

Abstract
Multiple genes (e.g., POMT1, POMT2, POMGnT1, ISPD, GTDC2, B3GALNT2, FKTN, FKRP, and LARGE) are known to be involved in the glycosylation pathway of α-dystroglycan (α-DG). Mutations of these genes result in muscular dystrophies with wide phenotypic variability. Abnormal glycosylation of α-DG with decreased extracellular ligand binding activity is a common biochemical feature of these genetic diseases. While it is known that LARGE overexpression can compensate for defects in a few aforementioned genes, it is unclear whether it can also rescue defects in FKRP function. We examined adeno-associated virus (AAV)-mediated LARGE or FKRP overexpression in two dystrophic mouse models with loss-of-function mutations: (1) Largemyd (LARGE gene) and (2) FKRPP448L (FKRP gene). The results agree with previous findings that overexpression of LARGE can ameliorate the dystrophic phenotypes of Largemyd mice. In addition, LARGE overexpression in the FKRPP448L mice effectively generated functional glycosylation (hyperglycosylation) of α-DG and improved dystrophic pathologies in treated muscles. Conversely, FKRP transgene overexpression failed to rescue the defect in glycosylation and improve the phenotypes of the Largemyd mice. Our findings suggest that AAV-mediated LARGE gene therapy may still be a viable therapeutic strategy for dystroglycanopathies with FKRP deficiency.
Introduction
Dystroglycanopathies are primary muscle diseases linked with progressive pathological changes in skeletal muscle. The diseases are characterized by mutations in a multitude of genes (e.g., POMT1, POMT2, POMGnT1, ISPD, GTDC2, B3GALNT2, FKTN, FKRP, and LARGE) that are associated with a broad spectrum of clinical and pathological severity (Toda et al., 2000; Yoshida et al., 2001; Beltrán-Valero de Bernabé et al., 2002; van Reeuwijk et al., 2005; Roscioli et al., 2012; Ogawa et al., 2013; Stevens et al., 2013). Specific to FKRP, mutations in the gene have been reported to cause mild limb-girdle muscular dystrophy type 2I to severe congenital muscular dystrophies (CMD) such as Walker–Warburg syndrome, muscle–eye–brain disease, or CMD type 1C with or without central nervous system involvement (Beltrán-Valero de Bernabé et al., 2004; Brown et al., 2004; Mercuri et al., 2006). Exact mechanisms for the variation in disease severity are not clearly understood, although several factors, including the site of the mutations, levels of gene expression, and genetic background, are considered important. The hallmark of all of these genetic mutations is the disruption of the dystrophin–glycoprotein complex (DGC).
The DGC is a multimeric transmembrane protein assemblage that provides a molecular link between the subsarcolemmal cytoskeleton and the extracellular matrix (Ervasti and Campbell, 1991, 1993). The exact function of the DGC as a whole has yet to be completely determined; however, it is deemed essential in contributing to the structural stability of the muscle cell membrane during the muscle contraction cycle (Campbell, 1995). In part, the integrity of muscle tissues is reliant on a central component known as dystroglycan (DG)—a glycoprotein encoded by the DAG1 gene that is posttranslationally cleaved into two noncovalently associated subunits: α-DG (N-terminus) and β-DG (C-terminus) (Ervasti et al., 1990; Yoshida and Ozawa, 1990; Ibraghimov-Beskrovnaya et al., 1992). Alpha-DG is extracellular and binds to laminin in the basement membrane, whereas β-DG is a transmembrane protein that binds to dystrophin within the cytoplasm (Ibraghimov-Beskrovnaya et al., 1992). Extensive posttranslational processing (glycosylation) of α-DG is critical for linking the DGC complex to extracellular matrix proteins (Gee et al., 1994; Talts et al., 1999). Alpha-DG is both N- and O-linked glycosylated, with only the O-linked glycans apparently essential for muscle membrane integrity—removal of the O-linked glycans eliminates the ligand binding capabilities (Ervasti et al., 1990; Combs and Ervasti, 2005). The process of creating ligand-binding glycan structures on α-DG is critically dependent on the function of the aforementioned genes that encode putative or known glycosyltransferases. POMT1 and POMT2 catalyze the initial O-mannosylation of proteins (Manya et al., 2004), while POMGnT1 acts as a protein O-mannose β-1,2-N-acetylglucosaminyltransferase (Yoshida et al., 2001). More recently, it was shown that LARGE could act as a bifunctional glycosyltransferase (xylosyltransferase and glucuronyltransferase activities) producing repeating units of [–3-xylose–α1,3-glucuronic acid-β1–] (Inamori et al., 2012). However, functions of both FKTN and FKRP in the pathway of α-DG glycosylation remain elusive, although they are likely to be involved in postphosphoryl modifications of the O-mannose (Kuga et al., 2012).
Among the known genes involved in glycosylation, LARGE appears to possess some of the more unique properties in the pathway of α-DG glycosylation. Early studies reported that LARGE could modify complex N- and mucine O-glycans on α-DG to induce laminin binding (Patnaik and Stanley, 2005). LARGE overexpression competes to modify GlcNAc terminals with galactosylation to generate the functional glycans on both O-linked and N-linked glycans (Hu et al., 2011). LARGE also facilitates functional phosphoryl glycosylation of O-linked mannose and N-linked glycans on proteins other than α-DG (Zhang and Hu, 2012). The unique role LARGE plays in the glycosylation of α-DG is further demonstrated by reports that LARGE overexpression can bypass defects in glycosylation of α-DG caused by mutations of FKTN, POMGnT1, and POMT1 in cell culture systems and in POMGnT1 knockout mouse models (Barresi et al., 2004; Yu et al., 2013). The interaction between LARGE and FKRP on the glycosylation of α-DG is not fully understood, and it is unclear whether overexpression of LARGE can overcome the defects in FKRP function and vice versa. In the current approach, we utilize adeno-associated virus (AAV)-mediated overexpression of LARGE and FKRP to investigate the functional role of the two genes for their ability to rescue the deficiency of the other.
Materials and Methods
Mouse models
Colonies of Largemyd and C57BL/6 (wild-type) mice were originally obtained from the Jackson Laboratory (Bar Harbor, ME). The wild-type mice were used as controls. FKRPP448L knock-in mice were generated by inGenious Targeting Laboratory (Stony Brook, NY). The targeting vector was engineered with a c.1343C>T point mutation in exon 3 of the mouse FKRP gene, resulting in an amino acid change from proline-to-leucine at position 448 (P448L) (Chan et al., 2010). A loxP/FRT-flanked neomycin phosphotransferase (Neor) gene expression cassette was inserted in intron 2 and subsequently removed in vivo by crossbreeding with C57BL/6 Flp mice and further backcrossed to C57BL/6 for 10 generations by inGenious Targeting Laboratory to create the homozygote FKRPP448L mice. All mice were housed in the vivarium of Carolinas Medical Center (CMC) according to animal care guidelines of the institute. All animal studies were approved by the Institutional Animal Care and Use Committee of CMC.
Gene construction and AAV production
Full-length human LARGE1 cDNA was amplified from human skeletal muscle tissue by RT-PCR. The 2320 bp fragment containing the 5′ NheI and 3′ BglII was cloned into pcDNA3.1/myc-His(−) A (Life Technologies, Carlsbad, CA). The resulting construct was named HL1/pcDNA3.1myc-His. For HL1/AAV production, the NheI/BglII fragment was subcloned into AAV serotype 9 (AAV9) vector under control of a chicken β-actin (CB) promoter and cytomegalovirus (CMV) enhancer. The final construct was named pXX-CB-LARGE-Myc (AAV9-LARGE in short) (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/hum).
Full-length human FKRP cDNA was codon optimized and synthesized for high expression in Mus musculus (mouse) (GeneArt; Life Technologies). The Myc tag coding sequence, N-EQKLISEEDL-C (1.2 kDa), was linked to the C-terminus of the FKRP and a Kozak sequence was placed at the N-terminus of the FKRP coding sequence. The synthetic fragment was cloned into pMK-RQ (kanR) using SfiI restriction sites. The resulting vector was named FKRP-pMK-RQ (GeneArt; Life Technologies). The NotI-SalI fragment of optimized FKRP-Myc was subcloned into the same restriction enzyme sites of the AAV9 vector previously used for LARGE. The final construct was named pXX-CB-FKRP-Myc (AAV9-FKRP in short) (Supplementary Fig. S1).
The recombinant AAV vector stocks were produced according to the three-plasmid cotransfection method reported previously (Xiao et al., 1998). The AAV vectors were subsequently purified using a second-generation CsCl protocol that incorporates differential precipitation of AAV particles by polyethylene glycol (Ayuso et al., 2010). Vector titers were determined by both dot-blot and real-time PCR methods using previously published protocols (Veldwijk et al., 2002; Rohr et al., 2005). The concentration of viral vectors was kept in the range of 2×1012 to 5×1012 vector genomes/ml and stored at −80°C until future use.
Cell culture and vector delivery to dystrophic mice
C2C12 myoblast cells (ATCC CRL-1772) were grown in Dulbecco's modified Eagle's medium (Gibco, Life Technologies) supplemented with 10% fetal bovine serum (FBS), 4 mM L-glutamine, and 100 μg/ml penicillin–streptomycin in a 96-well plate (Greiner Bio-One, Monroe, NC). For differentiation, cells were grown to about 90% confluence and the growth medium was then replaced with Skeletal Muscle Cell Media (Cell Applications, San Diego, CA). Two days after differentiation, AAV9-FKRP or AAV9-LARGE (2×1010 vector genomes/ml) was added into the culture medium to each 96-well, and continued culturing for 72 hr before harvesting. For Western blots, C2C12 cells were cultured in T-25 flasks under the same culture conditions, but infected with 5×1011 vector genomes/ml.
Largemyd mice were administered with AAV9-LARGE (5×1010 vector genomes/ml) in phosphate-buffered saline (PBS) via intramuscular injection into the tibialis anterior muscle at the age of 3 months (n=3); mice were sacrificed 1 week after injection. FKRPP448L mice were administered with AAV9-LARGE at a dose of 4×1010 vector genomes per gram body weight in PBS via a single intraperitoneal injection at the age of 5 weeks (n=3); mice were sacrificed 5 weeks after injection. Largemyd mice were administered with AAV9-FKRP at a dose of 4×1010 vector genomes per gram body weight in PBS via a single intraperitoneal injection at the age of 3 months (n=3); mice were sacrificed 5 weeks after injection. Untreated and wild-type control mice were 12 weeks and 3 months old for FKRPP448L and Largemyd cohorts, respectively. Blood and muscle samples were collected and stored accordingly for future analysis.
Histology and cross-sectional area analysis
Skeletal and heart muscle tissues were dissected and snap-frozen in liquid nitrogen-chilled isopentane. Tissues were cryosectioned (5 μm thickness) and positioned on glass microscope slides, and then stored at −80°C until future use. Tissue sections were stained with hematoxylin and eosin (H&E). Muscle cross-sectional fiber radii and percentage of myofibers with centrally located nuclei were determined from tibialis anterior muscles stained with H&E. Three random 20× magnification images per section per animal were used. Images were visualized using an Olympus BX51/BX52 fluorescence microscope (Opelco, Dulles, VA) and captured using the Olympus DP70 Digital Camera System (Opelco).
Antibodies
Antibodies used in this study were obtained as follows: mouse monoclonal α-DG (clone IIH6C4) and rabbit polyclonal GM130 from EMD Millipore (Billerica, MA); mouse monoclonal β-DG (7D11) and mouse monoclonal c-Myc (9E10) from Developmental Studies Hybridoma Bank (Iowa City, IA); rabbit polyclonal c-Myc and rabbit polyclonal laminin from Sigma (St. Louis, MO); rat monoclonal laminin α2 from Enzo Life Sciences (Farmingdale, NY); mouse monoclonal GM130-Alexa Fluor 488 from BD Biosciences (San Jose, CA); goat polyclonal LARGE (Y-14) from Santa Cruz Biotechnology (Dallas, TX); and rabbit polyclonal FKRP-STEM was a gift from Dr. Derek J. Blake (University of Oxford).
Immunohistochemical analysis
For the detection of LARGE, Myc tag, GM130, and FKRP, frozen cryosections (5 μm thick) were initially fixed with 4% paraformaldehyde for 12 min and then immediately washed with PBS three times, each for 5 min. Sections were blocked with 8% bovine serum albumin (BSA) and 10% FBS diluted in PBS for 30 min. Sections were then incubated with primary antibodies: LARGE (Y-14) (1:50); c-Myc (9E10) (1:15) or c-Myc (1:250); GM130-Alexa Fluor 488 or GM130 (1:50); FKRP-STEM (1:2500). For the detection of functionally glycosylated α-DG, frozen cryosections (5 μm thick) were initially fixed in a cold (−20°C) ethanol:acetic acid (1:1) mixture for 1 min and then immediately washed with PBS four times, each for 15 min. Sections were blocked with 8% BSA in PBS for 30 min. Sections were then incubated with the primary antibody: IIH6C4 (1:200). For the detection of laminin, frozen cryosections (5 μm thick) were initially washed with PBS. Sections were then blocked with 20% FBS/10% normal goat serum diluted in PBS for 30 min. Sections were then incubated with the primary antibody: laminin (1:60) or laminin α2 (1:200). Antigoat IgG, antimouse IgG, antirabbit IgG, antirat IgG, or antimouse IgM secondary antibodies tagged with Alexa Fluor 488 or Alexa Fluor 594 (Life Technologies) were applied at various dilutions for 1 hr. Sections were washed with PBS three times, each for 5 min, and mounted with Fluorescence Mounting Medium (Dako, Carpinteria, CA) containing 1×DAPI (4′,6-diamidino-2-phenylindole). Similar procedures were used for the staining of cultured cells. All steps were performed at room temperature unless specified otherwise. Images were visualized using an Olympus BX51/BX52 fluorescence microscope (Opelco) and captured using the Olympus DP70 Digital Camera System (Opelco). The percentage of IIH6C4-positive fibers was determined from tibialis anterior muscles. Three random 20× magnification images per section per animal were used.
Immunoprecipitation
Antibody/Protein G binding
About 25 μl of c-Myc (Sigma) was diluted in 200 μl PBS, incubated with Dynabeads Protein G (Life Technologies) for 1 hr at room temperature, and subsequently washed with PBS. Protein lysate was prepared with RIPA (Radio-IP Assay) buffer: 50 mM Tris-HCl, 150 mM NaCl, 0.1% sodium dodecyl sulfate, 0.1% sodium deoxycholate, and 1% Triton X-100, supplemented with a protease inhibitor cocktail (Sigma). One milligram of the cardiac muscle lysate in 500 μl volume from AAV9-FKRP-treated FKRPP448L mice or controls was incubated with the antibody/Protein G by swinging head-over-tail for overnight at 4°C. The mixture was then washed three times with RIPA buffer. Precipitates of the complex were then heated at 70°C for 10 min in Novex Tris-Glycine SDS (2×) (Life Technologies) and then subjected to Western blot analysis.
Western blot analysis and laminin overlay assay
Protein extraction and Western blot analysis were performed as reported previously (Chan et al., 2010). Initially, total proteins were extracted from tissues via homogenization in TX-100 buffer (1% Triton X-100; 50 mM Tris, pH 8.0; 150 mM NaCl; 0.1% sodium dodecyl sulfate) supplemented with protease inhibitor cocktail (Sigma). The supernatants were collected by centrifugation at 18,000×g for 15 min at 4°C. The clear lysates were then passed through Zeba desalting columns (Pierce, Rockford, IL). Protein concentrations were determined by a DC protein assay (Bio-Rad, Hercules, CA). Protein samples were electrophoresed on a 4–20% Tris-Glycine gel (Life Technologies) at 200 mA for 3 hr and then transferred to an Immun-Blot™ polyvinylidene difluoride (PVDF) membrane (Bio-Rad). For Western blot analysis, the PVDF membranes were blocked with Protein-Free T20 (TBS) Blocking Buffer (Pierce) overnight at 4°C and then incubated with primary antibodies—IIH6C4 (1:2000), β-DG (1:500), FKRP-STEM (1:20,000), or LARGE (Y-14) (1:500)—in 20 mM Tris (pH 7.4), 150 mM NaCl, 0.1% Tween 20, and 0.5% gelatin. Antigoat, antimouse, or antirabbit HRP-conjugated secondary antibodies (Santa Cruz Biotechnology) were applied to the membranes at a 1:1000–4000 dilution for 1 hr. For the laminin overlay assay, nitrocellulose membranes (Bio-Rad) were blocked with laminin overlay buffer (10 mM ethanolamine, 140 mM NaCl, 1 mM MgCl2, and 1 mM CaCl2, pH 7.4) containing 5% nonfat dry milk for 1 hr at 4°C followed by incubation with laminin from Engelbreth-Holm-Swarm murine sarcoma basement membrane (Sigma) at a 1:500 dilution overnight at 4°C in laminin overlay buffer. Membranes were washed and incubated with rabbit antilaminin antibody (Sigma) at a 1:1500 dilution followed by antirabbit HRP-conjugated secondary antibody (Santa Cruz Biotechnology) applied at a 1:3000 dilution. All blots were developed by electrochemiluminescence (ECL) immunodetection (PerkinElmer, Waltham, MA), exposed to BioMax Light film (Sigma), and processed by an LAS-4000 imaging system (Fujifilm, Valhalla, NY).
Results
AAV vector construction and gene expression in vitro
AAV vectors were generated for the expression of LARGE (AAV9-LARGE) or FKRP (AAV9-FKRP). The full-length human LARGE or FKRP coding sequence was optimized for high expression in a mouse model. The resulting construct was subcloned into an AAV9 vector under control of a CB promoter and CMV enhancer. AAV9 was chosen because it has been shown to have widespread transduction in multiple tissues, including skeletal and cardiac muscles (Inagaki et al., 2006; Pacak et al., 2006).
AAV-mediated LARGE expression was initially examined in mouse C2C12 myoblasts and myotubes by immunohistochemical analysis. Transduced C2C12 cultures were labeled with antibodies to LARGE and the Myc epitope—with the Myc-specific monoclonal antibody 9E10 (Evan et al., 1985). LARGE expression was visualized as punctuated signals regularly distributed throughout the cytoplasm and along the edges of the nuclei (Supplementary Fig. S2). The expression of LARGE increased throughout the differentiation of C2C12 myoblasts into myotubes, consistent with a higher infection efficiency to myotubes utilizing AAV. In transduced myotubes, the Myc tag also showed colocalization with GM130—a cis-Golgi matrix protein (Supplementary Fig. S2), confirming the localization of LARGE in the Golgi apparatus (Brockington et al., 2005; Grewal et al., 2005). The same series of experiments was carried out for AAV9-mediated FKRP expression, elucidating similar results reported previously (Xu et al., 2013) (Supplementary Fig. S3).
To investigate whether LARGE overexpression was able to alter glycosylation of α-DG, we conducted Western blot analysis on C2C12 differentiated myotubes (Supplementary Fig. S2) using the IIH6C4 antibody—which recognizes the ligand-binding carbohydrate epitope of α-DG (Campanelli et al., 1994; Gee et al., 1994). Immunoblots detected glycosylated α-DG as a broad band with a molecular weight of 150–300 kDa in AAV9-LARGE-treated cultures. In contrast, only a single narrow band with a molecular weight of about 170 kDa was observed in the same cells without AAV transfection. These results confirm the AAV9-mediated LARGE overexpression and its effect of hyperglycosylation on α-DG.
LARGE expression restores functional glycosylation of α-DG in muscle of Largemyd mice
Next, we examined LARGE expression in vivo using the Largemyd mouse model. Largemyd mice possess a spontaneous deletion of exons 5–7 of the gene (in front of the catalytic domains), which causes a frame-shift and a premature stop codon, leading to a loss of function and a severe muscle pathology (Grewal et al., 2005). AAV9-LARGE (5×1010 vector genomes/ml) was administered via intramuscular injection to the tibialis anterior muscles of the mice (3 months of age). Immunohistochemistry was unable to detect endogenous LARGE expression with the LARGE (Y-14) antibody against the C-terminus of human LARGE in the untreated Largemyd muscles (Fig. 1A). As expected, functional glycosylation of α-DG was also undetectable in the untreated Largemyd muscles using the IIH6C4 antibody (Fig. 1A). In contrast, muscles of the Largemyd mice treated with the AAV9-LARGE showed strong signals for LARGE expression, as indicated by punctuated dots distributed throughout the sarcoplasm and along the sarcolemma (Fig. 1A). Immunohistochemistry also confirmed functionally glycosylated α-DG with the IIH6C4 antibody in the AAV9-LARGE-treated muscle fibers (Fig. 1A). The coexpression of the LARGE and IIH6C4 epitopes in the same fibers was confirmed by the staining of serial sections (Fig. 1A). The signal intensity for both antibodies suggests that a strong expression of LARGE correlates positively with a strong expression of the IIH6C4 epitope.
FIG. 1.
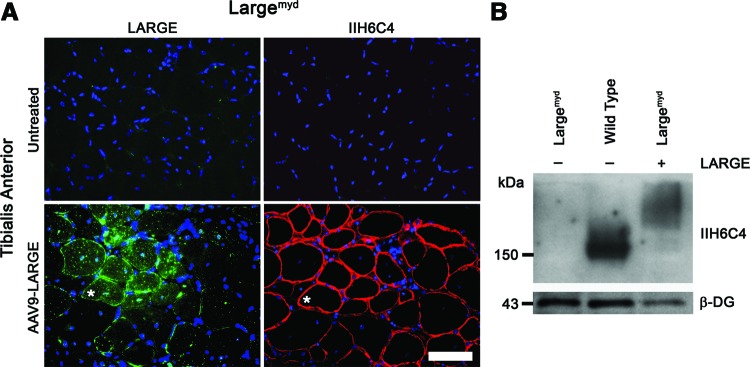
LARGE expression and rescue of functional α-DG in skeletal muscle of Largemyd mouse administered by intramuscular injection. (A) Immunohistochemistry of tibialis anterior muscle sections either untreated (top) or treated with AAV9-LARGE (bottom). LARGE expression was detected with LARGE (Y-14) for the untreated or Myc tag for the treated. Functional α-DG was detected with IIH6C4 antibody. Green or red signals indicate positive signals. Asterisks indicate the same fiber in serial sections. Cellular nuclei were counterstained with DAPI (blue). Scale bar: 50 μm. (B) Western blot analysis of protein lysates from skeletal muscle sections either untreated (−) or treated (+) with AAV9-LARGE using IIH6C4 and β-DG antibodies. AAV, adeno-associated virus; α-DG, α-dystroglycan. Color images available online at www.liebertpub.com/hgtb
Western blot analysis using the IIH6C4 antibody (Fig. 1B) showed that the AAV9-LARGE-treated muscle produced positive signals representing heavily glycosylated forms (≥200 kDa) of α-DG, which were significantly larger than that detected in the same muscle type of the control (wild-type) sample (∼160 kDa). As expected, IIH6C4 immunoreactivity was undetectable in the untreated Largemyd sample. The detection of β-DG confirmed that a similar amount of protein was loaded for each sample. Collectively, these results show that LARGE overexpression can result in hyperglycosylation of α-DG in Largemyd mice.
LARGE generates functional α-DG in FKRPP448L mice
We next investigated whether the overexpression of LARGE could also rescue the functional defects of the putative glycosyltransferase FKRP—a hypothesis proposed by Dr. Campbell's group (Barresi et al., 2004). For these experiments, we used the mouse model containing a knock-in proline-to-leucine missense mutation in the FKRP gene (FKRPP448L). This model revealed moderate skeletal muscle dystrophic phenotypes from as early as 2–3 weeks, but lacked clear histological defects in the central nervous system along with normal breeding activity and a near-normal life span (Blaeser et al., 2013).
The AAV9-LARGE vector was intraperitoneally injected into the FKRPP448L mice (5 weeks of age) with a single dose of 4×1010 viral genomes per gram body weight, and its effect was examined 5 weeks postinjection. Immunohistochemical staining revealed LARGE expression in the heart, diaphragm, and skeletal muscles (Fig. 2A). Fluorescence signals as punctuated dots, similar to that observed in the skeletal muscle of Largemyd mouse, were observed within the sarcoplasm and often concentrated at the sarcolemma with regular spacing in the majority of muscle fibers. The levels of LARGE varied considerably from fiber-to-fiber within the same muscles; there were very few fibers with signals fused into big patches within the sarcoplasm, and a portion of fibers remained negative for LARGE expression, suggesting partial transduction of the AAV9-LARGE vector with the current dosage or vector system. To determine whether AAV9-LARGE expression could generate functional α-DG, we tested the IIH6C4 immunoreactivity of the transduced tissues (Fig. 2A). Approximately 50% or more fibers in all the muscle examined were detected with positive signal by the antibody with the lowest percentage of positive fibers in the tibialis anterior and the highest in cardiac muscle (70–80%). This was in high contrast to the untreated FKRPP448L tissues, where only a few (maximum of 15%) tibialis anterior muscle fibers spontaneously expressed glycosylated α-DG, most of them with weak signal (Supplementary Fig. S4). This suggests that LARGE overexpression was able to restore functional glycosylation of α-DG in FKRP mutant muscles. This is further supported by the fact that almost all fibers expressing LARGE showed clearly detectable restoration of functionally glycosylated α-DG, and the levels of the two proteins are well correlated, although variation in signal intensity existed. Most fibers without a detectable signal for LARGE were also negative for IIH6C4 antibody staining, indicating that there may exist some threshold levels of LARGE expression that are required to restore detectable levels of glycosylated α-DG. As expected, there was a strong IIH6C4 signal for all tissues in the wild-type mice (Supplementary Fig. S4). Laminin expression (pan-laminin and laminin α2) was detected in muscles of all mice groups (Supplementary Fig. S5). The signals for both laminin antibodies were homogenous in wild-type muscles with the intensity similar to that detected in the majority of muscle fibers of the FKRPP448L mice with or without AAV9-LARGE treatment. However, clearly stronger signals were observed in small-caliber and mostly centrally nucleated (regenerating) fibers, but this was the case for both AAV9-LARGE-treated and untreated FKRPP448L tissues, although the number of such fibers are smaller in the treated than in untreated muscles. These results therefore suggest that stronger laminin α2 expression is related to the process of muscle regeneration rather than LARGE overexpression in our model system.
FIG. 2.

LARGE overexpression generates functional α-DG in FKRPP448L mice. (A) Immunohistochemical staining with Myc tag (green, left panel) or IIH6C4 (red, right panel) antibodies for LARGE and functional α-DG, respectively. Asterisks indicate the same fiber in serial sections. Cellular nuclei were counterstained with DAPI (blue). Scale bar: 50 μm. (B) Western blot analysis of protein lysates from the heart (H), diaphragm (D), and tibialis anterior (TA) using IIH6C4, LARGE (Y-14), and β-DG antibodies and a laminin overlay assay.
Transgene LARGE expression was also confirmed by Western blot analysis with the LARGE (Y-14) antibody (Fig. 2B). Positive signals were detected only in the AAV9-LARGE-treated FKRPP448L mice, with much higher signals in the heart and diaphragm than in the tibialis anterior. This is likely because of the high tropism of the AAV9 to cardiac muscle and the route of intraperitoneal injection (to diaphragm). Restoration of functionally glycosylated α-DG in the heart, diaphragm, and other skeletal muscles was also confirmed by Western blot analysis (Fig. 2B). Positive signals were hardly detectable in the cardiac muscle, and at very low levels in the diaphragm and tibialis anterior muscles of the untreated FKRPP448L mice when compared with the normal (wild-type) muscles. Conversely, the AAV9-LARGE-treated FKRPP448L mice samples demonstrated a dramatic increase in IIH6C4 signals as broader molecular weight bands with size ranges similar to and significantly larger than that in wild-type tissues, indicating the expression of variably hyperglycosylated α-DG species. The laminin overlay assay (Fig. 2B), showed broader bands with molecular weights similar to that detected by the IIH6C4 antibody in the AAV9-LARGE-treated FKRPP448L muscles. Conversely, the laminin-binding activity of α-DG was nearly undetectable in the untreated FKRPP448L tissues. As expected, β-DG immunoreactivity was similar for all samples (Fig. 2B).
The pathology of the AAV9-LARGE-treated FKRPP448L mice improved after the 5-week treatment as compared with the untreated mice. As evident from the H&E staining (Fig. 3A), large areas of degeneration commonly observed in untreated FKRPP448L mice became only occasionally detected in the AAV9-LARGE-treated mice. There was a significant reduction in the number of small-caliber regenerating fibers, especially in the diaphragm and the tibialis anterior muscles (Fig. 3A). The overall percentage of centralized nucleation was reduced to 23.5±9.4% in skeletal muscles of the treated tibialis anterior muscles compared with 53.6±3.6% in untreated muscles (statistically significant). The improvement in pathology with the LARGE expression was also reflected by the fiber size distribution (Fig. 3B). The FKRPP448L untreated mice showed more variability in fiber size than the AAV9-LARGE-treated FKRPP448L mice. Histology in the heart was undistinguishable between the two groups with only small areas of increased interstitial tissue present in both AAV9-LARGE-treated and FKRPP448L untreated mice (Fig. 3A). Likewise, the difference in creatine kinase levels was not significant between the cohorts, 3958±1868 (U/L) in the AAV9-LARGE-treated FKRPP448L mice and 3961±1504 (U/L) in the untreated FKRPP448L mice.
FIG. 3.

Histopathology of the AAV9-LARGE-treated FKRPP448L tissues. (A) Hematoxylin and eosin staining of the heart, diaphragm, and tibialis anterior tissues from AAV9-LARGE-treated FKRPP448L, untreated FKRPP448L, and wild-type (C57) mice. Scale bar: 200 μm. (B) Fiber size distribution of tibialis anterior muscles from each experimental group (n=3, mean±SEM, Student's t-test, *p<0.05). Color images available online at www.liebertpub.com/hgtb
FKRP overexpression is unable to restore functional glycosylation of α-DG in Largemyd mice
We have previously constructed an AAV9-FKRP recombinant vector expressing wild-type codon-optimized FKRP (Xu et al., 2013). Local and systemic delivery of AAV9-FKRP mediated effective expression of FKRP in both skeletal and, more notably, cardiac muscle of the FKRPP448L mutant mice. To examine if FKRP overexpression has any effect on the functional glycosylation of α-DG in the Largemyd mice (3 months of age), the AAV9-FKRP vector was administered via intraperitoneal injection, and the effect was examined 5 weeks postinjection. Expression of FKRP in Largemyd mice was clearly detected in all tissues examined, including the heart, diaphragm, and tibialis anterior muscles by immunostaining with the FKRP-STEM antibody (Fig. 4A). The distribution of FKRP expression in the Largemyd mouse was similar to that of the LARGE expression in the FKRPP448L mice described above. As expected, there was no signal detection in untreated mice (data not shown). Conversely, functionally glycosylated α-DG was undetectable by the IIH6C4 antibody in any fiber clearly expressing FKRP (Fig. 4A). Western blot analysis confirmed these findings and showed that there was no correlation between levels of FKRP expression and rescue of functional α-DG (Fig. 4B). As a control, functional glycosylation of α-DG was restored in skeletal muscles of the FKRPP448L mouse with the same batch of AAV9-FKRP (Supplementary Fig. S6). Histologically, the percentage of centrally nucleated fibers was similar between the AAV9-FKRP-treated (53.1±3.1%) and untreated (65.7±1.5%) Largemyd tibialis anterior muscles and there was no significant improvement in pathology with the FKRP expression as reflected by the H&E staining and fiber size distribution (Supplementary Fig. S7). Serum creatine kinase levels also remained unaffected between the treated and untreated Largemyd mice (data not shown).
FIG. 4.

Expression of FKRP and functional α-DG in Largemyd mice after AAV9-FKRP treatment. (A) Immunohistochemical staining of AAV9-FKRP-treated tissues with FKRP-STEM (red, left panel) or IIH6C4 (green, right panel) antibodies. Asterisks indicate the same fiber in serial sections. Cellular nuclei were counterstained with DAPI (blue). Scale bar: 50 μm. (B) Western blot analysis of protein lysates from the heart (H), diaphragm (D), and tibialis anterior (TA) using IIH6C4, FKRP-STEM, and β-DG antibodies. Asterisk indicates nonspecific bands observed in all samples. Color images available online at www.liebertpub.com/hgtb
FKRP forms a complex with LARGE
LARGE and FKRP are both known to modulate the postphosphoryl modification of α-DG, and mutations in either of the genes can result in the same defect in monoester-linked phosphorylated α-DG. Our results show that both proteins colocalized within the Golgi apparatus (Supplementary Fig. S8). As a result, we speculate that the two proteins might work in a complex for the completion of the postphosphoryl modification of α-DG. To test this theory, we examined the two proteins by immunoprecipitation (Fig. 5). Samples from cardiac muscle were chosen for tissue analysis, mainly because previous reports revealed higher transgene expression of LARGE in this tissue (Brockington et al., 2010). However, because antibodies directed against both proteins are ineffective in immunoprecipitating the targeted protein, we were forced to use the c-Myc (rabbit polyclonal) antibody for immunoprecipitation of vector-expressed FKRP in FKRPP448L mice. Examination of the immunoprecipitate by Western blot confirmed expression of FKRP (Fig. 5). The same immunoprecipitate was re-probed with LARGE (Y-14) antibody and showed a specific band of approximately 88 kDa, matching the size of endogenous LARGE (Fig. 5). It should be noted that this band was nearly undetectable in the void portion of the tissue lysate or in whole tissue lysate without immunoprecipitation. Overall, these results support the notion that FKRP and LARGE are likely parts of a protein complex working in collaboration, although whether the two proteins contact directly or indirectly has yet to be determined.
FIG. 5.

Immunoprecipitation for the detection of FKRP and LARGE in cardiac muscle. Lanes were either treated (+) or untreated (−) with the respective vector, protein, and/or antibody. Lane 1 (far left), void portion after immunoprecipitation; lane 2, immunoprecipitated portion; lane 3, AAV9-LARGE-infected tissue without immunoprecipitation; lanes 4 and 5, void and immunoprecipitated portion of control tissue, respectively; lane 6, whole tissue lysate of the AAV9-FKRP-treated tissue.
Discussion
Among the known genes involved in the functional glycosylation of α-DG, LARGE has a different regulatory role in α-DG glycosylation. This is most convincingly demonstrated by the fact that overexpression of LARGE was able to restore the functional glycosylation of α-DG not only in cells with LARGE mutations, but also with FKTN and POMGnT1 mutations in vitro and in vivo (Barresi et al., 2004). However, overexpression of POMGnT1 was unable to compensate for the loss of LARGE function. In this study, we further demonstrated that LARGE overexpression is able to rescue the functional defect of FKRP in the FKRPP448L mouse, leading to the restoration of functional α-DG in skeletal muscles as well as cardiac muscle. Furthermore, we showed that, as the result of LARGE overexpression, dystrophic pathology of the mutant muscles was improved with a significant reduction in the number of regenerating and degenerating fibers. The number of centrally nucleated fibers was also reduced when compared with the controls. In contrast, overexpression of FKRP was unable to correct the glycosylation defects and improve pathology in muscles of the Largemyd mice. Our results further support the notion that overexpression of LARGE could serve to rescue a wide range of dystroglycanopathies caused by defects in a multitude of genes, including FKRP.
To assess the efficacy of LARGE overexpression as a viable gene therapy approach for dystroglycanopathies, Muntoni's group studied LARGE transgenic models in a normal background and reported that overexpression of LARGE induced hyperglycosylation of α-DG (Brockington et al., 2010). The group also noticed that LARGE expression in the heart was regionally confined and variable—only approximately 25% of the cardiomyocytes showed positive signal with predominantly transmural distribution in all LARGE transgenic lines. Transgenic expression in the cardiomyocytes correlated well with hyperglycosylation of α-DG. Reasons for variable expression are not fully understood, but the observation suggests potential difficulty in inducing homogeneous vector-induced LARGE expression in the target tissue. On the contrary, our short-term study shows that the majority of cardiac muscle can be effectively induced to express the LARGE transprotein, although with considerable variation in expression levels. As a result, functionally glycosylated α-DG was detected in the majority of the cardiac muscle fibers, again with variation in signal levels. Therefore, it is possible that the limited expression of LARGE and associated hyperglycosylated α-DG in cardiac muscle in transgenic models could be related to developmental regulation specific to the LARGE transgene. The difference in levels of LARGE expression between the AAV9 vector and chromosomally located transgene could also be a contributing factor.
While some available evidence thus far has clearly demonstrated the potential of using LARGE as an effective transgene to treat a broader range of dystroglycanopathies, various potential concerns of LARGE overexpression have been noted in the study of LARGE transgenics. LARGE transgenic mice with a normal background tend to exhibit normal development and muscle histology; however, the aged LARGE transgenic mice show an increase in susceptibility to contraction-induced injury and a 30% decline in force generation when compared with age-matched controls (Brockington et al., 2010). More recently, Dr. Brown's group further reported that contrary to expectation, transgenic expression of LARGE in a dystrophic mouse with FKRP mutations resulted in a worsening of the muscle pathology, implying that any future strategies based upon LARGE upregulation require careful management (Whitmore et al., 2013). These results are apparently in contradiction to the observation from the current study, which showed a clear pathological benefit from the LARGE-induced functional glycosylation of α-DG in FKRP mutant mice. Both studies demonstrated the LARGE overexpression in muscles, but the time of expression is different and levels of expression cannot be compared. Therefore, the detrimental effect of LARGE overexpression could be limited to the transgenic model only. However, the current study has only examined the effect of LARGE overexpression for a short term, and as a result, a long-term efficacy study in adult FKRP mutant mice is required to determine the potential of LARGE upregulation as gene therapy for FKRP dystroglycanopathies.
Virus-mediated gene therapy has the advantage of simplicity, because it may only require one treatment to achieve long-lasting effects. However, it also creates difficulty in establishing optimal levels of transgene expression. This could be particularly important for LARGE as a therapeutic transgene for dystroglycanopathies. LARGE overexpression induced mainly abnormal glycosylation of α-DG, explicitly characterized by a larger molecular mass. Currently, glycosylated α-DG detected by IIH6C4 antibody is considered to be the functional variant of the protein; however, it is probable that glycosylated α-DG with larger molecular weights could function differentially as a molecular bridge between the cell membrane and extracellular proteins, thereby providing different levels of protection for membrane stability. We hypothesize that hyperglycosylated α-DG could be less effective in providing protection and may be related to the observed detrimental effect, especially when the amount of functional glycan epitopes exceeds certain levels. Because LARGE overexpression also produces glycosylated α-DG with a size similar to that of normal (wild-type) muscles, optimal levels of LARGE expression could be the key for achieving glycosylation of α-DG with normal functions. Clearly, carefully designed experiments are required to fully investigate these concerns. In contrast, FKRP overexpression produces the functionally glycosylated α-DG with the same size as those in normal muscles and thus less a concern if FKRP gene therapy is considered for the FKRP-related dystroglycanopathies.
Supplementary Material
Acknowledgments
This work was supported by the Carolinas Muscular Dystrophy Research Endowment at the Carolinas HealthCare Foundation and CMC, Charlotte, NC. This work is also partly supported by National Institute of Neurological Disorders and Stroke (1R01N5082536-01). The authors would like to thank Traci Williamson (Animal Facility, CMC); Jane Ingram and Tracy Walling (Histology Core, CMC); and Tonya Bates, Kris Bennett, Judy Vachris, and Nury Steuerwald (Molecular Biology Core, CMC) for their technical help. The c-Myc (9E10) antibody was developed by J. Michael Bishop and the β-DG antibody was developed by Glenn E. Morris; both were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by Department of Biology, The University of Iowa, Iowa City, IA.
Author Disclosure Statement
No competing financial interests exist.
References
- Ayuso E., Mingozzi F., Montane J., et al. (2010). High AAV vector purity results in serotype- and tissue-independent enhancement of transduction efficiency. Gene Ther. 17, 503–510 [DOI] [PubMed] [Google Scholar]
- Barresi R., Michele D.E., Kanagawa M., et al. (2004). LARGE can functionally bypass α-dystroglycan glycosylation defects in distinct congenital muscular dystrophies. Nat. Med. 10, 696–703 [DOI] [PubMed] [Google Scholar]
- Beltrán-Valero De Bernabé D., Currier S., Steinbrecher A., et al. (2002). Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am. J. Hum. Genet. 71, 1033–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltrán-Valero De Bernabé D., Voit T., Longman C., et al. (2004). Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker–Warburg syndrome. J. Med. Genet. 41, e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaeser A., Keramaris E., Chan Y., et al. (2013). Mouse models of fukutin-related protein mutations show a wide range of disease phenotypes. Hum. Genet. 132, 923–934 [DOI] [PubMed] [Google Scholar]
- Brockington M., Torelli S., Prandini P., et al. (2005). Localization and functional analysis of the LARGE family of glycosyltransferases: significance for muscular dystrophy. Hum. Mol. Genet. 14, 657–665 [DOI] [PubMed] [Google Scholar]
- Brockington M., Torelli S., Sharp P.S., et al. (2010). Transgenic overexpression of LARGE induces α-dystroglycan hyperglycosylation in skeletal and cardiac muscle. PLoS ONE 5, e14434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown S.C., Torelli S., Brockington M., et al. (2004). Abnormalities in α-dystroglycan expression in MDC1C and LGMD2I muscular dystrophies. Am. J. Pathol. 164, 727–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanelli J.T., Roberds S.L., Campbell K.P., and Scheller R.H. (1994). A role for dystrophin-associated glycoproteins and utrophin in agrin-induced AChR clustering. Cell 77, 663–674 [DOI] [PubMed] [Google Scholar]
- Campbell K.P. (1995). Three muscular dystrophies: loss of cytoskeleton-extracellular matrix linkage. Cell 80, 675–679 [DOI] [PubMed] [Google Scholar]
- Chan Y.M., Keramaris-Vrantsis E., Lidov H.G., et al. (2010). Fukutin-related protein is essential for mouse muscle, brain and eye development and mutation recapitulates the wide clinical spectrums of dystroglycanopathies. Hum. Mol. Genet. 19, 3995–4006 [DOI] [PubMed] [Google Scholar]
- Combs A.C., and Ervasti J.M. (2005). Enhanced laminin binding by α-dystroglycan after enzymatic deglycosylation. Biochem. J. 390, 303–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervasti J.M., and Campbell K.P. (1991). Membrane organization of the dystrophin-glycoprotein complex. Cell 66, 1121–1131 [DOI] [PubMed] [Google Scholar]
- Ervasti J., and Campbell K. (1993). A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J. Cell Biol. 122, 809–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ervasti J.M., Ohlendieck K., Kahl S.D., et al. (1990). Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 345, 315–319 [DOI] [PubMed] [Google Scholar]
- Evan G.I., Lewis G.K., Ramsay G., and Bishop J.M. (1985). Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol. Cell. Biol. 5, 3610–3616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee S.H., Montanaro F., Lindenbaum M.H., and Carbonetto S. (1994). Dystroglycan-α, a dystrophin-associated glycoprotein, is a functional agrin receptor. Cell 77, 675–686 [DOI] [PubMed] [Google Scholar]
- Grewal P.K., Mclaughlan J.M., Moore C.J., et al. (2005). Characterization of the LARGE family of putative glycosyltransferases associated with dystroglycanopathies. Glycobiology 15, 912–923 [DOI] [PubMed] [Google Scholar]
- Hu Y., Li Z.-F., Wu X., and Lu Q. (2011). Large induces functional glycans in an O-mannosylation dependent manner and targets GlcNAc terminals on alpha-dystroglycan. PLoS ONE 6, e16866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibraghimov-Beskrovnaya O., Ervasti J.M., Leveille C.J., et al. (1992). Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature 355, 696–702 [DOI] [PubMed] [Google Scholar]
- Inagaki K., Fuess S., Storm T.A., et al. (2006). Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol. Ther. 14, 45–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inamori K.-I., Yoshida-Moriguchi T., Hara Y., et al. (2012). Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science 335, 93–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuga A., Kanagawa M., Sudo A., et al. (2012). Absence of post-phosphoryl modification in dystroglycanopathy mouse models and wild-type tissues expressing non-laminin binding form of α-dystroglycan. J. Biol. Chem. 287, 9560–9567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manya H., Chiba A., Yoshida A., et al. (2004). Demonstration of mammalian protein O-mannosyltransferase activity: coexpression of POMT1 and POMT2 required for enzymatic activity. Proc. Natl. Acad. Sci. USA 101, 500–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercuri E., Topaloglu H., Brockington M., et al. (2006). Spectrum of brain changes in patients with congenital muscular dystrophy and FKRP gene mutations. Arch. Neurol. 63, 251–257 [DOI] [PubMed] [Google Scholar]
- Ogawa M., Nakamura N., Nakayama Y., et al. (2013). GTDC2 modifies O-mannosylated α-dystroglycan in the endoplasmic reticulum to generate N-acetyl glucosamine epitopes reactive with CTD110.6 antibody. Biochem. Biophys. Res. Commun. 440, 88–93 [DOI] [PubMed] [Google Scholar]
- Pacak C.A., Mah C.S., Thattaliyath B.D., et al. (2006). Recombinant adeno-associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ. Res. 99, e3–e9 [DOI] [PubMed] [Google Scholar]
- Patnaik S.K., and Stanley P. (2005). Mouse Large can modify complex N- and mucin O-glycans on α-dystroglycan to induce laminin binding. J. Biol. Chem. 280, 20851–20859 [DOI] [PubMed] [Google Scholar]
- Rohr U.-P., Heyd F., Neukirchen J., et al. (2005). Quantitative real-time PCR for titration of infectious recombinant AAV-2 particles. J. Virol. Methods 127, 40–45 [DOI] [PubMed] [Google Scholar]
- Roscioli T., Kamsteeg E.-J., Buysse K., et al. (2012). Mutations in ISPD cause Walker-Warburg syndrome and defective glycosylation of α-dystroglycan. Nat. Genet. 44, 581–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens E., Carss Keren j., Cirak S., et al. (2013). Mutations in B3GALNT2 cause congenital muscular dystrophy and hypoglycosylation of α-dystroglycan. Am. J. Hum. Genet. 92, 354–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talts J.F., Andac Z., Gohring W., et al. (1999). Binding of the G domains of laminin α1 and α2 chains and perlecan to heparin, sulfatides, α-dystroglycan and several extracellular matrix proteins. EMBO J. 18, 863–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda T., Kobayashi K., Kondo-Iida E., et al. (2000). The Fukuyama congenital muscular dystrophy story. Neuromuscul. Disord. 10, 153–159 [DOI] [PubMed] [Google Scholar]
- Van Reeuwijk J., Janssen M., Van Den Elzen C., et al. (2005). POMT2 mutations cause α-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J. Med. Genet. 42, 907–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldwijk M.R., Topaly J., Laufs S., et al. (2002). Development and optimization of a real-time quantitative PCR-based method for the titration of AAV-2 vector stocks. Mol. Ther. 6, 272–278 [DOI] [PubMed] [Google Scholar]
- Whitmore C., Fernandez-Fuente M., Booler H., et al. (2013). The transgenic expression of LARGE exacerbates the muscle phenotype of dystroglycanopathy mice. Hum. Mol. Genet. 23, 1842–1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X., Li J., and Samulski R.J. (1998). Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 72, 2224–2232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L., Lu P.J., Wang C.-H., et al. (2013). Adeno-associated virus 9 mediated FKRP gene therapy restores functional glycosylation of α-dystroglycan and improves muscle functions. Mol. Ther. 10, 1832–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M., and Ozawa E. (1990). Glycoprotein complex anchoring dystrophin to sarcolemma. J. Biochem. (Tokyo) 108, 748–752 [DOI] [PubMed] [Google Scholar]
- Yoshida A., Kobayashi K., Manya H., et al. (2001). Muscular dystrophy and neuronal migration disorder caused by mutations in a glycosyltransferase, POMGnT1. Dev. Cell 1, 717–724 [DOI] [PubMed] [Google Scholar]
- Yu M., He Y., Wang K., et al. (2013). Adeno-associated viral-mediated LARGE gene therapy rescues the muscular dystrophic phenotype in mouse models of dystroglycanopathy. Hum. Gene Ther. 24, 317–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P., and Hu H. (2012). Differential glycosylation of α-dystroglycan and proteins other than α-dystroglycan by like-glycosyltransferase. Glycobiology 22, 235–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.