those used in the study by Chopra et al. were from Novartis), the potential differences in behavioral protocols [8], and the route of administration and dosages of C2-8 used in the study. With regard to the drug administration and dosage regimen, we have provided strong evidence that sufficient levels of C2-8 reached the brain (PK data) to reduce mHTT aggregation volumes in the R6/2 model (Fig. 6), therefore our study at least supports the interpretation that C2-8 mediated reduction of mHTT nuclear aggregate volume alone is insufficient to ameliorate disease phenotypes in R6/2 mice.
Our study raises several questions related to the strategy and preclinical study of mHTT aggregation reducing agents in HD therapy. First, polyQ-expanded mHTT and its proteolytic fragments are known to misfold and form small and large aggregates in affected neurons, but the precise toxic mHTT species in HD remain elusive. There is evidence to suggest the large nuclear and cytoplasmic inclusions may not be necessary for disease pathogenesis and may even be neuroprotective in HD and related polyQ disorders [9–11]. Our study that shows reduction of the size of NIs in R6/2 mice, but lack phenotypic improvement, is consistent with this view. Second, existing mouse models of HD, expressing either mHTT N-terminal polyglutamine fragments (R6/2) or full-length mHTT (e.g. murine Htt knock-in models, YAC128, or BACHD) exhibit different load and subcellular distribution of mHTT aggregates, as well as different severity of disease [12, 13]. Moreover, differences in genetic background of the murine models of HD could confer phenotype variability beyond these models. By large, mHTT fragment models have high levels of nuclear and cytoplasmic mHTT aggregates and more aggressive, often lethal disease, while full-length mHTT models have much lower aggregate load and slowly progressive behavioral and neurodegenerative phenotypes. Therefore, a compound that could not ameliorate the more aggressive disease phenotype in a fragment model may still have efficacy in full-length mHTT mouse models with milder disease phenotypes. To this end, it will be valuable to assess whether chronic administration of C2-8 in one of the full-length mHTT mouse models (e.g. BACHD, YAC128 or zQ175) [12–14] could lead to aggregate reduction and amelioration of behavioral deficits and brain atrophy in these mice. In general, the use of both a mHTT fragment model and a full-length mHTT mouse model, and using well-defined (either inbred of F1) genetic background for each model system, are considered an optimal strategy to evaluate the preclinical efficacy in HD mice [12]. Finally, our study supports the strategy to have independent research groups test a given preclinical compound or other therapeutic agents in mouse models of neurodegenerative disorders. Such a strategy is similar to the multi-center clinical trials in human patients and will help to identify phenotypic effects that are readily replicable and others that may be variable across labs. Such studies will help to standardize preclinical studies and to identify molecules/reagents with the most potent effects for further clinical development.
In conclusion, our independent preclinical study of C2-8 in R6/2 mice supports the role of this compound in reducing mHTT aggregation size in vivo, but is not able to support its role in ameliorating behavioral deficits in R6/2 mouse model of HD. Future preclinical studies using C2-8 in R6/2 and a full-length mHTT mouse model are needed to further determine the preclinical therapeutic potential of this compound for HD.
Supplementary Material
Acknowledgments
This research is supported by NINDS Supplement (3R01NS049501-07S1) to X. W. Yang. We would like to thank Drs. Margaret Sutherland and Shai D. Silberberg (NINDS) for support and discussion throughout the project. We also would like to thank Drs. Steven Hersch (MGH/Harvard), Robert Ferrante (Univ. Pittsburgh), and Aleksey Kazantsev (MGH/Harvard) for helpful discussion and suggestion related to the current study. Finally, we would like to thank Drs. Mohammad Khanfar and Richard B. Silverman (Northwestern Univ.) for providing us the C2-8 compound.
Footnotes
Conflict of Interest: The authors have no conflict of interest to report.
Supplementary Material: Supplementary figures can be found here: http://dx.doi.org/10.3233/JHD-130074.
References
- 1.Zhang X, Smith DL, Meriin AB, Engemann S, Russel DE, Roark M, Washington SL, Maxwell MM, Marsh JL, Thompson LM, Wanker EE, Young AB, Housman DE, Bates GP, Sherman MY, Kazantsev AG. A potent small molecule inhibits polyglutamine aggregation in Huntington's disease neurons and suppresses neurodegeneration in vivo. Proc Natl Acad Sci U S A. 2005;102:892–7. doi: 10.1073/pnas.0408936102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chopra V, Fox JH, Lieberman G, Dorsey K, Matson W, Waldmeier P, Housman DE, Kazantsev A, Young AB, Hersch S. A small-molecule therapeutic lead for Huntington's disease: Preclinical pharmacology and efficacy of C2-8 in the R6/2 transgenic mouse. Proc Natl Acad Sci U S A. 2007;104:16685–9. doi: 10.1073/pnas.0707842104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 4.Landis SC, Amara SG, Asadullah K, Austin CP, Blumenstein R, Bradley EW, Crystal RG, Darnell RB, Ferrante RJ, Fillit H, Finkelstein R, Fisher M, Gendelman HE, Golub RM, Goudreau JL, Gross RA, Gubitz AK, Hesterlee SE, Howells DW, Huguenard J, Kelner K, Koroshetz W, Krainc D, Lazic SE, Levine MS, Macleod MR, McCall JM, Moxley RT, 3rd, Narasimhan K, Noble LJ, Perrin S, Porter JD, Steward O, Unger E, Utz U, Silberberg SD. A call for transparent reporting to optimize the predictive value of preclinical research. Nature. 2012;490:187–91. doi: 10.1038/nature11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gray M, Shirasaki DI, Cepeda C, André VM, Wilburn B, Lu XH, Tao J, Yamazaki I, Li SH, Sun YE, Li XJ, Levine MS, Yang XW. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J Neurosci. 2008;28:6182–95. doi: 10.1523/JNEUROSCI.0857-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Menalled L, El-Khodor BF, Patry M, Suárez-Fariñas M, Orenstein SJ, Zahasky B, Leahy C, Wheeler V, Yang XW, MacDonald M, Morton AJ, Bates G, Leeds J, Park L, Howland D, Signer E, Tobin A, Brunner D. Systematic behavioral evaluation of Huntington's disease transgenic and knock-in mouse models. Neurobiol Dis. 2009;35:319–36. doi: 10.1016/j.nbd.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turner PV, Brabb T, Pekow C, Vasbinder MA. Administration of substances to laboratory animals: Routes of administration and factors to consider. J Am Assoc Lab Anim Sci. 2011;50:600–13. [PMC free article] [PubMed] [Google Scholar]
- 8.Lu XH, Fleming SM, Meurers B, Ackerson LC, Mortazavi F, Lo V, Hernandez D, Sulzer D, Jackson GR, Maidment NT, Chesselet MF, Yang XW. Bacterial artificial chromosome transgenic mice expressing a truncated mutant parkin exhibit age-dependent hypokinetic motor deficits, dopaminergic neuron degeneration, and accumulation of proteinase K-resistant alpha-synuclein. J Neurosci. 2009;29:1962–76. doi: 10.1523/JNEUROSCI.5351-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HT. Ataxin-1 nuclear localization and aggregation: Role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- 10.Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- 11.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–10. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 12.Truant R, Atwal RS, Desmond C, Munsie L, Tran T. Huntington's disease: Revisiting the aggregation hypothesis in polyglutamine neurodegenerative diseases. Febs J. 2008;275:4252–62. doi: 10.1111/j.1742-4658.2008.06561.x. [DOI] [PubMed] [Google Scholar]
- 13.Menalled LB, Kudwa AE, Miller S, Fitzpatrick J, Watson-Johnson J, Keating N, Ruiz M, Mushlin R, Alosio W, McConnell K, Connor D, Murphy C, Oakeshott S, Kwan M, Beltran J, Ghavami A, Brunner D, Park LC, Ramboz S, Howland D. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington's disease: zQ175. PLoS One. 2012;7:e49838. doi: 10.1371/journal.pone.0049838. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.