

Abstract
The acquisition of an androgen-independent phenotype by prostate cancer cells is presently a death sentence for patients. In order to have a realistic chance of changing this outcome, an understanding of what drives the progression to androgen independence is critical. We review here a working hypothesis based on the position that the development of androgen-independent epithelial cells is the result of a series of cellular and molecular events within the whole tissue that culminates in the loss of normal tissue-maintained growth control. This tissue includes the epithelial and stromal cells, the supporting extracellular matrix and circulating hormones. This review discusses the characteristics of these malignant cells, the role of stromal cells involved in growth and the differentiation of epithelial cells, and the role of the extracellular matrix as a mediator of the phenotypes of stromal and epithelial cells. In addition, environmental, neuroendocrine and immune factors that may contribute to disturbance of the fine balance of the epithelial–stromal–extracellular matrix connection are considered. While the goal of many therapeutic approaches to prostate cancer has been androgen ablation or targeting the androgen receptor (AR) of epithelial cells, these therapies become ineffective as the cells progress beyond dependence on androgen for growth control. Twenty years ago Sir David Smithers debated that cancer is the result of loss of tolerance within tissues and the organizational failure of normal growth-control mechanisms. This is precipitated by prolonged or abnormal demands for regeneration or repair, rather than of any inherent disorder peculiar to each of the individual components involved. He wrote ‘It is not the cell itself that is disorderly, but its relationship with the rest of the tissue’. We have gained significantly large amounts of precise data on the effects of androgenic ablation on cancerous prostate cells and on the role of the AR in prostate cancer. The need has come to compile this information towards a perspective of dysregulation of tissue as a whole, and to develop experimental systems to address this broader perspective to find and develop therapies for treatment and prevention.
Introduction
Normal prostate epithelial cells depend on a critical level of androgen stimulation for growth and survival. Androgen ablation is the most common prostate cancer therapy, aiming to devoid the androgen-receptive cells of their growth stimulus. Yet androgen ablation therapy is not curative for metastatic prostate cancer, because of a portion of prostate cancer cells that do not depend on androgen for growth and survival. These cancer cells have become androgen independent. Prostate epithelial cells can also become androgen sensitive, growing in the presence of androgen but not dying when androgen is removed. Androgen regulates the total prostatic cell number by continually stimulating the rate of cell proliferation while at the same time inhibiting the rate of cell death (Isaacs 1984, 1994). Treatment of prostate cancers containing these androgen-independent and androgen-sensitive cells by androgen ablation therapy sometimes does not produce a clinically detectable response (Eisenberger et al. 1998). The therapy may have no effect or only slow the growth of the cancer. This cancer is defined clinically as hormone refractory and unresponsive.
Where do these androgen-independent cells come from? How do these androgen-independent cells differ from normal prostatic epithelial cells? How does the tissue microenvironment affect the progression and expression of these cells? What other factors in this process besides the cancer cells become targets for prevention or therapy of prostate cancer? We first need a clear understanding of how normal prostatic epithelial cells are organized within the tissue and how they respond to androgen.
Prostate tissue architecture
In the prostate gland, the stratified epithelium is divided into two layers, the basal layer made of low cuboidal cells, and a layer of columnar secretory cells (luminal cells). The basal cells next to the basement membrane include the stem cells that form the proliferative compartment of the prostate epithelium. These androgen-independent basal cells give rise to an intermediate cell type called transitional cells, which are androgen-responsive, pluripotent cells that in turn generate basal cells, differentiated glandular luminal cells, and possibly the neuroendocrine cells (Isaacs & Coffey 1989). The secretory glandular cells of the prostate are derived from the transitional cell population. These luminal cells synthesize and secrete the products of the seminal plasma, including prostatic-specific antigen (PSA) and prostate-specific acid phosphatase, polyamines and prostaglandins (Coffey 1992). They express androgen receptors (AR) and are dependent on androgenic stimulation for their viability and secretory ability (Isaacs 1999). If androgen is removed, these glandular cells undergo apoptosis and the prostate gland becomes regressive. Alternatively, the basal cells only rarely express the AR (Nakada et al. 1993). These cells are not affected by withdrawal of androgens and do not undergo apoptosis (Isaacs 1994).
The glands are surrounded by a basement membrane that separates them from the prostatic fibromuscular stroma, containing AR-positive smooth muscle cells and fibroblasts, and additional endothelial cells, nerves, and infiltrating lymphocytes and mast cells. This surrounding stroma not only physically supports the glandular epithelium but also contributes to the endocrine and paracrine microenvironment.
Androgens are the major regulators of proliferation and death for the normal prostate
In the normal prostate epithelial compartment, the expanding stem cell units are hierarchically organized as described in Fig. 1. There are a limited number of androgen-independent stem cells that, besides maintaining their own limited numbers, give rise to a larger subset of progeny that differentiate into the androgen-independent but androgen-sensitive amplifying cells. If sufficient androgen is not present, such as in a castrated host or following androgen ablation, the amplifying cells are maintained with the rate of proliferation equal to the rate of cell death and do not expand and differentiate into transit cells. When physiologically normal levels of androgen are exogenously replaced in a previously castrated host, the majority of these androgen-sensitive amplifying cells differentiate into androgen-dependent transit glandular epithelial cells. Once the normal number of these androgen-dependent transit (glandular) cells is reached, their rate of cell proliferation balances their rate of cell death such that neither prostatic regression nor continuous glandular overgrowth occurs. Thus, in the presence of physiologic androgen, the normal prostate is in a steady-state, self-renewing maintenance condition, heterogeneously composed of androgen-independent, androgen-sensitive and androgen-dependent epithelial cells. Because of the clonally expansive nature of this hierarchical stem cell organization, the vast majority of the epithelial compartment is composed of androgen-dependent glandular cells, with lower numbers of androgen-sensitive basal cells and a limited number of androgen-independent basal stem cells.
Figure 1.

Stem cell model for the organization of the prostate epithelium. The prostate epithelial compartment is organized in a hierarchy of expanding stem cell units. There are a limited number of androgen-independent basal stem cells that, in dividing, create new basal stem cells as well as giving rise to a larger subset of androgen-sensitive amplifying basal cells. In the absence of androgen (as in the castrated prostate) the amplifying cells are in a steady-state with the rate of proliferation equal to the rate of cell death. If androgen is present, the majority of these androgen-sensitive amplifying cells differentiate into androgen-dependent transit glandular epithelial cells. Once the normal number of these androgen-dependent glandular cells is reached, the cell proliferation rate balances the cell death rate such that neither prostatic regression nor continuous glandular overgrowth occurs. Because of the clonally expansive nature of this hierarchical stem cell organization, the vast majority of the epithelial compartment is composed of androgen-dependent glandular cells, with lower numbers of androgen-sensitive basal cells and a limited number of androgen-independent basal stem cells.
Relationship between cell of origin for prostate cancer and androgen responsiveness
Based on stem cell organization, it is possible for prostate cancer to have three distinct cells of origin. The first alternative is that the prostate cancer is monoclonally derived from an androgen-independent stem cell. Even if the cell of origin is an androgen-independent stem cell, it is still possible for the resulting cancer to be responsive to androgen ablation. The malignant stem cell could retain the ability to progress down the hierarchical pathway described in Fig. 1, giving rise to larger subsets of androgen-sensitive amplifying and even larger numbers of androgen-dependent transit malignant cells. Such a heterogeneous cancer composed of these three cell types would respond to androgen ablation with the elimination of the largest subset of cancer cells (i.e. the androgen-dependent malignant transit cells) and a reduction in the growth rate of the next largest subset of cancer cells (i.e. the androgen-sensitive malignant amplifying cells). Such a response would not be curative because neither the malignant androgen-independent stem cell nor the androgen-sensitive malignant amplifying cells would be eliminated.
A second alternative is that the original prostate cancer is monoclonally derived from an androgen-sensitive amplifying basal cell. If this occurs, the cancer would again be androgen responsive because it is composed of androgen-sensitive malignant amplifying cells that retain the ability of differentiating into androgen-dependent transit cell progeny. Again, owing to clonal expansion, the major type of cancer cells present would be the androgen-dependent malignant transit cells. Such a heterogeneous cancer would be responsive to androgen ablation because of the elimination of the major subset of malignant transit cells; however, the cancer would not be cured by such therapy because the androgen-sensitive amplifying cells would not be eliminated.
A third alternative is that the original cancer is monoclonally derived from an androgen-dependent transit (glandular) cell. If this occurs, the cancer would initially be highly androgen responsive to androgen ablation. If no further malignant progression occurred, the cancer theoretically could be cured by such therapy; however, even if this third possibility occurs and the initial prostate cancer is homogeneously composed of androgen-dependent cancer cells, as these cells undergo sufficient cellular proliferation to produce clinically detectable prostate cancer (i.e. more than 39 population doublings), a series of mechanisms would eventually lead to the heterogeneous development of malignant clones of androgen-sensitive or androgen-insensitive prostate cancer cells, or both.
Development of androgen-independent prostate cancer cells
As subpopulations of malignant prostate epithelial cells begin to grow independent of androgen, basic genetic changes likely occur in the cancer cell genome. The tumor cells become increasingly genetically unstable, developing clones that can proliferate without the requirement for androgenic stimulation (Isaacs et al. 1982, Wake et al. 1982). Once these androgen-independent clones develop, they have a growth advantage following androgen ablation over all other newly developed tumor clones that retain androgen dependence. Other genetic changes may contribute to the progression to androgen independence. The frequency of expression of the apoptosis inhibitor, bcl-2, has been correlated with the progression from androgen dependence to the androgen-independent metastatic phenotype (Furuya et al. 1996). If cells over-express bcl-2 and are thereby protected from apoptotic stimuli, their resistance to androgen depletion is augmented, along with their ability to progress towards hormone-refractory tumors (Raffo et al. 1995).
The AR is found to be expressed in both androgen-dependent and androgen-independent cells. Interestingly, the chromosomal region containing the AR (Xq11-q13) was amplified following androgen ablation in 30% of recurrent prostate cancers. In these patients, the withdrawal of androgens may have even induced the clonal outgrowth of prostate cancer cells with increased AR expression. Additionally the AR may be dysfunctional due to mutations, which alter the receptor specificity and binding affinity (Choong et al. 1996). Androgen ablation may induce or preferentially select for cells with mutated AR, especially in metastases (Feldman & Feldman 2001). An additional mechanism of growth of androgen-independent cancer cells includes stimulation of proliferation by cortisol and cortisone using a double-mutated AR (Zhao et al. 2000). These studies suggest that in some patients with these AR mutations that have a high affinity for glucocorticoids, tumor growth could be promoted by physiological levels of cortisone and cortisol.
To understand how hormone-independent cells may develop, we need to know the mechanisms of androgen action within the different cells of the prostate tissue, the AR status of the cell types and the nature of the cofactors that may interact with the AR.
How androgen functions at molecular and cellular levels
Androgens function by binding to nuclear receptors, and inducing a conformation change in the occupied AR. This allows binding of AR to the androgen-responsive element, along with additional nuclear proteins, coactivator proteins and general transcription factors to induce transcription of androgen-induced genes (Brinkman et al. 1999, Brinkman 2001).
Experimental studies demonstrate a ‘cross talk’ between the AR and peptide growth factor-signaling pathways. These studies in androgen-independent prostate cancer cells show that peptide growth factors such as epidermal growth factor (EGF), transforming growth factor-α (TGF-α), keratinocyte growth factor (KGF) and insulin-like growth factor-I (IGF-I), stimulate the transcription of androgen-responsive genes in an androgen ligand-independent manner, but only if the AR is present (Culig et al. 1994, Nazareth & Weigel 1996). Additionally, heregulin was shown to activate the AR-signaling pathway by tyrosine kinase receptors acting synergistically with low concentrations of androgen to activate PSA transcription (Craft et al. 1999). During androgen deprivation, growth factors normally expressed by stromal cells can stimulate epithelial cell proliferation by activating the AR in a ligand-independent manner. These prostate epithelial cells have become independent of androgens for growth and survival yet, retaining the AR, can use paracrine (stromal-derived) or autocrine (epithelial-derived) peptide growth factors to activate the nuclear transcriptional machinery for androgen-responsive genes.
Additionally, SRC-1is a coactivator for AR. SRC-1family members enhance AR-dependent transactivation of nuclear genes. Regulation of the expression of these genes stimulates prostate cell proliferation and inhibits prostate apoptosis (Denmeade et al. 1996). These and other intracellular mechanisms involved in hormone-independent cell growth are described in detail by Feldman & Feldman (2001). The previous perspectives of hormone-independent prostate cancer has focused on abnormalities within the cancer cells. But these cells do not grow in a void. They are products of – and contributors to – the microenvironment of the surrounding tissue. The perspective will now focus outside of the cancer cells and consider exogenous and environmental factors, including the tissue microenvironment of stromal influences, and systemic factors that may induce or promote these internal cellular abnormalities in the progression towards hormone-independent prostate cancer cell growth.
Role of stromal cells in mediating epithelial cell function and androgen signals
Prostate cancer appears to begin with genetic alterations to the epithelial cells, yet there are no specific mutations associated with prostate cancer in humans. Only a minority (5–10%) of patients has a hereditary form of prostate cancer (Carter et al. 1993). Precancerous epithelial lesions may additionally be promoted by alterations in the epithelial cell communications with the tissue microenvironment, including the stromal cells, resident or migrating immune cells and the extracellular matrix (ECM). The epithelial cells are dependent on the stromal environment for control of growth and differentiation. In steroid target tissues, stromal cells appear to play a vital role in mediating the responsiveness of epithelial cells to steroid hormones (Cunha et al. 1983, Cooke et al. 1986, Bigsby 1989). While sex steroids can act directly on the epithelium via epithelial steroid receptors (Brenner et al. 1990), there are many instances where sex steroids appear to act indirectly via stromal cell steroid receptors and the elaboration of paracrine mediators. In both developing and mature prostate, for example, it has been shown that the stroma mediates many androgenic effects seen in the epithelium (Cunha et al. 1985). Stromal cells secrete paracrine peptide growth factors such as EGF, fibroblast growth factor, KGF, nerve growth factor (NGF) and IGF (for reviews see Cunha 1994, Wong & Wang 2000). These paracrine factors can diffuse from the stromal compartment to the epithelial cells, influencing epithelial growth and differentiation.
Stromal-mediated androgenic effects on epithelial cells were elucidated in the now classic studies of Cunha and Lung (1978). These studies showed that male mice had a loss-of-function mutation in the AR gene resulting in testicular feminization (TFM). Urogenital sinus stromal and epithelial compartments were dissected, then experimentally recombined from normal and TFM male mice. The results showed that when epithelial cells from TFM mice were recombined with stroma from normal mice (with functional AR), they were induced to grow and undergo prostatic organogenesis in the presence of adequate androgen. Yet, if the TFM stroma (AR-negative) was combined with normal epithelium (AR-positive), there was no organogenesis even in the presence of adequate systemic androgen. These data indicate that the secondary factors produced by the androgen-induced stromal cells were sufficient for epithelial organogenesis.
The powerful inductive signals from the mesenchyme/stroma that are present in both embryonic and adult tissues may become disordered, leading to alterations in the complex signaling within the epithelium, stroma and ECM. It is commonly believed that many cancers arise because of genetic mutation followed by a long latency period where increased genetic instability in the target cells leads to the final manifestation of neoplasia (Cho & Vogelstein 1992). Disruption in the controlling signals may lead to dysregulation of growth and promote progression towards neoplasia (Kaufman & Arnold 1995). Additionally, the normal stroma and ECM microenvironment may play a regulatory role in the growth of cancers. This has been exemplified in recombination studies using the Dunning rat prostate adenocarcinoma, a slow growing but persistently tumorigenic carcinoma (Hayashi & Cunha 1991). Fragments of this tumor were grafted in the presence of normal mesenchyme under the renal capsule of male athymic mice. After 1month, the tumor tissue’s growth rate was inhibited and became more normal in morphology, with cystic ducts containing tall columnar secretory epithelium. These studies illustrate that while some tumor cells have overt abnormalities in genetic structure (mutations, chromosomal abnormalities etc.), the stromal environment may over-ride the effects of those defective genes. Conversely, the lack of stroma may contribute to the tumorigenic phenotype of the Dunning tumor cells and, with the addition of normal stromal signals, the carcinoma was induced to differentiate towards a secretory phenotype. In the progression of cancer, there may be additional alterations in the stroma that contribute to aberrant growth of the nascent cancer cell (Chung et al. 1992). The loss of regulatory or suppressive influence over epithelial proliferation may also be the result of aging of the stroma, or reversion to a more fetal phenotype, which contributes to the expansion of associated epithelial cells (Schor et al. 1985). In any case, such observations raise the possibility that some cancers are the result of the epithelial cells’ escape from normal stromal control.
A potential mechanism for the progression of cancer cells to androgen independence is that the cancer cells can become reprogrammed to synthesize and secrete their own peptide growth factors, in the absence of androgens or stromal cells. This autocrine reprogramming could be via epigenetic or genetic mechanisms.
Paracrine to autocrine shift in androgen regulation during malignant transformation of epithelial cells
Since AR is expressed in both epithelial and stromal cells, an androgen-stimulated growth response could involve either autocrine pathways initiated within the epithelial cells themselves or paracrine pathways initiated within the AR expression subset of stromal cells. The interactions between the stromal and epithelial components in the normal adult prostate are evident where stromal smooth muscle cells provide a homeostatic state in the presence of androgens. The smooth muscle cells maintain the epithelial cells in a differentiated and non-proliferative state. In the absence of androgen, as in castration, the smooth muscle cells dedifferentiate into fibroblasts, yet maintain the AR. This process is reversed with the addition of androgen, where the AR-positive fibroblasts promote growth of the epithelium which, in turn, induces smooth muscle differentiation of the fibroblasts (Cunha et al. 1996, Hayward et al. 1998).
The role of stromal cell AR in prostatic epithelial growth was explored in studies done in our laboratory (Gao et al. 2001). In these studies, AR-null/nude male mice were created from a cross-breed of immunodeficient (‘nude’) mice with mice carrying the testicular feminized mutation in the X-linked AR gene (TfM). These males, with no functional AR protein in prostatic stromal and epithelial cells, were used as recipients for transplantation of normal or cancerous epithelial cell lines. Normal prostatic glandular morphogenesis occurred when prostatic epithelial organoids were transplanted using Matrigel into intact or castrated wild-type hosts, independent of androgenic stimulation. The mouse mesenchymal cells migrated into the Matrigel transplants forming a histologically normal stromal interface for transplanted epithelial cells. At this stage, the cell types depended on a cross talk potentially involving the extracellular matrix of Matrigel. It was only in the presence of normal levels of androgen that these rudimentary glands underwent further cell proliferation and differentiation into secretory epithelial cells, producing PSA. The paracrine role of stromal cells was further elucidated when prostatic epithelial cells from either human or rat prostate were injected into intact versus castrated nu/AR-null male mice hosts. The host mouse stromal cells migrated and surrounded the rudimentary acini as before but were negative for AR expression. Consequentially, the epithelial cells only sporadically expressed AR and the glands did not grow in either the castrated AR-null male mice or in the intact animals, even in the presence of androgens. These results underline the importance of androgen-stimulated factors liberated from AR-positive stromal cells to mediate androgen-stimulated epithelial cell growth, morphogenesis and AR expression. Stromal cells would also contribute growth-inhibitory homeostatic factors. Conversely, both epithelial cells and androgen induce differentiation of mesenchyme but this cross talk is interrupted in cancers where malignant prostatic epithelial cells lose their stromal inductive ability.
Our study also demonstrated that when prostate cancer cell lines (i.e. human PC-82, LNCaP, LAPC-4 and rat R 3327G) were transplanted into the AR-null mice, tumor growth was stimulated in both AR-null and AR-wild-type nude male mice. Androgen-induced growth did not occur when normal prostate epithelial cells were transplanted into the AR-null mice. This indicates that a direct autocrine mechanism is responsible for the androgen-stimulated growth of malignant prostatic epithelial cells. The cancer cells relied neither on androgen nor on the androgen-induced stromal-mediated factors for growth. This is an example of the fundamental change in the mechanism for androgen-stimulated growth that occurs during the transformation from normal to malignant prostatic epithelial cells. These studies demonstrate that during transformation of androgen-responsive normal prostatic epithelial cells to malignant cancer cells, there is a shift in the AR axis from stromal cell-dependent paracrine pathways to autocrine-dependent pathways. At the same time, the prostatic cancer cells lose their ability to maintain epithelial growth regulation. When the cancer cells shift to the autocrine mechanism for androgen-stimulated proliferation it appears that AR protein can regulate a new series of genes for proliferation and survival, not normally expressed in prostate epithelial cells.
Other important factors that are shifted from paracrine to autocrine regulation are the neurotropins. NGF, neurotropin-3 and brain-derived growth factor are involved in the neuroendocrine regulation of prostatic function that may also contribute to the homeostasis of the tissue. NGF is secreted by stromal cells but is not a survival factor for the normal prostatic epithelium. In contrast, NGF is directly secreted by malignant prostatic epithelial cells to regulate an acquired survival pathway (Weeraratna et al. 2000). KGF is also a stromal-derived mediator of androgen signaling, which functions as an autocrine growth factor in prostate cancer and benign prostatic hypertrophy (Planz et al. 1999). The identification of additional growth and survival factors whose production by malignant prostatic cells is under AR control, and an understanding of the molecular mechanisms underlying this acquired expression, are critically needed.
Prostate cancer cells can alter stromal cell functioning
As we have seen, epithelial AR expression is lost in the progression to malignancy and epithelial cells become independent of androgen for growth, relying on autocrine-produced growth factors. What precedes the loss or mutation of the epithelial cell AR? Is the genetic instability in the epithelial cells a cause or an effect of the loss of stromal cell AR and regulation? When stromal–epithelial cross talk is interrupted, then signals from epithelial cells that regulate stromal AR and smooth muscle actin must be altered. The consequence of this is that the stroma no longer expresses AR and does not make proper growth and inhibitory factors to regulate homeostasis of epithelial cell function and growth.
How does the cross talk between the stromal and epithelial cells become obstructed? What is the role of ECM in stromal expression of AR? In another hormonally sensitive cell model, ECM (Matrigel) was shown to regulate stromal phenotype (Arnold et al. 2001). In this study, the co-culture of human endometrial stromal cells in contact with basement membrane proteins provided growth regulation and differentiation in the endometrial epithelial cells. Furthermore, the proper stromal and ECM environment was shown to restrict growth and induce a more differentiated phenotype in endometrial cancer cells (Arnold et al. 2002). These studies support the hypothesis that subepithelial stroma, nearest to basement membrane and to the epithelial cells, plays a unique role in the regulation of epithelial and cancer cells. Alterations in these periepithelial regulatory stromal cells or alterations in the ECM may contribute to the destabilization of the homeostatic cross talk between the stromal and epithelial compartments. Such changes occur due to signals from cancer cells to surrounding stromal cells, which alter stromal function and ECM production. For example, TGF-β1 from prostate tumor cells can induce stromal secretion of versican, an extracellular chondroitin sulfate proteoglycan (Sakko et al. 2001). In addition, malignant prostate epithelium may influence surrounding stromal AR expression. Henshall et al. (2001) demonstrated a loss of AR expression in stromal cells associated with malignant epithelium while the malignant glands had an overexpression of AR. These results coincided with higher clinical stage cancers with higher PSA levels and earlier relapse rates following radical prostatectomy. The loss of the AR in the periepithelial stroma was unique from the interstitial stroma where the AR was still present. The authors suggest that this alteration of the AR expression in the tumor-associated stroma may be another mechanism of achieving androgen-independent prostate epithelial cell proliferation, and that the regulation of tumor stroma AR expression is altered in prostate cancer.
A similar phenomenon was reported by Olapade-Olaopa et al. (1999) where a loss of AR expression was found in stromal cells surrounding prostatic cancer cells in malignant tissue taken directly from patients. The authors observed a ‘field effect’ of decreased AR expression in stroma and epithelial glands in the vicinity of cancer glands. They hypothesized that with the loss of AR, stromal cells cease production of androgen-mediated peptide growth factors early in the malignant transformation process. Interestingly, this altered AR expression was also found in stroma surrounding highgrade prostatic intraepithelial neoplasia, further indicating that this is an early event in prostate cancer progression. The surviving cancer cells adapt to the loss of stromal growth factors by switching to hormone-independent autocrine growth pathways. The authors suggest these original alterations in the stromal compartment contribute to the progression to hormone-refractory disease, preceding the genetic changes in the epithelial cells.
Hormone-induced changes in ECM and stroma were also found to affect prostatic growth and differentiation (Chang et al. 1999). By studying tissues from neonatal estrogen-exposed rat prostates, unique changes were noted in stromal cells and ECM surrounding the epithelial ducts. The ducts are normally surrounded by a single cell layer of fibroblasts, followed by a collar of laminin-positive, AR-positive, non-proliferative smooth muscle cells. With neonatal estrogen exposure, the fibroblast layer was found to be multiple cells thick, distancing the smooth muscle cells from the basement membrane and the epithelial cells. The fibroblasts were AR-negative and laminin-negative, yet proliferating. These estrogen-induced morphological disruptions of paracrine interactions and the mediating ECM were seen within the first 15 days of life, and may lead to the permanent prostatic dysfunction during adulthood, including a loss of epithelial AR and a reduced responsiveness to androgens. Current concerns about environmental estrogenic exposure support further investigations of this mechanism of altering the prostatic microenvironment (Bosland 2000).
It is not clear from the studies above what precedes the dysregulation of the AR expression in the stroma. Do the prostate cancer glands actively suppress the expression of the surrounding stromal (and non-cancerous epithelial) AR as a field effect? Or, as the tumors grow, does the dysregulated oncogenesis produce abnormal epithelial and stromal compartments, which are unable to sustain normal regulation or maintenance of AR in stroma? This cascade of dysregulation may not be apparent in analyses of histological sections, which are snapshots of the end result of these cell–cell interactions. Cell culture models that recombine the stromal and cancer cell types may provide a better understanding of factors involved in the dynamics of tissue regulation. This scenario of progression to hormone-independent prostate cancer has multiple elements, each dependent on each other. One dysregulated component may lead to further destabilization of homeostasis of the tissue (Fig. 2). The epithelial cells may have increased genetic instability; additionally, the stromal cells may have altered AR expression and switch phenotypes from smooth muscle cells to fibroblasts (Hayward et al. 1996). The ECM could be viewed as telegraph wires connecting the two cell types and mediating the transfer of proteins involved in communications. ECM abnormalities may stem from changes in the anionic charge, in protein deposition (Goulas et al. 2000), or incomplete removal of oxidatively damaged, relatively indigestible material including ‘cellular detritus’ (Terman 2001). These abnormalities may result in cellular signals being lost or rewired or otherwise blocked. Disruption of the delicate balance of signaling results in dramatic changes in the way cells interact with each other and with the ECM. In studies of tissue microenvironmental effects within the breast, Bissell & Hall (1987) hypothesized that the unit of function in higher organisms is not the individual cell, but the tissue itself. Tissue architecture is intimately involved in controlling the signaling processes within cells (Hansen & Bissell 2000). It is important to determine the unique combination of steroid hormonal, ECM and growth factor effects that contribute to the final fate of the prostatic tissue as has been defined for breast tissue.
Figure 2.
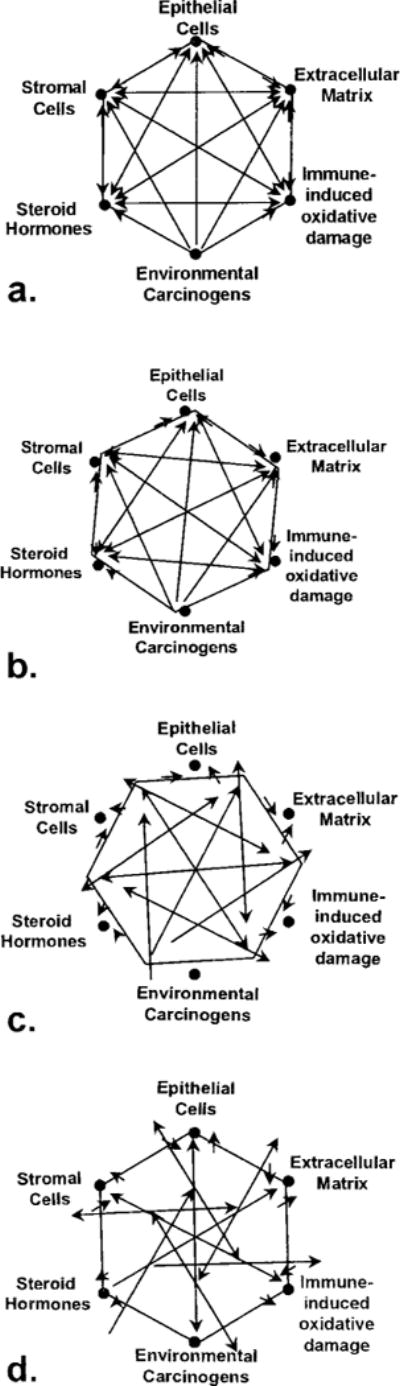
Representation of inter-relationships within the prostatic microenvironment. The interplay between epithelial cells, stromal cells, ECM, steroid hormones, oxidative damage and carcinogens contribute to the homeostasis of tissue function (a). Dysequilibrium within the tissue results from initial insults even within one component (b) and causes minor imbalance of regulation (arrows) between the rest of components. Additional insults (c and d) in the same or additional components lead to increasingly chaotic state with loss of regulation of growth and function within the tissue.
Both the epigenetic mechanism of altered AR expression in stroma associated with the tumor, along with genetic alterations within the epithelium, promote the development of androgen-independent prostate epithelial cell proliferation. If the ECM contributes to the stromal expression of AR, could any changes in the constitution of the ECM predict a change in stromal phenotype? If so, what would contribute to changes in ECM in prostate tissue? What would be the sustained effects from inflammatory responses in the prostate ECM? Inflammatory-induced oxidative damage in the prostate is of parallel importance in the dysregulation of the genetics of prostatic cells via DNA adduct formation and the tissue microenvironment (for reviews see Rowley 1998–1999, DeMarzo et al. 1999, DeWeese et al. 2001). Conversely, does the constitution of the ECM produced by tumor-associated stroma contribute to the dysregulation found in the early stages of neoplasia? These questions remain unanswered without appropriate models to recombine the components of the tissue microenvironment.
Cell culture models to understand AR function and cell communications
Efforts to model stromal and epithelial cell interactions in in vitro culture have helped elucidate the cross talk between cell types (Bayne et al. 1998, Habib et al. 2000). In vitro co-culture studies have produced conflicting results of cell regulation. Primary stromal cells were growth stimulatory to normal and malignant epithelial cells (Kabalin et al. 1989, Gleave et al. 1991), yet in other studies the stromal cells were inhibitory to epithelial growth (König et al. 1987, Kooitsra et al. 1995, Degeorges et al. 1996). Cell culture models of AR function in normal primary prostate epithelial cells have been limited. This is because the cells quickly lose the AR in cell culture (Grant et al. 1996). Even normal primary epithelial cells become ‘androgen-independent’, proliferating in cell culture without added androgens, if they are grown isolated from other tissue components. If stromal components and ECM are retained in early culture as in organ culture, the AR remains present (Nevalainen 1993). While this phenomenon is inconvenient for researchers wishing to isolate AR-positive epithelial cells, it underlies the importance of the proper stromal and ECM in expression of the AR.
We need cell culture models that can reconstruct stromal, epithelial and ECM components to define markers and mediating factors produced by the stroma and/or ECM that allow maintenance of AR expression in epithelial cells in culture. These models could elucidate the interplay of integrins, hormones, steroid receptors and growth factors involved in tissue regulation for the prostate and begin to dissect the vocabulary of cellular communications.
Endocrine-immune interactions and adrenal androgens
The next concentric layer of influence on the regulation of androgen-independent prostate cancer cells is from the hormonal milieu of the host, including serum levels of gonadal and adrenal androgens, cortisol and cytokines. Altered hormonal levels may represent underlying pathological or pre-pathological conditions that can be enough to offset the equilibrium within and between cell communications.
Of one concern is the over-the-counter use of dehydroxy-epiandrosterone (DHEA) for purported anti-aging therapy and boosting the immune system. DHEA is an adrenal androgen and a precursor to testosterone and estrogen. The sulfated form, DHEAS is present in high levels in the prostate, as well as the sulfatase that converts DHEAS to DHEA (Farnsworth 1973, Voigt & Bartsch 1986, Klein et al. 1988, 1989). DHEA is further metabolized to dihydrotestosterone (DHT) accounting for up to one-sixth of total prostate DHT (Geller et al. 1986). Although DHEA has effective mechanisms to inhibit glucose-6-phosphate dehydrogenase (Schwartz & Pashko 1995) and has been found to have chemopreventive activity in rodent models for the prostate or breast (Perkins et al. 1997, Lubet et al. 1998, McCormick & Rao 1999, Green et al. 2001), there is conflicting evidence on its effects in men with known or unknown cancer lesions. Several studies suggest that DHEA can activate mutant ARs (Culig et al. 1993, Tan et al. 1997). DHEA may be an additional prostate cancer-promoting factor for populations who use it as a supplement, unaware of any pre-existing cancer lesions.
There may be additional endocrine-immune complications, such as sulfatase activity, present in macrophages that are able to convert DHEAS to DHEA (Hennebold & Daynes 1994). Also, the cytokines interleukin (IL)-4 and IL-13 were found to induce 3β-hydroxysteroid dehydrogenase activity in normal prostate epithelial cells which converts the DHEA into testosterone and estrogen metabolites (El-Alfy et al. 1999, Simard & Gingras 2001). Either of these mechanisms could amplify the effects of DHEA within the prostate tissue. The growth and functioning of the prostate tissue is the end result of complex signaling between the stromal and epithelial cells along with resident and infiltrating immune cells all within the neuroendocrine hormonal milieu.
Environmental role in dysregulating prostatic tissue equilibrium
The final question is ‘What exogenous environmental factors may contribute to the original insults that initiate this cascade of dysregulation in the prostatic tissue, leading to androgen-independent prostate cancer? Ever present environmental carcinogens (Waalkes & Rehm 1994), hormones (Huff et al. 1996) and dietary factors (Weisberger 2000) are involved in both the initiation and promotion of prostate oncogenesis.
An important carcinogen in the etiology of prostate cancer is a heterocyclic amine, 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) and its metabolite, N-hydroxy PhIP. PhIP is found in the human diet and in cigarette smoke (Shirai et al. 1997). PhIP metabolism involves both phase I and phase II enzymes for bioactivation and detoxification. Cells respond to carcinogenic insults by upregulating the synthesis of detoxification enzymes, such as the phase II enzyme, glutathione-S-transferase (GST) (Rushmore & Pickett 1993). The presence of GST in the stromal and epithelial cells can provide protection from PhIP by detoxification of the metabolites (Nelson et al. 2001). The gene for the GST isoform PI (GSTPI) was shown to be silenced through hypermethylation of the dinucleotide CpG, which is found in nearly all prostate carcinomas (Lee et al. 1994, 1997). Interestingly, DeMarzo et al. (1999) report that epithelial cells in lesions of prostatic inflammatory atrophy are GSTPI(+), while the adjacent PIN (prostatic intraepithelial neoplasia) cells are GSTPI(−). Further progression of epithelial neoplastic lesions was documented by the loss of GSTPI. This loss of GST expression in PIN cells and consequently the loss of protective effects against DNA damage (such as adduct formation by PhIP) may lead to further instability in the genome of the PIN cells, allowing progression to a more cancerous phenotype. The stromal compartment may be involved in either the focal expression of epithelial carcinogen-metabolizing enzymes or detoxification enzymes such as GST. Consequently, if the stromal–ECM environment has been altered during the neoplastic process, the regulation of these enzymes may also be altered.
Conclusion
The progression of prostate cancer is a cascade of cellular and molecular events following initial perturbations of a finely balanced complex homeostasis (Fig. 2). Androgen-independent cells are able to proliferate due to loss of checks and balances on their growth. In normal functioning prostate epithelial cells, growth and death are controlled by androgen. In the development of androgen-independent autocrine pathways, the cells begin synthesizing their own growth factors, becoming more autonomous and are under less exogenous tissue microenvironmental control. Abnormalities on the level of the epithelial cells may be precipitated by a dysfunctional stroma. Potter (2001) revised the two stages of carcinogenesis previously defined as involving a mutating initiation event followed by progression towards cancer, to a definition including one ‘hit’ as mutations in epithelial cells which cause disruptions of cell function and structure and the other ‘hit’ is in the mesenchymal cell functioning resulting in disruption of tissue homeostasis. The stroma can no longer provide a growth-regulatory environment, therefore epithelial cells lapse into dysregulation, accumulating genetic changes, losing reciprocal control of stromal cells and continuing on a pathway of increasing progression towards cancer. This perspective has been eloquently described by Sir David Smithers (1983).
‘The complex interactions which control growth and regeneration are secured through a whole system of interlocking messages is not easily unraveled. The fine organizational process, with its alternation of calibration and feed-back adjustment, requires detailed study no less than the molecular changes it sets in motion. Genes do not impart disorder directly to an ordered setting any more than they confer regularity on random surroundings; they are part of a normally ordered system with which they interact. Finally, nothing in the realm of organizational response is simple; it is all too easy to elevate a mechanism to the distinction of a prime cause … Were we able to elucidate every genetic mechanism involved within the tumor cells this could only provide a partial explanation for there has to be a wider concern with influences which switch mechanisms on and off and with how the whole chain of reactions operates to disturb normal function and produce such a system disorder.’
Our challenge resulting from this broader perspective is in developing additional therapies or, more importantly, preventive regimens for prostate cancer. Therapies may be added to existing treatments that would target the stroma or ECM or factors involved in cell communications concerned within higher order tissue structure. These approaches would help prevent the early stages of prostate cancer, before tissues begin to lose their regulation.
References
- Arnold JT, Kaufman DG, Seppälä M, Lessey BA. Endometrial stromal cells regulate epithelial cell growth in vitro: a new co-culture model. Human Reproduction. 2001;16:836–845. doi: 10.1093/humrep/16.5.836. [DOI] [PubMed] [Google Scholar]
- Arnold JT, Lessey BA, Seppälä M, Kaufman DG. Effect of normal endometrial stroma on growth and differentiation in Ishikawa endometrial adenocarcinoma cells. Cancer Research. 2002;62:79–88. [PubMed] [Google Scholar]
- Bayne CW, Ross M, Donnelly F, Chapman K, Buck C, Bollina P, Habib FK. Selective interactions between prostate fibroblast and epithelial cells in co-culture maintain the BPH phenotype. Urologia Internationalis. 1998;61:1–7. doi: 10.1159/000030274. [DOI] [PubMed] [Google Scholar]
- Bigsby RM. Reciprocal tissue interactions in morphogenesis and hormonal responsiveness of the female reproductive tract. In: Lavia L, editor. Cellular Signals Controlling Uterine Function. New York: Plenum Press; 1989. pp. 11–29. [Google Scholar]
- Bissell MJ, Hall HG. Form and function in the mammary gland: the role of extracellular matrix. In: Neville MC, Daniel CW, editors. The Mammary Gland. New York: Plenum Publishing Corporation; 1987. pp. 97–146. [Google Scholar]
- Bosland MC. The role of steroid hormones in prostate carcinogenesis. Journal of the National Cancer Institute Monograph. 2000;27:39–66. doi: 10.1093/oxfordjournals.jncimonographs.a024244. [DOI] [PubMed] [Google Scholar]
- Brenner RM, West NB, McClellan MC. Estrogen and progestin receptors in the reproductive tract of male and female primates. Biology of Reproduction. 1990;42:11–19. doi: 10.1095/biolreprod42.1.11. [DOI] [PubMed] [Google Scholar]
- Brinkmann AO. Lessons to be learned from the androgen receptor. European Journal of Dermatology. 2001;11:301–303. [PubMed] [Google Scholar]
- Brinkmann AO, Blok LJ, de Ruiter PE, Doesburg P, Steketee K, Berrevoets CA, Trapman J. Mechanisms of androgen receptor activation and function. Journal of Steroid Biochemistry and Molecular Biology. 1999;69:307–313. doi: 10.1016/s0960-0760(99)00049-7. [DOI] [PubMed] [Google Scholar]
- Carter BS, Bova GS, Beaty TH, Steinberg GD, Childs B, Isaacs WB, Walsh PC. Hereditary prostate cancer: epidemiologic and clinical features. Journal of Urology. 1993;150:797–802. doi: 10.1016/s0022-5347(17)35617-3. [DOI] [PubMed] [Google Scholar]
- Chang WY, Wilson MJ, Birch L, Prins GS. Neonatal estrogen stimulates proliferation of periductal fibroblasts and alters the extracellular matrix composition in the rat prostate. Endocrinology. 1999;140:405–415. doi: 10.1210/endo.140.1.6401. [DOI] [PubMed] [Google Scholar]
- Cho KR, Vogelstein B. Suppressor gene alterations in the colorectal adenoma–carcinoma sequence. Journal of Cellular Biochemistry. 1992;16G(Suppl):137–141. doi: 10.1002/jcb.240501124. [DOI] [PubMed] [Google Scholar]
- Choong CS, Sturm MJ, Strophair JA, McCulloch RK, Tilley WD, Leedman PJ, Hurley DM. Partial androgen insensitivity caused by an androgen receptor mutation at amino acid 907 (Gly–>Arg) that results in decreased ligand binding affinity and reduced androgen receptor messenger ribonucleic acid levels. Journal of Clinical Endocrinology and Metabolism. 1996;81:236–243. doi: 10.1210/jcem.81.1.8550758. [DOI] [PubMed] [Google Scholar]
- Chung LWK, Wei L, Gleave ME, Hseih J, Wu HC, Sikes RA, Zhau HE, Bandyk MG, Logothetis CJ, Rubin JS, von Eshenbach AC. Human prostate cancer model: roles of growth factors and extracellular matrices. Journal of Cellular Biochemistry. 1992;16H(Suppl):99–105. doi: 10.1002/jcb.240501222. [DOI] [PubMed] [Google Scholar]
- Coffey DS. The molecular biology, endocrinology and physiology of the prostate and seminal vesicles. In: Walsh PC, Retik AB, Stamey TA, Vaughn ED, editors. Campbell’s Textbook of Urology. Philadelphia: Saunders; 1992. pp. 221–266. [Google Scholar]
- Cooke PS, Uchima FDA, Fujii DK, Bern HA, Cunha GR. Restoration of normal morphology and estrogen responsiveness in cultured vaginal and uterine epithelia transplanted with stroma. PNAS. 1986;83:2109–2113. doi: 10.1073/pnas.83.7.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft N, Shostak Y, Carey M, Sawyers CL. A mechanism for hormone-independent prostate cancer through modulation of androgen receptor signaling by the HER-2/neu tyrosine kinase. Nature Medicine. 1999;5:280–285. doi: 10.1038/6495. [DOI] [PubMed] [Google Scholar]
- Culig Z, Hobisch A, Cronauer MV, Cato AC, Hittmair A, Radmayr C, Eberle J, Bartsch G, Klocker H. Mutant androgen receptor detected in an advanced-stage prostatic carcinoma is activated by adrenal androgens and progesterone. Molecular Endocrinology. 1993;7:1541–1550. doi: 10.1210/mend.7.12.8145761. [DOI] [PubMed] [Google Scholar]
- Culig Z, Hobisch A, Cronauer MV, Radmayr C, Trapman J, Hittmair A, Bartsch G, Klocker H. Androgen receptor activation in prostatic tumor cell lines by insulin-like growth factor-I, keratinocyte growth factor, and epidermal growth factor. Cancer Research. 1994;54:5474–5478. [PubMed] [Google Scholar]
- Cunha GR. Role of mesenchymal–epithelial interactions in normal and abnormal development of the mammary gland and prostate. Cancer. 1994;74(Suppl 3):1030–1044. doi: 10.1002/1097-0142(19940801)74:3+<1030::aid-cncr2820741510>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Cunha GR, Lung B. The possible influences of temporal factors in androgenic responsiveness of urogenital tissue recombinants from wild-type and androgen insensitive (Tfm) mice. Journal of Experimental Zoology. 1978;205:181–193. doi: 10.1002/jez.1402050203. [DOI] [PubMed] [Google Scholar]
- Cunha CR, Chung LWK, Shannon JM, Taguchi O, Fujii H. Hormone-induced morphogenesis and growth: role of mesenchymal–epithelial interactions. Recent Progress in Hormone Research. 1983;39:559–598. doi: 10.1016/b978-0-12-571139-5.50018-5. [DOI] [PubMed] [Google Scholar]
- Cunha GR, Bigsby RM, Cooke PS, Sugimura Y. Stromal–epithelial interactions in the determination of hormonal responsiveness. In: McLachlan JA, editor. Estrogens in the Environment II. New York: Elsevier; 1985. pp. 273–287. [Google Scholar]
- Cunha GR, Hayward SW, Dahiya R, Foster BA. Smooth muscle–epithelial interactions in normal and neoplastic prostatic development. Acta Anatomica. 1996;155:63–72. doi: 10.1159/000147791. [DOI] [PubMed] [Google Scholar]
- Degeorges A, Tatoud R, Fauvel-Lafeve F, Podgorniak MP, Millot G, De Cremoux P, Calvo F. Stromal cells from human benign prostate hyperplasia produce a growth-inhibitory factor for LNCaP prostate cancer cells, identified as interleukin-6. International Journal for Cancer. 1996;68:207–214. doi: 10.1002/(SICI)1097-0215(19961009)68:2<207::AID-IJC12>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- DeMarzo AM, Marchi VL, Epstein JI, Nelson WG. Proliferative inflammatory atrophy of the prostate – implications for prostatic carcinogenesis. American Journal of Pathology. 1999;155:1985–1992. doi: 10.1016/S0002-9440(10)65517-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denmeade SR, Lin XS, Isaacs JT. Role of programmed (apoptotic) cell death during the progression and therapy for prostate cancer. Prostate. 1996;28:251–265. doi: 10.1002/(SICI)1097-0045(199604)28:4<251::AID-PROS6>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- DeWeese TL, Hruszkewycz AM, Marnett LJ. Oxidative stress in chemoprevention trials. Urology. 2001;57(4 Suppl 1):137–140. doi: 10.1016/s0090-4295(00)00959-6. [DOI] [PubMed] [Google Scholar]
- Eisenberger MA, Blumenstein BA, Crawford ED, Miller G, McLeod DG, Loehrer PJ, Wilding G, Sears K, Culkin DJ, Thompson IM, Jr, Bueschen AJ, Lowe BA. Bilateral orchiectomy with or without flutamide for metastatic prostate cancer. New England Journal of Medicine. 1998;339:1036–1042. doi: 10.1056/NEJM199810083391504. [DOI] [PubMed] [Google Scholar]
- El-Alfy M, Luu-The V, Huang XF, Berger L, Labrie F, Pelletier G. Localization of type 5 17beta-hydroxysteroid dehydrogenase, 3beta-hydroxysteroid dehydrogenase, and androgen receptor in the human prostate by in situ hybridization and immunocytochemistry. Endocrinology. 1999;140:1481–1491. doi: 10.1210/endo.140.3.6585. [DOI] [PubMed] [Google Scholar]
- Farnsworth WE. Human prostatic dehydroepiandrosterone sulfate sulfatase. Steroids. 1973;21:647–664. doi: 10.1016/0039-128x(73)90134-7. [DOI] [PubMed] [Google Scholar]
- Feldman BJ, Feldman D. The development of androgen-independent prostate cancer. Nature Reviews Cancer. 2001;1:34–45. doi: 10.1038/35094009. [DOI] [PubMed] [Google Scholar]
- Furuya Y, Krajewski S, Epstein JI, Reed JC, Isaacs JT. Expression of bcl-2 and the progression of human and rodent prostatic cancers. Clinical Cancer Research. 1996;2:389–398. [PubMed] [Google Scholar]
- Gao J, Arnold JT, Isaacs JT. Conversion from a paracrine to an autocrine mechanism of androgen-stimulated growth during malignant transformation of prostatic epithelial cells. Cancer Research. 2001;61:5038–5044. [PubMed] [Google Scholar]
- Geller J, Liu J, Albert J, Fay W, Berry CC. Effect of antiandrogen and/or antiestrogen blockade on human prostate epithelial and stromal cell protein synthesis. Journal of Steroid Biochemistry. 1986;25:759–763. doi: 10.1016/0022-4731(86)90305-5. [DOI] [PubMed] [Google Scholar]
- Gleave M, Hsieh JT, Gao C, Von Eschenbach AC, Chung LWK. Acceleration of human prostate cancer growth in vivo by factors produced by prostate and bone fibroblasts. Cancer Research. 1991;51:3753–3761. [PubMed] [Google Scholar]
- Goulas A, Hatzichristou DG, Karakiulakis G, Mirtsou-Fidani V, Kalinderis A, Papakonstantinou E. Benign hyperplasia of the human prostate is associated with tissue enrichment in chondroitin sulphate of wide size distribution. Prostate. 2000;44:104–110. doi: 10.1002/1097-0045(20000701)44:2<104::aid-pros2>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Grant ES, Batchelor KW, Habib FK. Androgen independence of primary epithelial cultures of the prostate is associated with a down-regulation of androgen receptor gene expression. Prostate. 1996;29:339–349. doi: 10.1002/(SICI)1097-0045(199612)29:6<339::AID-PROS1>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Green JE, Shibata MA, Shibata E, Moon RC, Anver MR, Kelloff G, Lubet R. 2-Difluoromethylornithine and dehydroepiandrosterone inhibit mammary tumor progression but not mammary or prostate tumor initiation in C3(1)/SV40 T/t-antigen transgenic mice. Cancer Research. 2001;61:7449–7455. [PubMed] [Google Scholar]
- Habib FK, Ross M, Bayne CW. Development of a new in vitro model for the study of benign prostatic hyperplasia. Prostate. 2000;9(Suppl):15–20. doi: 10.1002/1097-0045(2000)45:9+<15::aid-pros4>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Hansen RK, Bissell MJ. Tissue architecture and breast cancer: the role of extracellular matrix and steroid hormones. Endocrine-Related Cancer. 2000;7:95–113. doi: 10.1677/erc.0.0070095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi N, Cunha GR. Mesenchyme-induced changes in the neoplastic characteristics of the Dunning prostatic adenocarcinoma. Cancer Research. 1991;51:4924–4930. [PubMed] [Google Scholar]
- Hayward SW, Cunha GR, Dahiya R. Normal development and carcinogenesis of the prostate. Annals of the New York Academy of Sciences. 1996;784:50–62. doi: 10.1111/j.1749-6632.1996.tb16227.x. [DOI] [PubMed] [Google Scholar]
- Hayward SW, Haughney PC, Rosen MA, Greulich KM, Weier HU, Dahiya R, Cunha GR. Interactions between adult human prostatic epithelium and rat urogenital sinus mesenchyme in a tissue recombination model. Differentiation. 1998;63:131–140. doi: 10.1046/j.1432-0436.1998.6330131.x. [DOI] [PubMed] [Google Scholar]
- Hennebold JD, Daynes RA. Regulation of macrophage dehydroepiandrosterone sulfate metabolism by inflammatory cytokines. Endocrinology. 1994;135:67–75. doi: 10.1210/endo.135.1.8013393. [DOI] [PubMed] [Google Scholar]
- Henshall SM, Quinn DI, Lee CS, Head DR, Golovsky D, Brenner PC, Delprado W, Stricker PD, Grygiel JJ, Sutherland RL. Altered expression of androgen receptor in the malignant epithelium and adjacent stroma is associated with early relapse in prostate cancer. Cancer Research. 2001;61:423–427. [PubMed] [Google Scholar]
- Huff J, Boyd J, Barrett JC. Hormonal carcinogenesis and environmental influences: background and overview. Progress in Clinical Biological Research. 1996;394:3–23. [PubMed] [Google Scholar]
- Isaacs JT. Antagonistic effect of androgen on prostatic cell death. Prostate. 1984;5:545–557. doi: 10.1002/pros.2990050510. [DOI] [PubMed] [Google Scholar]
- Isaacs JT. Role of androgens in prostatic cancer. Vitamins and Hormones. 1994;49:433–502. doi: 10.1016/s0083-6729(08)61152-8. [DOI] [PubMed] [Google Scholar]
- Isaacs JT. The biology of hormone refractory prostate cancer: why does it develop? Urologic Clinics in North America. 1999;26:263–273. doi: 10.1016/s0094-0143(05)70066-5. [DOI] [PubMed] [Google Scholar]
- Isaacs JT, Coffey DS. Etiology and disease process of benign prostatic hyperplasia. Prostate. 1989;2(Suppl):33–50. doi: 10.1002/pros.2990150506. [DOI] [PubMed] [Google Scholar]
- Isaacs JT, Wake N, Coffey DS, Sandberg AA. Genetic instability coupled to clonal selection as a mechanism for tumor progression in the Dunning R-3327 rat prostatic adenocarcinoma system. Cancer Research. 1982;42:2353–2371. [PubMed] [Google Scholar]
- Kabalin JN, Peehl DM, Stamey TA. Clonal growth of human prostatic epithelial cells is stimulated by fibroblasts. Prostate. 1989;14:251–263. doi: 10.1002/pros.2990140306. [DOI] [PubMed] [Google Scholar]
- Kaufman DG, Arnold JT. Stromal–epithelial interactions in normal and neoplastic development. In: Sirica AE, editor. Cellular and Molecular Pathogenesis. Philadelphia: Lippincott-Raven Publishers; 1995. pp. 403–431. [Google Scholar]
- Klein H, Bressel M, Kastendieck H, Voigt KD. Quantitative assessment of endogenous testicular and adrenal sex steroids and of steroid metabolizing enzymes in untreated human prostatic cancerous tissue. Journal of Steroid Biochemistry. 1988;30:119–130. doi: 10.1016/0022-4731(88)90084-2. [DOI] [PubMed] [Google Scholar]
- Klein H, Molwitz T, Bartsch W. Steroid sulfate sulfatase in human benign prostatic hyperplasia: characterization and quantification of the enzyme in epithelium and stroma. Journal of Steroid Biochemistry. 1989;33:195–200. doi: 10.1016/0022-4731(89)90294-x. [DOI] [PubMed] [Google Scholar]
- König JJ, Romijn JC, Schröder FH. Prostatic epithelium inhibiting factor (PEIF): organ specificity and production by prostatic fibroblasts. Urology Research. 1987;15:145–149. doi: 10.1007/BF00254426. [DOI] [PubMed] [Google Scholar]
- Kooistra A, van Den Eijnden-van Raaij AJM, Klaij IA, Romijn JC, Schröder FH. Stromal inhibition of prostatic epithelial cell proliferation not mediated by transforming growth factor beta. British Journal of Cancer. 1995;72:427–434. doi: 10.1038/bjc.1995.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WH, Morton RA, Epstein JI, Brooks JD, Campbell PA, Bova GS, Hsieh WS, Isaacs WB, Nelson WG. Cytidine methylation of regulatory sequences near the pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. PNAS. 1994;91:11733–11737. doi: 10.1073/pnas.91.24.11733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WH, Isaacs WB, Bova GS, Nelson WG. CG island methylation changes near the GSTP1gene in prostatic carcinoma cells detected using the polymerase chain reaction: a new prostate cancer biomarker. Cancer Epidemiology Biomarkers and Prevention. 1997;6:443–450. [PubMed] [Google Scholar]
- Lubet RA, Gordon GB, Prough RA, Lei XD, You M, Wang Y, Grubbs CJ, Steele VE, Kelloff GJ, Thomas CF, Moon RD. Modulation of methylnitrosourea-induced breast cancer in Sprague Dawley rats by dehydroepiandrosterone: dose-dependent inhibition, effects of limited exposure, effects on peroxisomal enzymes, and lack of effects on levels of Ha-Ras mutations. Cancer Research. 1998;58:921–926. [PubMed] [Google Scholar]
- McCormick DL, Rao KV. Chemoprevention of hormone-dependent prostate cancer in the Wistar–Unilever rat. European Urology. 1999;35:464–467. doi: 10.1159/000019880. [DOI] [PubMed] [Google Scholar]
- Nakada SY, di Sant’Agnese PA, Moynes RA, Hiipakka RA, Liao S, Cockett AT, Abrahamsson PA. The androgen receptor status of neuroendocrine cells in human benign and malignant prostatic tissue. Cancer Research. 1993;53:1967–1970. [PubMed] [Google Scholar]
- Nazareth LV, Weigel NL. Activation of the human androgen receptor through a protein kinase A signaling pathway. Journal of Biological Chemistry. 1996;271:19900–19907. doi: 10.1074/jbc.271.33.19900. [DOI] [PubMed] [Google Scholar]
- Nelson CP, Kidd LC, Sauvageot J, Isaacs WB, DeMarzo AM, Groopman JD, Nelson WG, Kensler TW. Protection against 2-hydroxyamino-1-methyl-6-phenylimidazo[4,5-b]pyridine cytotoxicity and DNA adduct formation in human prostate by glutathione S-transferase P1. Cancer Research. 2001;61:103–109. [PubMed] [Google Scholar]
- Nevalainen MT, Harkonen PL, Valve EM, Ping W, Nurmi M, Martikainen PM. Hormone regulation of human prostate in organ culture. Cancer Research. 1993;53:5199–5207. [PubMed] [Google Scholar]
- Olapade-Olaopa EO, MacKay EH, Taub NA, Sandhu DP, Terry TR, Habib FK. Malignant transformation of human prostatic epithelium is associated with the loss of androgen receptor immunoreactivity in the surrounding stroma. Clinical Cancer Research. 1999;5:569–576. [PubMed] [Google Scholar]
- Perkins SN, Hursting SD, Haines DC, James SJ, Miller BJ, Phang JM. Chemoprevention of spontaneous tumorigenesis in nullizygous p53-deficient mice by dehydroepiandrosterone and its analog 16alpha-fluoro-5-androsten-17-one. Carcinogenesis. 1997;18:989–994. doi: 10.1093/carcin/18.5.989. [DOI] [PubMed] [Google Scholar]
- Planz B, Aretz HT, Wang Q, Tabatabaei S, Kirley SD, Lin CW, McDougal WS. Immunolocalization of the keratinocyte growth factor in benign and neoplastic human prostate and its relation to androgen receptor. Prostate. 1999;41:233–242. doi: 10.1002/(sici)1097-0045(19991201)41:4<233::aid-pros3>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Potter JD. Morphostats: a missing concept in cancer biology. Cancer Epidemiology Biomarkers and Prevention. 2001;10:161–170. [PubMed] [Google Scholar]
- Raffo AJ, Perlman H, Chen MW, Day ML, Streitman JS, Buttyan R. Overexpression of bcl-2 protects prostate cancer cells from apoptosis in vitro and confers resistance to androgen depletion in vivo. Cancer Research. 1995;55:4438–4445. [PubMed] [Google Scholar]
- Rowley DR. What might a stromal response mean to prostate cancer progression? Cancer Metastasis Review. 1998–1999;17:411–419. doi: 10.1023/a:1006129420005. [DOI] [PubMed] [Google Scholar]
- Rushmore TH, Pickett CB. Glutathione S-transferases, structure, regulation, and therapeutic implications. Journal of Biological Chemistry. 1993;268:11475–11478. [PubMed] [Google Scholar]
- Sakko AJ, Ricciardelli C, Mayne K, Tilley WD, Lebaron RG, Horsfall DJ. Versican accumulation in human prostatic fibroblast cultures is enhanced by prostate cancer cell-derived transforming growth factor beta1. Cancer Research. 2001;61:926–930. [PubMed] [Google Scholar]
- Schor SL, Schor AM, Durning P, Rushton G. Skin fibroblasts obtained from cancer patients display foetal-like behaviour on collagen gels. Journal of Cell Science. 1985;73:235–244. doi: 10.1242/jcs.73.1.235. [DOI] [PubMed] [Google Scholar]
- Schwartz AG, Pashko LL. Mechanism of cancer preventive action of DHEA. Role of glucose-6-phosphate dehydrogenase. Annals of the New York Academy of Sciences. 1995;774:180–186. doi: 10.1111/j.1749-6632.1995.tb17381.x. [DOI] [PubMed] [Google Scholar]
- Shirai T, Sano M, Tamano S, Takahashi S, Hirose M, Futakuchi M, Hasegawa R, Imaida K, Matsumoto K, Wakabayashi K, Sugimura T, Ito N. The prostate: a target for carcinogenicity of 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) derived from cooked foods. Cancer Research. 1997;57:195–198. [PubMed] [Google Scholar]
- Simard J, Gingras S. Crucial role of cytokines in sex steroid formation in normal and tumoral tissues. Molecular and Cellular Endocrinology. 2001;171:25–40. doi: 10.1016/s0303-7207(00)00387-7. [DOI] [PubMed] [Google Scholar]
- Smithers D. On some general concepts in oncology with special reference to Hodgkin’s disease. International Journal of Radiation Oncology-Biology-Physics. 1983;9:731–738. doi: 10.1016/0360-3016(83)90242-0. [DOI] [PubMed] [Google Scholar]
- Tan J, Sharief Y, Hamil KG, Gregory CW, Zang DY, Sar M, Gumerlock PH, deVere White RW, Pretlow TG, Harris SE, Wilson EM, Mohler JL, French FS. Dehydroepiandrosterone activates mutant androgen receptors expressed in the androgen-dependent human prostate cancer xenograft CWR22 and LNCaP cells. Molecular Endocrinology. 1997;11:450–459. doi: 10.1210/mend.11.4.9906. [DOI] [PubMed] [Google Scholar]
- Terman A. Garbage catastrophe theory of aging: imperfect removal of oxidative damage? Redox Reports. 2001;6:15–26. doi: 10.1179/135100001101535996. [DOI] [PubMed] [Google Scholar]
- Voigt KD, Bartsch W. Intratissular androgens in benign prostatic hyperplasia and prostatic cancer. Journal of Steroid Biochemistry. 1986;25:749–757. doi: 10.1016/0022-4731(86)90304-3. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Rehm S. Cadmium and prostate cancer. Journal of Toxicology and Environmental Health. 1994;43:251–269. doi: 10.1080/15287399409531920. [DOI] [PubMed] [Google Scholar]
- Wake N, Isaacs J, Sandberg AA. Chromosomal changes associated with progression of the Dunning R-3327 rat prostatic adenocarcinoma system. Cancer Research. 1982;42:4131–4142. [PubMed] [Google Scholar]
- Weeraratna AT, Arnold JT, George DJ, DeMarzo A, Isaacs JT. Rational basis for Trk inhibition therapy for prostate cancer. Prostate. 2000;45:140–148. doi: 10.1002/1097-0045(20001001)45:2<140::aid-pros8>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- Weisburger JH. Eat to live, not live to eat. Nutrition. 2000;16:767–773. doi: 10.1016/s0899-9007(00)00400-7. [DOI] [PubMed] [Google Scholar]
- Wong YC, Wang YZ. Growth factors and epithelial–stromal interactions in prostate cancer development. International Review of Cytology. 2000;199:65–116. doi: 10.1016/s0074-7696(00)99002-8. [DOI] [PubMed] [Google Scholar]
- Zhao XY, Malloy PJ, Krishnan AV, Swami S, Navone NM, Peehl DM, Feldman D. Glucocorticoids can promote androgen-independent growth of prostate cancer cells through a mutated androgen receptor. Nature Medicine. 2000;6:703–706. doi: 10.1038/76287. [DOI] [PubMed] [Google Scholar]