

Abstract
Cell cycle arrest in response to hypoxia is a fundamental physiological mechanism to maintain a balance between O2 supply and demand. Many of the cellular responses to reduced O2 availability are mediated through the transcriptional activity of hypoxia-inducible factor 1 (HIF-1). We report a role for the isolated HIF-1α subunit as an inhibitor of DNA replication, and this role was independent of HIF-1β and transcriptional regulation. In response to hypoxia, HIF-1α bound to Cdc6, a protein that is essential for loading of the minichromosome maintenance (MCM) complex (which has DNA helicase activity) onto DNA, and promoted the interaction between Cdc6 and the MCM complex. Although the interaction between Cdc6 and the MCM complex increased the association of the MCM proteins with chromatin, the binding of HIF-1α to the complex decreased phosphorylation and activation of the MCM complex by the kinase Cdc7. As a result, HIF-1α inhibited firing of replication origins, decreased DNA replication, and induced cell cycle arrest in various cell types. These findings establish a transcription-independent mechanism by which the stabilization of HIF-1α leads to cell cycle arrest in response to hypoxia.
INTRODUCTION
Hypoxia-inducible factor 1 (HIF-1) is a transcription factor that mediates adaptive responses to hypoxia. First identified in studies of erythropoietin gene expression (1), HIF-1 was subsequently shown to regulate oxygen homeostasis at both the cellular and systemic levels (2-4). HIF-1 is a heterodimer composed of HIF-1α and HIF-1β subunits (5). The abundance and activity of the HIF-1α subunit are regulated by O2-dependent hydroxylation (6). Proline hydroxylation targets HIF-1α for ubiquitination by the von Hippel-Lindau ligase complex and subsequent proteasomal degradation (7-9), whereas asparagine hydroxylation blocks interaction of HIF-1α with the coactivator p300 (10, 11). These posttranslational modifications couple HIF-1 activity to the cellular O2 concentration. Because the hydroxylases contain Fe(II) in their catalytic centers and use α-ketoglutarate (in addition to O2) as a substrate, their activity can be inhibited by iron chelators, such as desferrioxamine (DFX), and by competitive antagonists of α-ketoglutarate, such as dimethyloxalylglycine (DMOG) (6). HIF-1 regulates the expression of hundreds of target genes involved in angiogenesis, erythropoiesis, metabolism, autophagy, and other physiological responses to hypoxia (12). The HIF-2α protein shares sequence similarity and functional overlap with HIF-1α, but its distribution is restricted to certain cell types, and in some cases, it mediates distinct biological functions (13).
An imbalance between O2 supply and consumption that results in hypoxia will be exacerbated by an increased number of cells. Consequently, a fundamental adaptation to hypoxia that is mediated by HIF-1α is reduced cell proliferation. Induction of HIF-1α by hypoxia leads to G1-phase cell cycle arrest in multiple cell types including various cancer cell lines (14-17), fibroblasts (18), lymphocytes (18), and hematopoietic stem cells (19), and forced overexpression of HIF-1α, including under nonhypoxic conditions, is sufficient to inhibit cell proliferation (20). The role of HIF-2α in cell cycle regulation is less clear and may be cell type– and stimulus-specific. Previous studies have reported that HIF-2α either arrests proliferation in a manner similar to HIF-1α (20) or increases cell proliferation (17) in a context-dependent manner. Thus far, studies examining the molecular mechanism by which HIF-1α mediates cell cycle arrest have focused on the role of HIF-1α in regulating the expression of the genes encoding p21 and p27 (15, 17, 18), which inhibit the activity of cyclin-dependent kinases (CDKs).
The initiation of DNA replication is a tightly controlled process, the first steps of which are origin recognition, licensing, and activation, which involve formation (during the G1 phase) of a multiprotein pre–replication complex (pre-RC) that marks all potential origins of replication (21). Pre-RC formation begins with binding of the origin recognition complex (ORC), which is composed of six subunits (Orc1 to 6), to replication origins. ORC subsequently binds Cdc6 (22) and Cdt1 (23), leading to recruitment of the minichromosome maintenance (MCM) helicase (24), which is a hexamer consisting of MCM2 to 7, that functions to unwind DNA during replication (25). However, Cdc6 and Cdt1 inhibit activation of the MCM helicase until the start of S phase (26), when Cdc6 is phosphorylated by S phase CDKs, leading to its nuclear export and degradation (27, 28). Inactivation of Cdc6 and Cdt1 allows Cdc7 to phosphorylate the MCM helicase at the start of S phase (29), leading to its activation. Cdc45 subsequently binds to the helicase and recruits DNA polymerase α, which initiates DNA replication (30).
Here, we report a role for the HIF-1α protein as a regulator of DNA helicase loading and activation. HIF-1α interacted with Cdc6 and promoted nuclear localization of Cdc6 and interaction with MCM proteins. This led to enhanced MCM helicase loading, but blocked subsequent recruitment of Cdc7, leading to decreased Cdc7-mediated phosphorylation and decreased replication origin firing. Induction of HIF-1α blocked replication origin firing and DNA replication in multiple cell types, which led to decreased cell proliferation.
RESULTS
HIF-1α inhibits cell proliferation in the absence of p21
On the basis of our previous discovery of interactions between HIF-1α and MCM subunits (16), we hypothesized that HIF-1α may directly inhibit proliferation by effects on the pre-RC, independent of effects on p21 transcription. To test this hypothesis, we analyzed the cell cycle profiles of otherwise isogenic human HCT116 colorectal carcinoma cells that were either wild type or p21-null. G1 phase cell cycle arrest was induced by hypoxia (1% O2) in both wild-type and p21-null cells (Fig. 1A and fig. S1A). Similarly, chemical induction of HIF-1α by treatment of cells with DFX or DMOG led to a p21-independent decrease in bromodeoxyuridine (BrdU) incorporation (Fig. 1B).
Fig. 1.
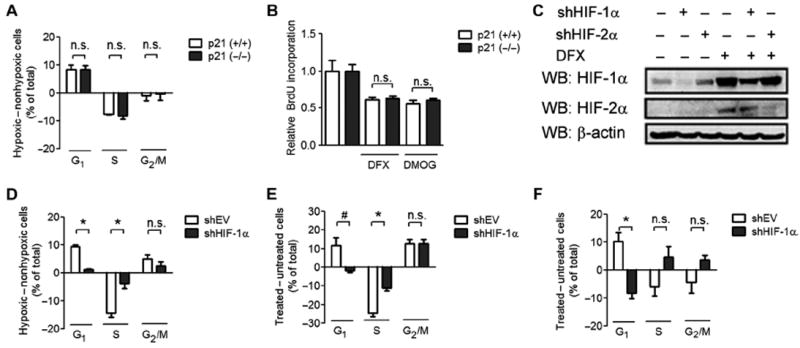
HIF-1α promotes cell cycle arrest that is independent of p21. (A) HCT116 p21 (+/+) and p21 (−/−) cells were exposed to nonhypoxic (20% O2) or hypoxic (1% O2) culture conditions. The difference in the percentage of cells at the indicated stage of the cell cycle between the hypoxic and the nonhypoxic states is shown as means ± SEM; n.s., difference is not significant. n = 3 to 4 independent replicates. (B) HCT116 p21 (+/+) cells (open bar) or p21 (−/−) cells (closed bar) were left untreated or treated with DFX or DMOG and then labeled with BrdU. BrdU labeling was measured, and data were normalized to the untreated control. n = 3 to 4 independent replicates. (C) HCT116 cells were infected with shEV or vector encoding shHIF-1α or shHIF-2α. Cells were treated with DFX or left untreated. Cell lysates were subjected to Western blot (WB) assays using antibodies against HIF-1α, HIF-2α, and β-actin. Data are representative of two independent experiments. (D to F) HCT116-shEV or HCT116–shHIF-1α cells were exposed to 20 or 1% O2 (D), treated with DMOG (E), or treated with DFX (F). The difference in the percentage of cells at the indicated stage of the cell cycle between the treated and the untreated states is shown as mean ± SEM. *P < 0.01; #P < 0.05; n.s., not significant. n = 3 to 4 independent replicates.
To verify that the cell cycle arrest induced by hypoxia, DMOG, or DFX was HIF-1α–dependent, we stably transfected HCT116 cells with either an empty short hairpin RNA (shRNA) vector (shEV) or vector encoding shRNA against HIF-1α (shHIF-1α). Immunoblot assays confirmed that HIF-1α abundance was decreased in cells expressing shHIF-1α, with no apparent effect on HIF-2α abundance (Fig. 1C). HIF-1α knockdown blocked the G1 phase cell cycle arrest induced by hypoxia (Fig. 1D and fig. S1B), DMOG (Fig. 1E and fig. S1C), or DFX (Fig. 1F and fig. S1D).
HIF-1α interacts with Cdc6
The rapid time scale in which DNA replication was inhibited after induction of HIF-1α suggested a direct effect on the DNA replication machinery. To understand the effect of HIF-1α on the cell cycle, we performed binding assays between HIF-1α and various components of the pre-RC, including Orc2, Cdc6, and Cdt1. Endogenous HIF-1α coimmunoprecipitated with endogenous Cdc6 in hypoxic cell lysates from 293T, HCT116, and HeLa cells (Fig. 2A). Similarly, endogenous Cdc6 coimmunoprecipitated with endogenous HIF-1α from hypoxic cell lysates (Fig. 2B). To ensure that the interaction was not an artifact of DNA bridging, we performed coimmunoprecipitations in the presence of the intercalating agent ethidium bromide (EtBr), which disrupts nonspecific protein interactions mediated by DNA (31). The interaction of Cdc6 with HIF-1α was resistant to EtBr treatment (Fig. 2C). Finally, we determined that Cdc6 bound to the N-terminal region of HIF-1α (residues 1 to 329) (Fig. 2D), which is essential for inhibition of the cell cycle (15); HIF-1α bound to the N-terminal half of Cdc6 (Fig. 2E).
Fig. 2.
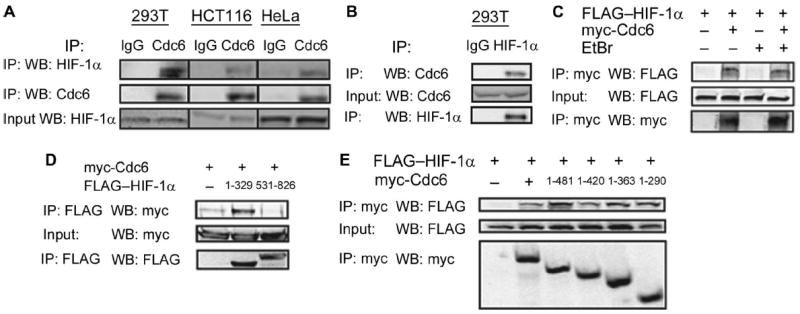
HIF-1α binds to Cdc6. (A) Immunoglobulin G (IgG) or Cdc6 immunoprecipitates (IP) from hypoxic 293T, HCT116, and HeLa cells were probed for either Cdc6 or HIF-1α. (B) IgG or HIF-1α immunoprecipitates from 293T cells exposed to 1% O2 were probed for HIF-1α and Cdc6. (C) Co-IP assay of HIF-1α in cells cotransfected with either empty vector (EV) or vector encoding myc-epitope–tagged Cdc6. IPs were performed in the presence or absence of EtBr. (D) Co-IP assay of 293T cells cotransfected with myc-Cdc6 expression vector and EV or vector encoding FLAG–HIF-1α (1 to 329) or FLAG–HIF-1α (531 to 826). (E) Co-IP assay of 293T cells cotransfected with FLAG–HIF-1α vector and EV or vector encoding the indicated myc-Cdc6 deletion mutant. All data are representative of two independent experiments.
HIF-1α promotes Cdc6 nuclear localization and chromatin association
Despite a readily detectable interaction between Cdc6 and HIF-1α, Cdc6 did not affect HIF-1α protein abundance (fig. S2A) or HIF-1 transcriptional activity (fig. S2B), as determined by immunoblot and reporter assays (32), respectively. However, confocal microscopy demonstrated a shift in Cdc6 localization from the cytoplasm to the nucleus in DMOG-treated cells (Fig. 3A). Exposure of cells to hypoxia or DMOG significantly increased the number of cells with Cdc6-positive nuclei (Fig. 3B). Similarly, induction of HIF-1α by exposure of cells to hypoxia or DMOG increased the abundance of endogenous Cdc6 in the chromatin fraction (Fig. 3, C and D). However, a similar effect on Cdt1 was not observed (Fig. 3, C and D). Knockdown of HIF-1α decreased the association of Cdc6 with chromatin upon DMOG treatment in HCT116 cells (Fig. 3E). Exposure of normal human foreskin fibroblasts to hypoxia appeared to have similar effects (Fig. 3F). Overexpression of HIF-1α was sufficient to increase the abundance of Cdc6 in the chromatin fraction of nonhypoxic HCT116 (Fig. 3G), HeLa (Fig. 3H), and NIH-3T3 (Fig. 3I) cells, which all arrest in response to hypoxia (Fig. 1D and fig. S3, A and B). The effect of HIF-1α on the association of Cdc6 with chromatin appeared to be maintained even in quiescent NIH-3T3 cells (Fig. 3J), suggesting that this finding was not a secondary effect of cell cycle arrest induced by HIF-1α overexpression.
Fig. 3.
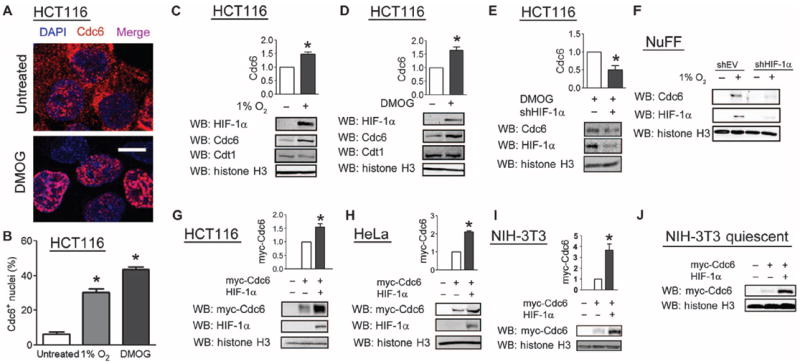
HIF-1α promotes the association of Cdc6 with chromatin. (A) HCT116 cells that were untreated, treated with DMOG, or exposed to 1% O2 were stained with an anti-Cdc6 antibody and DAPI (4′,6-diamidino-2-phenylindole) to mark nuclei and analyzed by confocal microscopy. Scale bar, 10 μm. (B) The percentage of nuclei that stained for Cdc6 was determined. Results are shown as means ± SEM. *P < 0.01. Fifty to 100 cells were analyzed per experiment. Data are representative of three independent experiments. (C and D) Immunoblot analysis was performed on the chromatin fraction from HCT116 cells that were exposed to hypoxic conditions (C) or DMOG (D). (E) Immunoblot analysis was performed on the chromatin fraction from HCT116 cells transfected with empty vector or vector encoding an shHIF-1α and treated with DMOG. (F) Immunoblot analysis was performed on the chromatin fraction from newborn human foreskin fibroblasts that were stably transfected with empty vector or shHIF-1α vector and exposed to 20 or 1% O2. Data are representative of two independent experiments. (G to J) Immunoblot analysis was performed on the chromatin fraction from HCT116 (G), HeLa (H), proliferating NIH-3T3 (I), or contact-inhibited NIH-3T3 (J) cells that were cotransfected with empty vector, myc-epitope–tagged Cdc6 vector, or HIF-1α expression vector. Western blot bands were quantified from three independent experiments in (C), (D), (E), (G), (H), and (I). Results are shown as means ± SEM. *P < 0.05.
HIF-1α promotes interaction of Cdc6 with the MCM helicase
Because a key function of Cdc6 is to recruit the MCM helicase to origins of replication in an inactive state (33), we examined the effect of HIF-1α on the interaction of Cdc6 with the MCM helicase. DMOG treatment of HCT116 cells increased the colocalization of Cdc6 with MCM5 (Fig. 4, A and B) and with MCM7 (Fig. 4C). Overexpression of HIF-1α in HeLa cells led to an increase in the colocalization of Cdc6 with MCM5 (Fig. 4D) and with MCM7 (Fig. 4E). Coimmunoprecipitation experiments demonstrated that despite an 80% reduction in total Cdc6 abundance, DMOG treatment appeared to enhance binding of endogenous Cdc6 to all members of the MCM complex that were analyzed (Fig. 4F). HIF-1α knockdown blocked the increase in Cdc6-MCM interactions induced by exposure to hypoxia or treatment with DMOG (Fig. 4G), directly implicating HIF-1α in promoting the interaction between Cdc6 and MCM proteins.
Fig. 4.
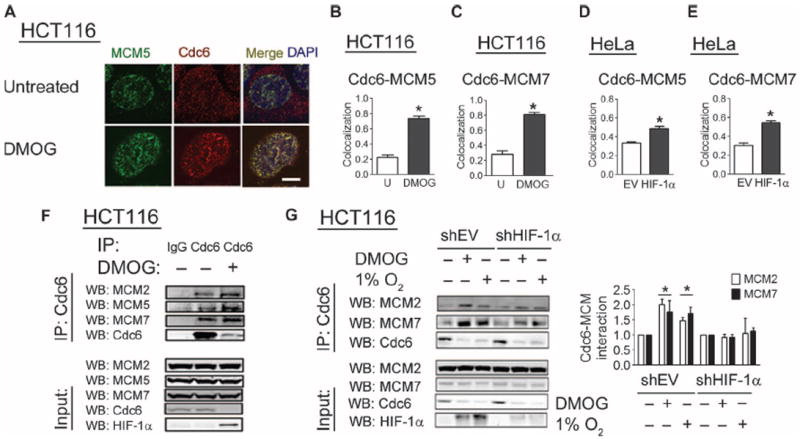
HIF-1α promotes interaction of Cdc6 with the MCM helicase. (A) HCT116 cells were untreated or treated with DMOG, stained with anti-Cdc6 and anti-MCM5 antibodies, and analyzed by confocal microscopy. Scale bar, 5 μm. (B and C) Pearson’s correlation coefficient between the Cdc6 and MCM5 signals (B) or between the Cdc6 and MCM7 signals (C) was determined as a measure of colocalization. (D and E) HeLa cells transfected with a green fluorescent protein (GFP) vector and either empty vector (EV) or HIF-1α vector were stained with Cdc6 and either MCM5 or MCM7 antibody, and GFP-positive cells were analyzed by confocal microscopy. Pearson’s correlation coefficient between the Cdc6 and MCM5 (D) or MCM7 (E) signal is indicated. *P < 0.01. Ten to 20 cells were examined from two independent experiments in (B) to (E). (F) Lysates (input; bottom panels) and IgG or Cdc6 immunoprecipitates (top panels) from untreated cells or cells treated with DMOG were probed with anti-Cdc6, MCM2, MCM5, and MCM7 antibodies. Data are representative of two independent experiments. (G) Lysates and immunoprecipitates from HCT116-shEV and HCT116–shHIF-1α cells that were untreated, treated with DMOG, or exposed to 1% O2 were probed with anti-Cdc6, MCM2, MCM7, and HIF-1α antibodies. Western blot data from three independent replicates were quantified. Results are shown as means ± SEM. DMOG treatment or 1% O2 exposure was compared to the untreated condition. *P < 0.05.
HIF-1α promotes loading of the MCM helicase
Consistent with an effect of HIF-1α on the association of Cdc6 with chromatin, confocal microscopy demonstrated an increase in MCM5 foci in the nuclei of cells treated with DMOG (Fig. 5A). Overexpression of HIF-1α in nonhypoxic HeLa cells was sufficient to increase chromatin loading of MCM2 (Fig. 5B) and MCM7 (Fig. 5C). We examined MCM chromatin loading in HCT116 (Fig. 5D), HeLa (fig. S4A), and NIH-3T3 (fig. S4B) cells, all of which showed increased chromatin loading of endogenous MCM proteins in response to HIF-1α induction by hypoxia, DFX, or DMOG. Hypoxia increased MCM chromatin loading even in quiescent NIH-3T3 cells (fig. S4C). Knockdown of HIF-1α reduced MCM chromatin loading in HCT116 cells (Fig. 5E) and normal human foreskin fibroblasts (Fig. 5F) exposed to 1% O2, further demonstrating that this is a physiologic response to hypoxia that is mediated by HIF-1α. Overexpression of HIF-1α or Cdc6 increased chromatin loading of MCM2 (Fig. 5G), MCM3 (Fig. 5H), and MCM7 (Fig. 5I) to comparable extents, and MCM loading was further increased by overexpression of both Cdc6 and HIF-1α (Fig. 5G). Analysis of NIH-3T3 cells revealed a combined effect of HIF-1α and Cdc6 on MCM loading (fig. S4, D and E) in that cell line as well.
Fig. 5.
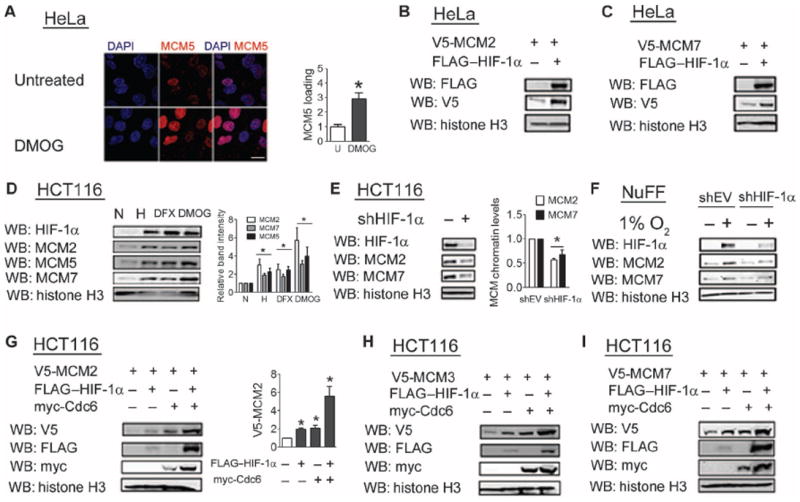
HIF-1α promotes chromatin loading of MCM helicase. (A) HeLa cells were left untreated or treated with DMOG, stained with MCM5 antibody, and analyzed by confocal microscopy. MCM5 staining was quantified, and results are shown as means ± SEM. *P < 0.01. n = 8 cells from two independent experiments. Scale bar, 20 μm. (B and C) The chromatin fraction from HeLa cells cotransfected with expression vector encoding V5-MCM2 (B) or V5-MCM7 (C) and either empty vector (EV) or FLAG–HIF-1α vector was probed with the indicated antibodies. Data are representative of two independent experiments. (D) The chromatin fraction from HCT116 cells exposed to 20% O2 (normoxia, N), 1% O2 (hypoxia, H), DFX, or DMOG was probed with the indicated antibodies. Data are compared to the untreated and nonhypoxic condition. (E and F) The chromatin fraction from HCT116 cells (E) or newborn foreskin fibroblasts (F) stably transfected with shEV or shHIF-1α vector and exposed to 1% O2 was probed with the indicated antibodies. (G to I) The chromatin fraction from HCT116 cells cotransfected with vector encoding V5-MCM2 (G), V5-MCM3 (H), or V5-MCM7 (I) and vector encoding FLAG–HIF-1α, myc-Cdc6, or both was probed with the indicated antibodies. Western blot data from three independent replicates were quantified for (D), (E), and (G). Results are shown as means ± SEM. *P < 0.05.
Effects of HIF-1α on MCM chromatin loading and activation are independent of transcription
The effect of HIF-1α overexpression was observed in cells that were co-transfected with a Cdc6 expression vector lacking any transcriptional or translational regulatory elements, which excluded a role for HIF-1α in increasing Cdc6 abundance through regulation of the Cdc6 promoter (Fig. 3, G to J). Hypoxia decreases Cdc6 mRNA expression in endothelial cells (34), and, consistent with this report, we observed a decrease in the abundance of both Cdc6 mRNA (fig. S5A) and protein (Fig. 4G and fig. S5B) upon exposure of cells to hypoxia or DMOG, whereas the abundance of another pre-RC component, Orc2, appeared to be unchanged (fig. S5B).
To determine whether HIF-1 transcriptional activity was required for the effects of HIF-1α on MCM chromatin loading, we generated two subclones that stably expressed shRNAs targeting two different sequences within HIF-1β mRNA, which were designated shHIF-1β-A and shHIF-1β-B. We confirmed that both shRNAs decreased HIF-1β mRNA abundance (fig. S6A). Both knockdown subclones showed impaired induction of HIF-1 reporter activity (fig. S6B) and impaired induction of the HIF-1 target gene VEGF (vascular endothelial growth factor) in response to hypoxia (fig. S6C). Enhanced loading of MCM proteins in response to hypoxia was preserved in both the shHIF-1β-A (Fig. 6A) and shHIF-1β-B (Fig. 6B) subclones. Similarly, enhanced loading of MCM7 in response to HIF-1α overexpression was preserved in the subclone expressing shHIF-1β-A (Fig. 6C) or shHIF-1β-B (Fig. 6D).
Fig. 6.
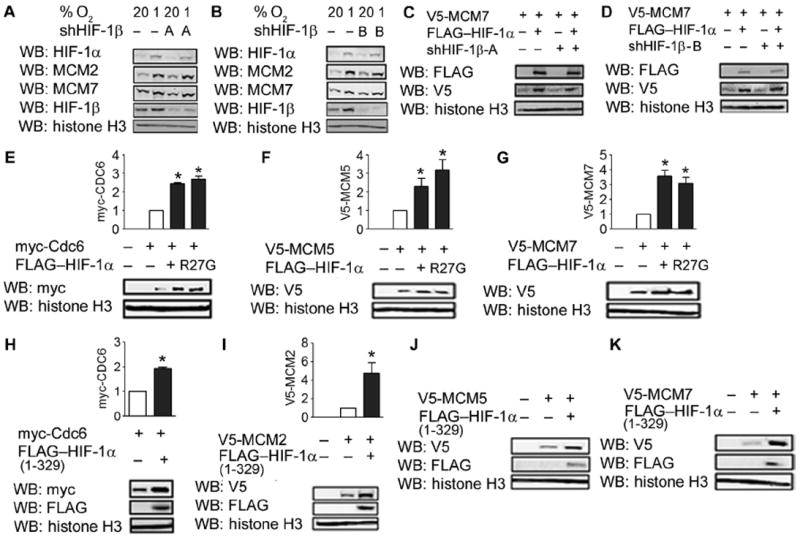
The effects of HIF-1α on Cdc6 and the MCM helicase are independent of transcriptional activity. (A and B) MCM loading in the chromatin fraction of shEV and shHIF-1β-A (A) or shHIF-1β-B (B) cells under non-hypoxic and hypoxic conditions was analyzed. Data are representative of two independent experiments. (C and D) V5-MCM7 in the chromatin fraction of shEV and shHIF-1β-A (C) or shHIF-1β-B (D) cells was analyzed. Data are representative of two independent experiments. (E) The chromatin fraction from HeLa cells transfected with EV, myc-Cdc6, and either the P402/564A double-mutant HIF-1α (DM) or HIF-1α (DM; R27G) vector was probed with the indicated antibodies. (F and G) The chromatin fraction from HeLa cells cotransfected with vector encoding V5-MCM5 (F) or V5-MCM7 (G) and either EV, HIF-1α (DM), or HIF-1α (DM; R27G) vector was probed with the indicated antibodies. (H) The chromatin fraction from HeLa cells transfected with vector encoding myc-Cdc6 and either EV or vector encoding HIF-1α (1 to 329) deletion mutant was probed with the indicated antibodies. (I to K) The chromatin fraction from cells cotransfected with vector encoding V5-MCM2 (I), V5-MCM5 (J), or V5-MCM7 (K) and either EV or HIF-1α (1 to 329) vector was probed with the indicated antibodies. Western blots from (J) and (K) are representative of two independent experiments. Western blot data from three independent replicates were quantified for (E) to (I). Results are shown as means ± SEM. *P < 0.05.
We also observed that an R27G mutation in HIF-1α that abolishes the DNA binding activity of HIF-1 (15) had no effect on the ability of HIF-1α to increase Cdc6 chromatin association (Fig. 6E). Similarly, the R27G mutation had no effect on the ability of HIF-1α to increase the association of MCM5 (Fig. 6F) and MCM7 (Fig. 6G) with chromatin. On the basis of these results, we concluded that the effects of HIF-1α on MCM chromatin loading were independent of HIF-1 transcriptional activity.
The N-terminal region of HIF-1α contains binding sites for Cdc6 (Fig. 2D) and MCM proteins (16). Expression of the HIF-1α (1 to 329) deletion fragment, which lacks the C-terminal transactivation domains, was sufficient to increase the chromatin localization of Cdc6 (Fig. 6H), as well as that of MCM2 (Fig. 6I), MCM5 (Fig. 6J), and MCM7 (Fig. 6K). We conclude that the Cdc6- and MCM-interacting binding domains in the N-terminal half of HIF-1α are sufficient to regulate MCM chromatin loading independent of transcriptional activation.
HIF-1α blocks Cdc7-mediated activation of the MCM helicase
Cdc6 inhibits helicase activation by blocking Cdc7-mediated phosphorylation of the MCM helicase (26). We hypothesized that the Cdc6-mediated increase in MCM chromatin loading was associated with an inhibition of helicase activation. Consistent with this hypothesis, we observed decreased abundance of chromatin-bound Cdc7 in hypoxic HCT116 cells, whereas no effect on total Cdc7 abundance was observed (Fig. 7A). In NIH-3T3 cells, DFX or DMOG treatment decreased the abundance of chromatin-bound Cdc7 (Fig. 7, B and C). Induction of HIF-1α by exposure to DMOG or hypoxia led to decreased Cdc7-mediated phosphorylation of MCM2 (Fig. 7D). Despite increased MCM2 chromatin loading upon exposure to hypoxia, phosphorylation of MCM2 in the chromatin fraction was decreased (Fig. 7E). Similar results were obtained in HeLa cells (Fig. 7F). In contrast, hypoxia did not affect the phosphorylation of MCM2 in WT8 cells (Fig. 7G), which lack HIF-1α and do not arrest in response to hypoxia (17).
Fig. 7.
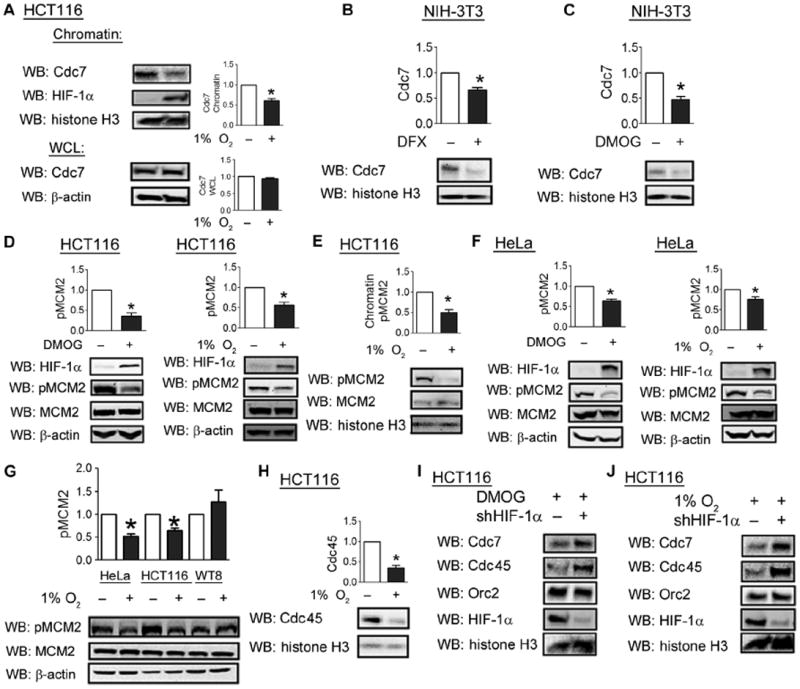
HIF-1α inhibits Cdc7-mediated phosphorylation and activation of the MCM helicase. (A) The chromatin fraction and whole-cell lysates of HCT116 cells exposed to 20 or 1% O2 were isolated and analyzed with anti-Cdc7, HIF-1α, or histone H3 antibodies. (B and C) NIH-3T3 cells were untreated or treated with DFX (B) or DMOG (C) for 24 hours, after which the chromatin fraction was isolated. (D) HCT116 cells were left untreated, treated with DMOG, or exposed to 1% O2 for 24 hours, and cell lysates were probed with the indicated antibodies. (E) The chromatin fraction from HCT116 cells exposed to 20 or 1% O2 was probed with indicated antibodies. (F) HeLa cells were processed as in (D). (G) Lysates from HeLa, HCT116, and WT8 cells exposed to either 20 or 1% O2 were probed with the indicated antibodies. (H) The chromatin fraction from HCT116 cells exposed to 20 or 1% O2 was probed with indicated antibodies. (I and J) HCT116 cells were transfected with either empty shRNA vector or vector encoding shRNA against HIF-1α and exposed to DMOG (I) or 1% O2 (J). The chromatin fraction was analyzed with the indicated antibodies. Data in (I) and (J) represent two independent experiments. Western blot data from three independent replicates were quantified for (A) to (H). Results are shown as means ± SEM. *P < 0.05.
Consistent with a decrease in MCM helicase activation, hypoxic HCT116 cells showed a decrease in the abundance of Cdc45 in the chromatin fraction, which serves as a direct indicator of active replication origins (Fig. 7H). Conversely, HIF-1α knockdown was sufficient to increase chromatin-bound Cdc7 abundance in HCT116 cells exposed to DMOG (Fig. 7I) or hypoxia (Fig. 7J). Similarly, the abundance of chromatin-bound Cdc45 was increased upon HIF-1α knockdown (Fig. 7, I and J). However, HIF-1α knockdown did not appear to alter the abundance of Orc2, an upstream component of the pre-RC (Fig. 7, I and J). Together, these results indicate that by increasing the abundance of chromatin-bound Cdc6, HIF-1α enhances chromatin loading of MCMs but inhibits Cdc7-mediated phosphorylation and activation of the MCM helicase.
HIF-1α inhibits replication origin firing
To investigate whether the observed effects of HIF-1α lead to altered replication origin firing, we pulse-labeled untreated or DMOG-treated cells with BrdU to visualize sites of DNA synthesis. DMOG treatment decreased not only the number of BrdU+ cells (Fig. 8A) but also the number of foci of DNA synthesis in BrdU+ cells (Fig. 8B). We next tested whether HIF-1α overexpression was sufficient to inhibit DNA synthesis. The fraction of cells undergoing DNA replication was significantly lower in cells transfected with an HIF-1α expression vector than in cells transfected with an empty vector (Fig. 8C), as was the number of foci of DNA synthesis in replicating cells (Fig. 8D).
Fig. 8.
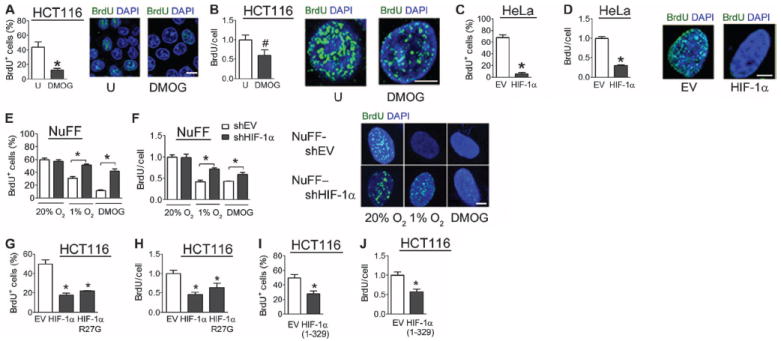
HIF-1α inhibits firing of replication origins and inhibits DNA synthesis. (A and B) Untreated or DMOG-treated HCT116 cells were exposed to a 5-min pulse of BrdU and analyzed by confocal microscopy. The percentage of BrdU+ cells was determined (A), and the number of sites of BrdU incorporation among replicating cells only was determined (B). n = 10 to 20 cells from two independent experiments. Scale bars, 10 μm (A) and 5 μm (B). (C and D) HeLa cells cotransfected with a GFP vector and either empty vector (EV) or HIF-1α vector were pulse-labeled with BrdU and analyzed by confocal microscopy. Transfected cells were analyzed for the percentage of BrdU+ cells (C) and BrdU incorporation among replicating cells only (D). n = 10 to 20 cells from two independent experiments. Scale bar, 5 μm. (E and F) Newborn human foreskin fibroblasts stably transfected with empty shRNA vector (NuFF-shEV) or vector encoding shRNA against HIF-1α (NuFF–shHIF-1α) were left untreated, treated with DMOG, or exposed to 1% O2, pulse-labeled with BrdU, and analyzed by confocal microscopy for sites of BrdU incorporation. The percentage of cells positive for BrdU incorporation (E) and BrdU incorporation among BrdU+ cells only (F) were determined. n = 10 to 20 cells from two independent experiments. Scale bar, 5 μm. (G and H) HCT116 cells transfected with EV or vector encoding HIF-1α or HIF-1α (R27G) were pulse-labeled with BrdU and analyzed. n = 15 to 30 cells from three independent experiments. (I and J) HCT116 cells transfected with either EV or FLAG–HIF-1α (1 to 329) vector were pulse-labeled with BrdU and analyzed. The percentage of cells positive for BrdU incorporation (I) and relative BrdU foci among BrdU+ cells only (J) were determined. n = 15 to 30 cells from three independent experiments. Results are shown as means ± SEM. *P < 0.01; #P < 0.05.
To complement the observations in cell lines, we examined the effect of HIF-1α on DNA synthesis in primary human newborn foreskin fibroblasts. Exposing cells to hypoxia or DMOG led to a decreased percentage of replicating cells (Fig. 8E) and a decrease in the number of foci of DNA synthesis in replicating cells (Fig. 8F). The effects of hypoxia and DMOG on DNA synthesis were blunted in HIF-1α knockdown cells (Fig. 8, E and F). Together, these results indicate that hypoxia inhibits DNA replication in mammalian cells and that increased HIF-1α protein abundance induces this response.
To exclude a role for HIF-1–mediated transcription, we analyzed the effect on BrdU incorporation using the R27G mutant that is defective in DNA binding and transcriptional activity. The R27G mutation did not affect the ability of HIF-1α to inhibit BrdU incorporation (Fig. 8, G and H). Similarly, overexpression of HIF-1α (1 to 329), which lacks the C-terminal transactivation domains, decreased the number (Fig. 8I) of BrdU+ cells and DNA synthesis in replicating cells (Fig. 8J). We conclude that HIF-1α serves a nontranscriptional role as a direct regulator of DNA replication and cell cycle progression.
DISCUSSION
The regulation of cell proliferation according to O2 availability is a fundamental physiological response of mammalian cells. Here, we have delineated a mechanism by which DNA replication is inhibited in response to hypoxia (Fig. 9). We have demonstrated that HIF-1α interacts with Cdc6, increases the abundance of chromatin-bound Cdc6, and promotes Cdc6 interaction with the MCM helicase, which is associated with increased chromatin loading of the MCM complex but decreased recruitment of the kinase Cdc7 as well as decreased phosphorylation and activation of the MCM helicase, leading to reduced replication origin firing.
Fig. 9.
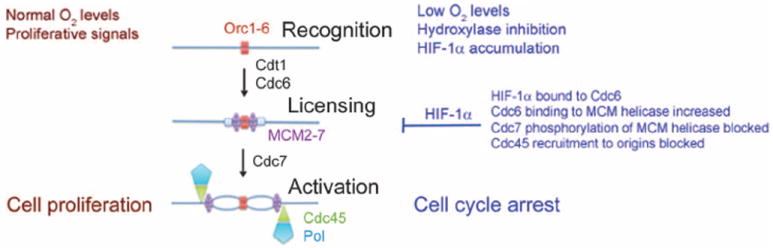
HIF-1α inhibits DNA replication in hypoxic cells. Under proliferative conditions, the ORC binds to replication origins. Cdc6 and Cdt1 subsequently recruit the MCM helicase complex but maintain it in an inactive state until they are phosphorylated and exported to the cytoplasm at the start of S phase. The subsequent recruitment of Cdc7 leads to phosphorylation and activation of the MCM helicase, leading to recruitment of Cdc45 and DNA polymerase a (Pol). Under hypoxic conditions, HIF-1α mediates increased Cdc6 binding to the MCM helicase, which leads to increased MCM helicase loading but decreased recruitment of Cdc7, thereby blocking DNA replication and cell cycle progression.
The mechanisms by which hypoxia affects cell proliferation have been controversial. Multiple studies in diverse cell lines, which implicated HIFs in this process, have focused mechanistically on effects mediated through increased expression of genes encoding the CDK inhibitors p21 (15, 17), p27 (35), or both (36). In certain cancer cell lines, a HIF-independent effect on cyclin D1 has also been reported (37, 38). Previous work has suggested that, in certain cases, the relative abundance of HIF-1α as opposed to HIF-2α determines the sensitivity of the cell cycle to hypoxia through transcriptional effects (17).
Our study delineates a mechanism by which HIF-1α can immediately block DNA replication by directly binding to components of the pre-RC. Multiple lines of experimental evidence indicate that this role of HIF-1α is independent of transcriptional activity. First, the effects on BrdU incorporation were detectable within 6 hours of HIF-1α induction, a time scale inconsistent with effects mediated through new gene transcription and translation. Second, the effects of HIF-1α overexpression were observed in cells transfected with Cdc6 and MCM expression vectors that lacked any transcriptional or translational regulatory elements. Hypoxia, in fact, decreased mRNA and protein abundance of Cdc6 and the MCM components (16), indicating that the increased Cdc6 and MCM chromatin loading that was mediated by HIF-1α did not result from changes in the abundance of these proteins. Third, the proliferation of HIF-1β knockdown cells was reduced in response to hypoxia. The abundance of HIF-1α increased in these cells after exposure to hypoxia, but induction of HIF-1 target genes, which requires activity of the HIF-1α/HIF-1β heterodimer, was impaired. Nonetheless, the effect of HIF-1α overexpression and hypoxic induction of HIF-1α on the MCM helicase was preserved in these cell lines. Fourth, a point mutant of HIF-1α with impaired DNA binding retained effects on the Cdc6 and MCM proteins. Finally, a HIF-1α deletion mutant retaining only the Cdc6- and MCM-interacting domains, but lacking transactivation domains, mediated the antiproliferative effects of full-length HIF-1α. Our data establish a paradigm in which HIF-1α, by binding and regulating the activity of protein complexes, may exert biological effects through rapid, nontranscriptional mechanisms.
We demonstrated the effects of HIF-1α on the MCM helicase in various cell types, including HCT116 colorectal carcinoma cells, HeLa cervical carcinoma cells, NIH-3T3 mouse fibroblasts, and newborn foreskin fibroblasts. However, certain cell types, including some cancer cells, maintain cell proliferation under hypoxic conditions. We previously demonstrated that, in addition to their role as a helicase, multiple MCM proteins bind to and enhance the degradation of HIF-1α (16), thereby providing a mechanism by which proliferative signals can override the effect of hypoxia on the cell cycle. Together with the data presented here, a model emerges in which a dynamic and mutually antagonistic balance between HIF-1α and MCM proteins, sensitive to both hypoxia and proliferative signals, regulates the cellular decision to arrest or maintain proliferation under hypoxic conditions.
While this manuscript was in preparation, another group reported that under conditions of severe hypoxia (<0.1% O2), during which MCM helicase loading is inhibited (39), an HIF-independent decrease in Cdc6 abundance becomes limiting (40). Previous work has established that severe hypoxia leads to increased activity of the kinase ATR (ataxia telangiectasia) (41), even in the absence of DNA damage (42). Increased kinase activity of ATR may underlie the observed decrease in Cdc6 abundance during severe hypoxia (40). We demonstrated a decrease in Cdc6 mRNA and protein abundance, even under more moderate hypoxia (fig. S5, A and B), which appeared to be hydroxylase-dependent but HIF-1α–independent (Fig. 4G). We observed that total MCM protein abundance also decreased after prolonged exposure to hypoxia (16). However, under moderate hypoxia, the decrease in total Cdc6 or MCM protein abundance was not sufficiently limiting to decrease Cdc6 or MCM abundance in chromatin. Together with our study, these results suggest that hypoxia regulates MCM helicase activity through multiple mechanisms depending on the duration and severity of the hypoxic stimulus. The acute response to moderate (physiological) hypoxia is reversible upon reoxygenation, whereas the response to chronic or severe (pathological) hypoxia is longer lasting because of the need for new synthesis of pre-RC components. Under physiological conditions, the mutual antagonism of HIFs and MCMs provides a potential mechanism for cell type–specific proliferative responses to hypoxia based on the relative balance between HIFs and MCMs. Despite dysregulated cell proliferation, many cancer cells still undergo cell cycle arrest in response to hypoxia and are therefore resistant to the effect of cytotoxic chemotherapy, which targets dividing cells. Our results suggest that drugs that inhibit HIF-1α accumulation (43) may render hypoxic cancer cells sensitive to chemotherapy.
MATERIALS AND METHODS
Tissue culture
The 293T, NIH-3T3, HeLa, and WT8 cell lines and newborn foreskin fibroblasts were cultured in Dulbecco’s modified Eagle’s medium, and HCT116 cells were cultured in McCoy Medium 5A, all supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Cells were maintained at 37°C in a 5% CO2, 95% air incubator. Cells were subjected to hypoxia by exposure to 1% O2/5% CO2/balance N2 at 37°C in a modular incubator chamber (Billups-Rothenberg). DFX (Sigma) was used at a dose of 100 μM, and DMOG (Sigma) was used at a dose of 1 mM.
Immunoprecipitation and immunoblot assays
Cells were lysed in phosphate-buffered saline (PBS) with 0.1% Tween 20, 1 mM dithiothreitol (DTT), protease inhibitor cocktail, 1 mM Na3VO4, and 10 mM NaF, followed by gentle sonication. For immunoprecipitation assays, 30 μl of anti-V5 agarose beads (Sigma) was incubated with 2.5 mg of cell lysate overnight at 4°C. Beads were washed four times in lysis buffer. Proteins were eluted in SDS sample buffer and fractionated by SDS–polyacrylamide gel electrophoresis. The following antibodies were used in immunoblot and immunoprecipitation assays: histone H3, Cdc6, and β-actin (Santa Cruz Biotechnology); HIF-1α (BD Biosciences); FLAG (Sigma); and MCM2, phospho-MCM2 (Ser41), MCM5, MCM7, Cdt1, Cdc6, Cdc7, Cdc45, Orc2, and Myc epitope (Novus Biologicals).
Reverse transcription–quantitative real-time polymerase chain reaction assay
Total RNA was extracted from 293T cells using TRIzol (Invitrogen) and was treated with DNase I (Ambion). Total RNA (1 μg) was used for firststrand synthesis with the iScript cDNA Synthesis system (Bio-Rad). Real-time polymerase chain reaction (PCR) was performed with iQ SYBR Green Supermix and iCycler Real-Time PCR Detection System (Bio-Rad). Expression of target mRNA relative to 18S ribosomal RNA (rRNA) was calculated on the basis of the threshold cycle (CT) for amplification as 2−ΔCT, where ΔCT = CT,target – CT,18S. Primer sequences used were as follows: 18S rRNA, 5′-cggcgacgacccattcgaac-3′ and 5′-gaatcgaaccctgattccccgtc-3′; VEGF mRNA, 5′-cttgccttgctgctctac-3′ and 5′-tggcttgaagatgtactgg-3′; and HIF-1β, 5′-ctgccaaccccgaaatgacat-3′ and 5′-gccgcttaatagccctctgg-3′.
Immunofluorescence assay
Immunofluorescence was performed as previously described (44). HCT116 cells were plated on gelatin-coated glass-bottomed plates (LiveAssay, LA-RD1.5UNC) and exposed to hypoxia or hydroxylase inhibitor for 18 hours. For immunocytochemistry, samples were washed with ice-cold PBS, fixed with ethanol for 20 min at room temperature, permeabilized with 0.05% Triton X-100 for 15 min, washed twice with PBS, and blocked with 10% goat serum/1% AlbuMAX (Invitrogen) for 1 hour. Samples were incubated with primary antibody for 1 hour, washed, and incubated with Alexa Fluor–conjugated secondary antibody (Invitrogen) for 1 hour. Samples were washed and mounted to microscopy slides with a drop of SlowFade (Invitrogen) and sealed with medical adhesive (Hollister).
Microscopy
Phase contrast microscopy was performed with Nikon SPOT 2.0. Images were acquired using SPOT Insight 2MP camera with SPOT Imaging software (Diagnostic Instruments) and processed using Photoshop (Adobe Systems). Epifluorescence microscopy was performed with an Axiovert 2000 (Zeiss) using 10×, 20×, and 40× apochromat objective lenses with 1.4 numerical aperture (NA). Lambda LS xenon arc lamp (Sutter Instrument) was used for excitation, and acquisition was performed with a Cascade 512BII camera (Roper Scientific). Images were acquired and processed using Slidebook 4.2.0 (Intelligent Imaging Innovations), and processing was performed with Adobe Photoshop. Confocal microscopy was performed with an LSM 510 Meta CLSM (Zeiss) using 20× and 40× apochromat objective lens with 1.4 NA. Pinholes were adjusted to 1 μm. For excitation, a blue diode (405 nm), a green argon laser (488 nm), and a red HeNe laser (568 nm) were used. Images were processed using Meta software version 3.5 (Zeiss). Computational analysis was performed with custom software written using MATLAB 6.0.
Cell cycle analysis
Otherwise isogenic wild-type and p21-null HCT116 cells (45) were fixed in 70% ethanol, stained with propidium iodide, and analyzed by fluorescence-activated cell sorting. Data were acquired in an LSR II flow cytometer (Becton Dickinson). A minimum of 10,000 events per sample were recorded. Analysis of cell cycle phases was modeled using the Dean-Jett-Fox algorithm (46).
Chromatin isolation
Chromatin fraction was isolated as previously described (47). Briefly, cells were washed with PBS, pelleted, and lysed with cytoskeleton buffer [20 mM Hepes (pH 7.8), 10 mM KCl, 2 mM EDTA, 300 mM sucrose, 0.5% Triton X-100, with protease inhibitors and phenylmethylsulfonyl fluoride]. After incubation on ice for 10 min, samples were centrifuged, and the pellet was isolated. This process was repeated twice, after which the pellets were suspended in 10 mM Hepes (pH 7.8), 2 mM EDTA, 0.3 mM EGTA, and 1 mM DTT. Samples were sonicated, and protein concentrations were normalized before immunoblot assay.
Statistical analysis
Data are presented as means ± SEM, except where otherwise noted. Differences between two conditions were analyzed using Student’s t test. Normalized Western blot data were analyzed using a one-sample t test against a theoretical mean of 1. Differences between three variables were analyzed by two-way analysis of variance (ANOVA) with Bonferroni correction.
Supplementary Material
Fig. S1. Flow cytometry data analyzed in Fig. 1.
Fig. S2. Cdc6 has no effect on HIF-1α protein abundance or HIF-1 activity.
Fig. S3. Cell cycle arrest in response to hypoxia.
Fig. S4. HIF-1α promotes MCM loading in HeLa and NIH-3T3 cells.
Fig. S5. Hypoxia decreases total Cdc6 abundance.
Fig. S6. HIF-1β knockdown cells have decreased HIF-1 transcriptional activity.
Acknowledgments
We thank P. Coulombe and G. Tomaselli (Johns Hopkins University) for helpful advice throughout the course of this study; B. Vogelstein (Johns Hopkins University) for the gift of HCT116 wild-type and p21-null subclones; and K. Padgett (Novus Biologicals Inc.) for providing antibodies against phospho-MCM2, MCM2, MCM5, MCM7, Cdt1, Cdc6, Cdc7, Cdc45, Orc2, and Myc epitope. Funding: This work was supported by Public Health Service contracts N01-HV28180 and HHS-N268201000032c, postdoctoral training grant T32-HL007525, and predoctoral training grant T32-GM008752 from the NIH; predoctoral fellowship 10PRE4160120 from the American Heart Association; postdoctoral fellowship KG111254 from the Susan G. Komen Foundation; a fellowship from the Fowler Foundation for Advanced Research in the Medical Sciences; and funds from the Johns Hopkins Institute for Cell Engineering. G.L.S. is the C. Michael Armstrong Professor at Johns Hopkins University School of Medicine and an American Cancer Society Research Professor.
Footnotes
Author contributions: M.E.H. and G.L.S. designed the experiments, analyzed the data, and wrote the paper. M.E.H. performed most of the experiments. K. collected and analyzed microscopy data and contributed to experimental design. S.R. analyzed flow cytometry data. D.M.G., C.C.W., and W.L. contributed reagents. C.V.D., D.-H.K., and A.L. provided intellectual input.
Competing interests: The authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
REFERENCES AND NOTES
- 1.Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–5454. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang GL, Semenza GL. Characterization of hypoxia-inducible factor 1 and regulation of DNA binding activity by hypoxia. J Biol Chem. 1993;268:21513–21518. [PubMed] [Google Scholar]
- 3.Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, Sham JS, Wiener CM, Sylvester JT, Semenza GL. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1α. J Clin Invest. 1999;103:691–696. doi: 10.1172/JCI5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic–helix–loop–helix–PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. C. elegans EGL-9 and mammalian homologs define a family of di-oxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 7.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr HIFα targeted for VHL-mediated destruction by proline hydroxylation: Implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 8.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 9.Yu F, White SB, Zhao Q, Lee FS. HIF-1α binding to VHL is regulated by stimulus-specific proline hydroxylation. Proc Natl Acad Sci U S A. 2001;98:9630–9635. doi: 10.1073/pnas.181341498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466–1471. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science. 2002;295:858–861. doi: 10.1126/science.1068592. [DOI] [PubMed] [Google Scholar]
- 12.Semenza GL. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology. 2009;24:97–106. doi: 10.1152/physiol.00045.2008. [DOI] [PubMed] [Google Scholar]
- 13.Patel SA, Simon MC. Biology of hypoxia-inducible factor-2α in development and disease. Cell Death Differ. 2008;15:628–634. doi: 10.1038/cdd.2008.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Box AH, Demetrick DJ. Cell cycle kinase inhibitor expression and hypoxia-induced cell cycle arrest in human cancer cell lines. Carcinogenesis. 2004;25:2325–2335. doi: 10.1093/carcin/bgh274. [DOI] [PubMed] [Google Scholar]
- 15.Koshiji M, Kageyama Y, Pete EA, Horikawa I, Barrett JC, Huang LE. HIF-1α induces cell cycle arrest by functionally counteracting Myc. EMBO J. 2004;23:1949–1956. doi: 10.1038/sj.emboj.7600196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hubbi ME, Luo W, Baek JH, Semenza GL. MCM proteins are negative regulators of hypoxia-inducible factor 1. Mol Cell. 2011;42:700–712. doi: 10.1016/j.molcel.2011.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF-2α promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell. 2007;11:335–347. doi: 10.1016/j.ccr.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goda N, Ryan HE, Khadivi B, McNulty W, Rickert RC, Johnson RS. Hypoxia-inducible factor 1α is essential for cell cycle arrest during hypoxia. Mol Cell Biol. 2003;23:359–369. doi: 10.1128/MCB.23.1.359-369.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takubo K, Goda N, Yamada W, Iriuchishima H, Ikeda E, Kubota Y, Shima H, Johnson RS, Hirao A, Suematsu M, Suda T. Regulation of the HIF-1α level is essential for hematopoietic stem cells. Cell Stem Cell. 2010;7:391–402. doi: 10.1016/j.stem.2010.06.020. [DOI] [PubMed] [Google Scholar]
- 20.Hackenbeck T, Knaup KX, Schietke R, Schödel J, Willam C, Wu X, Warnecke C, Eckardt KU, Wiesener MS. HIF-1 or HIF-2 induction is sufficient to achieve cell cycle arrest in NIH3T3 mouse fibroblasts independent from hypoxia. Cell Cycle. 2009;8:1386–1395. doi: 10.4161/cc.8.9.8306. [DOI] [PubMed] [Google Scholar]
- 21.Sclafani RA, Holzen TM. Cell cycle regulation of DNA replication. Annu Rev Genet. 2007;41:237–280. doi: 10.1146/annurev.genet.41.110306.130308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saha P, Chen J, Thome KC, Lawlis SJ, Hou ZH, Hendricks M, Parvin JD, Dutta A. Human CDC6/Cdc18 associates with Orc1 and cyclin-cdk and is selectively eliminated from the nucleus at the onset of S phase. Mol Cell Biol. 1998;18:2758–2767. doi: 10.1128/mcb.18.5.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramachandran N, Hainsworth E, Bhullar B, Eisenstein S, Rosen B, Lau AY, Walter JC, LaBaer J. Self-assembling protein microarrays. Science. 2004;305:86–90. doi: 10.1126/science.1097639. [DOI] [PubMed] [Google Scholar]
- 24.Cook JG, Park CH, Burke TW, Leone G, DeGregori J, Engel A, Nevins JR. Analysis of Cdc6 function in the assembly of mammalian prereplication complexes. Proc Natl Acad Sci U S A. 2002;99:1347–1352. doi: 10.1073/pnas.032677499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Labib K, Tercero JA, Diffley JF. Uninterrupted MCM2-7 function required for DNA replication fork progression. Science. 2000;288:1643–1647. doi: 10.1126/science.288.5471.1643. [DOI] [PubMed] [Google Scholar]
- 26.Kundu LR, Kumata Y, Kakusho N, Watanabe S, Furukohri A, Waga S, Seki M, Masai H, Enomoto T, Tada S. Deregulated Cdc6 inhibits DNA replication and suppresses Cdc7-mediated phosphorylation of Mcm2–7 complex. Nucleic Acids Res. 2010;38:5409–5418. doi: 10.1093/nar/gkq262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jiang W, Wells NJ, Hunter T. Multistep regulation of DNA replication by Cdk phosphorylation of HsCdc6. Proc Natl Acad Sci U S A. 1999;96:6193–6198. doi: 10.1073/pnas.96.11.6193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petersen BO, Lukas J, Sørensen CS, Bartek J, Helin K. Phosphorylation of mammalian CDC6 by cyclin A/CDK2 regulates its subcellular localization. EMBO J. 1999;18:396–410. doi: 10.1093/emboj/18.2.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lei M, Kawasaki Y, Young MR, Kihara M, Sugino A, Tye BK. Mcm2 is a target of regulation by Cdc7–Dbf4 during the initiation of DNA synthesis. Genes Dev. 1997;11:3365–3374. doi: 10.1101/gad.11.24.3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Owens JC, Detweiler CS, Li JJ. CDC45 is required in conjunction with CDC7/DBF4 to trigger the initiation of DNA replication. Proc Natl Acad Sci U S A. 1997;94:12521–12526. doi: 10.1073/pnas.94.23.12521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lai JS, Herr W. Ethidium bromide provides a simple tool for identifying genuine DNA-independent protein associations. Proc Natl Acad Sci U S A. 1992;89:6958–6962. doi: 10.1073/pnas.89.15.6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A. Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem. 1996;271:32529–32537. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- 33.Borlado LR, Méndez J. CDC6: From DNA replication to cell cycle checkpoints and oncogenesis. Carcinogenesis. 2008;29:237–243. doi: 10.1093/carcin/bgm268. [DOI] [PubMed] [Google Scholar]
- 34.Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG, Semenza GL. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105:659–669. doi: 10.1182/blood-2004-07-2958. [DOI] [PubMed] [Google Scholar]
- 35.Gardner LB, Li Q, Park MS, Flanagan WM, Semenza GL, Dang CV. Hypoxia inhibits G1/S transition through regulation of p27 expression. J Biol Chem. 2001;276:7919–7926. doi: 10.1074/jbc.M010189200. [DOI] [PubMed] [Google Scholar]
- 36.Mack FA, Patel JH, Biju MP, Haase VH, Simon MC. Decreased growth of Vhl-/- fibrosarcomas is associated with elevated levels of cyclin kinase inhibitors p21 and p27. Mol Cell Biol. 2005;25:4565–4578. doi: 10.1128/MCB.25.11.4565-4578.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Q, Gu J, Li L, Liu J, Luo B, Cheung HW, Boehm JS, Ni M, Geisen C, Root DE, Polyak K, Brown M, Richardson AL, Hahn WC, Kaelin WG, Jr, Bommi-Reddy A. Control of cyclin D1 and breast tumorigenesis by the EglN2 prolyl hydroxylase. Cancer Cell. 2009;16:413–424. doi: 10.1016/j.ccr.2009.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lim JH, Lee YM, Chun YS, Park JW. Reactive oxygen species-mediated cyclin D1 degradation mediates tumor growth retardation in hypoxia, independently of p21cip1 and hypoxia-inducible factor. Cancer Sci. 2008;99:1798–1805. doi: 10.1111/j.1349-7006.2008.00892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pires IM, Bencokova Z, Milani M, Folkes LK, Li JL, Stratford MR, Harris AL, Hammond EM. Effects of acute versus chronic hypoxia on DNA damage responses and genomic instability. Cancer Res. 2010;70:925–935. doi: 10.1158/0008-5472.CAN-09-2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Martin L, Rainey M, Santocanale C, Gardner LB. Hypoxic activation of ATR and the suppression of the initiation of DNA replication through cdc6 degradation. Oncogene. 2012;31:4076–4084. doi: 10.1038/onc.2011.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hammond EM, Denko NC, Dorie MJ, Abraham RT, Giaccia AJ. Hypoxia links ATR and p53 through replication arrest. Mol Cell Biol. 2002;22:1834–1843. doi: 10.1128/MCB.22.6.1834-1843.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bencokova Z, Kaufmann MR, Pires IM, Lecane PS, Giaccia AJ, Hammond EM. ATM activation and signaling under hypoxic conditions. Mol Cell Biol. 2009;29:526–537. doi: 10.1128/MCB.01301-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Onnis B, Rapisarda A, Melillo G. Development of HIF-1 inhibitors for cancer therapy. J Cell Mol Med. 2009;13:2780–2786. doi: 10.1111/j.1582-4934.2009.00876.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kshitiz, Hubbi ME, Ahn EH, Downey J, Afzal J, Kim DH, Rey S, Chang C, Kundu A, Semenza GL, Abraham RM, Levchenko A. Matrix rigidity controls endothelial differentiation and morphogenesis of cardiac precursors. Sci Signal. 2012;5:ra41. doi: 10.1126/scisignal.2003002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995;55:5187–5190. [PubMed] [Google Scholar]
- 46.Fox MH. A model for the computer analysis of synchronous DNA distributions obtained by flow cytometry. Cytometry. 1980;1:71–77. doi: 10.1002/cyto.990010114. [DOI] [PubMed] [Google Scholar]
- 47.Dominguez-Sola D, Ying CY, Grandori C, Ruggiero L, Chen B, Li M, Galloway DA, Gu W, Gautier J, Dalla-Favera R. Non-transcriptional control of DNA replication by c-Myc. Nature. 2007;448:445–451. doi: 10.1038/nature05953. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Flow cytometry data analyzed in Fig. 1.
Fig. S2. Cdc6 has no effect on HIF-1α protein abundance or HIF-1 activity.
Fig. S3. Cell cycle arrest in response to hypoxia.
Fig. S4. HIF-1α promotes MCM loading in HeLa and NIH-3T3 cells.
Fig. S5. Hypoxia decreases total Cdc6 abundance.
Fig. S6. HIF-1β knockdown cells have decreased HIF-1 transcriptional activity.