

Abstract
Chemotherapy for patients with metastatic colorectal cancer (CRC) is the standard of care, but ultimately nearly all patients develop drug resistance. Understanding the mechanisms that lead to resistance to individual chemotherapeutic agents may help identify novel targets and drugs that will, in turn, improve therapy. Oxaliplatin is a common component combination therapeutic regimen for use in patients with metastatic CRC, but is also used as a component of adjuvant therapy for patients at risk for recurrent disease. In this study, unbiased microRNA array screening revealed that the miR‐203 microRNA is up‐regulated in three of three oxaliplatin‐resistant CRC cell lines, and therefore we investigated the role of miR‐203 in chemoresistance. Exogenous expression of miR‐203 in chemo‐naïve CRC cells induced oxaliplatin resistance. Knockdown of miR‐203 sensitized chemoresistant CRC cells to oxaliplatin. In silico analysis identified ataxia telangiectasia mutated (ATM), a primary mediator of the DNA damage response, as a potential target of miR‐203. ATM mRNA and protein levels were significantly down‐regulated in CRC cells with acquired resistance to oxaliplatin. Using TCGA database, we identified a significant reverse correlation of miR‐203 and ATM expression in CRC tissues. We validated ATM as a bona fide target of miR‐203 in CRC cells. Mutation of the putative miR‐203 binding site in the 3′ untranslated region (3′UTR) of the ATM mRNA abolished the inhibitory effect of miR‐203 on ATM. Furthermore, stable knockdown of ATM induced resistance to oxaliplatin in chemo‐naïve CRC cells. This is the first report of oxaliplatin resistance in CRC cells induced by miR‐203‐mediated suppression of ATM.
Keywords: Chemoresistance, miR‐203, ATM, DNA damage response, Oxaliplatin
Highlights
miR‐203 was up‐regulated in oxaliplatin‐resistant colorectal cancer (CRC) cells.
Knockdown of miR‐203 sensitized chemoresistant CRC cells to oxaliplatin.
ATM levels were significantly down‐regulated in oxaliplatin‐resistant CRC cells.
Stable knockdown of ATM induced resistance to oxaliplatin in chemo‐naïve CRC cells.
Oxaliplatin resistance in CRC cells induced by miR‐203‐mediated suppression of ATM.
1. Introduction
Oxaliplatin, a third‐generation platinum compound, is the first platinum‐based compound to show efficacy in the treatment of colorectal cancer (CRC) (Meyerhardt and Mayer, 2005). Oxaliplatin's use in combination with 5‐fluorouracil plus leucovorin (FOLFOX) and biologics as first‐line therapy for metastatic CRC has led to response rates >50% and a median survival time approaching 2 years (Alberts et al., 2005; Cassidy et al., 2008). FOLFOX has also been found to be effective in the adjuvant setting, leading to an increase in the number of patients who are disease free after surgical resection of stage II/III CRCs in comparison with the use of 5‐fluorouracil plus leucovorin alone (Andre et al., 2004). This finding attests to the clinical significance of oxaliplatin‐containing regimens in management of CRC. Despite this demonstrated efficacy, virtually all metastatic CRCs eventually become resistant to oxaliplatin; the median time to disease progression is ∼8 months (Goldberg et al., 2004).
Oxaliplatin is known to induce formation of intra‐strand guanine–guanine and guanine–adenine DNA links, cell cycle arrest, and death of proliferating cells (Wang and Lippard, 2005). Resistance to oxaliplatin is multifactorial. Hypothetical drug resistance mechanisms include inefficient cellular drug uptake and accumulation (Hector et al., 2001), activation of the antioxidant glutathione system for detoxication (Landriscina et al., 2009; Sau et al., 2010), enhancement of DNA repair (Ahmad, 2010), and up‐regulation of anti‐apoptosis pathways (Dai et al., 2004; Gourdier et al., 2002; Huang and Hung, 2009; Yang et al., 2009). To understand the molecular mechanisms of oxaliplatin resistance in CRC cells, our laboratory established an in vitro chemoresistant CRC cell line model by chronic exposure of human CRC cell lines (HT29, HCT116, and RKO) to increasing doses of oxaliplatin. The selected resistant cells are stably resistant to oxaliplatin and show cross‐resistance to other chemotherapeutic agents. We have used these cell lines to study mechanisms of oxaliplatin resistance. Our previous studies showed that induction of epithelial–mesenchymal transition increased insulin‐like growth factor signaling and that changes in cell metabolism are involved in the development of resistance to oxaliplatin in CRC cells (Bose et al., 2011; Dallas et al., 2009; Yang et al., 2006; Zhou et al., 2012).
MicroRNAs (miRNAs) have been reported to play important roles in tumorigenesis, tumor growth, metastasis, angiogenesis, and drug resistance in both hematopoietic and solid tumors (Calin et al., 2004; Fish et al., 2008; Spizzo et al., 2009; Tokarz and Blasiak, 2012; Volinia et al., 2006; Zhai et al., 2012). The functions of individual miRNAs are highly dependent on tissue and cell context. Aberrant miRNA expression has been reported for several types of malignancies, including CRC (Gottardo et al., 2007; Iorio et al., 2007; Liu et al., 2008; Mathe et al., 2009; Zhai and Ju, 2011). However, the mechanisms of miRNA involvement in the development of acquired drug resistance in CRC cells are largely unknown. It is likely that miRNAs, in response to genotoxic stress, regulate key DNA damage response pathways that mediate survival and escape of cancer cells from drug‐induced apoptosis, thereby making the cells chemoresistant. We hypothesized that deregulation of miRNAs under chemotoxic stress plays a role in oxaliplatin resistance in CRC cells. In this study, using unbiased genome‐wide miRNA array profiling, we identified a single miRNA, miR‐203, that was significantly overexpressed in all three oxaliplatin‐resistant cell lines compared with its expression levels in the chemo‐naïve parental cells. In silico miR‐203 target analysis identified a number of regulators of the DNA damage response pathway. Ataxia telangiectasia mutated kinase (ATM), a central regulator of the DNA damage response pathway, was down‐regulated in the oxaliplatin‐resistant cells. We further investigated the roles of miR‐203 and ATM in inducing an acquired chemoresistant phenotype in CRC cells.
2. Materials and methods
2.1. Cell lines and in vitro chemoresistance model
Human CRC cell lines HT29, RKO, and HCT116 were obtained from the American Type Culture Collection (ATCC, Manassas, VA). Oxaliplatin‐resistant cell lines HT29‐OxR, RKO‐OxR, and HCT116‐OxR were developed in our laboratory as previously described (Yang et al., 2006). Oxaliplatin‐resistant cells were continuously cultured in 2 μM oxaliplatin unless otherwise indicated. In vitro experiments were carried out in triplicate at 70% cell confluence. All cell lines were authenticated by short‐tandem‐repeat sequencing and matched with 100% accuracy to the ATCC database.
2.2. RNA isolation and miRNA microarray profiling
RNAs from parental and resistant CRC cells were extracted using TRIzol reagent (Invitrogen, Carlsbad, CA). miRNA microarray profiling was performed as previously described (Liu et al., 2008) with modifications. Briefly, 5 μg of total RNA was labeled and hybridized to each miRNA microarray (Sequencing and Microarray Facility, The University of Texas MD Anderson Cancer Center, Houston TX) containing quadruplicates of ∼1000 human miRNA probes. Parental and resistant CRC cell RNAs were profiled simultaneously. Slides were scanned with a PerkinElmer ScanArray LX5K scanner (PerkinElmer, Waltham, MA).
2.3. miRNA microarray analysis
miRNA array analyses were conducted as previously described (Schetter et al., 2008). Data were preprocessed by the statistical software R 2.5.0 (R Foundation for Statistical Computing, Vienna, Austria) to remove probes with higher background intensities than foreground and probes with inconsistent measurements across the quadruplicates. The data were normalized by locally weighted scatterplot smoothing and imported into Biometric Research Branch (BRB) array tools 3.5.0 (http://linus.nci.nih.gov/BRB‐ArrayTools.html). After probes with values missing from more than 20% of the arrays were removed from the analysis, 230 probes were left. This filtering method was chosen a priori to eliminate probes whose miRNA expression levels were thought to be unreliable. Class comparison analysis using paired t tests identified miRNAs that were differentially expressed in tumors (P < 0.001). Class prediction algorithms in BRB array tools were used to determine whether miRNA microarray expression patterns could accurately differentiate between resistant and parental CRC cells.
2.4. miR‐mRNA target prediction and pathway analysis
Several algorithms for predicting miRNAs targets and binding sites were developed and are publically available: http://www.microrna.org for miRanda algorithm, http://www.targetscan.org for TargetScan algorithm, http://genie.weizmann.ac.il/pubs/mir07 for PITA algorithm, https://cm.jefferson.edu/rna22v1.0/for RNA22 algorithm, http://diana.cslab.ece.ntua.gr/microT for microT algorithm, and http://dorina.mdc‐berlin.de/rbp_browser/dorina.html for PicTar algorithm. Common mRNAs predicted by at least four out of all the above algorithms to be targeted by miR‐203 in the 3′UTR were further analyzed. Integrated function and pathway analysis were performed using DAVID bioinformatics resources (http://david.abcc.ncifcrf.gov/) and significant features of predicted miRNA targets were clustered.
2.5. qPCR
miR‐203 and ATM gene expression levels in the parental and resistant cells were analyzed by quantitative real‐time PCR (qPCR) using prevalidated TaqMan primers (Applied Biosystems, Carlsbad, CA). U6 small non‐coding RNA was used as an internal control. Total RNA was extracted from tumor cells grown at 60–70% confluence. Reverse transcription PCR was performed with the First‐Strand RT‐PCR kit (Invitrogen). qPCR was performed with the PCR Master Mix (Roche, Branchburg, NJ) on the ABI‐7500 platform (Applied Biosystems).
2.6. Lipofectamine transfection of pre‐miR‐203 and a miR‐203 inhibitor
Precursor miR‐203 (pre‐miR‐203) and anti‐miR‐203 oligonucleotides (for overexpression and down‐regulation of miR‐203, respectively) and their negative controls (all purchased from Exiqon, Woburn, MA) were transfected at a final concentration of 40 nM with Lipofectamine (Invitrogen) according to the manufacturer's protocol. Briefly, 120 pmol of an oligonucleotide and 16 μl of Lipofectamine were mixed in 500 μl of Opti‐MEM medium (Life Technologies, Carlsbad, CA). After 20 min of incubation, the mixture was added to cells at 70% confluence, and the sample was plated on a 6‐well plate (2 ml final volume). Fresh medium was added to cells after 6 h. Twenty‐four hours after transfection, cells were harvested for RNA extraction. Treatment was continued in other cells with oxaliplatin for an additional 48–72 h to assess protein expression levels as described below and the growth‐inhibitory and/or cytotoxic effect of oxaliplatin (MTT assay).
2.7. Lentivirus transduction
Lentiviruses were produced from 293 cells transfected with empty control vector or vector expressing ATM short hairpin RNA (shRNA) (GeneCopoeia, Rockville, MD). Both vectors express green fluorescent protein (GFP) as a marker for positive transduction. Concentrated virus particles and polybrene (10 μl/ml) were added to cells at 50% confluence in 6‐well plates for 48 h. After cell expansion for one passage, GFP‐positive cells were subjected to fluorescence‐activated cell sorting to get pooled clones of cells stably expressing GFP.
2.8. MTT assay
Cell growth inhibition was determined by MTT assay in 96‐well plates. First, cells were seeded at 3000/well in 100 μl medium and kept overnight to allow attachment. The next day, 100 μl of a stock solution of oxaliplatin at a 2× final concentration was added to the cell suspension. After a 72‐hr drug incubation, 40 μl of MTT (3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide; 3 mg/ml) was added to each well, and incubated for 2–4 h. After the supernatant was removed, the formazan precipitates in the cells were dissolved in 100 μl of dimethyl sulfoxide. Absorbance was measured with a Multiskan plate reader (Thermo LabSystems, Beverly, MA) at 570 nm. Fractional survival was plotted against the logarithm of the drug dose, and 50% inhibitory concentrations were calculated by Prism software (GraphPad Software, La Jolla, CA).
2.9. Site‐directed mutagenesis and luciferase assays
The wild‐type 3′ untranslated region (UTR) of ATM (3592 bp) was amplified from a commercial vector (HmiT011736) purchased from GeneCopoeia and cloned into the XbaI site of the pRL vector (Promega, Fitchburg, WI). Based on the pRL‐ATM‐3'UTR construct, mutant ATM 3′UTRs were generated by deleting two regions of 6 nucleotides each (1935–1941 bp and 2297–2302 bp) that are recognized by miR‐203 in silico. Lentiviral miR‐203 and miR‐203 expression inhibitor constructs were obtained from GeneCopoeia. 293 cells were transfected with either miR‐203 or the miR‐203 expression inhibitor construct, the pRL‐ATM‐3'UTR construct, and a control Renilla luciferase construct by using the X‐tremeGENE HP transfection reagent (Roche). Cells were incubated for 24 hr, and luciferase activity was measured with the Dual‐Luciferase System (Promega) according to the manufacturer's instructions.
2.10. Western blot and antibodies
The antibodies to cleaved PARP and cleaved caspase‐3 were purchased from Cell Signaling Technology (Danvers, MA). ATM antibody was purchased from Bethyl Laboratories (Montgomery, TX). β‐Actin antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Whole‐cell lysates were collected from cells cultured at 70% confluence for standard Western blotting.
2.11. Cell viability quantification by trypan blue staining
Cells at the exponential growth stage were treated with oxaliplatin at 20 μM for 48 h. Cell viability was quantified by trypsinizing the cells and staining them with trypan blue dye. Floating cells in the culture medium were collected together with attached cells for cell viability measurement by a Vi‐Cell XR analyzer (Beckman Coulter, Brea, CA).
2.12. Statistical analysis
We downloaded clinical data and mRNA, and miRNA expression data from TCGA (The Cancer Genome Atlas) available through the associated files of the publication (Network, 2012). The Spearman's rank‐order correlation test was applied to measure the strength of the association between miR‐203 levels and mRNA levels of ATM. The analysis was performed in R (version 2.14.2) (http://www.r‐project.org/).
For all in vitro experiments, statistical analyses used Student's t test (Prism). All statistical tests were two‐sided, and P values <0.05 were considered significant. From MTT assay, 50% inhibitory concentrations of oxaliplatin on CRC cells were calculated by Prism software (GraphPad Software, La Jolla, CA).
3. Results
3.1. miR‐203 is overexpressed in oxaliplatin‐resistant CRC cells
We compared the miRNA expression profiles of the three parental (chemo‐naïve) CRC cell lines HT29, HCT116, and RKO with those of their oxaliplatin‐resistant derivative cells by using miRNA microarray chip analysis with ∼1200 pre‐miRNA and mature human miRNA probes (Suppl. Table S1–S3). Figure 1A lists the 10 miRNAs expressed in oxaliplatin‐resistant cells at levels different from their expression levels in parental cells. Of note, miR‐203 was expressed at elevated (∼2–6 fold) levels in all three oxaliplatin‐resistant cell lines. Using different sets of biological samples, we validated the increased expression levels of miR‐203 by qPCR in all three chemoresistant cell lines compared with those in the parental cells (Figure 1B). Since miR‐203 was the only miRNA up‐regulated in all three oxaliplatin‐resistant CRC cell lines, we hypothesized that miR‐203 plays a role in development of acquired resistance to oxaliplatin in CRC cells.
Figure 1.
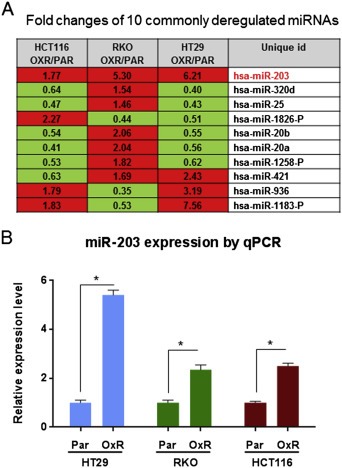
miR‐203 is overexpressed in oxaliplatin‐resistant (OXR) CRC cells. A. The 10 common miRNAs differentially expressed in all three OXR cell lines compared with their expression levels in the parental (PAR) cell lines. miR‐203 is up‐regulated in all three OXR cell lines. B. qPCR validation of miR‐203 expression levels significantly higher in oxaliplatin‐resistant (OXR) cells. *P < 0.05. Results are representative of three experiments.
3.2. miR‐203 induces oxaliplatin resistance in CRC cells
To confirm the functional role of miR‐203 in inducing chemoresistance, we altered the levels of miR‐203 in parental HT29 and HT29‐OxR cells by introducing pre‐miR‐203 oligonucleotides and anti‐miR‐203 oligonucleotides, respectively. Increased levels of miR‐203 protected the chemo‐naïve parental HT29 cells from oxaliplatin treatment, as evidenced by a right shift of the growth inhibition curve (Figure 2A and B) and reduced levels of cleaved caspase‐3 and PARP (Figure 2C). Conversely, inhibition of miR‐203 resensitized HT29‐OxR cells to oxaliplatin treatment, as evidenced by a left shift of the growth inhibition curve (Figure 3A and B) and increased cleavage of caspase‐3 and PARP (Figure 3C).
Figure 2.
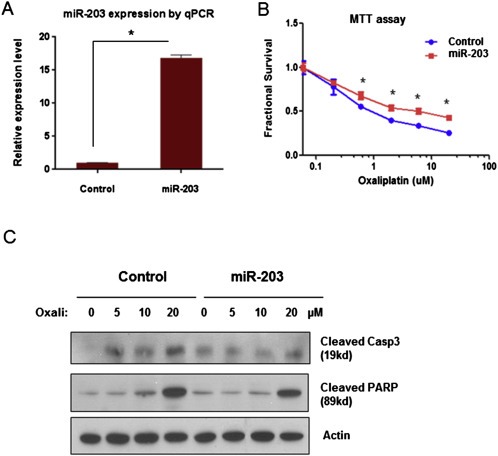
Pre‐miR‐203 induced oxaliplatin resistance in HT29 cells. A. miR‐203 expression level was significantly increased after transient transfection of pre‐miR‐203 in HT29 cells as measured by qPCR. B. Overexpression of miR‐203 induced by pre‐miR‐203 transfection significantly decreased the growth‐inhibitory effect of oxaliplatin in HT29 cells, as measured by MTT assay. C. The apoptosis‐inducing effect of oxaliplatin (Oxali) at 48 h post treatment, as reflected by induction of cleaved PARP and caspase‐3 (Casp3) by Western blotting. The effect was partially blocked by overexpression of miR‐203 through transient transfection of pre‐miR‐203 in HT29 cells. *P < 0.05. Results are representative of three experiments.
Figure 3.
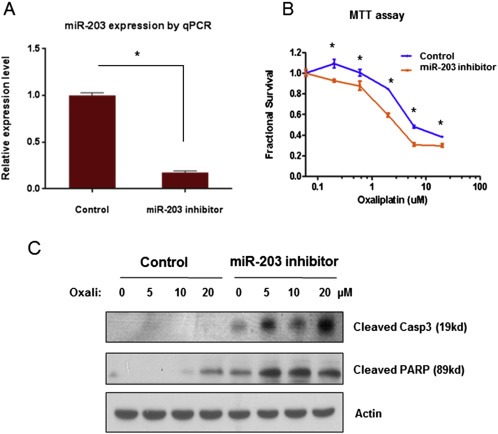
Anti‐miR‐203 reversed oxaliplatin resistance in HT29‐OxR cells. A. miR‐203 expression level was significantly decreased after transient transfection of anti‐miR‐203 in HT29‐OxR cells as measured by qPCR. B. Down‐regulation of miR‐203 induced by anti‐miR‐203 transfection significantly slowed down HT29‐OxR cells growth under oxaliplatin treatment by MTT assay. C. Down‐regulation of miR‐203 induced by transient transfection of anti‐miR‐203 resensitized HT29‐OxR cells to oxaliplatin (Oxali) at 48 h post treatment, as demonstrated by induction of cleaved PARP and caspase‐3 by Western blotting. *P < 0.05. Results are representative of three experiments.
3.3. Correlation of ATM and miR‐203 levels in oxaliplatin‐resistant cells
Using six in silico prediction software and a functional pathway analysis database (DAVID, http://david.abcc.ncifcrf.gov), we identified a number of potential miR‐203 targets (Suppl. Table S4). Among the predicted miR‐203 targets, two of which are in the 3′UTR region of Ataxia telangiectasia mutated (ATM), a central regulator of the DNA damage response pathway. The potential target sites for interaction between miR‐203 and ATM were retrieved from the data bases and presented in Figure 4A. During the process of developing chemoresistance to oxaliplatin, cancer cells are likely to acquire an enhanced ability to repair damaged DNA or to suppress activation of the DNA damage response pathway and thereby avoid apoptosis. This mechanistic link between the DNA damage response pathway and chemoresistance prompted us to examine whether ATM protein expression is correlated with miR‐203 levels in oxaliplatin‐resistant cells. We found that both the mRNA levels and the protein expression levels of ATM are down‐regulated in HT29‐OxR and HCT116‐OxR cells compared with the levels in their parental, chemo‐naïve cells (Figure 4B and C). Thus, ATM is inhibited at both the transcriptional level and the translational level when chemoresistance occurs. We also observed that miR‐203 overexpression inhibited the oxaliplatin‐induced formation of γ‐H2AX foci in HT‐29 cells, similar to what was observed in the cells treated with ATM inhibitor (Supplemental Figure). To investigate the impact of miR‐203‐ATM interactions on clinical outcome of colon cancer patients, we used the Spearman Rank correlation test and identified an inverse relationship between miR‐203 and ATM gene expression (coefficient = − 0.21, p‐value = 0.017) in tumor samples of the patients included in the TCGA colon and rectum study (Network, 2012). The list of tumor samples used for the correlation analysis is provided in the Supplementary Table 5.
Figure 4.
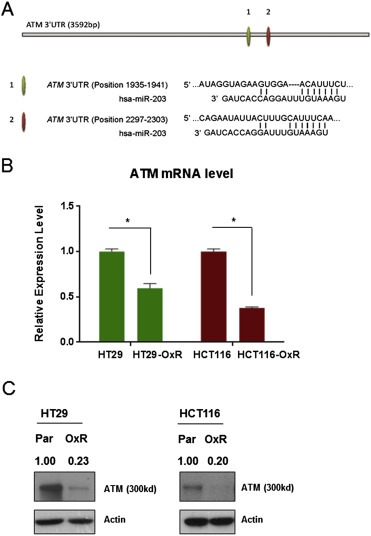
ATM expression is suppressed in oxaliplatin‐resistant CRC cells. A. Illustration of the two in silico predicted miR‐203 targeting sites in the ATM 3′UTR region. B. HT29‐OxR and HCT116‐OxR cells express significantly decreased ATM mRNA levels compared with the levels expressed by their chemo‐naïve parental cell lines as measured by qPCR. C. HT29‐OxR and HCT116‐OxR cells express decreased ATM protein levels compared from those expressed by their chemo‐naïve parental (Par) cell lines by Western blotting. *P < 0.05. Results are representative of three experiments.
3.4. miR‐203 regulates ATM protein expression by binding to the ATM 3′UTR
To confirm that miR‐203 regulates ATM protein expression by directly binding to the ATM 3′UTR, we generated two luciferase reporter constructs containing a specific deletion mutation at each putative miR‐203 binding site (Figure 5A). We first transiently transfected 293 cells with either the mutant or the wild‐type ATM 3′UTR‐luciferase plasmid. Next, we co‐transfected these 293 cells with either pre‐miR‐203 or anti‐miR‐203 oligonucleotides to test the functional link between miR‐203 and the ATM 3′UTR by the luciferase assay. As shown in Figure 5B, when cells were transfected with the wild‐type ATM 3′UTR (isoform b), co‐transfection of pre‐miR‐203 inhibited luciferase activity and co‐transfection of anti‐miR‐203 enhanced it. In contrast, the effects of pre‐miR‐203 and anti‐miR203 were eliminated in cells transfected with a mutant ATM 3′UTR (mutant 2). However, the ATM 3′UTR (isoform a) containing another putative miR‐230 binding site (mutant 1) did not show similar luciferase activity after co‐transfection with either the pre‐miR‐203 or the anti‐miR‐203 oligonucleotide. These findings suggest that miR‐203 binds directly to one of the two putative ATM 3′UTR regions (isoform b), as predicted by the in silico model. Transient transfection of pre‐miR‐203 decreased ATM protein expression in HT29 cells, and transfection of anti‐miR‐203 restored ATM protein expression in HT29‐OxR cells.
Figure 5.
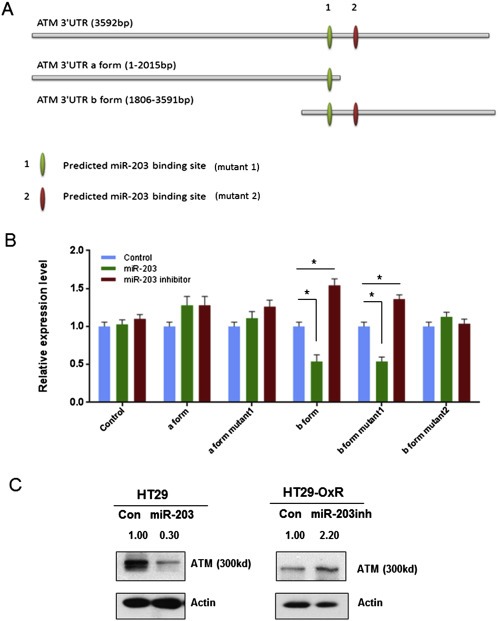
miR‐203 regulates ATM protein expression by directly binding to the ATM mRNA 3′UTR. A. Site‐directed mutagenesis targeting two potential miR‐203 binding sites (mutants 1 and 2) on an ATM mRNA 3′UTR‐luciferase construct. The ATM 3′UTR was studied in two forms (a and b) to cover the full length of the 3′UTR region. B. Luciferase activities of ATM mRNA 3′UTR‐luciferase constructs containing either the wild type or a deletion mutant for miR‐203 binding sites after co‐transfection of either pre‐miR‐203 or anti‐miR‐203 in 293 cells. miR‐203 significantly decreased the ATM 3′UTR (form b) luciferase activity, and miR‐203 inhibitor significantly enhanced the ATM 3′UTR (form b) luciferase activity. Both effects were blocked by mutant 1, but not by mutant 2. C. ATM protein expression levels are regulated directly by miR‐203, as reflected by decreased ATM expression in HT29 cells after transient transfection of pre‐miR‐203 and increased ATM expression in HT29‐OxR cells after miR‐203 inhibitor transfection. *P < 0.05. Results are representative of three experiments.
3.5. Suppression of ATM expression induces oxaliplatin resistance in chemo‐naïve CRC cells
To further confirm that ATM is one of the downstream effectors of miR‐203‐mediated oxaliplatin resistance, we used lentivirus transduction with HT29 cells and a construct expressing human ATM shRNA to generate HT29‐ATMsh‐GFP cells, in which ATM is stably knocked down. ATM protein expression was significantly down‐regulated in HT29‐ATMsh‐GFP cells (Figure 6A), and these cells were less sensitive to oxaliplatin‐induced growth inhibition than HT29‐GFP cells, as reflected by a right shift of the growth inhibition curve (Figure 6B). Suppression of ATM blocked oxaliplatin‐induced apoptosis in HT29‐ATMsh‐GFP cells, as evidenced by decreased levels of cleaved PARP and caspase‐3 (Figure 6C). Cell survival assays showed significantly higher survival of HT29‐ATMsh‐GFP cells than that of HT29‐GFP cells after oxaliplatin treatment for 48 h. These results indicate that ATM knockdown induced chemoresistance to oxaliplatin in chemo‐naïve HT29 cells.
Figure 6.
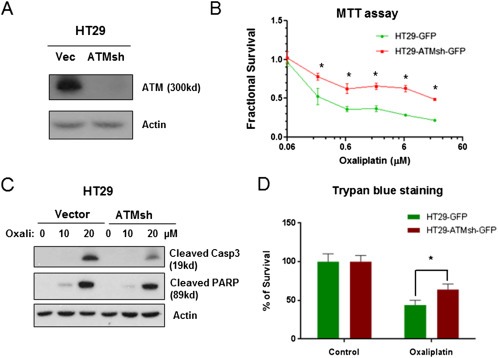
ATM knockdown induced resistance to oxaliplatin in chemo‐naïve CRC cells. A. Western blotting showed a stable knockdown of ATM expression in HT29 cells. B. A lentiviral small interfering RNA‐induced knockdown of ATM led to increased resistance to oxaliplatin, as shown by a significantly right‐shift of the growth inhibition curve of oxaliplatin in HT29 cells 72 h post treatment (MTT assay). C. Knockdown of ATM (Sh‐ATM) induced resistance to oxaliplatin (Oxali), as shown by decreased levels of cleaved PARP and caspase‐3 (Casp3) at 48 h post treatment with 20 μM oxaliplatin. D. ATM knockdown cells (HT29‐ATMsh‐GFP) had significantly lower percentage of cell death than control cells (HT29‐GFP) at 48 h post treatment with 20 μM oxaliplatin. Quantification of cell death was by trypan blue staining. *P < 0.05. Results are representative of three experiments.
4. Discussion
Metastatic CRC leads to the death of ∼50,000 patients a year in the U.S. due to primary or acquired resistance to chemotherapy (Davies and Goldberg, 2008). It is critical to delineate drug resistance mechanisms to commonly used therapeutic agents such as oxaliplatin if we are to improve survival in patients with CRC. The mechanisms underlying the cytotoxic effects of oxaliplatin have not been completely elucidated. Our study established a link between miR‐203 and oxaliplatin resistance. This link likely involves an ATM‐mediated mechanism. First, by miRNA microarray profiling, we showed that only miR‐203 is commonly overexpressed in three CRC cell lines with acquired resistance to oxaliplatin. Second, we showed that overexpression of miR‐203 protects chemo‐naïve CRC cells from cell death due to oxaliplatin and that suppression of miR‐203 sensitizes resistant CRC cells to oxaliplatin. Third, we demonstrated that miR‐203 regulates the DNA damage response mediator ATM by directly binding to the ATM 3′UTR site. Finally, we showed that ATM knockdown leads to oxaliplatin resistance in chemo‐naïve CRC cells.
miR‐203 promotes epidermal differentiation by restricting proliferative potential (Yi et al., 2008). Its expression is regulated by ZEB1, similar to the miR‐200 family, which is known to play an important role in regulation of epithelial–mesenchymal transition (EMT) by suppression of several key regulators of EMT such as ZEB1 and SP1 (Gregory et al., 2008, 2011). miR‐203 has a physiological role in skin cell differentiation by promoting the cell cycle exit and suppression of self‐renewal of the skin progenitor cells. In vivo targets of miR‐203 are highly enriched in regulation of cell cycle and cell division, as well as in response to DNA damage (Jackson et al., 2013; Viticchie et al., 2012). Previous studies showed that miR‐203 expression is negatively associated with survival in tumors arising from the oral cavity, esophagus, pancreas, colon, and ovary (Greither et al., 2010; Iorio et al., 2007; Mathe et al., 2009), suggesting that miR‐203 is involved in tumor growth and metastasis. The role of miR‐203 is disease and tissue‐type specific; it has anti‐oncogenic effect in prostate cancer (Boll et al., 2013; Viticchie et al., 2011) and melanoma (Noguchi et al., 2012). Our findings in this study indicate that miR‐203 induces drug resistance through an ATM‐mediated mechanism that has not been previously reported.
The DNA damage response plays critical roles in tumorigenesis and drug resistance (Bartkova et al., 2005; Gilbert and Hemann, 2010; Gorgoulis et al., 2005). Oxaliplatin causes genotoxic stress and apoptosis by inducing DNA damages primarily in the form of single‐strand and double‐strand DNA breaks. Once the DNA damage response occurs, one of the major mediators, ATM, could be activated to induce cell cycle arrest for cell fate determination: to repair the DNA damage or induce apoptosis. Recent studies showed that miRNAs contribute to the DNA damage response by regulating their target gene expression. For example, miR‐24 inhibits the expression of the histone variant H2AX, which plays a key role in the repair of double‐strand breaks (Lal et al., 2009). In addition, DNA damage can also regulate miRNA expression at the transcriptional and posttranscriptional levels (Wan et al., 2013; Zhang et al., 2011). Our data showed that miR‐203 negatively regulates ATM by binding to a conserved site of the ATM 3′UTR. The deregulation of the miR‐203/ATM pathway in CRC cells is mechanistically involved in the resistance to oxaliplatin.
Given the complexity of miRNA regulation on gene expression and biological function in physiological and pathological conditions, drug resistance mechanisms are unlikely to be caused by deregulation of a single miRNA. In our study, we identified multiple microRNA deregulations in oxaliplatin‐resistant CRC cells (Figure 1A). miR‐203 is the most significant one among this group of genes. Other candidate miRNAs may also play roles in oxaliplatin resistance in an independent or collaborative manner. For example, it is reported that overexpression of miR‐421 induces resistance to apoptosis in nasopharyngeal carcinoma (Chen et al., 2013). We observed miR‐421 is up‐regulated ∼2‐fold in two of the three oxaliplatin‐resistant CRC cells suggesting a possible role of this miRNA in chemoresistance (Figure 1A). Another example is that there is a reverse correlation of miR‐20a and miR‐203 in two of the three oxaliplatin resistant cancer cell lines, which is consistent with a report on the inverse correlation of these two miRNAs in cervical cancer (Zhao et al., 2013). Interestingly, miR‐203 was reported to play a role in reversing drug resistance to paclitaxel through an AKT‐mediated mechanism in chemo‐naïve HT29 cells (Li et al., 2011). Paclitaxel is a cell cycle‐specific chemotherapeutic drug that primarily induces M‐phase associated apoptosis (Jordan and Wilson, 2004). miR‐203 is known to induce cell growth delay and senescence, thus may function as a chemosensitization factor to cell cycle‐specific drugs such as paclitaxel. Our study used a different model and focused on a DNA‐damaging agent that is more clinically relevant, as nearly all patients with metastatic CRC will receive oxaliplatin. We previously observed a significantly slower cell proliferation in the oxaliplatin‐resistant cells compared with the chemo‐naïve cells (Zhou et al., 2012), which is consistent with the role of miR‐203 in inducing cell‐cycle arrest and senescence. In this study, we specifically identified a mechanistic link of miR‐203 to oxaliplatin resistance through ATM‐mediated mechanism.
In summary, our study showed that miR‐203 overexpression is a critical factor in the mechanism by which CRC cells resist the cytotoxicity of oxaliplatin. Our findings have significant translational potential for identifying novel approaches to overcoming drug resistance related to miRNA deregulation. In addition, miR‐203 has the potential as a predictive biomarker for therapy in regimens utilizing oxaliplatin.
Financial support
X.L. was supported by the National Institutes of Health R01 grant CA136549. L.M.E. was supported by the William C. Liedtke, Jr., Chair in Cancer Research. The work described was supported by MD Anderson Cancer Center Support Grant CA016672.
Supporting information
The following is the supplementary data related to this article:
Supplementary data
Acknowledgments
We thank Li (Lily) Huang (Subcloning Core Facility, Department of Cancer Biology) for assistance with lentivirus particle production, Arthur Gelmis (Department of Scientific Publications) and Rita Hernandez (Department of Surgical Oncology) for editorial assistance.
Supplementary data 1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2013.09.004.
Zhou Yunfei, Wan Guohui, Spizzo Riccardo, Ivan Cristina, Mathur Rohit, Hu Xiaoxiao, Ye Xiangcang, Lu Jia, Fan Fan, Xia Ling, Calin George A.,Ellis Lee M. and Lu Xiongbin, (2014), miR‐203 induces oxaliplatin resistance in colorectal cancer cells by negatively regulating ATM kinase, Molecular Oncology, 8, doi: 10.1016/j.molonc.2013.09.004.
Contributor Information
Lee M. Ellis, Email: lellis@mdanderson.org
Xiongbin Lu, Email: xlu2@mdanderson.org.
References
- Ahmad, S. , 2010. Platinum-DNA interactions and subsequent cellular processes controlling sensitivity to anticancer platinum complexes. Chem. Biodiversity. 7, 543–566. [DOI] [PubMed] [Google Scholar]
- Alberts, S.R. , Horvath, W.L. , Sternfeld, W.C. , Goldberg, R.M. , Mahoney, M.R. , Dakhil, S.R. , Levitt, R. , Rowland, K. , Nair, S. , Sargent, D.J. , Donohue, J.H. , 2005. Oxaliplatin, fluorouracil, and leucovorin for patients with unresectable liver-only metastases from colorectal cancer: a North Central Cancer Treatment Group phase II study. J. Clin. Oncol. Official J. Am. Soc. Clin. Oncol.. 23, 9243–9249. [DOI] [PubMed] [Google Scholar]
- Andre, T. , Boni, C. , Mounedji-Boudiaf, L. , Navarro, M. , Tabernero, J. , Hickish, T. , Topham, C. , Zaninelli, M. , Clingan, P. , Bridgewater, J. , Tabah-Fisch, I. , de Gramont, A. , 2004. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N. Engl. J. Med.. 350, 2343–2351. [DOI] [PubMed] [Google Scholar]
- Bartkova, J. , Horejsi, Z. , Koed, K. , Kramer, A. , Tort, F. , Zieger, K. , Guldberg, P. , Sehested, M. , Nesland, J.M. , Lukas, C. , Orntoft, T. , Lukas, J. , Bartek, J. , 2005. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 434, 864–870. [DOI] [PubMed] [Google Scholar]
- Boll, K. , Reiche, K. , Kasack, K. , Morbt, N. , Kretzschmar, A.K. , Tomm, J.M. , Verhaegh, G. , Schalken, J. , von Bergen, M. , Horn, F. , Hackermuller, J. , 2013. MiR-130a, miR-203 and miR-205 jointly repress key oncogenic pathways and are downregulated in prostate carcinoma. Oncogene. 32, 277–285. [DOI] [PubMed] [Google Scholar]
- Bose, D. , Zimmerman, L.J. , Pierobon, M. , Petricoin, E. , Tozzi, F. , Parikh, A. , Fan, F. , Dallas, N. , Xia, L. , Gaur, P. , Samuel, S. , Liebler, D.C. , Ellis, L.M. , 2011. Chemoresistant colorectal cancer cells and cancer stem cells mediate growth and survival of bystander cells. Br. J. Cancer.. 105, 1759–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calin, G.A. , Sevignani, C. , Dumitru, C.D. , Hyslop, T. , Noch, E. , Yendamuri, S. , Shimizu, M. , Rattan, S. , Bullrich, F. , Negrini, M. , Croce, C.M. , 2004. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. U S A. 101, 2999–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy, J. , Clarke, S. , Diaz-Rubio, E. , Scheithauer, W. , Figer, A. , Wong, R. , Koski, S. , Lichinitser, M. , Yang, T.S. , Rivera, F. , Couture, F. , Sirzen, F. , Saltz, L. , 2008. Randomized phase III study of capecitabine plus oxaliplatin compared with fluorouracil/folinic acid plus oxaliplatin as first-line therapy for metastatic colorectal cancer. J. Clin. Oncol. Official J. Am. Soc. Clin. Oncol.. 26, 2006–2012. [DOI] [PubMed] [Google Scholar]
- Chen, L. , Tang, Y. , Wang, J. , Yan, Z. , Xu, R. , 2013. miR-421 induces cell proliferation and apoptosis resistance in human nasopharyngeal carcinoma via downregulation of FOXO4. Biochem. Biophys. Res. Commun.. 435, 745–750. [DOI] [PubMed] [Google Scholar]
- Dai, Z. , Huang, Y. , Sadee, W. , 2004. Growth factor signaling and resistance to cancer chemotherapy. Curr. Top. Med. Chem.. 4, 1347–1356. [DOI] [PubMed] [Google Scholar]
- Dallas, N.A. , Xia, L. , Fan, F. , Gray, M.J. , Gaur, P. , van Buren, G. , Samuel, S. , Kim, M.P. , Lim, S.J. , Ellis, L.M. , 2009. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res.. 69, 1951–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, J.M. , Goldberg, R.M. , 2008. First-line therapeutic strategies in metastatic colorectal cancer. Oncol. (Williston Park, N.Y). 22, 1470–1479. [PubMed] [Google Scholar]
- Fish, J.E. , Santoro, M.M. , Morton, S.U. , Yu, S. , Yeh, R.F. , Wythe, J.D. , Ivey, K.N. , Bruneau, B.G. , Stainier, D.Y. , Srivastava, D. , 2008. miR-126 regulates angiogenic signaling and vascular integrity. Develop. Dev. Cell.Cell. 15, 272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert, L.A. , Hemann, M.T. , 2010. DNA damage-mediated induction of a chemoresistant niche. Cell. 143, 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg, R.M. , Sargent, D.J. , Morton, R.F. , Fuchs, C.S. , Ramanathan, R.K. , Williamson, S.K. , Findlay, B.P. , Pitot, H.C. , Alberts, S.R. , 2004. A randomized controlled trial of fluorouracil plus leucovorin, irinotecan, and oxaliplatin combinations in patients with previously untreated metastatic colorectal cancer. J. Clin. Oncol. Official J. Am. Soc. Clin. Oncol.. 22, 23–30. [DOI] [PubMed] [Google Scholar]
- Gorgoulis, V.G. , Vassiliou, L.V. , Karakaidos, P. , Zacharatos, P. , Kotsinas, A. , Liloglou, T. , Venere, M. , Ditullio, R.A. , Kastrinakis, N.G. , Levy, B. , Kletsas, D. , Yoneta, A. , Herlyn, M. , Kittas, C. , Halazonetis, T.D. , 2005. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 434, 907–913. [DOI] [PubMed] [Google Scholar]
- Gottardo, F. , Liu, C.G. , Ferracin, M. , Calin, G.A. , Fassan, M. , Bassi, P. , Sevignani, C. , Byrne, D. , Negrini, M. , Pagano, F. , Gomella, L.G. , Croce, C.M. , Baffa, R. , 2007. Micro-RNA profiling in kidney and bladder cancers. Urologic. Oncol.. 25, 387–392. [DOI] [PubMed] [Google Scholar]
- Gourdier, I. , Del Rio, M. , Crabbe, L. , Candeil, L. , Copois, V. , Ychou, M. , Auffray, C. , Martineau, P. , Mechti, N. , Pommier, Y. , Pau, B. , 2002. Drug specific resistance to oxaliplatin is associated with apoptosis defect in a cellular model of colon carcinoma. FEBS Lett.. 529, 232–236. [DOI] [PubMed] [Google Scholar]
- Gregory, P.A. , Bert, A.G. , Paterson, E.L. , Barry, S.C. , Tsykin, A. , Farshid, G. , Vadas, M.A. , Khew-Goodall, Y. , Goodall, G.J. , 2008. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell. Biol.. 10, 593–601. [DOI] [PubMed] [Google Scholar]
- Gregory, P.A. , Bracken, C.P. , Smith, E. , Bert, A.G. , Wright, J.A. , Roslan, S. , Morris, M. , Wyatt, L. , Farshid, G. , Lim, Y.Y. , Lindeman, G.J. , Shannon, M.F. , Drew, P.A. , Khew-Goodall, Y. , Goodall, G.J. , 2011. An autocrine TGF-beta/ZEB/miR-200 signaling network regulates establishment and maintenance of epithelial-mesenchymal transition. Mol. Biol. Cell. 22, 1686–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greither, T. , Grochola, L.F. , Udelnow, A. , Lautenschlager, C. , Wurl, P. , Taubert, H. , 2010. Elevated expression of microRNAs 155, 203, 210 and 222 in pancreatic tumors is associated with poorer survival. Int. J. Cancer. 126, 73–80. [DOI] [PubMed] [Google Scholar]
- Hector, S. , Bolanowska-Higdon, W. , Zdanowicz, J. , Hitt, S. , Pendyala, L. , 2001. In vitro studies on the mechanisms of oxaliplatin resistance. Cancer. Chemother. Pharmacol.. 48, 398–406. [DOI] [PubMed] [Google Scholar]
- Huang, W.C. , Hung, M.C. , 2009. Induction of Akt activity by chemotherapy confers acquired resistance. J. Formos. Medical Assoc. – Taiwan yi zhi. 108, 180–194. [DOI] [PubMed] [Google Scholar]
- Iorio, M.V. , Visone, R. , Di Leva, G. , Donati, V. , Petrocca, F. , Casalini, P. , Taccioli, C. , Volinia, S. , Liu, C.G. , Alder, H. , Calin, G.A. , Menard, S. , Croce, C.M. , 2007. MicroRNA signatures in human ovarian cancer. Cancer Res.. 67, 8699–8707. [DOI] [PubMed] [Google Scholar]
- Jackson, S.J. , Zhang, Z. , Feng, D. , Flagg, M. , O'Loughlin, E. , Wang, D. , Stokes, N. , Fuchs, E. , Yi, R. , 2013. Rapid and widespread suppression of self-renewal by microRNA-203 during epidermal differentiation. Development. 140, 1882–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan, M.A. , Wilson, L. , 2004. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer.. 4, 253–265. [DOI] [PubMed] [Google Scholar]
- Lal, A. , Pan, Y. , Navarro, F. , Dykxhoorn, D.M. , Moreau, L. , Meire, E. , Bentwich, Z. , Lieberman, J. , Chowdhury, D. , 2009. miR-24-mediated downregulation of H2AX suppresses DNA repair in terminally differentiated blood cells. Nature Struct. Mol. Biol.. 16, 492–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landriscina, M. , Maddalena, F. , Laudiero, G. , Esposito, F. , 2009. Adaptation to oxidative stress, chemoresistance, and cell survival. Antioxid. Redox. Signal.. 11, 2701–2716. [DOI] [PubMed] [Google Scholar]
- Li, J. , Chen, Y. , Zhao, J. , Kong, F. , Zhang, Y. , 2011. miR-203 reverses chemoresistance in p53-mutated colon cancer cells through downregulation of Akt2 expression. Cancer. Lett.. 304, 52–59. [DOI] [PubMed] [Google Scholar]
- Liu, C.G. , Calin, G.A. , Volinia, S. , Croce, C.M. , 2008. MicroRNA expression profiling using microarrays. Nature Protocol.. 3, 563–578. [DOI] [PubMed] [Google Scholar]
- Mathe, E.A. , Nguyen, G.H. , Bowman, E.D. , Zhao, Y. , Budhu, A. , Schetter, A.J. , Braun, R. , Reimers, M. , Kumamoto, K. , Hughes, D. , Altorki, N.K. , Casson, A.G. , Liu, C.G. , Wang, X.W. , Yanaihara, N. , Hagiwara, N. , Dannenberg, A.J. , Miyashita, M. , Croce, C.M. , Harris, C.C. , 2009. MicroRNA expression in squamous cell carcinoma and adenocarcinoma of the esophagus: associations with survival. Clin Cancer Res.. 15, 6192–6200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerhardt, J.A. , Mayer, R.J. , 2005. Systemic therapy for colorectal cancer. N. Engl. J. Med.. 352, 476–487. [DOI] [PubMed] [Google Scholar]
- Network, C.G.A. , 2012. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 487, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi, S. , Mori, T. , Otsuka, Y. , Yamada, N. , Yasui, Y. , Iwasaki, J. , Kumazaki, M. , Maruo, K. , Akao, Y. , 2012. Anti-oncogenic microRNA-203 induces senescence by targeting E2F3 protein in human melanoma cells. J. Biol. Chem.. 287, 11769–11777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sau, A. , Pellizzari Tregno, F. , Valentino, F. , Federici, G. , Caccuri, A.M. , 2010. Glutathione transferases and development of new principles to overcome drug resistance. Arch. Biochem. Biophys.. 500, 116–122. [DOI] [PubMed] [Google Scholar]
- Schetter, A.J. , Leung, S.Y. , Sohn, J.J. , Zanetti, K.A. , Bowman, E.D. , Yanaihara, N. , Yuen, S.T. , Chan, T.L. , Kwong, D.L. , Au, G.K. , Liu, C.G. , Calin, G.A. , Croce, C.M. , Harris, C.C. , 2008. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. Jama. 299, 425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spizzo, R. , Nicoloso, M.S. , Croce, C.M. , Calin, G.A. , 2009. SnapShot: MicroRNAs in cancer. Cell. 137, 586 e581 [DOI] [PubMed] [Google Scholar]
- Tokarz, P. , Blasiak, J. , 2012. The role of MiRNA in metastatic colorectal cancer and its significance in cancer prognosis and treatment. Acta. Biochim. Pol.. 59, 467–474. [PubMed] [Google Scholar]
- Viticchie, G. , Lena, A.M. , Cianfarani, F. , Odorisio, T. , Annicchiarico-Petruzzelli, M. , Melino, G. , Candi, E. , 2012. MicroRNA-203 contributes to skin re-epithelialization. Cell Death Dis.. 3, e435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viticchie, G. , Lena, A.M. , Latina, A. , Formosa, A. , Gregersen, L.H. , Lund, A.H. , Bernardini, S. , Mauriello, A. , Miano, R. , Spagnoli, L.G. , Knight, R.A. , Candi, E. , Melino, G. , 2011. MiR-203 controls proliferation, migration and invasive potential of prostate cancer cell lines. Cell Cycle. 10, 1121–1131. [DOI] [PubMed] [Google Scholar]
- Volinia, S. , Calin, G.A. , Liu, C.G. , Ambs, S. , Cimmino, A. , Petrocca, F. , Visone, R. , Iorio, M. , Roldo, C. , Ferracin, M. , Prueitt, R.L. , Yanaihara, N. , Lanza, G. , Scarpa, A. , Vecchione, A. , Negrini, M. , Harris, C.C. , Croce, C.M. , 2006. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. U S A. 103, 2257–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan, G. , Zhang, X. , Langley, R.R. , Liu, Y. , Hu, X. , Han, C. , Peng, G. , Ellis, L.M. , Jones, S.N. , Lu, X. , 2013. DNA-damage-induced nuclear export of precursor microRNAs is regulated by the ATM-AKT pathway. Cell Rep. 3, 2100–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, D. , Lippard, S.J. , 2013. Cellular processing of platinum anticancer drugs. Nat. Rev. Drug. Discov.. 4, 307–320. [DOI] [PubMed] [Google Scholar]
- Yang, A.D. , Fan, F. , Camp, E.R. , van Buren, G. , Liu, W. , Somcio, R. , Gray, M.J. , Cheng, H. , Hoff, P.M. , Ellis, L.M. , 2006. Chronic oxaliplatin resistance induces epithelial-to-mesenchymal transition in colorectal cancer cell lines. Clin Cancer Res.. 12, 4147–4153. [DOI] [PubMed] [Google Scholar]
- Yang, S.Y. , Sales, K.M. , Fuller, B. , Seifalian, A.M. , Winslet, M.C. , 2009. Apoptosis and colorectal cancer: implications for therapy. Trends. Mol. Med.. 15, 225–233. [DOI] [PubMed] [Google Scholar]
- Yi, R. , Poy, M.N. , Stoffel, M. , Fuchs, E. , 2008. A skin microRNA promotes differentiation by repressing ‘stemness’. Nature. 452, 225–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai, H. , Fesler, A. , Ju, J. , 2012. MicroRNA: a third dimension in autophagy. Cell Cycle. 12, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai, H. , Ju, J. , 2011. Implications of microRNAs in colorectal cancer development, diagnosis, prognosis, and therapeutics. Front. Genet.. 2, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, X. , Wan, G. , Berger, F.G. , He, X. , Lu, X. , 2011. The ATM kinase induces microRNA biogenesis in the DNA damage response. Mol. Cell. 41, 371–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, S. , Yao, D.S. , Chen, J.Y. , Ding, N. , 2013. Aberrant expression of miR-20a and miR-203 in cervical cancer. Asian Pac. J. Cancer Prevent.: APJCP. 14, 2289–2293. [DOI] [PubMed] [Google Scholar]
- Zhou, Y. , Tozzi, F. , Chen, J. , Fan, F. , Xia, L. , Wang, J. , Gao, G. , Zhang, A. , Xia, X. , Brasher, H. , Widger, W. , Ellis, L.M. , Weihua, Z. , 2008. Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells. Cancer Res.. 72, 304–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Supplementary data