

Abstract
RDH12 has been suggested to be one of the retinol dehydrogenases (RDH) involved in the vitamin A recycling system (visual cycle) in the eye. Loss of function mutations in the RDH12 gene were recently reported to be associated with autosomal recessive childhood-onset severe retinal dystrophy. Here we show that RDH12 localizes to the photoreceptor inner segments and that deletion of this gene in mice slows the kinetics of all-trans-retinal reduction, delaying dark adaptation. However, accelerated 11-cis-retinal production and increased susceptibility to light-induced photoreceptor apoptosis were also observed in Rdh12−/− mice, suggesting that RDH12 plays a unique, nonredundant role in the photoreceptor inner segments to regulate the flow of retinoids in the eye. Thus, severe visual impairments of individuals with null mutations in RDH12 may likely be caused by light damage1.
11-cis-Retinal, the chromophore of rhodopsin and cone pigments in photoreceptor cells, is essential for vision (1, 2). Photoactivation of rhodopsin and cone pigments causes isomerization of 11-cis-retinal to all-trans-retinal (2, 3), which is recycled in a pathway termed the visual (retinoid) cycle (4, 5). The importance of retinol recycling is emphasized by the large number of retinal diseases associated with mutations in genes involved in the visual cycle (6). Loss of function mutations in the retinol dehydrogenase 12 gene (RDH12) were recently reported to be associated with severe, early-onset autosomal recessive retinal dystrophy in several independent pedigrees (7, 8). RDH12 is a member of a novel subfamily of four retinol dehydrogenases (RDH11–14) active toward all-trans- and cis-retinals with C (15) pro-R specificity (9) as well as other aldehydes (10). Based on in situ hybridization, RDH12 expression was observed in photoreceptor cells (9). Thus, in the visual cycle, RDH12 can catalyze reduction of all-trans-retinal and 11-cis-retinal to their corresponding retinols. In vitro studies suggest that decreased 11-cis-retinal production due to disruption of the visual cycle (4, 5) can be a cause of the degeneration with RDH12 mutations (8, 11, 12). Deletion of RDH8 (also known as prRDH), another all-trans-RDH present in photoreceptor cells (13), caused only a mild phenotype exhibiting delayed dark adaptation (14). No RDH8 mutations have yet been identified in patients with retinal diseases. Thus, RDH12 is possibly a key enzyme in the visual cycle pathway.
MATERIALS AND METHODS
Rdh12−/− Mice, Genotyping, and PCR
Rdh12−/− mice were generated by standard procedures (Ingenious Targeting, Inc., Rochester, NY). The targeting vector was constructed by replacing exons 1, 2, and 3 with the neo cassette (supplemental Fig. S1). The Rdh12−/− genotype was maintained in a mixed background of C57BL/6J and 129Sv/Ev mice, and siblings were used for the majority of experiments. Genotyping was carried out by PCR using primers RDH12U5, 5′-GCTGAGCCACTTTCCCTGCCCT-3′, and RDH12d5, 5′-AGAGCCGCCAGAGCACAGCCT-3′, for wild type (751 bp) and RDH12U5 and neoa1, 5′-GCCCCGACTGCATCTGCGTGTT-3′, for targeted deletion (468 bp). PCR products were cloned and sequenced to verify their identity. 129Sv/Ev (Leu-450; RPE65)2 and C57Bl/6J (Met-450; RPE65) mice were purchased from Taconic Inc. and The Jackson Laboratory. The RPE65 variant was verified by direct sequencing. Rdh12−/− mice were fertile, developed normally, and reached similar body weights in both sexes. All animal experiments employed procedures approved by the Case Western Reserve University Animal Care Committee and conformed to both the recommendations of the American Veterinary Medical Association Panel on Euthanasia and the Association of Research for Vision and Ophthalmology. Mice were maintained in a 12-h light/12-h dark (6 a.m./6 p.m.) cycle and were reared under less than 10 lux white light. All manipulations were done under dim red light employing a Kodak No. 1 safelight filter (transmittance >560 nm).
Total RNA was isolated from 3-month-old Rdh12−/−, Rdh8−/−, and Rdh12+/+ mouse retinas and the RPE. First-strand cDNA was synthesized by using SuperScript First-Strand Synthesis system for reverse transcription-PCR (Invitrogen). Primers for mouse RDH12 (Mm01307472_m1) and RDH8 (Mm01178944_m1) were ordered from Applied Biosystems. All samples were triple-loaded and 18 S rRNA was used as the internal control. The results are presented with S.E., and n was between 3 and 5.
Immunoblots and Antibodies
Immunoblotting was done according to standard protocols using Immobilon-P to adsorb proteins (polyvinylidene difluoride; Millipore Corp.) (15). Mouse polyclonal antibodies against bacterially expressed full-length mouse RDH12 were raised in BALB/c mice as described (15). Antibody specificity was verified by using bacterially or Sf9-expressed RDH12 protein (data not shown). Monoclonal and polyclonal antibodies againstRDH8were generated against bacterially expressed protein as previously described (16), and anti-β-actin antibody was purchased from Santa Cruz Biotechnology, Inc. Alkaline phosphatase-conjugated goat anti-mouse IgG or goat anti-rabbit IgG (Promega) were used as secondary antibodies. Protein bands were visualized with 5-bromo-4- chloro-3-indolyl phosphate/nitro blue tetrazolium color development substrate (Promega).
Histology, Immunocytochemistry, and Electroretinography (ERG)
For histology, eyecups were fixed in 2% glutaraldehyde, 4% paraformaldehyde for 18 h, incubated in 20% sucrose, and then embedded in 50% optimal cutting temperature (OCT) compound (Miles) diluted with 20% sucrose in sodium phosphate buffer, pH 7.5. Sections were cut at 5 μm, stained with Harris-modified hematoxylin solution (Sigma), and viewed with Nomarski optics (labophot-2, Nikon). Procedures for immunocytochemistry were described previously (9). Sections were analyzed with a Leica DM6000 B microscope. Digital images were captured as described before (9). For tunnel staining, which indicates apoptosis, eyecups were processed as described above with two minor modifications. Paraformaldehyde (4%) in sodium phosphate buffer, pH 7.5, was used for fixation, and sections were cut at 10 μm. Slides were post-fixed for 30 min at room temperature in 4% paraformaldehyde in sodium phosphate buffer, pH 7.5, followed by a second post-fixation/permeabilization in precooled ethanol:acetic acid (2:1) for 5 min at −20 °C. Apoptosis staining was carried out with the ApopTag Peroxidase in Situ Apoptosis detection Kit (Chemicon International) for tissue cryosections or adherent cultured cells. Images were captured as described above. ERG employing anesthetized mice were recorded as previously reported (14, 17).
Retinoid Analysis, A2E Analysis, Preparation of Mouse Rod Outer Segments (ROS), Rhodopsin Measurement, and All-trans- RDH Activity
All experimental procedures related to extraction, derivatization, and separation of retinoids from dissected mouse eyes were carried out as described previously (17). Mouse A2E analysis was done according to published procedures (14). Quantification of A2E was performed with a known concentration of pure synthetic A2E (18). Preparation of osmotically intact ROS from 15 mice was done as reported previously (14). The concentration of rhodopsin was determined after the sample illumination by the decrease in absorption at 500 nm using the molar extinction coefficient, ε = 42,000 m−1 cm−1. Typically, two mouse eyes were used. All-trans-RDH activities of the retina and ROS were assayed by monitoring the production of all-trans-retinol (reduction of all-trans- retinal) as previously reported (14).
Light Damage
LD was induced as previously published (19) but with slight modifications. Mice were dark-adapted for 48 h, and LD was induced after pupil dilation with 0.1% atropine (Sigma) by exposure to 3,000 or 10,000 lux diffuse white fluorescent light (150 watt spiral lamp, Commercial Electric) (lights on at 11:00 a.m.).
DNA Fragmentation
Genomic DNA was extracted from two dissected retinas with a DNeasy kit (Qiagen). DNAsamples (1 μg) were loaded on 2% agarose gels containing ethidium bromide. Electrophoresis was carried out at 50 V, and gels were visualized under UV light.
RESULTS
Disruption of the Rdh12 Gene in Mice and RDH Activity
Here we sought to elucidate the physiological role of RDH12 in vision by understanding how disruption of its function leads to retinal degeneration in a knock-out mouse model. By targeted recombination, we generated an Rdh12−/− mouse in which exons 1–3 of the mouse Rdh12 gene were replaced by a neo cassette (supplemental Fig. S1). The expression of RDH12 was abolished in the retina of Rdh12−/− mice as determined by immunoblotting (Fig. 1A), immunocytochemistry (Fig. 1B), and quantitative PCR (Table 1); RDH8 expression was unchanged in Rdh12−/− mice. Immunofluorescence microscopy showed uniform labeling of RDH12 in the photoreceptor inner segment layer, suggesting the RDH12 is localized to both cone and rod inner segments. More extensive double immunolabeling studies will be needed to reveal the detailed localization of RDH12 in cone photoreceptor cells.
FIGURE 1. Characterization of the Rdh12 knock-out mice.
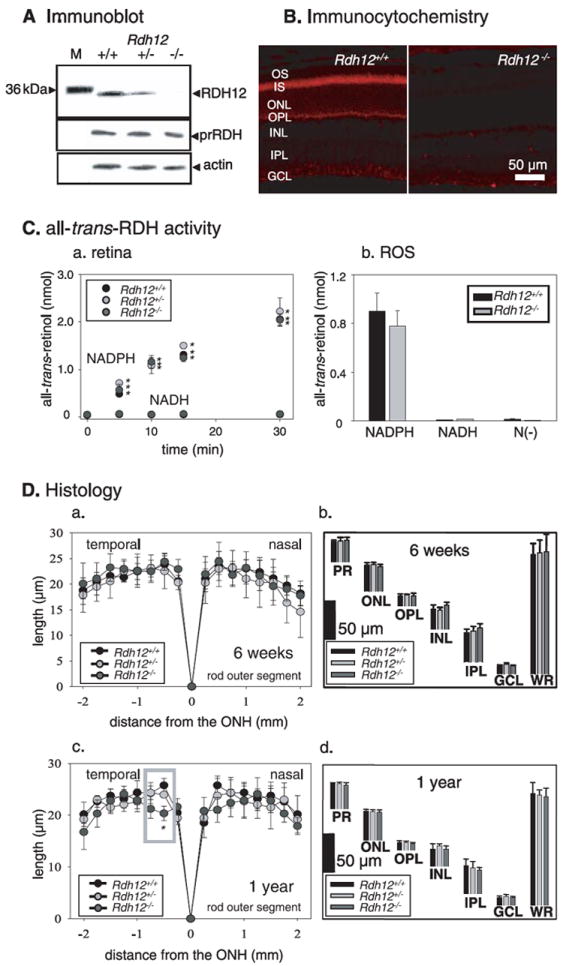
A, immunoblots of Rdh12+/+, Rdh12+/−, and Rdh12−/− retina extracts probed with anti-RDH12, anti-RDH8 (prRDH), and anti-actin polyclonal antibodies. M, molecular mass standards. B, immunocytochemical localization of RDH12 (red) in 8-week-old Rdh12+/+ and Rdh12−/− retina frozen sections. C, all-trans-RDH activity in the retina (NADPH versus NADH) (a) and ROS from Rdh12+/+, Rdh12+/−, and Rdh12−/− mice (b). The bars indicate the S.E. of the mean (n > 5). RDH activities of the retina and ROS (homogenates with the reaction buffer) of mice were assayed by monitoring the production of all-trans-retinol (reduction of all-trans-retinal). The reaction mixture (100 μl of 10 mm sodium phosphate buffer, pH 7.0, containing 100 mm NaCl and 1 mm n-dodecyl-β-maltoside) contained one retina or 5 μg mouse ROS membrane in the presence of NAD(P)H (1 mm) or absence of dinucleotide (N(–)). The reaction was initiated by the addition of 20 μm all-trans-retinal, incubated at 37 °C, and then terminated with 400 μl of CH3OH, and the retinoids were extracted with twice 500 μl of hexane. The hexane solution was analyzed by HPLC using 10% ethyl acetate in hexane to measure the production of all-trans-retinol. D, retina histology of Rdh12+/+, Rdh12+/−, and Rdh12−/− mice. ROS length (in μm) is plotted as a function of distance (in mm) from the optic nerve head (ONH). The ages of mice were 6 weeks (a) or 1 year (c). Quantification of the thickness of different layers of the retina from Rdh12−/−, Rdh12+/−, and Rdh12+/+ mice were measured at 1.25mmsuperior to the optic nerve head. The ages of mice were 6 weeks (b) or 1 year (d). Error bars indicate the S.E. of the mean (n > 3). OS, outer segment; IS, inner segment; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer; WR, whole retina (*, p < 0.001).
TABLE 1.
Differences and similarities between dark-adapted mice from different Rdh12 genetic backgrounds
| Rdh12+/+ | Rdh12+/− | Rdh12−/− | |
|---|---|---|---|
| pmol/eye | pmol/eye | pmol/eye | |
| Rhodopsina | |||
| 6-week-old | 480.3 ± 25.8 | 493.3 ± 25.9 | 487.4 ± 28.8 |
|
| |||
| Rdh12+/+ | Rdh12+/− | Rdh12−/− | |
|
| |||
| pmol/eye | pmol/eye | pmol/eye | |
| A2Eb | |||
| 6-week-old | NDc | ND | ND |
| 6-month-old | 5.89 ± 0.48 | 8.11 ± 1.13 | 10.51 ± 1.78 |
| 12-month-old | 11.00 ± 0.17 | 17.74 ± 0.20 | 22.03 ± 1.10 |
|
| |||
| Rdh12+/+ | Rdh12+/− | Rdh12−/− | |
|
| |||
| min | min | ||
| Meta II decayd | |||
| 6-week-old | 30.6 | 27.8 | |
|
| |||
| Expression level (retina)e | Rdh12+/+ | Rdh12−/− | Rdh8−/− |
|
| |||
| Relative quantification | Relative quantification | Relative quantification | |
| RDH12 | 1 | 0.00 | 1.00 |
| RDH8 | 1 | 1.01 | 0.00 |
Mice were dark-adapted for more than 48 h. The results are presented with S.E., and n was between 3 and 5. Rhodopsin and retinoids were measured as described under “Materials and Methods.”
A2E was measured as described under “Materials and Methods.” The results are presented with S.E., and n was between 3 and 5.
ND, not detected.
Meta II decay (n = 3) was examined as described under “Materials and Methods.”
Quantitative PCR was carried out in the DNA Sequencing and Real-time PCR Core (Department of Orthopedics, Case Western Reserve University).
First, we determined rhodopsin levels in Rdh12+/+, Rdh12+/−, and Rdh12−/− mice and found that they were not significantly different (Table 1). Next, we measured how disruption of the Rdh12 gene affected reduction of all-trans-retinal. All-trans-retinal was applied exogenously to the dissected retina, and all-trans-RDH activity was determined from the rate of all-trans-retinol production (see “Materials and Methods”). Retinas from Rdh12+/+, Rdh12+/−, and Rdh12−/− mice showed comparable NADPH-specific all-trans-RDH activities (Fig. 1Ca). The all-trans-RDH activity of expressed RDH8 and RDH12 in Sf9 cells also showed NADPH dependence (not shown) (9). Isolated ROS from Rdh12+/+ and Rdh12−/− mice displayed similar activities in the presence of NADPH (Fig. 1Cb). Thus, deletion of RDH12 did not affect the all-trans-RDH activity in the retina and ROS and showed that other enzymes are sufficient to reduce a majority of all-trans-retinal in mouse retina. This observation does not mean that RDH12 is a minor enzyme in certain specific subcellular structures, like inner segments. However, RDH12 is a minor activity relative to the total RDH activity in the retina. To determine the influence of the lack of RDH12 on decay of photoactivated rhodopsin, Meta II, we used an assay based on Trp fluorescence. When the data were fitted to a first-order reaction, comparable τ = 30.6 min and τ = 27.8 min for the decay of Meta II were observed in ROS membranes from Rdh12+/+ and Rdh12−/− mice, respectively (Table 1; supplemental Fig. S2). This finding suggested that RDH12 does not directly facilitate removal of the chromophore from the binding site in rhodopsin. These observations are also consistent with RDH12 localization to photoreceptor inner segments (Fig. 1B).
Histology and ERG of Rdh12−/− Mice
Light microscopy revealed no apparent abnormalities in the retinas of 6-week-old Rdh12−/− mice raised in 12-h dark/12-h light conditions (data not shown). The ROS were similar in length in Rdh12−/−, Rdh12+/−, and Rdh12+/+ mice (Fig. 1 Da). The thickness of each major layer in the retina was also similar in all three genetic strains (Fig. 1Db). In 1-year-old animals, light microscopy revealed that the ROS around the central area of the retina (500 μm from the optic nerve head) were reduced in length in Rdh12−/− mice (Fig. 1Dc). In contrast, the thickness of each major layer in the retina at 1250 μm from the optic nerve head (superior area of the retina) was similar in the three genetic strains (Fig. 1Dd).
To evaluate rod- and cone-mediated light responses, we studied Rdh12−/− mice using ERG. The amplitudes of the a and b waves were not significantly different in dark- and light-adapted Rdh12−/−, Rdh12+/−, and Rdh12+/+ mice (p > 0.2, one-way analysis of variance) (Fig. 2, A and B). Flicker ERG responses also were similar for dark- and light-adapted Rdh12 mice (supplemental Fig. S3). Thus, RDH12 deletion did not have a significant effect on the ability of rods and cones to generate light responses. Recovery of the ERG response (dark adaptation) after bleach was then also measured by monitoring the amplitude of the a-wave after exposure to intense constant illumination (500 cd·m−2) for 3 min. Recovery of the responses was substantially slower in Rdh12−/− mice compared with Rdh12+/+ mice (p < 0.0001, Fig. 2C). Rdh12+/− mice showed slower dark adaptation in the first 20 min, but their adaptation kinetics became similar to Rdh12+/+ mice by 30 min after the bleach. Amounts of all-trans-retinal and 11-cis-retinal after the bleach indicated that the delay was correlated with the amount of accumulated all-trans-retinal (Fig. 2D). This suggested that clearance of all-trans-retinal in Rdh12−/− mice is slower than production of 11-cis-retinal during dark adaptation.
FIGURE 2. Full field ERG responses of Rdh12+/+, Rdh12+/−, and Rdh12−/− mice with RPE65 (Met-450).
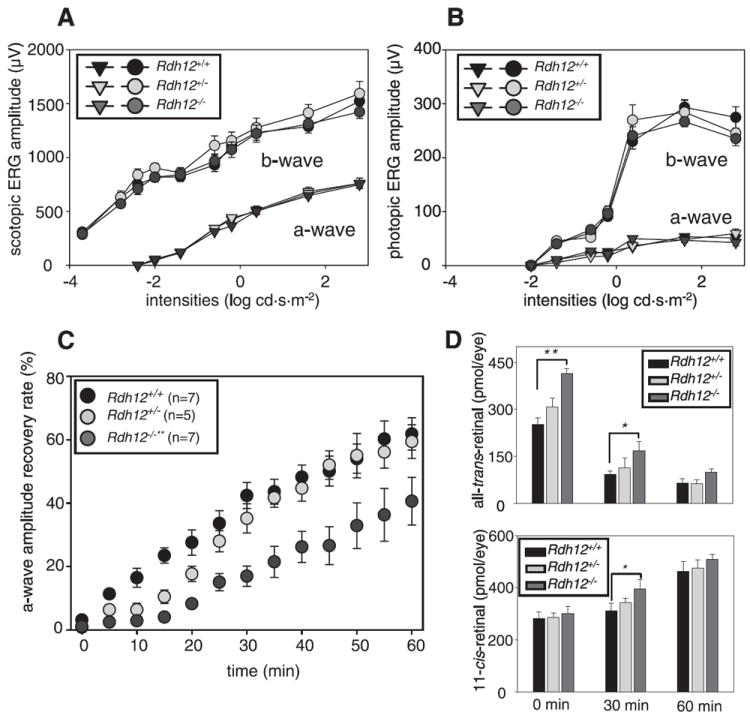
A and B, the amplitudes of a-wave and b-wave were plotted as a function of light intensity under dark- and light-adapted conditions, respectively. C, recovery of a-wave amplitudes after constant light exposure. The dark-adapted mice were exposed to intense illumination (500 cd·m−2) for 3 min, and the recovery of a-wave amplitudes was monitored with single-flash ERG (−0.2 cd·s·m−2) every 5 min for 60 min. The recovery rate was significantly attenuated in Rdh12−/− (**, p < 0.0001) compared with Rdh12−/−mice (n = 5 each), whereas Rdh12+/− mice were slightly attenuated showing a lag phase for the first 30 min. D, amounts of all-trans-retinal (upper) and 11-cis-retinal (lower) were analyzed with HPLC at several points of dark adaptation after intense constant illumination (500 cd·m−2) for 3 min. Error bars indicate the S.E. of the mean(n > 3; **, p < 0.0001; *, p < 0.001).
Flow of Retinoids in Rdh12−/− Mice Exposed to a Single Flash
To understand how loss of RDH12 affected the retinoid flow throughout the visual cycle, we analyzed retinoids by HPLC at various times after an intense flash (bleaching ~35% rhodopsin). As expected, bleaching caused the formation of all-trans-retinal, levels of which decreased with time (Fig. 3A) because it was reduced to all-trans-retinol and esterified. The reduction kinetics were slower in Rdh12−/− than in Rdh12+/+ retinas. Slower reduction was also observed in Rdh12+/− retinas (data not shown). Mice lacking an alternative enzyme, RDH8, also showed slow reduction kinetics, and the delay in Rdh8−/− mice was statistically more significant compared with that in Rdh12−/− mice (14).
FIGURE 3. Kinetics of all-trans-retinal reduction and 11-cis-retinal regeneration in Rdh12+/+ and Rdh12−/− mice with RPE65 (Met-450).
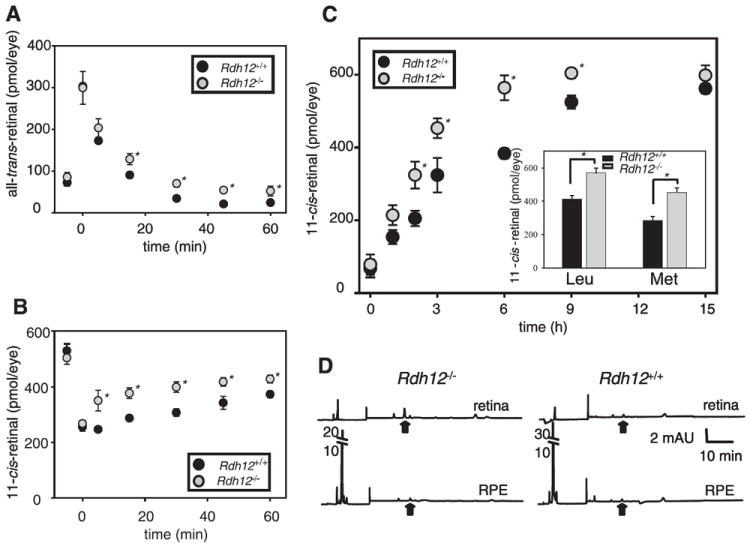
Retinoids were quantified by HPLC on samples collected at different time points after a flash that bleached ~35% of the visual pigment for the pigmented mice. A, changes in the all-trans-retinal levels. B, changes in the 11-cis-retinal levels. Error bars indicate the S.E. of the mean (n > 3). Mice were reared under 12 h/12h dark/light cycle conditions. (*, p < 0.001). C, dark-adapted mice were exposed to background light of 500 cd·m−2 for 24 min (~90% rhodopsin bleach) and returned to the dark until retinoid analysis by HPLC. Amounts of 11-cis-retinal at several time points after the bleach were plotted. Inset, amount of 11-cis-retinal was examined at 3 h of dark adaptation after the light of 500 cd·m−2 for 24 min in Rdh12−/− and Rdh12+/+ mice with Leu and Met at position 450 of RPE65. Error bars indicate the S.E. of the mean (n > 3) (*, p < 0.001). D, dark-adapted mice were exposed to background light of 500 cd·m−2 for 48 min (~98% rhodopsin bleach) and returned to the dark. Retinoid analysis of the dissected retina and RPE was performed separately immediately after the bleach. There was unavoidable cross-contamination of the RPE with retina and vice versa as measured by the presence of retinyl esters in the retinal fraction and 11-cis-retinal in the RPE. Representative chromatograms are shown (n > 3). Black arrows indicate elution times for syn- all-trans-retinyl isomer.
The kinetics of 11-cis-retinal formation were faster in Rdh12−/− than in Rdh12+/+ mice (Fig. 3B), indicating that although the reduction of the conversion rate of all-trans-retinal to all-trans-retinol in Rdh12−/− mice was slightly reduced, 11-cis-retinal formation in the visual cycle was accelerated. Rdh8−/− mice with slower all-trans-retinal reduction did not show accelerated 11-cis-retinal production (14). Total amounts of retinoids in the eye were not changed before and after illumination in Rdh12+/+, Rdh12+/−, and Rdh12−/− mice (data not shown).
Recovery of the Chromophore in Rdh12−/− Mice Exposed to an Intense Bleach
We employed different light conditions to analyze the process of 11-cis-retinal formation in Rdh12−/− mice further. After intense light that bleached ~90% of rhodopsin, accelerated 11-cis-retinal production was observed in Rdh12−/− mice (half-life of the 11-cis-retinal recovery was 2 h compared with 3 h in Rdh12+/+ mice) (Fig. 3C). Retinoid analysis performed in the dissected retina and the RPE after 48 min of illumination that bleached ~98% of rhodopsin is shown in Fig. 3D. All-trans-retinal was detected in the retina, and more accumulated in Rdh12−/− mice (arrow in Fig. 3D). Increased retinoid amounts in the retina and in the eye confirmed that slower reduction of all-trans-retinal was detectable in Rdh12−/− mice (data not shown).
Retinal Damage in Rdh12−/− Mice Exposed to Intense Light
RPE65 is another component of the visual cycle involved in the isomerization of all-trans-retinyl esters to 11-cis-retinol (20-22). It also is a modifier of light-induced retinal degeneration (LD) because RPE65 null mice show resistance to LD (23). An RPE65 variation in which Leu-450 is replaced by Met (M450L) shows less efficient production of 11-cis-retinal, and indeed mice carrying this variation are also resistant to LD (19). The inset of Fig. 3C shows the amount of 11-cis-retinal after the intense light (bleaching ~90% rhodopsin) in Rdh12−/− back-crossed to mice with Leu or Met at position 450 of RPE65. Mice with the Leu-450 variation showed higher levels of 11-cis-retinal as reported previously (19). However, deletion of RDH12 clearly showed accelerated 11-cis-retinal production after illumination in both RPE65 variants. Similar experiments to those shown in Fig. 3 were also done with Leu-450 background mice, confirming that 11-cis-retinal regeneration was accelerated (data not shown). The regeneration rate of 11-cis-retinal regulates susceptibility to LD, and the M450L RPE65 variation affects its sensitivity due to reduced 11-cis-retinal regeneration. Because Rdh12−/− mice showed accelerated 11-cis-retinal regeneration (Fig. 3, B and C), susceptibility to LD was examined. When exposed to 3000 lux white light for 48 h, the nuclear layer of Rdh12−/− around the central area (500 μm from the optic nerve head), where the strongest intensity light penetrates, was reduced, and ROS were shortened (Fig. 4, A and B), whereas no changes were observed in the same area of control Rdh12+/+. No significant changes in the peripheral retina were detected in either genotype. Rhodopsin amounts in illuminated Rdh12−/− mice were lowered to half those present in non-illuminated mice, whereas rhodopsin amounts in Rdh12+/+ mice after illumination were similar to those before illumination (Fig. 4C). When total retinoids in illuminated eyes were analyzed by HPLC, Rdh12−/− mice had less retinoids compared with Rdh12+/+ mice (Fig. 4D), suggesting that broad photoreceptor outer segment loss occurred in Rdh12−/− mice after intense light.
FIGURE 4. Light induced photoreceptor cell death in Rdh12−/− mice with RPE65 (Leu-450).
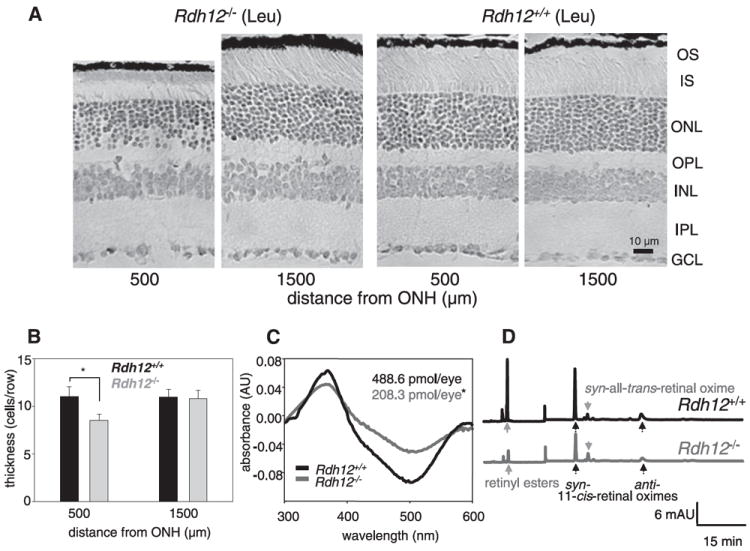
LD was induced in Rdh12−/− and Rdh12+/+ mice with dilated pupils by 48 h exposure to 3000 lux of diffuse white fluorescent light as described under “Materials and Methods” A, montage of cross-sections of the retina from mice that were exposed to light for 48 h and then dark-adapted for 24 h. ONH, optic nerve head; OS, outer segment; IS, inner segment; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. B, number of nuclei in a row of the outer nuclear layer of Rdh12−/− and Rdh12+/+ mice exposed to the light. Error bars indicate the S.E. of the mean (n > 5; *, p < 0.001). C, rhodopsin concentration after the light exposure was measured as described under “Materials and Methods.” Representative difference spectra are shown from Rdh12−/− and Rdh12+/+ mice, and numbers display averaged rhodopsin concentrations from 5 mice. Error bars indicate the S.E. of the mean (n > 5; *, p < 0.001). D, HPLC elution profile indicating the retinoid content in Rdh12−/− and Rdh12+/+ mice exposed to light.
A stronger light intensity (10,000 lux) was also employed to induce LD in Rdh12−/− mice. In Rdh12−/− mice with Leu-450 in RPE65, apoptotic cells were detected after 20 min of illumination, whereas 2 h of illumination were needed for Rdh12+/+ mice with Leu-450 in RPE65 (Fig. 5A and supplemental Fig. S4). LD was induced in only a few cells in Rdh12+/+ mice with Met-450 RPE65 after 2 h of illumination. By contrast, extensive LD over a large area was evident in Rdh12−/− mice (supplemental Fig. S3) and was accompanied by DNA fragmentation (Fig. 5B). ERG showed significant reduction in Rdh12−/− mice with 24 h of illumination (Fig. 5C), also suggesting that Rdh12−/− mice are more susceptible to LD, with their photoreceptors more broadly impaired. These experimental light conditions are equivalent to daylight brightness. During summer in Cleveland the typical shade illumination is 7,000 lux, whereas during a sunny day at noon illumination approaches 80,000 lux.
FIGURE 5. Light induced retinal degeneration under various light exposure durations.
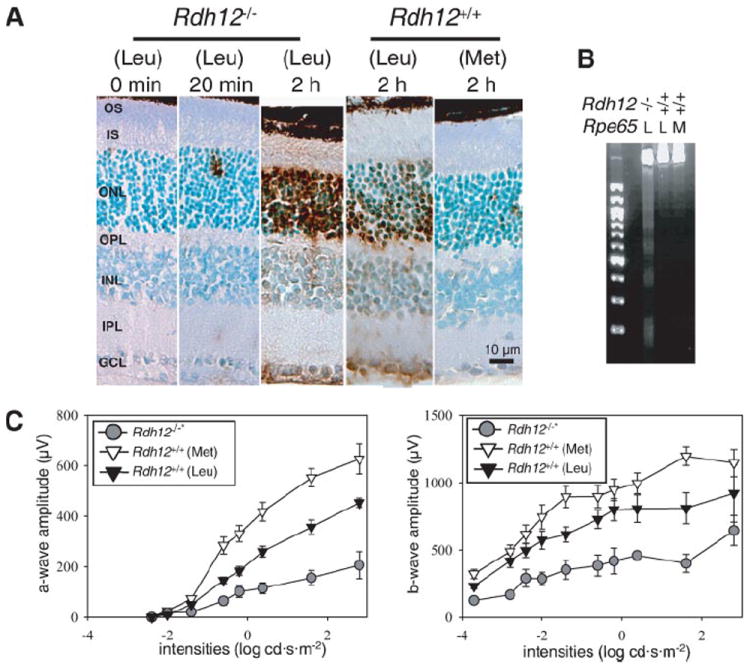
LD was induced in Rdh12−/− and Rdh1+/+ mice with Leu or Met at position 450 of RPE65 mice with dilated pupils by exposure to 10,000 lux of diffuse white fluorescent light as described under “Materials and Methods”. A, apoptotic cells were detected by terminal dUTP nick-end labeling stain after various exposure times. Representative retinal histology 500 μm from the optic nerve head is presented. B, formation of a DNA ladder in agarose gel electrophoresis was examined 24 h after 2 h illumination. L, Leu; M, Met. C, the level of retinal damage was evaluated with full-field ERG 24 h after intense light exposure. Both a- and b-wave amplitudes were attenuated significantly in Rdh12−/− mice expressing RPE65 (Leu-450) compared with Rdh12+/+ mice expressing RPE65 (Met-450) (*, p < 0.001).
A2E Fluorophore in the Retina of Rdh12−/− Mice
Free all-trans- retinal can condense into an A2E fluorescent product as observed in Abca4−/− mice (24). A2E was detected in Rdh12−/− and Rdh12+/− mice at levels higher than in Rdh12+/+ mice but was still several times lower than in Abca4−/− mice (Table 1) (24). A2E accumulated in Rdh12−/− mice is unlikely to be the cause of observed LD because Abca4−/− mice with higher accumulations of A2E manifest only mild photoreceptor degeneration (24). Interestingly, Abca4−/− (24), Rdh8−/− (14), and Rdh12−/− mice (this study) showed distinct patterns in clearing all-trans-retinal from their photoreceptors, each representing a unique model to study degenerative processes in the retina, where A2E is hypothesized to be one of the toxic agents to RPE cells (25). But it should be noted that A2E is not necessarily the only compound that might induce toxic effects. For example, aldehyde groups of small organic compounds are very reactive and could produce toxic effects on mitochondria by blocking redox reactions.
DISCUSSION
RDHs in the Retina
Several dehydrogenases were identified in the retina that may contribute to the redox reaction of the retinoid (visual) cycle (26). RDHs constitute two groups of enzymes that belong to microsomal and soluble short-chain dehydrogenase/reductase (9, 10, 27-29) and alcohol dehydrogenase families that possess RDH activity (30). In addition to biochemical assays confirming the specificity of various dehydrogenases (9, 31, 32), mouse genetic studies promise to provide a powerful approach to elucidate their function (14, 32-34). These enzymes may have specific localizations in tissues and, like RDH12, may contribute only minimally to total RDH activity. Yet they may also have a profound effect on a particular retinoid transformation. But the genetic approach to elucidate RDH functions has limitations due to the hydrophobic membrane diffusible properties of retinoids. Moreover, short-chain dehydrogenases can to a large degree substitute for each other and utilize a broad range of substrates including steroids (35), thereby complicating straightforward interpretations. Indeed, its catalytic properties suggest that RDH12 primarily contributes to the reduction of all-trans-retinal only at saturating concentrations of aldehyde substrate in cells undergoing oxidative stress (10). RDH12 might also play a role in the detoxification of lipid peroxidation products. Hence, RDH12 may be involved in degradation of A2E and lipid peroxidation products, which could be a cause of photoreceptor degeneration. So our results must be considered with these caveats in mind. Nonetheless, the direct or indirect roles of RDH8 and RDH12 in the retinoid cycle in vivo have become better understood by use of genetically modified mice. An advantage of the visual system also is that the examined processes are initiated by photoactivation of rhodopsin (36, 37).
RDH8 Versus RDH12; Similarities and Differences
It appears that both RDH8 and RDH12 are involved in the reduction of all-trans-retinal released from photoactivated pigments but by two distinct processes. RDH8 (also known as prRDH) is present in ROS and reduces all-trans-retinal released from photoactivated rhodopsin (4, 5, 26). Thus, the clearance of all-trans- retinal in Rdh8-null mice is tightly coupled to the formation of 11-cis-retinal and regeneration of the visual pigments. In contrast to RDH8, RDH12 localizes to inner segments and likely contributes to reduction of all-trans-retinal by different mechanisms (Fig. 1B).
As diffusible agents, retinoids not only are present in oxidized and reduced forms depending on the redox state of the cells, but these hydrophobic substrates also partition differently into different membranes. Free all-trans-retinal can contribute to changes in the physiological responses of rod cells by activating ligand-free opsin (38-40) or cyclic GMP-dependent cation channels of the plasma membrane (41, 42). Thus, physiological responses such as these measured by ERG could be disconnected from the results of chemical analyses, e.g. measurements of total retinoid content.
Mechanisms of vectorial retinoid transport in the retinoid cycle are unknown, although our observations suggest, among other possibilities, that reduction of all-trans-retinal in the photoreceptor inner segment is required for normal function of the visual cycle. Based on the results of Rdh12- and Rdh8-null mice, it appears that all-trans-retinal, if not cleared, blocks the regeneration process. When all-trans-retinal diffuses to the inner segments, where it is normally reduced by RDH12, regeneration is accelerated because ROS is filled with unliganded opsin, whereas the excess retinoids present in the RPE produce 11-cis-retinal. Thus, regeneration of rhodopsin and 11-cis-retinal formation are two different processes. Because total retinoid amounts were unchanged in the Rdh12-null mice, they probably are not derived from accelerated incorporation of retinol from the circulation. Thus, our results suggest an unexpected role of RDH12 for clearance of all-trans-retinal in the retinoid cycle.
RDH12 and Light Damage
In the Rdh12−/− mice, all-trans-retinal present in the inner segments where the RDH12 enzyme normally resides cannot be quickly cleared. The presence of all-trans-retinal in the inner segments would be a sensitizer for LD far beyond what is observed in wild type mice. Photoreceptor inner segments are rich in mitochondria and play a major role in protein synthesis and energy production. It is probably essential to clear the inner segments of toxic all-trans-retinal. By supplying chromophore to rhodopsin, accelerated 11-cis-retinal production in Rdh12−/− mice can also contribute to the aberrant accumulation of all-trans-retinal in the inner segments under intense light illumination.
Leber Congenital Amaurosis and RDH12
Several independent studies revealed that mutations in the human RDH12 gene are responsible for severe forms of blindness termed early-onset autosomal recessive Leber congenital amaurosis (7, 8, 12). The mouse phenotype presented in this study is drastically different from previously characterized mice carrying disruption of the retinoid cycle genes encoding enzymes such as lecithin, retinol acyltransferase (43, 44), or the retinoid isomerase (RPE65) (45, 46). Based on the mouse model with a disrupted RDH12 gene, the retina may not lack the chromophore but, rather, is susceptible to the toxic effects of light. Most rodents are nocturnal in nature and, thus, are exposed to lower illumination levels. Their visual cycle processes retinoid slowly in hours after intense bleach, and most rodents produce completely regenerated visual pigments (47). In contrast, the human visual cycle displays much faster regeneration kinetics (48). The human retina is twice as rich in cone pigments as rodent retina (49, 50) and, thus, could be more susceptible to LD due to a more robust flux of retinoids. Thus, protection of the retina from intense illumination may reduce the rate of degeneration in patients carrying a RDH12 null allele. Studies of affected patients carrying disabling mutations in the RDH12 gene could provide additional support for this hypothesis.
Supplementary Material
Acknowledgments
We thank Dr. Leslie Webster for comments on the manuscript.
Footnotes
This research was supported by National Institutes of Health Grants EY09339 and P30 EY11373, a grant from the National Neurovision Research Institute (to A. M.), a center grant from the Foundation Fighting Blindness to the University of Utah, and by Landelijke stichting Blinden en Slechtzienden, Gelderse Blinden Stichting, Stichting OOG, Stichting Blindenhulp Rotterdamse Vereniging Blindenbelangen, and Stichting Ooglijders/Stichting het Hooykaas La Lau Fonds. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4.
The abbreviations used are: RPE, retinal pigment epithelium; A2E, (2-[2,6- dimethyl-8-(2,6,6-trimethyl-1-cyclohexen-1-yl)-1E,3E,5E,7E-octatetraenyl]- 1-(2-hydroxyethyl)-4-[4-methyl-6-(2,6,6-trimethyl-1-cyclohexen-1- yl)-1E,3E,5E-hexatrienyl]-pyridinium); ERG, electroretinogram; HPLC, high pressure liquid chromatography; LD, light damage; RDH, retinol dehydrogenase; ROS, rod outer segment(s); cd, candela.
References
- 1.Matthews RG, Hubbard R, Brown PK, Wald G. J Gen Physiol. 1963;47:215–240. doi: 10.1085/jgp.47.2.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palczewski K. Annu Rev Biochem. 2006;75:743–767. doi: 10.1146/annurev.biochem.75.103004.142743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Filipek S, Stenkamp RE, Teller DC, Palczewski K. Annu Rev Physiol. 2003;65:851–879. doi: 10.1146/annurev.physiol.65.092101.142611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McBee JK, Palczewski K, Baehr W, Pepperberg DR. Prog Retin Eye Res. 2001;20:469–529. doi: 10.1016/s1350-9462(01)00002-7. [DOI] [PubMed] [Google Scholar]
- 5.Lamb TD, Pugh EN., Jr Prog Retin Eye Res. 2004;23:307–380. doi: 10.1016/j.preteyeres.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Thompson DA, Gal A. Prog Retin Eye Res. 2003;22:683–703. doi: 10.1016/s1350-9462(03)00051-x. [DOI] [PubMed] [Google Scholar]
- 7.Janecke AR, Thompson DA, Utermann G, Becker C, Hubner CA, Schmid E, McHenry CL, Nair AR, Ruschendorf F, Heckenlively J, Wissinger B, Nurnberg P, Gal A. Nat Genet. 2004;36:850–854. doi: 10.1038/ng1394. [DOI] [PubMed] [Google Scholar]
- 8.Perrault I, Hanein S, Gerber S, Barbet F, Ducroq D, Dollfus H, Hamel C, Dufier JL, Munnich A, Kaplan J, Rozet JM. Am J Hum Genet. 2004;75:639–646. doi: 10.1086/424889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haeseleer F, Jang GF, Imanishi Y, Driessen CA, Matsumura M, Nelson PS, Palczewski K. J Biol Chem. 2002;277:45537–45546. doi: 10.1074/jbc.M208882200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Belyaeva OV, Korkina OV, Stetsenko AV, Kim T, Nelson PS, Kedishvili NY. Biochemistry. 2005;44:7035–7047. doi: 10.1021/bi050226k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yzer S, Leroy BP, De Baere E, de Ravel TJ, Zonneveld MN, Voesenek K, Kellner U, Ciriano JP, de Faber JT, Rohrschneider K, Roepman R, den Hollander AI, Cruysberg JR, Meire F, Casteels I, van Moll-Ramirez NG, Allikmets R, van den Born LI, Cremers FP. Investig Ophthalmol Vis Sci. 2006;47:1167–1176. doi: 10.1167/iovs.05-0848. [DOI] [PubMed] [Google Scholar]
- 12.Thompson DA, Janecke AR, Lange J, Feathers KL, Hubner CA, McHenry CL, Stockton DW, Rammesmayer G, Lupski JR, Antinolo G, Ayuso C, Baiget M, Gouras P, Heckenlively JR, den Hollander A, Jacobson SG, Lewis RA, Sieving PA, Wissinger B, Yzer S, Zrenner E, Utermann G, Gal A. Hum Mol Genet. 2005;14:3865–3875. doi: 10.1093/hmg/ddi411. [DOI] [PubMed] [Google Scholar]
- 13.Rattner A, Smallwood PM, Nathans J. J Biol Chem. 2000;275:11034–11043. doi: 10.1074/jbc.275.15.11034. [DOI] [PubMed] [Google Scholar]
- 14.Maeda A, Maeda T, Imanishi Y, Kuksa V, Alekseev A, Bronson JD, Zhang H, Zhu L, Sun W, Saperstein DA, Rieke F, Baehr W, Palczewski K. J Biol Chem. 2005;280:18822–18832. doi: 10.1074/jbc.M501757200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohguro H, Chiba S, Igarashi Y, Matsumoto H, Akino T, Palczewski K. Proc Natl Acad Sci U S A. 1993;90:3241–3245. doi: 10.1073/pnas.90.8.3241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gorczyca WA, Polans AS, Surgucheva IG, Subbaraya I, Baehr W, Palczewski K. J Biol Chem. 1995;270:22029–22036. doi: 10.1074/jbc.270.37.22029. [DOI] [PubMed] [Google Scholar]
- 17.Maeda T, Van Hooser JP, Driessen CA, Filipek S, Janssen JJ, Palczewski K. J Neurochem. 2003;85:944–956. doi: 10.1046/j.1471-4159.2003.01741.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Parish CA, Hashimoto M, Nakanishi K, Dillon J, Sparrow J. Proc Natl Acad Sci U S A. 1998;95:14609–14613. doi: 10.1073/pnas.95.25.14609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wenzel A, Reme CE, Williams TP, Hafezi F, Grimm C. J Neurosci. 2001;21:53–58. doi: 10.1523/JNEUROSCI.21-01-00053.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moiseyev G, Chen Y, Takahashi Y, Wu BX, Ma JX. Proc Natl Acad Sci U S A. 2005;102:12413–12418. doi: 10.1073/pnas.0503460102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Redmond TM, Poliakov E, Yu S, Tsai JY, Lu Z, Gentleman S. Proc Natl Acad Sci U S A. 2005;102:13658–13663. doi: 10.1073/pnas.0504167102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jin M, Li S, Moghrabi WN, Sun H, Travis GH. Cell. 2005;122:449–459. doi: 10.1016/j.cell.2005.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grimm C, Wenzel A, Hafezi F, Yu S, Redmond TM, Reme CE. Nat Genet. 2000;25:63–66. doi: 10.1038/75614. [DOI] [PubMed] [Google Scholar]
- 24.Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, Travis GH. Cell. 1999;98:13–23. doi: 10.1016/S0092-8674(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 25.Mata NL, Tzekov RT, Liu X, Weng J, Birch DG, Travis GH. Investig Ophthalmol Vis Sci. 2001;42:1685–1690. [PubMed] [Google Scholar]
- 26.Travis GH, Golczak M, Moise AR, Palczewski K. Annu Rev Pharmacol Toxicol. 2007;47:1–44. doi: 10.1146/annurev.pharmtox.47.120505.105225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu BX, Moiseyev G, Chen Y, Rohrer B, Crouch RK, Ma JX. Investig Ophthalmol Vis Sci. 2004;45:3857–3862. doi: 10.1167/iovs.03-1302. [DOI] [PubMed] [Google Scholar]
- 28.Song MS, Chen W, Zhang M, Napoli JL. J Biol Chem. 2003;278:40079–40087. doi: 10.1074/jbc.M304910200. [DOI] [PubMed] [Google Scholar]
- 29.Driessen C, Winkens H, Haeseleer F, Palczewski K, Janssen J. Vision Res. 2003;43:3075–3079. doi: 10.1016/s0042-6989(03)00483-8. [DOI] [PubMed] [Google Scholar]
- 30.Martras S, Alvarez R, Martinez SE, Torres D, Gallego O, Duester G, Farres J, de Lera AR, Pares X. Eur J Biochem. 2004;271:1660–1670. doi: 10.1111/j.1432-1033.2004.04058.x. [DOI] [PubMed] [Google Scholar]
- 31.Jang GF, McBee JK, Alekseev AM, Haeseleer F, Palczewski K. J Biol Chem. 2000;275:28128–28138. doi: 10.1074/jbc.M004488200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jang GF, Van Hooser JP, Kuksa V, McBee JK, He YG, Janssen JJ, Driessen CA, Palczewski K. J Biol Chem. 2001;276:32456–32465. doi: 10.1074/jbc.M104949200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Driessen CA, Winkens HJ, Hoffmann K, Kuhlmann LD, Janssen BP, Van Vugt AH, Van Hooser JP, Wieringa BE, Deutman AF, Palczewski K, Ruether K, Janssen JJ. Mol Cell Biol. 2000;20:4275–4287. doi: 10.1128/mcb.20.12.4275-4287.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim TS, Maeda A, Maeda T, Heinlein C, Kedishvili N, Palczewski K, Nelson PS. J Biol Chem. 2005;280:8694–8704. doi: 10.1074/jbc.M413172200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Biswas MG, Russell DW. J Biol Chem. 1997;272:15959–15966. doi: 10.1074/jbc.272.25.15959. [DOI] [PubMed] [Google Scholar]
- 36.Palczewski K, Jager S, Buczylko J, Crouch RK, Bredberg DL, Hofmann KP, Asson-Batres MA, Saari JC. Biochemistry. 1994;33:13741–13750. doi: 10.1021/bi00250a027. [DOI] [PubMed] [Google Scholar]
- 37.Saari JC, Garwin GG, Van Hooser JP, Palczewski K. Vision Res. 1998;38:1325–1333. doi: 10.1016/s0042-6989(97)00198-3. [DOI] [PubMed] [Google Scholar]
- 38.Palczewski K, Saari JC. Curr Opin Neurobiol. 1997;7:500–504. doi: 10.1016/s0959-4388(97)80029-3. [DOI] [PubMed] [Google Scholar]
- 39.Fain GL, Matthews HR, Cornwall MC, Koutalos Y. Physiol Rev. 2001;81:117–151. doi: 10.1152/physrev.2001.81.1.117. [DOI] [PubMed] [Google Scholar]
- 40.Fain GL. Prog Brain Res. 2001;131:383–394. doi: 10.1016/s0079-6123(01)31031-2. [DOI] [PubMed] [Google Scholar]
- 41.Horrigan DM, Tetreault ML, Tsomaia N, Vasileiou C, Borhan B, Mierke DF, Crouch RK, Zimmerman AL. J Gen Physiol. 2005;126:453–460. doi: 10.1085/jgp.200509387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCabe SL, Pelosi DM, Tetreault M, Miri A, Nguitragool W, Kovithvathanaphong P, Mahajan R, Zimmerman AL. J Gen Physiol. 2004;123:521–531. doi: 10.1085/jgp.200409011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Imanishi Y, Batten ML, Piston DW, Baehr W, Palczewski K. J Cell Biol. 2004;164:373–383. doi: 10.1083/jcb.200311079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Batten ML, Imanishi Y, Maeda T, Tu DC, Moise AR, Bronson D, Possin D, Van Gelder RN, Baehr W, Palczewski K. J Biol Chem. 2004;279:10422–10432. doi: 10.1074/jbc.M312410200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Redmond TM, Yu S, Lee E, Bok D, Hamasaki D, Chen N, Goletz P, Ma JX, Crouch RK, Pfeifer K. Nat Genet. 1998;20:344–351. doi: 10.1038/3813. [DOI] [PubMed] [Google Scholar]
- 46.Rohrer B, Goletz P, Znoiko S, Ablonczy Z, Ma JX, Redmond TM, Crouch RK. Investig Ophthalmol Vis Sci. 2003;44:310–315. doi: 10.1167/iovs.02-0567. [DOI] [PubMed] [Google Scholar]
- 47.Green DG, Dowling JE, Siegel IM, Ripps H. J Gen Physiol. 1975;65:483–502. doi: 10.1085/jgp.65.4.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alpern M. J Physiol (Lond) 1971;217:447–471. doi: 10.1113/jphysiol.1971.sp009580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jeon CJ, Strettoi E, Masland RH. J Neurosci. 1998;18:8936–8946. doi: 10.1523/JNEUROSCI.18-21-08936.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiao M, Hendrickson A. J Comp Neurol. 2000;425:545–559. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.