

Abstract
Background
Syntrichia caninervis is a desiccation-tolerant moss and the dominant bryophyte of the Biological Soil Crusts (BSCs) found in the Mojave and Gurbantunggut deserts. Next generation high throughput sequencing technologies offer an efficient and economic choice for characterizing non-model organism transcriptomes with little or no prior molecular information available.
Results
In this study, we employed next generation, high-throughput, Illumina RNA-Seq to analyze the poly-(A) + mRNA from hydrated, dehydrating and desiccated S. caninervis gametophores. Approximately 58.0 million paired-end short reads were obtained and 92,240 unigenes were assembled with an average size of 493 bp, N50 value of 662 bp and a total size of 45.48 Mbp. Sequence similarity searches against five public databases (NR, Swiss-Prot, COSMOSS, KEGG and COG) found 54,125 unigenes (58.7%) with significant similarity to an existing sequence (E-value ≤ 1e-5) and could be annotated. Gene Ontology (GO) annotation assigned 24,183 unigenes to the three GO terms: Biological Process, Cellular Component or Molecular Function. GO comparison between P. patens and S. caninervis demonstrated similar sequence enrichment across all three GO categories. 29,370 deduced polypeptide sequences were assigned Pfam domain information and categorized into 4,212 Pfam domains/families. Using the PlantTFDB, 778 unigenes were predicted to be involved in the regulation of transcription and were classified into 49 transcription factor families. Annotated unigenes were mapped to the KEGG pathways and further annotated using MapMan. Comparative genomics revealed that 44% of protein families are shared in common by S. caninervis, P. patens and Arabidopsis thaliana and that 80% are shared by both moss species.
Conclusions
This study is one of the first comprehensive transcriptome analyses of the moss S. caninervis. Our data extends our knowledge of bryophyte transcriptomes, provides an insight to plants adapted to the arid regions of central Asia, and continues the development of S. caninervis as a model for understanding the molecular aspects of desiccation-tolerance.
Keywords: Biological soil crust, Desiccation, Physcomitrella, Stress, Syntrichia, Transcriptome
Background
Biological soil crusts (BSCs) form at the surface of desert soils [1] and play an important role in arid and semi-arid ecosystems around the world [2,3]. BSCs are comprised of a number of organisms including cyanobacteria, green algae, fungi, liverworts, lichens and mosses [4]. Organisms found in BSCs have developed a suite of adaptive mechanisms that permit the avoidance of water loss and/or the survival of complete dehydration (i.e. desiccation) [3-7]. Desiccation-tolerant organisms have been observed among the three domains of life (Archaea, Bacteria and Eukarya) and the phenomenon has been extensively studied in cyanobacteria and plants [3,8,9]. Among land plants, desiccation-tolerance is rare in angiosperms and common in mosses [8-10]. More than 200 moss species have been experimentally verified to be desiccation-tolerant [9] and Tortula ruralis (=Syntrichia ruralis) is the model species for understanding the molecular aspects of vegetative desiccation-tolerance in mosses [6-11]. Desiccation-tolerant mosses are a key component of BSCs and Syntrichia ssp. are the dominant moss of both the Mojave [12] and Gurbantunggut deserts [13]. The Gurbantunggut desert (Xinjiang, China) is one of the major arid regions of central Asia with an area of 48.8 thousand km2, an average yearly precipitation of ~80 mm and mean annual pan evaporation of ~2607 mm [13-15]. Our research groups are interested in Syntrichia caninervis, a desiccation-tolerant moss and the dominant bryophyte of the Gurbantunggut desert BSC.
S. caninervis undergoes unpredictable cycles of dehydration and rehydration, and is frequently exposed to elevated temperature and higher amounts of UV-irradiation [16]. S. caninervis, like T. ruralis, can lose 90% of their protoplasmic water and subsequently rehydrate with no evidence of damage to the plasma membranes or chloroplasts [14,17]. A common feature of desiccation-tolerant mosses is the rapid recovery of photosynthesis and the rapid re-establishment of a positive carbon balance following rehydration [18,19]. Similar to other desiccation-tolerant mosses, rehydrated S. caninervis rapidly restores PSII activity in the context of enhanced Chl synthesis and the reorganization of PSII [16,19]. Following rehydration, S. caninervis has been shown to rapidly adjust leaf angle thereby maximizing net photosynthetic gain and minimizing water loss [20]. Successive cycles of rehydration/dehydration also have been shown to augment the surface wax content in S. caninervis leaves [21]. In addition to desiccation-tolerance, S. caninervis gametophores are extremely tolerant to heat and are able to regenerate following exposure to 120°C for 30 min [22].
The moss Physcomitrella patens is an important experimental model [23] and was the first published genome from a non-angiosperm, land plant [24]. The P. patens genome sequence allowed genome-wide analysis and demonstrated the utility of bryophyte genomes for the identification and characterization of plant genes [25-28]. However, P. patens is a mesic moss [9] and cannot survive desiccation [29]. As a key BSC species, S. caninervis is an attractive model for the study of desiccation tolerance and good candidate for -omic sequencing and analysis. Transcriptome sequencing is one of the most important tools for gene discovery and the identification of expression patterns [30-33]. Bryophyte transcriptomes has been generated and characterized from a number of species including Marchantia polymorpha[34], Pohlia nutans[35], T. ruralis[36,37] and P. patens[32,33]. Next generation high throughput sequencing technologies offer an efficient and economic choice for characterizing non-model organism transcriptomes with little or no prior molecular information available. Next generation sequencing platforms, such as Illumina, Roche 454 and SOLiD [38], have dramatically improved the efficiency of gene discovery and make it possible to detect low abundant transcripts [39,40].
In this study, we generated a global transcriptome assembly from S. caninervis using the Illumina HiSeq™ 2000 sequencing platform. Preliminary gene annotations of function, classification and metabolic pathways were obtained by searching public protein databases. We performed GO-based comparison with P. patens, comprehensive annotation of transcription factors and generated a MapMan metabolic pathway. Analysis of the de novo assembled transcriptome will provide a better understanding of the mechanisms associated with dehydration, the phenomena of vegetative desiccation-tolerance and identify a core set of abiotic stress-related transcripts.
Results and discussion
Illumina paired-end sequencing and de novo transcriptome assembly
To generate a broad survey of transcripts associated with the S. caninervis dehydration/rehydration cycle, a cDNA library was constructed from mRNAs extracted from various dehydration and rehydration stages. Raw Illumina sequencing reads were quality and adapter trimmed to yield a total of 58,031,432 paired-end short reads comprising of 4.64 Gb of nucleotide data from a single sequencing run. The Q20 percentage was 97.55% and the GC content was 55.09% when assessing the cleaned reads with no ambiguous bases. De novo transcriptome assembly was performed using Trinity [41] generating 162,865 contigs with an average length of 288 bp and the N50 value was 429 bp. After final paired-end read mapping and clustering, 92,240 unigenes were assembled with an average size of 493 bp, N50 of 662 bp and a total size of 45.48 Mbp (Table 1). The transcript abundance of unigenes was evaluated relative to sequence length and RPKM value (Figure 1A, 1B). All the assembled unigenes longer than or equal to 150 bp were retained for further analysis. An average sequencing depth of 51× for the final unigene assembly was achieved (Figure 1C).
Table 1.
Summary of sequence assembly after Illumina sequencing
| Sequenced reads |
Total number |
58,031,432 |
| Total read length (bp) |
4,642,514,580 |
|
| Reads length |
90 + 70 |
|
| GC content |
55.09% |
|
| Q20 persentage |
97.55% |
|
| Contigs |
Total number |
162,865 |
| Total length (bp) |
46,952,370 |
|
| Mean length (bp) |
288 |
|
| Contig N50 (bp) |
429 |
|
| Unigenes | Total number |
92,240 |
| Total length (bp) |
45,480,162 |
|
| Mean length (bp) |
493 |
|
| Unigene N50 (bp) |
662 |
|
| Minimum length (bp) |
150 |
|
| Maximum length (bp) | 4,909 |
Figure 1.
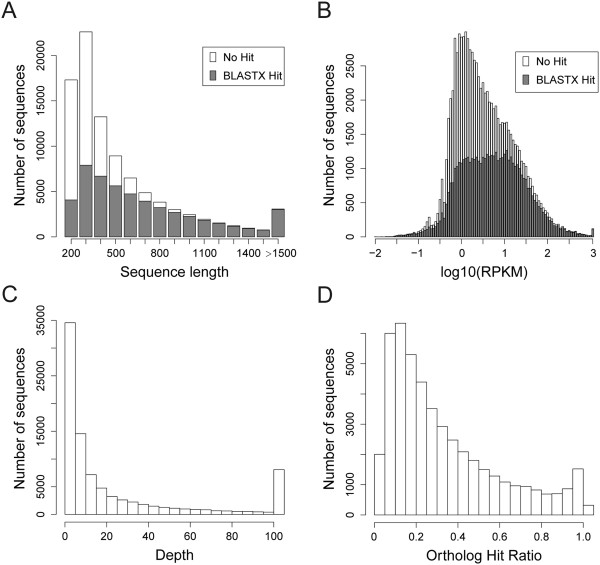
Overview of the S. caninervis transcriptome sequencing and assembly. (A) Histogram of the length of unigenes that returned BLASX hits in public protein databases and the sequences with no hit. (B) Distribution of unigene RPKM values for identified protein coding sequences and putative non-coding sequences. (C) Histogram of the average read depth for unigenes. Sequencing depth values above 100× was binned. (D) Ortholog hit ratio analysis for S. caninervis unigene sequences.
Using BLASTX, we compared the number of base pairs in the unigene hit region to the total length of the open reading frame [42] from the best-matching protein sequence from P. patens[43]. The overall distribution of the ortholog hit ratio (OHR) for the assembled S. caninervis unigenes is depicted in Figure 1D. A total of 47,559 S. caninervis unigenes returned BLASTX hits with P. patens proteins with an OHR average of 33%. 4,116 unigenes (8.7%) had an OHR ≥ 0.8 and 10,762 unigenes (22.6%) had an OHR ≥ 0.5. Using this metric, more than one fifth of the putative P. patens orthologs captured within the S. caninervis transcriptome covered at least 50% of the predicted ORF. Similar analysis of Coelacanthiformes [44], Cypriniformes [45], Hemiptera [46] and Lepidoptera [42] have obtained ortholog hit ratios that range from 35-to- 72%. Similar to BLASTX hit ratios (Figure 1A), the ortholog hit ratio is also strongly governed by the length of unigenes. For unigenes longer than 1,500 bp average OHR = 0.68 while for unigenes shorter than 200 bp average OHR = 0.14. This data suggests that the sequencing data is suitable for further analysis.
Annotation of the S. caninervis transcriptome
For annotation and classification of the assembled unigenes, we conducted sequence similarity searches against five public protein databases: NCBI nr protein database (NR), Swiss-Prot, COSMOSS v1.6 (P. patens proteins) [43], Kyoto Encyclopedia of Genes and Genomes (KEGG) and Clusters of Orthologous Groups (COG). Of all the 92,240 unigenes with length ≥ 150 bp, 54,125 (58.7%) revealed significant similarity (E-value ≤ 1e-5) with existing records in at least one of the five databases (Figure 2 and Additional file 1). Among the annotated unigenes, 51,938 (96%) unigenes returned a positive BLASTX hit with deduced polypeptides within either the NR or Swiss-Prot database. However, 38,115 unigenes (41.3%) have no significant match with any deposited sequence and are considered unknowns. Transcriptome annotation in other bryophyte species have obtained similar results with the percentage of unknown sequences greater than 40%: T. ruralis (40.3%) [36], P. patens (42%) [47], Marchantia polymorpha (43%) [34] and Pohlia nutans (82%) [35]. 14, 057 S. caninervis transcripts had significant similarity to deduced polypeptides in each of the five databases. 11,663 had significant similarity to sequences from both the NR and moss-specific COSMOSS databases. 4,884 unigenes had significant similarity to a single database: COSMOSS (1984 unigenes), NR (1672 unigenes), Swiss-Prot (1116 unigenes), KEGG (45 unigenes) and COG (67 unigenes).
Figure 2.
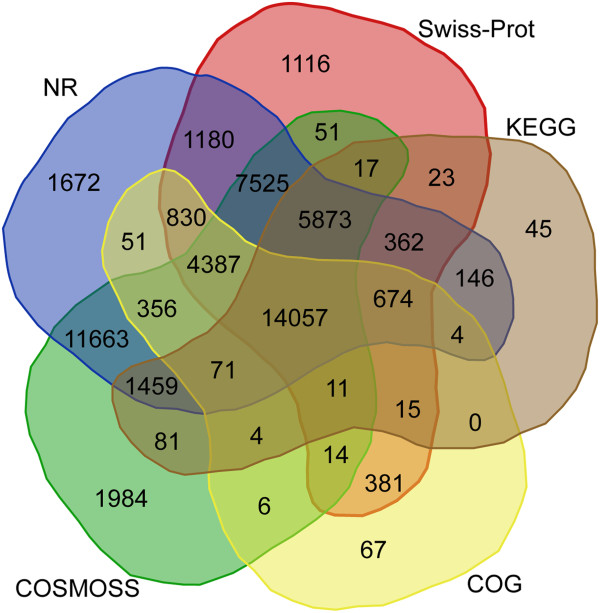
Venn diagram showing the BLASTX results of the S. caninervis transcriptome against five protein databases. Using BLASTX search, de novo reconstructed unigene sequences were queried against the following public databases: NCBI-NR, Swiss-Prot, COSMOSS, KEGG and COG. The number of transcripts that have significant hits (E-value ≤ 1e-5) against the five databases is shown in each intersection of the Venn diagram.
Sorting the BLASTX hits by species revealed the top three species to be P. patens (11,136), A. thaliana (8733) and O. sativa (8173) (Figure 3). P. patens accounted for 22% of the identified unigenes and the three top-hit species accounted for more than 55% of the identified unigenes. The remaining 45% is distributed among members of the algae, ferns and angiosperms. It is important to note that several stress-related sequences have been identified and extensively studied in the closely related species T. ruralis[36]; however, due to the limited number of deduced polypeptide sequences present in the public databases, T. ruralis does not appear within the top hit species. These results indicate that a large number of moss-specific genes are present in the S. caninervis transcriptome (Figures 2 and 3). The presence of lineage-specific genes was a striking and dominant feature revealed in the reannotation of the P. patens genome (v1.6) [43]. 48% of all P. patens loci were clustered into P. patens-only clusters and about 22% (7,169) of all loci within P. patens-only clusters have no detectable homolog in any databases [43]. Among the annotated unigenes, 20,928 (40.1%) unigenes were assigned to the 25 COG categories (Table 2). Since some transcripts could be assigned to multiple COG functional categories, 39,756 total functional annotations were produced and all identified transcripts were grouped into one of the COG categories. “General function prediction”, “translation, ribosomal structure and biogenesis” were the two most represented categories (24% of all annotations), followed by “cell wall/membrane/envelope biogenesis” (9%), “transcription” (7%), “replication, recombination and repair” (7%) and “lipid transport and metabolism” (6%). Among the transcripts classified into specific COG functional categories, 2,618 unigenes (13%) were identified as “function unknown”. We hypothesize that these unknown and unclassified (Table 2) transcripts might represent species- and/or lineage-specific genes for adaptive innovations.
Figure 3.
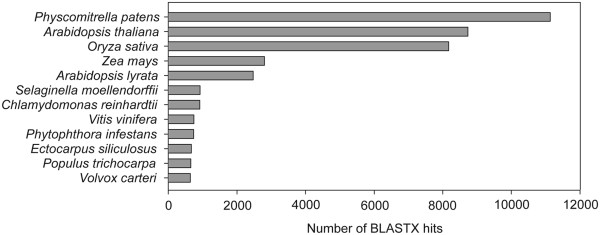
Species distribution of the top BLASTX hits obtained using the S. caninervis transcriptome. Top scoring BLASTX hits against the NCBI-NR protein database are depicted. The number of BLAST hits per species is shown on the x-axis. The 12 most represented species with proportions of more than 1% are shown in this graph.
Table 2.
COG functional classification of S. caninervis transcripts
| Description | Code | Unigenes (n) |
|---|---|---|
|
Information storage and processing |
|
|
| RNA processing and modification |
A |
125 |
| Chromatin structure and dynamics |
B |
342 |
| Translation, ribosomal structure and biogenesis |
J |
4,202 |
| Transcription |
K |
2,927 |
| Replication, recombination and repair |
L |
2,876 |
|
Cellular processing and signaling |
|
|
| Cell cycle control, cell division, chromosome partitioning |
D |
1,812 |
| Cell wall/membrane/envelope biogenesis |
M |
1,774 |
| Cell motility |
N |
348 |
| Posttranslational modification, protein turnover, chaperones |
O |
2,825 |
| Signal transduction mechanisms |
T |
2,069 |
| Intracellular trafficking, secretion, and vesicular transport |
U |
1,249 |
| Defense mechanisms |
V |
477 |
| Extracellular structures |
W |
19 |
| Nuclear structure |
Y |
9 |
| Cytoskeleton |
Z |
496 |
|
Metabolism |
|
|
| Energy production and conversion |
C |
1,782 |
| Amino acid transport and metabolism |
E |
1,584 |
| Nucleotide transport and metabolism |
F |
447 |
| Carbohydrate transport and metabolism |
G |
2,573 |
| Coenzyme transport and metabolism |
H |
685 |
| Lipid transport and metabolism |
I |
1,247 |
| Inorganic ion transport and metabolism |
P |
1,126 |
| Secondary metabolites biosynthesis, transport and catabolism |
Q |
932 |
|
Poorly characterized |
|
|
| General function prediction only |
R |
5,211 |
| Function unknown | S | 2,619 |
n = number of unigenes.
Gene ontology annotation and comparison with the P. patens genome
Gene ontology (GO) terms were assigned to each S. caninervis transcripts based on its best BLASTX hits in the NR database using the Blast2GO pipeline [48]. Of the 50,310 unigenes with NR annotation, 24,183 unigenes (48%) were assigned to one of three GO terms: Biological Process, Cellular Component or Molecular Function (Figure 4 and Additional file 2). Previous GO annotation of the P. patens genome assigned a functional annotation to 41% of the transcripts [49] and a subsequent reannotation increased the functional annotation to 58% [43]. GO comparison between P. patens and S. caninervis demonstrated similar sequence enrichment across all three GO categories. Transcripts were enriched for both P. patens and S. caninervis within the Cellular Component category (“cell”, “cell part” and “organelle”), the Molecular Function category (“binding” and “catalytic”) and the Biological Process category (“metabolic processes” and “cellular processes”, “response to stimulus”, “localization” and “establishment of localization”). Although enriched in both moss species, the “response to stimulus” category was significantly more enhanced in S. caninervis relative to P. patens. (2,775 vs. 1,335 unigenes) (Figure 4 and Additional file 2). Detailed analysis of the “response to stimulus” category revealed significant differences between the S. caninervis transcriptome compared with P. patens genome. Unigenes related to “response to radiation”, “osmotic stress”, “detection of abiotic stimulus” and “response to starvation” were significantly more represented in the S. caninervis transcriptome. Notably, the “translation regulator” category is significantly more represented in S. caninervis, while the “transcription regulator” category is less significantly represented.
Figure 4.
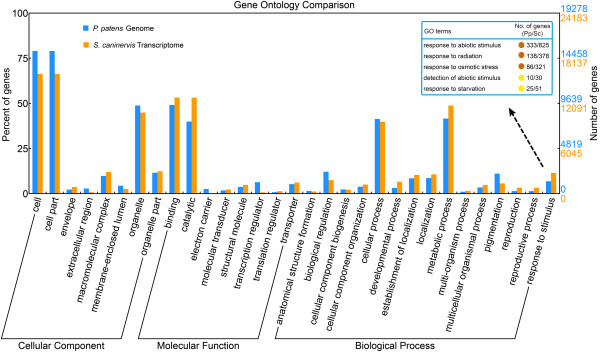
Gene ontology classification and comparison between the mosses P. patens and S. caninervis. Gene ontology annotation results of the genes from the P. patens genome and S. caninervis transcriptome were mapped to categories within the second level of GO terms. GO terms that contain more than 1% of total genes were included in this graph. Abiotic stress related subcategories of the term “response to stimulus” were shown in the box.
Additionally, a hypergeometric statistical test was employed to identify over-represented (p-value < 0.05) GO categories present in the most abundantly expressed genes (RPKM ≥ 100) (Figure 1B). GO enrichment analysis on the most abundant unigenes demonstrated transcripts clustered in the “response to stress”, “abiotic stimulus”, “lipid metabolic process”, “generation of precursor metabolites and energy”, “membrane”, “thylakoid”, “plastid” and “ribosome” were significantly overrepresented as compared to the entire S. caninervis transcriptome (Additional file 3). Studies into the response to desiccation and rehydration of desiccation-tolerant mosses such as S. caninervis and T. ruralis have proposed that a constitutive protective mechanism and an active rehydration-induced recovery mechanism is employed [14,17]. Annotation of the S. caninervis transcriptome supports the hypothesis that transcripts related to transcriptional gene control, response to abiotic stress, photosynthesis, membrane integrity and translational regulation play an important role rehydration/dehydration cycle.
Protein family assignment and transcription factor prediction
Identifying conserved domains present within a deduced polypeptide can provide insight into the function, regulation and/or localization of the predicted protein. An ORF was predicted for each S. caninervis unigene (see Methods) and the deduced polypeptide sequences were queried for the presence of protein motifs using the Pfam database [50]. 29,370 deduced polypeptide sequences were assigned Pfam domain information and categorized into 4,212 Pfam domains/families (Additional file 4). Pfam domains/families were ranked according to the frequency of occurrence of S. caninervis transcripts. The top 10 abundant domains/families are depicted in Figure 5A. The majority of the domains/families contained a small number of transcripts (i.e. 10 or less) and ranged from 1 to more than 700 transcripts per family (Figure 5B). “Protein kinase”, “protein kinase-tyrosine”, “WD40”, “leucine rich repeat”, “P450”, “ABC-containing” and “HSP70” are among the most abundant domain/families in S. caninervis. Protein kinases are known to function as an on/off switch and play a role in a multitude of cellular processes, such as metabolism and cell division [51]. WD40-repeat proteins are known to coordinate multi-protein complex assemblies and the proteins containing this domain are implicated in a variety of functions ranging from signal transduction and transcription regulation to cell cycle control, autophagy and apoptosis [52]. Proteins with leucine-rich repeat domains are frequently involved in the formation of protein–protein interactions [53]. P450 domain containing enzymes incorporate oxygen into small lipophilic compounds and play an important role in the biosynthesis of biopolymers and defense chemicals [54]. ABC-containing peptides are membrane-localized transport proteins that mediate the exchange of a broad range of molecules including metals, hormones lipids and secondary metabolites [55,56].
Figure 5.
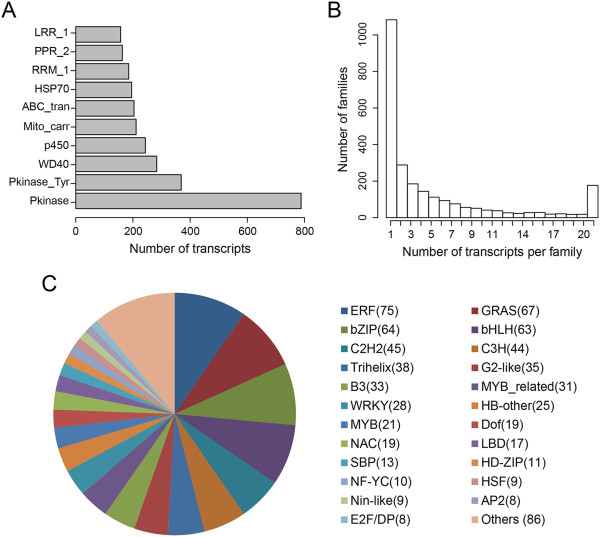
Protein families and transcription factors in the S. caninervis transcriptome. (A) The 10 most abundant protein families in the S. caninervis transcriptome. (B) Relationship between the occurrence of S. caninervis transcripts and the number of Pfam families in the S. caninervis transcriptome. (C) The 23 most abundant predicted transcription factor protein families. The number of members in each TF family is presented within the brackets. A total of 778 TFs were predicted and classified into 49 TF families (Additional file 1).
In order to more fully understand gene control and regulation in S. caninervis, all transcription factors were predicted according to the family assignment rules illustrated in PlantTFDB [57]. 778 unigenes were predicted to be involved in the regulation of transcription and were classified into 49 transcription factor families (Figure 5C and Additional file 1). The ERF (AP2-domain) transcription factor family is the most abundant TF families in S. caninervis and similar results were observed in P. patens as recorded in PlantTFDB [57]. AP2/ERF proteins have important functions in the transcriptional regulation of a variety of biological processes related to growth and development, abiotic stress tolerance and response(s) to a variety of environmental stimuli [58]. The remainder of the top 10 TFs are: GRAS, bZIP, bHLH, C2H2, C3H, Trihelix, G2-like, B3 and MYB-related. Transcription factor families with low gene number may play a unique role in gene control. S. caninervis contains a single VOZ (Vascular plant One Zinc finger protein) transcript and two CO (CONSTANS) transcripts. VOZ was characterized in Arabidopsis and homologues have been identified in the moss P. patens[59] but not in the liverwort M. polymorpha[34]. In Arabidopsis, VOZ genes have been implicated in the regulation of flowering time [60] and respond to both biotic and abiotic stress [61]. CO was also characterized in Arabidopsis[62] and plays an important role in photoperiod-regulated flowering. CO and CO-like genes are plant-specific and homologues have been identified in all flowering plants as well as in P. patens[63,64].
Metabolic pathways
To survey genes involved in important metabolic pathways, annotated S. caninervis transcripts were mapped to the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. A total of 22,842 unigenes (44%) were mapped to 119 KEGG metabolic pathway maps (Additional file 5). Among the 119 KEGG pathways, the pathways most represented by unigenes were “metabolic pathways” (5,730; 25%), “biosynthesis of secondary metabolites” (2,674; 12%), “ribosome” (1,891; 8%) and “plant-pathogen interaction” (1,310; 6%). These results are consistent with the hypothesis that moss transcriptomes reflect an enhanced versatility and mediate a variety of alternative metabolic pathways not observed in tracheophytes [25]. Examination of the colored KEGG maps demonstrated that we have captured almost all the genes required for the citrate cycle, photosynthesis, carbon fixation in photosynthetic organisms, flavonoid biosynthesis and the biosynthesis of unsaturated fatty acids. In addition, S. caninervis transcripts were annotated with MapMan bins and subsequently classified according to their major metabolic pathways and normalized expression level (Additional file 6). MapMan analysis showed that most biochemical pathways have been captured and demonstrated the diversity and completeness of the transcriptome. Consistent with the GO classification, the MapMan functional classification of metabolism showed high numbers and high expression levels of transcripts involved in lipids metabolism and the light reactions of photosynthesis.
Orthologous relationship with model plants
Prior to analysis of Plant Ortholog Group (Plant OG) membership, proteins inferred from the S. caninervis transcriptome were clustered to construct a reference protein dataset comprised of 41,530 representative protein sequences. Clustering reduces the presence of redundant sequences and base miscall errors, but can also eliminate highly similar homeoalleles. To further investigate proteomic similarity with P. patens and A. thaliana, we clustered the three proteomes into protein families using OrthoMCL [65] (Figure 6A). Most (~80%) of the OrthoMCL-defined protein families in S. caninervis are in common with P. patens. Approximately 44% of the protein families from the tracheophyte, Arabidopsis, are in common with the two bryophytes. 65% of the protein families are present in Arabidopsis, 54% present in P. patens and 45% are present in S. caninervis, and 35% are found in common between P. patens and S. caninervis. S. caninervis has significantly more OGs in common with P. patens as compared to Arabidopsis. BLASTP comparison of the deduced polypeptides between S. caninervis and P. patens (Sc-Pp), and S. caninervis and Arabidopsis (Sc-At) is presented in Figure 6B. In both cases the sequence identity ranged from 20% to 100%. The Sc-Pp mean is 71% and the Sc-At mean is 56%. S. caninervis has not only significantly more OGs in common with P. patens but also greater sequence identity with P. patens as compared to Arabidopsis.
Figure 6.
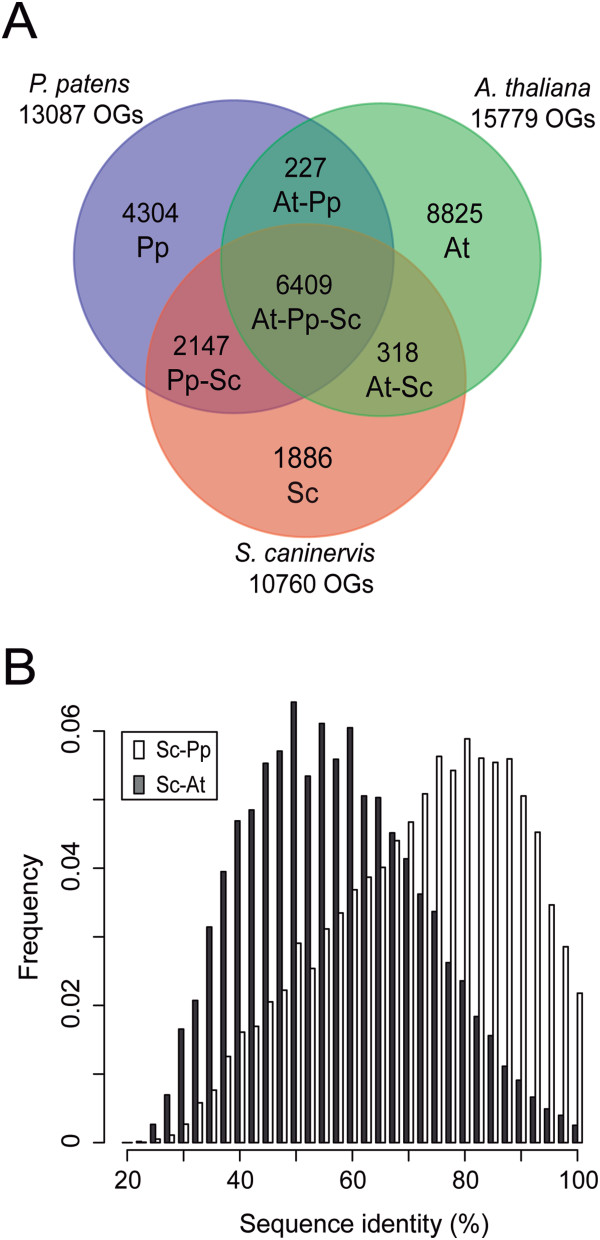
Detection of homologous genes in mosses and Arabidopsis: comparison of orthologous gene groups and protein sequence identity. (A) OrthoMCL was used to identify orthologous groups (OGs) among S. caninervis (Sc), A. thaliana (At) and P. patens (Pp). (B) Density plot of the protein identity between S. caninervis and the model plants.
The data identifies several categories of transcripts: 1) sequences shared in common between mosses and an angiosperm, 2) sequences found in Arabidopsis, 3) sequences found in mosses, 4) sequences found in P. patens and 5) sequences found in S. caninervis. Our particular interest in desert plants led to a comparison of the S. caninervis and T. ruralis transcriptomes. T. ruralis is a model organism for studying stress-responsive gene control and a large number of cDNAs/ESTs/transcripts have been isolated and characterized [36,37]. We have mined the current S. caninervis transcriptomic database and identified homologues (>95% identical at the amino acid level) to many previously characterized T. ruralis transcripts including Tr155[66] and Tr288[67], Elipa and Elipb[68], ALDH7B6[69], ALDH21A1[70], Vac1[71], TrDr1 and TrDr2[72] and the moss-retained TrDr3[73]. Identification of “desiccation related” homologues in both T. ruralis and S. caninervis support our long-standing hypothesis that desiccation-tolerant mosses proffer novel genes and gene products [37]. Further research is required to confirm the features and functions of these putative moss-specific and stress-tolerance related genes.
Conclusions
In this study we present a global characterization of the S. caninervis transcriptome using next-generation, RNA-Seq technology. Using high-throughput sequencing technology, we have captured most of the transcripts expressed in moss gametophores during rehydration and desiccation. De novo transcriptome assembly generated 92,240 unigene sequences. GO annotation of the S. caninervis transcriptome and comparison with the P. patens genome demonstrates the enrichment of sequences related to transcriptional gene control, response to abiotic stress, and translational regulation. Our data extends our knowledge of bryophyte transcriptomes, provides an insight to plants adapted to the arid regions of central Asia, and continues the development of S. caninervis as a model for understanding the molecular aspects in desiccation-tolerance.
Methods
Sample collection, cDNA library construction and Illumina sequencing
S. caninervis gametophytes were collected from the Gurbantunggut Desert of Xinjiang Uygur Autonomous Region of China (44° 32′ 30″ N, 88° 6′ 42″ E) and harvested and stored as described previously [14]. Since 2003, this sand dune has been identified as a permanent research site. In this study, patches of S. caninervis inhabiting the biological soil crusts were collected in petri dishes and stored in an air-dried state for at least 1 week at room temperature. All samples used in the experiment were collected from the same site within a 10 m2 plot. Voucher specimens are maintained in the Department of Plant Biology, Southern Illinois University (Carbondale, IL). To obtain a comprehensive transcriptome assembly of S. caninervis transcripts during the dehydration-rehydration process, an equal mixture of total RNAs isolated from various dehydration and rehydration time points were used to construct the sequencing library. Dried gametophyte tissue samples were placed on filter paper in petri dishes and rehydrated using purified water for 24 hours. Gametophores were harvested after 24 h of rehydration. Gametophores were subsequently allowed to dry on an open bench (ca 25°C, RH =25%) [74] and samples (i.e. 100 mg FW) were harvested at 0.5, 1, 1.5, 2, 4, 6, 8, 10, 12 and 24 h.
Total RNAs isolated from all samples were quality and purity assessed and pooled together for RNA-Seq [75]. Total RNAs were extracted from S. caninervis gametophyte tissue samples using Trizol Reagent (Invitrogen, USA). The resulting samples were treated with DNase I to remove any genomic DNAs. RNAs were quantified using an Agilent 2100 Bioanalyzer and checked for RNA integrity using denaturing agarose gel electrophoresis. The cDNA library was created and sequenced according to the manufacturer’s instructions (Illumina) and sequencing was performed at Beijing Genome Institute (BGI) in Shenzhen, China. Briefly, beads with Oligo(dT) were used to isolate poly(A) + mRNA after total RNA was obtained. Fragmentation buffer was added for interrupting mRNA into short fragments. First-strand cDNA was synthesized using these short fragments as templates, along with reverse transcriptase and random hexamer primer. And the second-strand cDNA was synthesized using buffer, dNTPs, RNaseH and DNA polymerase I. The resulting double stranded cDNA was then subjected to end-repair using T4 DNA polymerase, DNA polymerase I Klenow fragment, and T4 polynucleotide kinase, and ligated to adapters using T4 DNA ligase. Short fragments were purified with QIAquick PCR purification kit and eluted with EB buffer. After agarose gel electrophoresis, the suitable fragments (200 ± 50 bp) were selected as templates for bridged PCR amplification. The Illumina cBOT was used for cluster generation following the manufacturer’s instructions, and the clustered flow cell was loaded onto the sequencing machine. cDNA library products were sequenced on an Illumina HiSeq™ 2000 system.
Data filtering and de novo transcriptome assembly
Before assembly, clean reads of high quality were generated from the raw reads by removing adapter sequences, low-quality reads with ambiguous bases (‘N’), and reads with more than 10% of Q-values < 20 bases. All subsequent analyses were based on the clean reads. The quality of clean reads was further evaluated using NGS QC toolkit [76]. De novo transcriptome assembly was performed using Trinity [41] (release 2011-07-13). At the first step all clean reads were randomly clipped into 25-mers for assembly using de Bruijn graph algorithm, and we got the longest assembled fragments called contigs. Then paired-end reads were mapped back to contigs, using paired reads it is able to detect contigs from the same transcript as well as merge them. Finally, we obtained the sequences which cannot be extended on either end, and the resulting sequences were defined as unigenes. Only the unigene sequences longer than or equal to 150 bp were reserved for further analysis. Following assembly, unigenes were assigned an RPKM value [39] based on the number of uniquely mapped reads aligning to each unigene using SOAP [77] software (release 2.21).
Evaluation of transcripts integrity
The “Ortholog Hit Ratio” method proposed by O’Neil et al. was used to determine how closely our sequences approached full-length transcripts [42]. The “ortholog hit ratio” metric computes the length of the putative coding region found in the newly assembled sequence divided by the full length of its top BLAST hit. Thus, an ortholog hit ratio of 1.0 may imply that a transcript has been assembled to its true full length. P. patens is the most well-studied bryophyte with a complete reference genome and comprehensively annotated gene sequences [43]. A blastx search against protein sequences inferred from P. patens genome (COSMOSS v1.6) was used to evaluate the quality of our assembled transcripts. For the purposes of this study, we consider each unigene and its best P. patens BLASTX hit with an E-value ≤ 1e-5 to be a putative ortholog. All top hits for S. caninervis transcripts were parsed and used to calculate ortholog hit ratios. If there are relative insertions in best hit P. patens proteins, this will tend to lower ortholog hit ratios, while ratios bigger than 1.0 likely indicate insertions in the query sequence relative to its top BLAST hit.
Gene annotation and classification
For annotation and classification of the transcriptome, all assembled unigene sequences were subjected to BLASTX (E-value ≤ 1e-5) as queries to search against the following protein databases: NCBI nr, Swiss-Prot, COSMOSS, KEGG and COG database. Because the unigene sequences have no annotated open reading frames, proteins with highest ranks in BLASTX results were taken to decide the direction and coding region of the assembled transcripts. The best potential coding region of unigenes with no hit in these protein databases were predicted using ESTScan [78] with parameters trained on the annotated unigenes. Subsequently, all predicted coding regions from unigenes were translated into protein sequences using the standard codon table.
According to the NR annotation results, the top BLASTX hits were used to identify putative homologous proteins and annotate each unigene sequence with gene ontology (GO) terms using Blast2GO [48] program. The GO annotation information of P. patens genome was downloaded from the Physcomitrella patens computational biology resource site (http://www.cosmoss.org). GO classification and comparison with P. patens was performed using WEGO [79] according to molecular function, biological process, and cellular component ontologies.
Gene ontology enrichment analysis for the selected gene set with RPKM values [39] above 100 was performed and visualized in Cytoscape (v2.8.3). The cytoscape plugin BiNGO (v2.44) [80] was customized with the S. caninervis transcriptome GO annotation results and was then used to perform the hypergeometric statistical test of significance (corrected p-value < 0.05) to assess GO term enrichment. All GO-slim terms found within the gene list more often than expected by chance were highlighted in BiNGO. To adjust for multiple hypotheses testing, a Benjamini & Hochberg False Discovery Rate (FDR) correction was performed.
For Pfam domain/family annotation, the predicted protein sequences were submitted to search against HMM profiles contained in the Pfam database (version 27.0) [50] using HMMER v3.0 [81,82]. To resolve complex overlapping protein domains, only the most significant (lowest E-value) match within the clan was reported. The perl script PfamScan.pl downloaded from the Pfam database ftp server was used for the annotation and Linux tools was applied to parse the annotation results.
The S. caninervis transcription factors were predicted using PlantTFDB v3.0 [57]. The putative transcription factors in S. caninervis were initially identified, including proteins that contain a DNA binding domain (inferred from Pfam annotation) or gave a positive Blastp hit (E-value ≤ 1e-5) with recorded P. patens or A. thaliana transcription factors. Deduced polypeptide sequences were then submitted to the PlantTFDB prediction server (planttfdb.cbi.pku.edu.cn/prediction.php) for further classification and validation.
To investigate the metabolic pathway annotation of unigenes, unigenes were aligned using the KEGG database [83], enzyme codes were acquired for each sequence and EC accession numbers were used to color and retrieve the corresponding KEGG pathway maps. Scrutiny of transcript diversity and abundance was performed with MapMan [84,85]. The deduced polypeptide sequences were submitted to Mercator webserver [86] to classify them into MapMan functional plant categories. For color-coded representation (heat map) in MapMan, the log2 transformed of the RPKM-normalized expression counts was used. Deduced polypeptide sequences shorter than 100 amino acids or belonging to the least 5% abundant expressed transcripts (RPKM < 0.17) were not used to generate the MapMan metabolic pathway maps.
For comparison of gene models with A. thaliana and P. patens, protein-coding gene models in TAIR10 (http://www.arabidopsis.org) and COSMOSS v1.6 (http://www.cosmoss.org) were used. Prior to analysis of PlantOG (Plant Ortholog Groups) memberships, proteins inferred from the S. caninervis transcriptome were subjected to CD-HIT [87] (identity ≥0.95 and coverage ≥0.9) to eliminate redundancy. All protein sequences shorter than 50 amino acids were discarded. Protein sequences were clustered using OrthoMCL [65].
Abbreviations
BSC: Biological soil crust; GO: Gene ontology; RPKM: Reads per kilobase per million mapped reads; TF: Transcription factor.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BG performed the bioinformatics analyses and drafted the manuscript. DZ conceived the study and provided financial support for the project. XL contributed to the data analysis. HY participated in plant preparations. AW contributed to data interpretation and revised the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Comprehensive annotation of S. caninervis unigenes. Including BLASTX search hits in NCBI-Nr, Swiss-Prot, COG, KEGG and COSMOSS databases and all annotated transcription factors using PlantTFDB.
GO comparison results of P. patens genome and S. caninervis transcriptome.
Enrichment network based on GO of the most abundantly expressed transcripts (RPKM > 100) in the transcriptome. Significantly overrepresented (p-value < 0.05) GO terms based on GO-slim were visualized in Cytoscape. The node size is in proportional to the number of unigenes in the GO category. The color represents the enrichment significance. Nodes with white color are not enriched but show the essential hierarchical relationship among the enriched GO-slim terms.
Pfam domain/family annotation and statistics of S. caninervis sequences (2 sheets).
Statistics of KEGG pathways annotated in S. caninervis transcriptome.
MapMan overview of S. caninervis cellular metabolism. Individual assembled transcripts are represented by colored squares. The color code scale is based on the log2 of the RPKM values of each unigene. The greater intensity of red is associated with higher transcript abundance. Green highlighted metabolic pathways are biosynthetic while pink highlighted metabolic pathways are degradative.
Contributor Information
Bei Gao, Email: gaobei06@gmail.com.
Daoyuan Zhang, Email: zhangdy@ms.xjb.ac.cn.
Xiaoshuang Li, Email: xiaoshuangliok@sina.com.
Honglan Yang, Email: yanghonglan@ms.xjb.ac.cn.
Andrew J Wood, Email: wood@plant.siu.edu.
Acknowledgements
This research was supported by the National Basic Research Program of China (2014CB954203) and the National Natural Science Foundation of China (U1170304).
References
- Belnap J. The world at your feet: desert biological soil crusts. Front Ecol Environ. 2003;1(4):181–189. [Google Scholar]
- Pointing SB, Belnap J. Microbial colonization and controls in dryland systems. Nat Rev Microbiol. 2012;10(8):551–562. doi: 10.1038/nrmicro2831. [DOI] [PubMed] [Google Scholar]
- Rajeev L, da Rocha UN, Klitgord N, Luning EG, Fortney J, Axen SD, Shih PM, Bouskill NJ, Bowen BP, Kerfeld CA, Garcia-Pichel F, Brodie EL, Northen TR, Mukhopadhyay A. Dynamic cyanobacterial response to hydration and dehydration in a desert biological soil crust. ISME J. 2013;7(11):2178–2191. doi: 10.1038/ismej.2013.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belnap J, Büdel B, Lange OL. In: Biological Soil Crusts: Structure, Function, and Management, vol. 150. Belnap J, Lange O, editor. Berlin Heidelberg: Springer; 2003. Biological soil crusts: characteristics and distribution; pp. 3–30. [Google Scholar]
- Belnap J, Gillette DA. Vulnerability of desert biological soil crusts to wind erosion: the influences of crust development, soil texture, and disturbance. J Arid Environ. 1998;39(2):133–142. [Google Scholar]
- Oliver MJ, Velten J, Mishler BD. Desiccation tolerance in bryophytes: a reflection of the primitive strategy for plant survival in dehydrating habitats? Integr Comp Biol. 2005;45(5):788–799. doi: 10.1093/icb/45.5.788. [DOI] [PubMed] [Google Scholar]
- Proctor MCF, Oliver MJ, Wood AJ, Alpert P, Stark LR, Cleavitt NL, Mishler BD. Desiccation-tolerance in bryophytes: a review. Bryologist. 2007;110(4):595–621. [Google Scholar]
- Oliver M, Tuba Z, Mishler B. The evolution of vegetative desiccation tolerance in land plants. Plant Ecol. 2000;151(1):85–100. [Google Scholar]
- Wood AJ. The nature and distribution of vegetative desiccation-tolerance in hornworts, liverworts and mosses. Bryologist. 2007;110(2):163–177. [Google Scholar]
- Bewley JD. Physiological aspects of desiccation tolerance. Annu Rev Plant Physiol. 1979;30(1):195–238. [Google Scholar]
- Oliver MJ, Derek Bewley J. Horticultural Reviews. Oxford, UK: John Wiley & Sons, Inc; 2010. Desiccation-tolerance of plant tissues: a mechanistic overview; pp. 171–213. [Google Scholar]
- Stark LR, Brinda JC, McLetchie DN, Oliver MJ. Extended periods of hydration Do Not elicit dehardening to desiccation tolerance in regeneration trials of the moss Syntrichia caninervis. Int J Plant Sci. 2012;173(4):333–343. [Google Scholar]
- Zhang YM, Chen J, Wang L, Wang XQ, Gu ZH. The spatial distribution patterns of biological soil crusts in the Gurbantunggut Desert, Northern Xinjiang, China. J Arid Environ. 2007;68(4):599–610. [Google Scholar]
- Wu N, Zhang YM, Downing A, Zhang J, Yang C. Membrane stability of the desert moss Syntrichia caninervis Mitt. during desiccation and rehydration. J Bryol. 2012;34(1):1–8. [Google Scholar]
- Zheng Y, Xu M, Zhao J, Zhang B, Bei S, Hao L. Morphological adaptations to drought and reproductive strategy of the moss Syntrichia caninervis in the gurbantunggut desert. China Arid Land Res Manag. 2011;25(2):116–127. [Google Scholar]
- Li Y, Wang Z, Xu T, Tu W, Liu C, Zhang Y, Yang C. Reorganization of photosystem II is involved in the rapid photosynthetic recovery of desert moss Syntrichia caninervis upon rehydration. J Plant Physiol. 2010;167(16):1390–1397. doi: 10.1016/j.jplph.2010.05.028. [DOI] [PubMed] [Google Scholar]
- Platt KA, Oliver MJ, Thomson WW. Membranes and organelles of dehydrated Selaginella and Tortula retain their normal configuration and structural integrity. Protoplasma. 1994;178(1–2):57–65. [Google Scholar]
- Proctor MCF, Ligrone R, Duckett JG. Desiccation tolerance in the moss polytrichum formosum: physiological and fine-structural changes during desiccation and recovery. Ann Bot. 2007;99(1):75–93. doi: 10.1093/aob/mcl246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor MCF, Smirnoff N. Rapid recovery of photosystems on rewetting desiccation-tolerant mosses: chlorophyll fluorescence and inhibitor experiments. J Exp Bot. 2000;51(351):1695–1704. doi: 10.1093/jexbot/51.351.1695. [DOI] [PubMed] [Google Scholar]
- Wu N, Zhang Y-m, Downing A, Aanderud ZT, Tao Y, Williams S. Rapid adjustment of leaf angle explains how the desert moss, Syntrichia caninervis, copes with multiple resource limitations during rehydration. Funct Plant Biol. 2014;41(2):168. doi: 10.1071/FP13054. [DOI] [PubMed] [Google Scholar]
- Xu S-J, Jiang P-A, Wang Z-W, Wang Y. Crystal structures and chemical composition of leaf surface wax depositions on the desert moss Syntrichia caninervis. Biochem Syst Ecol. 2009;37(6):723–730. [Google Scholar]
- Stark LR, McLetchie DN, Roberts SP. Gender differences and a new adult eukaryotic record for upper thermal tolerance in the desert moss Syntrichia caninervis. J Therm Biol. 2009;34(3):131–137. [Google Scholar]
- Cove DJ, Perroud P-F, Charron AJ, McDaniel SF, Khandelwal A, Quatrano RS. The moss Physcomitrella patens: a novel model system for plant development and genomic studies. Cold Spring Harb Protoc. 2009;2009(2):pdb.emo115. doi: 10.1101/pdb.emo115. [DOI] [PubMed] [Google Scholar]
- Rensing SA, Lang D, Zimmer AD, Terry A, Salamov A, Shapiro H, Nishiyama T, Perroud PF, Lindquist EA, Kamisugi Y, Tanahashi T, Sakakibara K, Fujita T, Oishi K, Shin IT, Kuroki Y, Toyoda A, Suzuki Y, Hashimoto S, Yamaguchi K, Sugano S, Kohara Y, Fujiyama A, Anterola A, Aoki S, Ashton N, Barbazuk WB, Barker E, Bennetzen JL, Blankenship R. et al. The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science. 2008;319(5859):64–69. doi: 10.1126/science.1150646. [DOI] [PubMed] [Google Scholar]
- Wood AJ, Duff RJ. The Aldehyde Dehydrogenase (ALDH) gene Superfamily of the Moss Physcomitrella patens and the algae Chlamydomonas reinhardtii and Ostreococcus tauri. Bryologist. 2009;112(1):1–11. [Google Scholar]
- Carretero-Paulet L, Galstyan A, Roig-Villanova I, Martinez-Garcia JF, Bilbao-Castro JR, Robertson DL. Genome-wide classification and evolutionary analysis of the bHLH family of transcription factors in Arabidopsis, poplar, rice, moss, and algae. Plant Physiol. 2010;153(3):1398–1412. doi: 10.1104/pp.110.153593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koduri PK, Gordon GS, Barker EI, Colpitts CC, Ashton NW, Suh DY. Genome-wide analysis of the chalcone synthase superfamily genes of Physcomitrella patens. Plant Mol Biol. 2010;72(3):247–263. doi: 10.1007/s11103-009-9565-z. [DOI] [PubMed] [Google Scholar]
- Barker EI, Ashton NW. A parsimonious model of lineage-specific expansion of MADS-box genes in Physcomitrella patens. Plant Cell Rep. 2013;32(8):1161–1177. doi: 10.1007/s00299-013-1411-8. [DOI] [PubMed] [Google Scholar]
- Koster K, Balsamo R, Espinoza C, Oliver M. Desiccation sensitivity and tolerance in the moss Physcomitrella patens: assessing limits and damage. Plant Growth Regul. 2010;62(3):293–302. [Google Scholar]
- Liu M, Shi J, Lu C. Identification of stress-responsive genes in Ammopiptanthus mongolicus using ESTs generated from cold- and drought-stressed seedlings. BMC Plant Biol. 2013;13(1):88. doi: 10.1186/1471-2229-13-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Tan L, Dang C, Zhang H, Wu Q, An L. Deep-sequencing transcriptome analysis of chilling tolerance mechanisms of a subnival alpine plant. Chorispora bungeana. BMC Plant Biol. 2012;12(1):222. doi: 10.1186/1471-2229-12-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Wang H, Wan P, Kuang T, He Y. Genome-wide transcriptome analysis of gametophyte development in Physcomitrella patens. BMC Plant Biol. 2011;11:177. doi: 10.1186/1471-2229-11-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Zhang L, Yang G, Zhu H, He Y. Transcriptome of protoplasts reprogrammed into stem cells in Physcomitrella patens. PLoS One. 2012;7(4):e35961. doi: 10.1371/journal.pone.0035961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N, Bhalla PL, Singh MB. Transcriptome-wide profiling and expression analysis of transcription factor families in a liverwort. Marchantia polymorpha. BMC Genomics. 2013;14:915. doi: 10.1186/1471-2164-14-915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Wang N, Zhang P, Cong B, Lin X, Wang S, Xia G, Huang X. Next-generation sequencing-based transcriptome profiling analysis of Pohlia nutans reveals insight into the stress-relevant genes in Antarctic moss. Extremophiles. 2013;17(3):391–403. doi: 10.1007/s00792-013-0528-6. [DOI] [PubMed] [Google Scholar]
- Oliver M, Dowd S, Zaragoza J, Mauget S, Payton P. The rehydration transcriptome of the desiccation-tolerant bryophyte Tortula ruralis: transcript classification and analysis. BMC Genomics. 2004;5(1):89. doi: 10.1186/1471-2164-5-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood AJ, Duff RJ, Oliver MJ. Expressed sequence tags (ESTs) from desiccated Tortula ruralis identify a large number of novel plant genes. Plant Cell Physiol. 1999;40(4):361–368. doi: 10.1093/oxfordjournals.pcp.a029551. [DOI] [PubMed] [Google Scholar]
- Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11(1):31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Meth. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10(1):57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotech. 2011;29(7):644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neil S, Dzurisin J, Carmichael R, Lobo N, Emrich S, Hellmann J. Population-level transcriptome sequencing of nonmodel organisms Erynnis propertius and Papilio zelicaon. BMC Genomics. 2010;11(1):310. doi: 10.1186/1471-2164-11-310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer A, Lang D, Buchta K, Rombauts S, Nishiyama T, Hasebe M, Van de Peer Y, Rensing S, Reski R. Reannotation and extended community resources for the genome of the non-seed plant Physcomitrella patens provide insights into the evolution of plant gene structures and functions. BMC Genomics. 2013;14(1):498. doi: 10.1186/1471-2164-14-498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallavicini A, Canapa A, Barucca M, Alf Ldi J, Biscotti MA, Buonocore F, De Moro G, Di Palma F, Fausto AM, Forconi M, Gerdol M, Makapedua DM, Turner-Meier J, Olmo E, Scapigliati G. Analysis of the transcriptome of the Indonesian coelacanth Latimeria menadoensis. BMC Genomics. 2013;14(1):538. doi: 10.1186/1471-2164-14-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Ji P, Wang B, Zhao L, Wang J, Zhao Z, Zhang Y, Li J, Xu P, Sun X. Transcriptome sequencing and analysis of wild Amur Ide (Leuciscus waleckii) inhabiting an extreme alkaline-saline lake reveals insights into stress adaptation. PLoS One. 2013;8(4):e59703. doi: 10.1371/journal.pone.0059703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewen-Campen B, Shaner N, Panfilio KA, Suzuki Y, Roth S, Extavour CG. The maternal and early embryonic transcriptome of the milkweed bug Oncopeltus fasciatus. BMC Genomics. 2011;12:61. doi: 10.1186/1471-2164-12-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama T, Fujita T, Shin-I T, Seki M, Nishide H, Uchiyama I, Kamiya A, Carninci P, Hayashizaki Y, Shinozaki K, Kohara Y, Hasebe M. Comparative genomics of Physcomitrella patens gametophytic transcriptome and Arabidopsis thaliana: Implication for land plant evolution. Proc Natl Acad Sci. 2003;100(13):8007–8012. doi: 10.1073/pnas.0932694100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Lang D, Zimmer AD, Rensing SA, Reski R. Exploring plant biodiversity: the Physcomitrella genome and beyond. Trends Plant Sci. 2008;13(10):542–549. doi: 10.1016/j.tplants.2008.07.002. [DOI] [PubMed] [Google Scholar]
- Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer EL, Eddy SR, Bateman A, Finn RD. The Pfam protein families database. Nucleic Acids Res. 2012;40(D1):D290–D301. doi: 10.1093/nar/gkr1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehti-Shiu MD, Shiu S-H. Diversity, classification and function of the plant protein kinase superfamily. Philos Trans R Soc B: Biol Sci. 2012;367(1602):2619–2639. doi: 10.1098/rstb.2012.0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Min J. Structure and function of WD40 domain proteins. Protein Cell. 2011;2(3):202–214. doi: 10.1007/s13238-011-1018-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torii KU. Leucine-rich repeat receptor kinases in plants: structure, function, and signal transduction pathways. Int Rev Cytol. 2004;234:1–46. doi: 10.1016/S0074-7696(04)34001-5. [DOI] [PubMed] [Google Scholar]
- Mizutani M, Ohta D. Diversification of P450 genes during land plant evolution. Annu Rev Plant Biol. 2010;61:291–315. doi: 10.1146/annurev-arplant-042809-112305. [DOI] [PubMed] [Google Scholar]
- Kang J, Park J, Choi H, Burla B, Kretzschmar T, Lee Y, Martinoia E. Plant ABC transporters. The Arabidopsis Book/American Society of Plant Biologists. 2011;9:e0153. doi: 10.1199/tab.0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzschmar T, Burla B, Lee Y, Martinoia E, Nagy R. Functions of ABC transporters in plants. Essays Biochem. 2011;50(1):145–160. doi: 10.1042/bse0500145. [DOI] [PubMed] [Google Scholar]
- Jin J, Zhang H, Kong L, Gao G, Luo J. PlantTFDB 3.0: a portal for the functional and evolutionary study of plant transcription factors. Nucleic Acids Res. 2014;42(D1):D1182–D1187. doi: 10.1093/nar/gkt1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano T, Suzuki K, Fujimura T, Shinshi H. Genome-wide analysis of the ERF gene family in Arabidopsis and rice. Plant Physiol. 2006;140(2):411–432. doi: 10.1104/pp.105.073783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuda N, Hisabori T, Takeyasu K, Sato MH. VOZ; isolation and characterization of novel vascular plant transcription factors with a one-zinc finger from Arabidopsis thaliana. Plant Cell Physiol. 2004;45(7):845–854. doi: 10.1093/pcp/pch101. [DOI] [PubMed] [Google Scholar]
- Yasui Y, Mukougawa K, Uemoto M, Yokofuji A, Suzuri R, Nishitani A, Kohchi T. The phytochrome-interacting vascular plant one-zinc finger1 and VOZ2 redundantly regulate flowering in Arabidopsis. Plant Cell. 2012;24(8):3248–3263. doi: 10.1105/tpc.112.101915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai Y, Nakahira Y, Sumida H, Takebayashi K, Nagasawa Y, Yamasaki K, Akiyama M, Ohme-Takagi M, Fujiwara S, Shiina T, Mitsuda N, Fukusaki E, Kubo Y, Sato MH. Vascular plant one-zinc-finger protein 1/2 transcription factors regulate abiotic and biotic stress responses in Arabidopsis. Plant J. 2013;73(5):761–775. doi: 10.1111/tpj.12069. [DOI] [PubMed] [Google Scholar]
- Putterill J, Robson F, Lee K, Simon R, Coupland G. The CONSTANS gene of Arabidopsis promotes flowering and encodes a protein showing similarities to zinc finger transcription factors. Cell. 1995;80(6):847–857. doi: 10.1016/0092-8674(95)90288-0. [DOI] [PubMed] [Google Scholar]
- Griffiths S, Dunford RP, Coupland G, Laurie DA. The evolution of CONSTANS-like gene families in barley, rice, and Arabidopsis. Plant Physiol. 2003;131(4):1855–1867. doi: 10.1104/pp.102.016188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zobell O, Coupland G, Reiss B. The family of CONSTANS-like genes in Physcomitrella patens. Plant Biol (Stuttg) 2005;7(3):266–275. doi: 10.1055/s-2005-865621. [DOI] [PubMed] [Google Scholar]
- Fischer S, Brunk BP, Chen F, Gao X, Harb OS, Iodice JB, Shanmugam D, Roos DS, Stoeckert CJ. , JrUsing OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new ortholog groups. Current protocols in bioinformatics/editoral board, Andreas DB[et al] 2011. Chapter 6:Unit 6 12 11-19. [DOI] [PMC free article] [PubMed]
- Oliver M, Wood A, O’Mahony P. How some plants recover from vegetative desiccation: a repair based strategy. Acta Physiol Plant. 1997;19(4):419–425. [Google Scholar]
- Velten J, Oliver MJ. Tr288, a rehydrin with a dehydrin twist. Plant Mol Biol. 2001;45(6):713–722. doi: 10.1023/a:1010659120984. [DOI] [PubMed] [Google Scholar]
- Zeng Q, Chen X, Wood AJ. Two early light-inducible protein (ELIP) cDNAs from the resurrection plant Tortula ruralis are differentially expressed in response to desiccation, rehydration, salinity, and high light. J Exp Bot. 2002;53(371):1197–1205. doi: 10.1093/jexbot/53.371.1197. [DOI] [PubMed] [Google Scholar]
- Chen X, Zeng Q, Wood AJ. Aldh7B6 encodes a turgor-responsive aldehyde dehydrogenase homologue that is constitutively expressed in tortula ruralis gametophytes. Bryologist. 2002;105(2):177–184. [Google Scholar]
- Chen X, Qin Z, Wood AJ. The stress-responsive Tortula ruralis gene ALDH21A1 describes a novel eukaryotic aldehyde dehydrogenase protein family. J Plant Physiol. 2002;159(7):677–684. [Google Scholar]
- Chen X, Kanokporn T, Zeng Q, Wilkins TA, Wood AJ. Characterization of the V-type H((+))-ATPase in the resurrection plant Tortula ruralis: accumulation and polysomal recruitment of the proteolipid c subunit in response to salt-stress. J Exp Bot. 2002;53(367):225–232. doi: 10.1093/jexbot/53.367.225. [DOI] [PubMed] [Google Scholar]
- Triwitayakorn K, Wood A. Characterisation of two desiccation-stress related cDNAs TrDr1 and TrDr2 in the resurrection moss Tortula ruralis. S Afr J Bot. 2002;68(4):545–548. [Google Scholar]
- Peng CA, Oliver MJ, Wood AJ. Is the Rehydrin TrDr3 from Tortula ruralis associated with tolerance to cold, salinity, and reduced pH? Physiological evaluation of the TrDr3-orthologue, HdeD from Escherichia coli in response to abiotic stress. Plant Biol (Stuttg) 2005;7(3):315–320. doi: 10.1055/s-2005-865653. [DOI] [PubMed] [Google Scholar]
- Yang H, Zhang D, Wang J, Wood AJ, Zhang Y. Molecular cloning of a stress-responsive aldehyde dehydrogenase gene ScALDH21 from the desiccation-tolerant moss Syntrichia caninervis and its responses to different stresses. Mol Biol Rep. 2012;39(3):2645–2652. doi: 10.1007/s11033-011-1017-6. [DOI] [PubMed] [Google Scholar]
- Zhang J, Wu K, Zeng S, Teixeira da Silva J, Zhao X, Tian C-E, Xia H, Duan J. Transcriptome analysis of Cymbidium sinense and its application to the identification of genes associated with floral development. BMC Genomics. 2013;14(1):279. doi: 10.1186/1471-2164-14-279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel RK, Jain M. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS One. 2012;7(2):e30619. doi: 10.1371/journal.pone.0030619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Yu C, Li Y, Lam T-W, Yiu S-M, Kristiansen K, Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25(15):1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- Iseli C, Jongeneel CV, Bucher P. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc/Int Conf Intell Syst Mol Biol; ISMB Int Conf Intell Syst Mol Biol. 1999. pp. 138–148. [PubMed]
- Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L, Wang J. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34(Web Server issue):W293–W297. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maere S, Heymans K, Kuiper M. BiNGO: a Cytoscape plugin to assess overrepresentation of Gene Ontology categories in Biological Networks. Bioinformatics. 2005;21(16):3448–3449. doi: 10.1093/bioinformatics/bti551. [DOI] [PubMed] [Google Scholar]
- Eddy SR. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009;23(1):205–211. [PubMed] [Google Scholar]
- Eddy SR. Accelerated Profile HMM Searches. PLoS Comput Biol. 2011;7(10):e1002195. doi: 10.1371/journal.pcbi.1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2012;40(Database issue):D109–D114. doi: 10.1093/nar/gkr988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, Selbig J, Müller LA, Rhee SY, Stitt M. MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004;37(6):914–939. doi: 10.1111/j.1365-313x.2004.02016.x. [DOI] [PubMed] [Google Scholar]
- Usadel B, Nagel A, Thimm O, Redestig H, Blaesing OE, Palacios-Rojas N, Selbig J, Hannemann J, Piques MC, Steinhauser D, Scheible WR, Gibon Y, Morcuende R, Weicht D, Meyer S, Stitt M. Extension of the visualization tool MapMan to allow statistical analysis of arrays, display of corresponding genes, and comparison with known responses. Plant Physiol. 2005;138(3):1195–1204. doi: 10.1104/pp.105.060459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse M, Nagel A, Herter T, May P, Schroda M, Zrenner R, Tohge T, Fernie AR, Stitt M, Usadel B. Mercator: a fast and simple web server for genome scale functional annotation of plant sequence data. Plant, cell & environment. 2014;37(5):1250–1258. doi: 10.1111/pce.12231. [DOI] [PubMed] [Google Scholar]
- Li W, Godzik A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22(13):1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Comprehensive annotation of S. caninervis unigenes. Including BLASTX search hits in NCBI-Nr, Swiss-Prot, COG, KEGG and COSMOSS databases and all annotated transcription factors using PlantTFDB.
GO comparison results of P. patens genome and S. caninervis transcriptome.
Enrichment network based on GO of the most abundantly expressed transcripts (RPKM > 100) in the transcriptome. Significantly overrepresented (p-value < 0.05) GO terms based on GO-slim were visualized in Cytoscape. The node size is in proportional to the number of unigenes in the GO category. The color represents the enrichment significance. Nodes with white color are not enriched but show the essential hierarchical relationship among the enriched GO-slim terms.
Pfam domain/family annotation and statistics of S. caninervis sequences (2 sheets).
Statistics of KEGG pathways annotated in S. caninervis transcriptome.
MapMan overview of S. caninervis cellular metabolism. Individual assembled transcripts are represented by colored squares. The color code scale is based on the log2 of the RPKM values of each unigene. The greater intensity of red is associated with higher transcript abundance. Green highlighted metabolic pathways are biosynthetic while pink highlighted metabolic pathways are degradative.