

Abstract
Purpose
CEP-701 is a potent inhibitor of trk receptors that causes cell death in prostate cancer (PC) models. CEP-701 binds to serum proteins and a preprostatectomy study was performed to assess prostate tissue penetration and clinical response to CEP-701.
Methods
Growth assays and Western blot analyses were performed to evaluate CEP-701 kinase inhibition. In a preprostatectomy study, patients received CEP-701 for five days prior to prostatectomy and prostate tissue analyzed for CEP-701 levels. A phase II dose escalation study was performed in patients with hormone refractory PC with rising PSA and no metastases. Endpoints included PSA response and safety.
Results
CEP-701 binds to serum proteins limiting tissue penetration. An oral dose of 40 mg bid of CEP-701 for five days produced levels of 219 ± 38 nM in prostate at time of prostatectomy. No patients in the Phase II study met the primary response criteria of >50% PSA decline. Seven/9 patients had increase in PSA slope on CEP-701 compared to PSA slope prestudy. Five/9 patients had a decrease in PSA levels after stopping CEP-701. Laboratory studies showed increased PSA production by CEP-701 growth arrested human PC cells in vitro and in vivo.
Conclusions
Evaluation of PSA response is an inadequate indicator of response in CEP-701 treated PC patients. Therefore, the effectiveness of CEP-701 as treatment for prostate cancer has not been adequately tested. Based on a strong preclinical rationale, further clinical studies with CEP-701 using alternative endpoints are indicated.
Keywords: prostate cancer, PSA, kinase, peceptor, CEP-701
INTRODUCTION
Androgen ablation has been the mainstay of treatment for metastatic prostate cancer for the past 65 years and, therefore, can be considered the “grandfather” of signal transduction therapy.1,2 Androgen ablation results in significant palliation for the majority of patients with advanced prostate cancer and to this date represents the most successful anti-cancer signal transduction therapy yet developed. However, all patients eventually relapse on androgen ablative therapy because androgen ablation does not induce the apoptotic death of the nonandrogen-dependent cells because these latter cells activate survival pathways that do not require androgenic stimulation.3–6 It is the continuing survival and proliferation of these nonandrogen-dependent prostate cancer cells that eventually kills, no matter how complete the androgen ablation is within the prostate cancer patient.7 Therefore, therapies that can eliminate these nonandrogen-dependent cancer cells are urgently needed.
Recent studies have demonstrated a survival advantage of several months in men treated with docetaxel based regimens compared to mitoxantrone and prednisone.8,9 There is, however, a relatively small therapeutic index for the cytotoxic response induced by agents such as docetaxel between slowly proliferating prostate cancer cells and rapidly proliferating normal gut, skin, and blood cells.10 This lack of cancer cell specificity leads to host toxicity, which limits both the dose and total length of treatment with such nontargeted cytotoxic agents. Therefore, what is needed is a method for selectively targeting the apoptotic death of nonandrogen-dependent prostate cancer cells without inducing such death in host normal cells. One method to accomplish this goal is to inhibit additional signal transduction pathways to which the survival of these prostate cancer cells are restrictively sensitive.
Over the past two decades, a myriad of growth factors have been identified that promote survival and/or proliferation of hormone-refractory prostate cancer cells and could thus represent targets for therapy. In addition, the signal transduction pathways affected by these growth factors that are responsible for survival and proliferation of hormone refractory prostate cancer cells have begun to be elucidated. In a recent study, we evaluated activation and requirement of akt and mitogen activated protein kinase (erk, p38 and jnk) signaling for survival and proliferation of five malignant androgen independent prostate cancer cell lines and compared these with normal prostate epithelial cells.11 We documented that normal prostate cells require simultaneous erk and jnk signaling for survival plus akt signaling for proliferation.11 In malignant cells, however, only jnk inhibition as monotherapy produced consistent apoptotic response in all prostate cancer cell lines. However, only the combinatorial inhibition of MAP kinases (i.e., jnk, erk, p38) and akt induced sufficient apoptosis to produce a therapeutic effective response in all of the androgen independent cell lines tested.11 These results demonstrated that prostate cancer progression to a lethal androgen-independent state involves acquisition of an enhanced redundancy in downstream survival signaling. In a recent study we have demonstrated that, in addition to trk, CEP-701 can also directly inhibit Mek and jnk and activation of erk and akt (unpublished data). These results also suggest that CEP-701 could represent effective therapy for androgen independent prostate cancer through its ability to inhibit multiple kinases that are important for the growth and survival of prostate cancer cells.
Previous studies have shown that signal transduction induced by neurotrophin binding to its high-affinity trk receptors initiates restricted survival pathways for malignant prostate cells but not for normal host cells.6,12,13 Neurotrophins bind with high affinity to a specific subtype of the Mw 140,000 neurotrophin receptor family known as the trk receptors, each of which is encoded by a separate gene. For NGF, the specific receptor is trk A; for BDNF and neurotrophin-4/5, it is trk B and for NT-3, it is trk C.14 It has been demonstrated that more than 90% of human prostate cancers express at least one of the trk receptors.12 Additional studies have demonstrated that during the progression of prostate cancer to a metastatic state, these malignant cells also acquire the ability to synthesize and secrete various neurotrophin ligands in addition to expressing their cognate trk receptors.6
These studies served to validate the trk pathway as a putative prostate cancer selective signal transduction target and led to the preclinical development of the potent indolocarbarbazole trk receptor inhibitor CEP-701 (IC50 4 nM) by Cephalon, Inc. (Westchester, PA). This compound, however, is not a pure trk inhibitor. CEP-701 can also inhibit VEGFR2 (IC50 65 nM), PDGFRb (IC50 226 nM), and RET (IC50 50–150 nM in whole cells).12 CEP-701 is also a potent inhibitor of Flt-3 (IC50 3.2 nM) and, as such, is under clinical development as treatment for refractory acute myelogenous leukemia (AML). In vivo studies documented that CEP-701 inhibited the growth by 40–80% of five different models of human (i.e., LNCaP, DU145, PC-3, TSU, PC-82) and four rodent (Dunning G, MATLu, AT-2 and H) prostate cancers independent of their state of differentiation, androgen sensitivity, metastatic ability, or growth rate.12,13 On this basis, the orally active compound CEP-701 entered clinical development as therapy for androgen independent prostate cancer. Here we report on recent studies in our laboratory suggesting that the antitumor effects seen with CEP-701 against prostate cancer cells may not be solely due to inhibition of trk, but may be due to CEP-701’s ability to inhibit multiple kinases that are important for survival and proliferation of prostate cancer cells. In addition, here we also report the results of a preprostatectomy trial conducted to determine prostate tissue levels at the phase I recommended dose of 40 mg bid and a phase II dose escalation study to examine effect of CEP-701 in men with hormone-refractory prostate cancer and biochemical (i.e., PSA) recurrence.
MATERIALS AND METHODS
In vitro studies
LNCaP cancer cell line was obtained from ATCC (Rockville, MD) and maintained by serial passage in appropriate media containing indicated amount of fetal bovine serum (FBS). Cytotoxic response to CEP-701 was determined using the Promega Cell Titer 96 Non-Radioactive Cell Proliferation Assays, Promega Corp (Madison, WI) according to the manufacturer’s instructions as described previously.6 PSA concentrations were determined in the Clinical Chemistry lab at Johns Hopkins using an ELISA-based assay (Beckman-Coulter Hybritech assay).
Animal studies
Four to six-week-old nude mice (Harlan Sprague Dawley, Indianapolis, IN) were maintained five per cage and given a commercial diet and water ad libitum. Animals were housed under humidity and temperature controlled conditions. All of the animal studies were performed according to animal protocols approved by Cephalon, Inc. and the Johns Hopkins School of Medicine Animal Care and Use Committees. Animals were inoculated with 2 × 106 LNCaP cells in the left flank under sterile conditions. Tumor volumes were assessed biweekly using vernier calipers. Animal weights were also assessed at these times. When tumor volumes reached ~0.2 cc in size animals were randomized so starting average tumor volume were approximately equal. Animals were injected subcutaneously with either 20 mg/kg CEP-701 or vehicle control. CEP-701 was formulated in a vehicle containing 40% polyethylene glycol (Spectrum, Los Angeles, CA) 10% Povidone C-30 (ISP, Bound Brook, NJ) 2% benzyl alcohol (Spectrum) and water (milli-Q or high-performance liquid chromatography grade; Fisher Scientific, Malvern, PA). Blood for PSA determination was obtained from the tail veins of anesthetized mice (i.m. injection of ketamine (4.2 mg/100 g body weight) plus Xylazine (0.85 mg/100 g body weight). Animals with tumors >2 cc or with >15% body weight loss were euthanized by CO2 overdose.
Trk inhibition in whole cells
Varying concentrations of CEP-701 (10 nM–10 μM) were incubated in serum free medium or 75% human plasma (Harlan) for two hours at 37°C. These media were then added to trkC transfected NIH3T3 cells for two hours. TrkC phosphorylation was induced by adding 10 ng/ml NT-3 (R&D Systems) for 5 min. Western blot analysis of trkC phosphorylation was performed as previously described.6
Patient population
Patients with pathologically confirmed adenocarcinoma of the prostate were eligible for both the preprostatectomy and Phase II studies if they were older than 18 years; had an Eastern Cooperative Oncology Group (ECOG) performance status less than 2; and had adequate bone marrow, renal and hepatic function. For the preprostatectomy trial patients had to have met one of the following criteria: a Gleason score initially of ≥7 or a baseline PSA ≥10 ng/ml or clinical stage T2b, T2c or T3a. Due to slow accrual, the protocol was amended to allow patients with Gleason score ≥5 and PSA ≥4 ng/ml.
For the phase II study patients had to have hormone refractory prostate cancer with increasing PSA concentrations on adequate hormone suppression via either surgical castration or LHRH agonist; had to have undergone withdrawal from antiandrogen therapy; had no detectable metastatic disease as assessed by bone scan and computed tomography (CT); had no prior nonhormonal systemic anticancer therapy or investigational drug within one month of screening.
For both studies, written informed consent was obtained from all patients as approved by the Johns Hopkins Institutional Review Board. Patients agreed to collection of additional blood solely for research purposes. Patients on the preprostatectomy study agreed to use of harvested prostate tissue solely for research purposes.
Study design
Preprostatectomy study
This was an open-label trial of single agent CEP-701 administered for five days prior to scheduled prostatectomy. Patients were treated at a dose of 40 mg orally twice a day. CEP-701 was supplied in 20 ml amber glass vials as a clear yellow oral solution at a concentration of 25 mg/ml in polysorbate 80 NF (10 ml) and polypropylene glycol USP (10 ml). Individual doses were prepared for patient by the pharmacy and patients were instructed to dilute the study drug in 30 ml of juice prior to oral administration. Patients were instructed to take the last scheduled dose of CEP-701 ~12 hours before scheduled prostatectomy. Venous blood samples for determination of CEP-701 trough plasma levels were drawn in the morning within one hour of prostatectomy. After the pathologist completed evaluation of the prostate specimen post-surgery, pieces of normal prostate were harvested and frozen in liquid nitrogen for determination of intraprostatic CEP-701 levels. Tissues and plasma levels were determined using a previously validated HPLC-based assay performed by Cephalon, Inc., the study sponsors.
Phase II dose escalation study
This was a limited phase II open-label dose escalation trial of single agent CEP-701. Eligible patients were treated at an initial dose of 60 mg orally twice a day for 28 days. If patients tolerated this dose level, the dose was increased to 80 mg orally twice a day for an additional eight weeks. CEP-701 was prepared for administration as described above. PSA levels were obtained every two weeks on study and all patients remained on study even with a rising PSA. Although eligibility was limited to patients with rising PSA only, two patients were allowed on study with a single site of metastases (one bone, one lymph node). Grade I–II nausea was treated with antiemetics (Compazine, Reglan). Patients with Grade II nausea on the 60 mg dose were taken off drug for one week, patients on the 80 mg dose were decreased back to the 60 mg dose. Patients with refractory nausea were taken off study. The study design called for study termination if no PSA response (>50% decrease from baseline) was observed in the first nine patients (stage 1) and an enrollment of an additional 21 patients (stage 2) if one of nine patients had a PSA response in the first stage.
Statistics
For in vitro and in vivo preclinical studies results are expressed as the mean ± standard error with Students’ t-test was used to compare groups. For the Phase II study, the study size of 30 patients was chosen based on the two-stage, optimum design by Simon15 using the following assumptions: the probability of accepting a drug for development with a response probability of 5% is 0.05; the probability of rejecting a drug for development with a response probability of 25% is 0.10. Under these assumptions, if the true response rate is 5% the probability of early termination of the study was determined to be 0.63.
Assessment of toxicity and response
Patients on the preprostatectomy trial were assessed for toxicity after prostatectomy. Patients on the phase II study had a baseline PSA, bone scan and CT scan. Patients were monitored at baseline, day 8, 15 and then every two weeks until day 85. Measurements included hematologic tests, biochemical tests for renal and hepatic function, coagulation tests, PSA levels, physical examination, safety assessments for adverse effects, and observation of vital signs. Toxicity was graded according to the Common Toxicity Criteria version 2.0 of the National Cancer Institute. PSA levels were obtained and results shared with patients but overall response was not assessed until day 85 when patients underwent bone scan, CT scan of chest abdomen and pelvis and had review of PSA levels. Primary endpoint in this phase II study was PSA response. Patients were considered responders with >50% decrease in PSA compared to baseline value confirmed after four weeks without clinical or radiographic evidence of disease progression. Secondary endpoints were duration of PSA response (time from first PSA sample showing a 50% or greater decline to the time PSA returns to more than 50% of the baseline value). Additional secondary endpoints were safety and tolerability.
RESULTS
Determination of levels of CEP-701 in prostate tissue
The preclinical results with CEP-701 against prostate cancer xenografts provided a strong preclinical rationale for the evaluation of CEP-701 as treatment for prostate cancer. On the basis of these collective results, CEP-701 entered clinical testing. Such clinical testing has been complicated by the fact that indolocarbazoles such as UCN-01 (i.e., 7-hydroxystaurosporine) can bind with high affinity to the abundant serum protein human alpha-1 acidic glycoprotein (AGP).16 In this regard, while the IC50 value for inhibition of purified Trks A–C in aqueous buffer is ~5 nM the IC50 for inhibition of purified Trks in human plasma ranges between 900–2600 ng/ml (2100–6200 nM) (unpublished data). While total concentration of CEP-701 can be measured via HPLC analysis, assays for free concentration of CEP-701 are not available. To assess differences in trk inhibition using whole cells, an assay was developed using 3T3 cells transfected with trk C. The addition of 10 ng/ml NT-3 results in an increase in trk C phosphorylation by these cells that can be assayed by Western blot using a phosphotyrosine antibody. In this assay CEP-701 inhibited trk C phosphorylation with an IC50 of ~10 nM (Fig. 1A). In contrast, in the presence of 75% human plasma, the IC50 for inhibition of trkC phosphorylation is shifted ~100-fold (Fig. 1B). These studies confirm that CEP-701 binds tightly to plasma proteins and suggest that plasma levels of CEP-701 measured as part of pharmacokinetic analyses may not necessarily reflect levels present in tissue.
Figure 1.
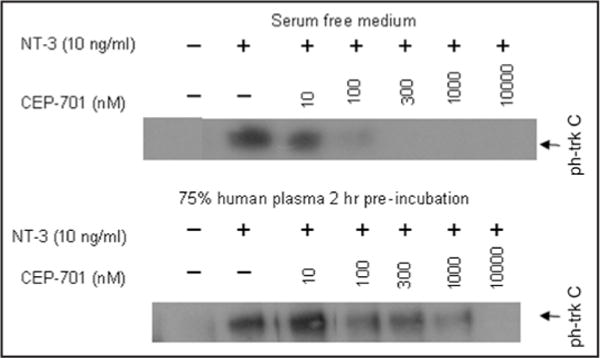
Western blot of phosphorylated trk C expression in trk C transfected 3T3 cells following exposure to trk C ligand NT-3 ± increasing doses of CEP-701. Dose responsive inhibition of trk C phosphorylation by CEP-701 in (A) serum free medium; (B) 75% human plasma.
To address this protein binding issue, a preprostatectomy study was performed to compare tissue levels of CEP-701 in the human prostate to plasma levels following oral administration. In this study, patients received 40 mg of CEP 701 orally twice daily for five days prior to scheduled prostatectomy. The 40 mg bid dosing level was chosen based on data from an earlier phase I study performed in patients with advanced cancers.17 In this preprostatectomy study, the last dose of CEP-701 was taken by patients ~12 hours prior to scheduled surgery. Therefore, nadir levels of CEP-701 were determined. A total of eight patients were enrolled. The five-day treatment course was completed by all patients and toxicity consisted of grade I–II gastrointestinal complaints (nausea, bloating). Using a previously validated, HPLC-based assay, nadir plasma levels of CEP-701 were determined to be 916 ± 150 nM. Nadir prostate tissue levels were 219 ± 38 nM. The prostate/plasma ratio was 0.25 ± 0.04. These results suggest that free CEP-701 within the plasma can enter tissue to produce nadir prostate tissue levels of CEP-701 following five days of dosing that are ~60-fold higher than the IC50 for inhibition of purified trks but only ~2-fold higher than the in vitro IC50 for inhibition of growth of human prostate cancer cell lines.13
Phase II study assessing PSA response to CEP-701 therapy
While this preprostatectomy study was in progress, a multi-institutional phase II study was performed using a discontinuation study design in which men with hormone-refractory prostate cancer were treated at a dose of 40 mg of CEP-701 twice daily and evaluated for PSA response. This study, the results of which have not been published to date, had a planned enrollment of 400 patients but was terminated early due to failure to meet study endpoints with ~5% PSA response in 159 evaluable patients (personal communication, Cephalon, Inc). In addition, in this study 47% of patients came off drug early due to mostly gastrointestinal toxicity.
Upon completion of these studies with CEP-701 in prostate cancer patients, Levis et al. demonstrated that CEP-701 was also an inhibitor of Flt-3 kinase and was cytotoxic to leukemia cells in vitro and in vivo.18 Subsequently, Smith et al. reported results of single-agent CEP-701 in treatment of patients with relapsed or refractory AML.19 This group utilized an ex vivo bioassay in which cells from a human AML cell line transfected with mutant FLT3 expressing a constitutively phosphorylated FLT3 were incubated with plasma from the patients treated with CEP-701 and then assayed for FLT3 autophosphorylation.19 This study demonstrated that high level inhibition of FLT3 activity (i.e., >85% in ex vivo assay) correlated with patient response. Such inhibition was not observed in patients at a treatment dose of 40 mg twice daily, but was increasingly observed at doses of 60 and 80 mg twice daily.19 The twice daily dose of 80 mg CEP-701 was well tolerated by these patients.
On the basis of these studies in the leukemia population and the preprostatectomy data showing marginal CEP-701 levels in prostate tissue at the 40 mg bid dose, a second phase II study of CEP-701 was designed to assess the activity of higher doses of CEP-701 in patients with hormone refractory recurrent prostate cancer with rising PSA only and no evidence of disease on bone or CT scan. The study consisted of two phases. The study was designed to be stopped if no response (i.e., PSA decline >50% from baseline) was observed in any of nine patients after 12 weeks of therapy. Patient characteristics are listed in Table 1. Patients received CEP-701 at a dose of 60 mg twice daily for 28 days. If the drug was well tolerated, the dose was then increased to 80 mg twice daily for eight additional weeks. PSA levels were determined every two weeks, but patients were expected to remain on study for a full twelve weeks, regardless of change in PSA level.
Table 1.
Characteristics of patients treated in phase II dose escalation study
| Characteristic
|
||
| Average Age, years | 67 | (52–87) |
| Initial Therapy | ||
| Prostatectomy | 2 | |
| External Beam Radiation | 4 | |
| No Local Therapy | 3 | |
| Median Gleason Score | 8 | (7–10) |
| Initial PSA (ng/ml) | 26.6 | (1.1–94.6) |
| Performance Score (ECOG) | ||
| 0 | 8 | |
| 1 | 1 | |
A total of nine men were enrolled on the study from March to September 2004. Two men withdrew early due to toxicity while on a dose of 80 mg twice daily (study day 42 and day 56 respectively). One withdrew due to complaints of emotional lability, crying spells and decreased appetite. The second patient withdrew due to nausea, increased fatigue and tremulousness. Both patients symptoms resolved on cessation of the study drug. Most patients noted grade I–II gastrointestinal complaints (i.e., loose stools, nausea and “bloating”).
None of the nine patients met the primary criteria for response of 50% decline in PSA from baseline. Seven of the nine patients had an increase in PSA of >50% from baseline over the course of the study while the two remaining patients had less than a 50% increase in PSA over the course of treatment. In the seven patients (78%), comparison of the rate of change in PSA serum level over time (i.e., PSA slope) prior to starting CEP-701 to the PSA slope after 12 weeks on drug revealed an increase in slope that ranged from 1.5 to 14-fold over baseline (Fig. 2A). Based on this observation, we reevaluated results from patients on the earlier CEP-701 Phase II discontinuation study made available by Cephalon, Inc. From this study, only 19 of 159 evaluable patients had two screening PSA’s recorded prior to entering the study. These two prestudy PSA levels were used to calculate the PSA slope. Change in PSA serum levels over the subsequent four weeks on 40 mg bid CEP-701 was used to determine PSA slope on drug. These analyses revealed that 14 out of the 19 patients (74%) had a similar increase in PSA slope (Fig. 2B).
Figure 2.
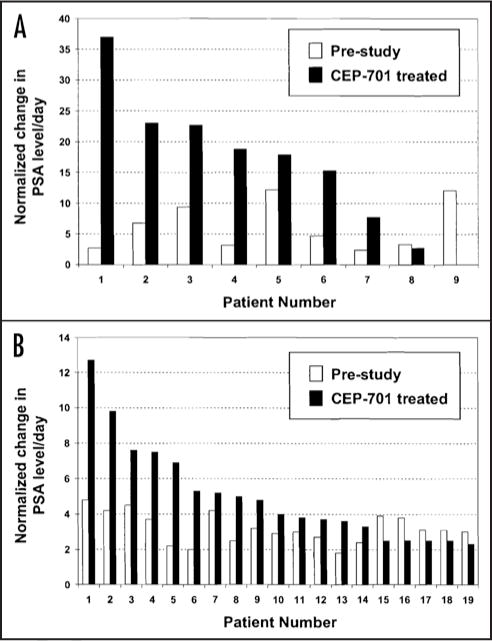
Change in PSA level/day (PSA slope) before starting CEP-701 and during treatment with CEP-701. (A) Patients treated in dose escalation study. Pre PSA slope determined from best fit line of all available PSA levels pre study (n = 4–8 PSA levels) and while on study (n = 5–7 PSA levels); (B) Evaluable patients treated on discontinuation phase II study. Patient with two screening PSA’s available for calculation of PSA slope (19 of 159 total patients) were included in analysis. PSA slope on CEP-701 calculated from change in PSA from PSA level on day 0 to PSA level obtained after four weeks on CEP-701. In each evaluation, patient PSA level on day 0 was normalized to one with subsequent normalization of pre and on-study PSA levels. Normalized change in PSA level/day determined by dividing normalized PSA slope by number of total days before starting (i.e., prestudy) or after starting (i.e., on-study) CEP-701.
Six of the nine patients completed the full 12 weeks of therapy with CEP-701. One patient with stable PSA continued on study for an additional ten weeks and was removed from study when his PSA had increased >50% from baseline. As noted, two patients came off study early due to toxicity. All nine patients had multiple PSA determinations once off study. Interestingly, five of nine patients (55%) had decreases in serum PSA following cessation of CEP-701 (Fig. 3). The duration of this PSA decrease ranged from two weeks to six months. PSA decreased by an average of 38 ± 9% (range 15–60%) from the peak PSA level achieved on the last day of CEP-701 therapy. One patient’s PSA decreased sequentially over a six month period almost back to his baseline PSA level measured at the start of CEP-701 therapy (Fig. 3).
Figure 3.
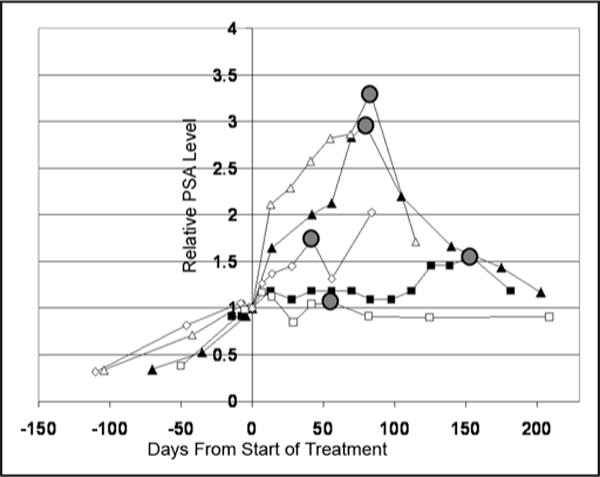
Decrease PSA levels in five patients after stopping CEP-701. Lines represent Normalized PSA levels plotted vs. time for each patient. Day that CEP-701 was stopped is by the gray circle. Two patients completed study on scheduled day 84, two patients ended study early due to toxicity and one patient continued on study until day 154 due to stable PSA (i.e., <50% increase in PSA).
Effect of CEP-701 on PSA-producing prostate cancer cell in vitro and in vivo
The increase in PSA slope observed in the majority of evaluable patients in these two studies suggested that CEP-701 may induce increased expression of PSA by prostate cancer cells. To evaluate this possibility, PSA-producing human LNCaP prostate cancer cells were treated with CEP-701 at a concentration of 200 nM. Cells were counted and PSA concentration in the conditioned media measured daily for four days. At this concentration CEP-701 completely inhibited the growth of LNCaP cells (Fig. 6A). PSA production per cell increased significantly compared to control cells over the four day exposure period (Fig. 4A).
Figure 4.
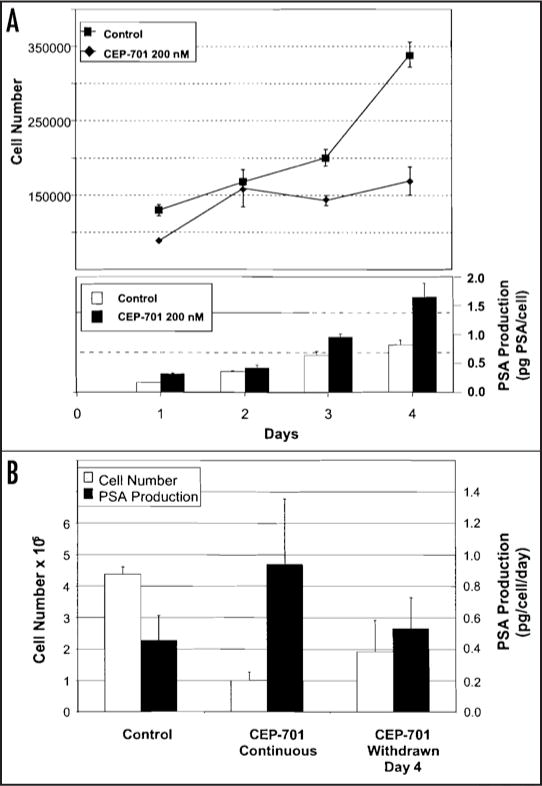
(A) Effect of continuous four-day exposure to CEP-701 (200 nM) on growth and PSA production by PSA-producing LNCaP cells. On indicated days, media (RPMI 1640, 10% fetal calf serum) was removed for determination of PSA levels and viable cells determined by MTT cell viability assay. Cell counts determined from standard curve of absorbance of known numbers of cells assayed by MTT. Experiment performed in triplicate flasks and data shown represent average ± standard error. (B) Effect of withdrawal of CEP-701 on growth and PSA production of LNCaP cells. Control cells were exposed for seven days to media alone with media exchange at day four. CEP-701 continuous received continuous exposure to 200 nM CEP-701 for seven days with media exchange at day four. Third group of cells were exposed to CEP-701 for four days and then placed into CEP-701 free media.
To evaluate effects of CEP-701 withdrawal on PSA production, LNCaP cells were exposed to continuous CEP-701 or had CEP-701 withdrawn after four day exposure (Fig. 4B). At the end of seven days, cells were counted and PSA levels in the conditioned media were determined. In this study, PSA production per cell per day in the continuously treated cells was ~2.5-fold higher than controls after seven days while cell counts were 4.5-fold lower (Fig. 4B). In contrast, PSA production per cell returned to control levels in cells in which CEP-701 exposure was discontinued.
To evaluate effects of CEP-701 on PSA production in vivo, nude mice bearing LNCaP xenografts received subcutaneous administration of 20 mg/kg CEP-701 daily × five consecutive days for two cycles as indicated. Control animals received vehicle subcutaneously according to the same schedule. As an additional control, tumor bearing animals were treated intravenously with 12.5 mg/kg docetaxel every three days for a total of three doses. Tumors volumes were measured using calipers and blood drawn on day 12 post initiation of dosing for determination of PSA levels. At this dose level, CEP-701 significantly inhibited growth of LNCaP tumors compared to controls with T/C ratio of ~33% at day 30 post treatment (Fig. 5A). Day 12 PSA serum levels were lowest in the docetaxel treated group and highest in the CEP-701 treated group (Fig. 5B). However, on normalization per gram of tumor, PSA levels in the docetaxel treated group were not statistically different than that of the control treated animals (Fig. 5B). In contrast, PSA levels/gram of tumor were ~60% higher in the CEP-701 treated group compared to control (p = 0.02) consistent with CEP-701’s positive effect on PSA production observed in vitro (Fig. 5B).
Figure 5.
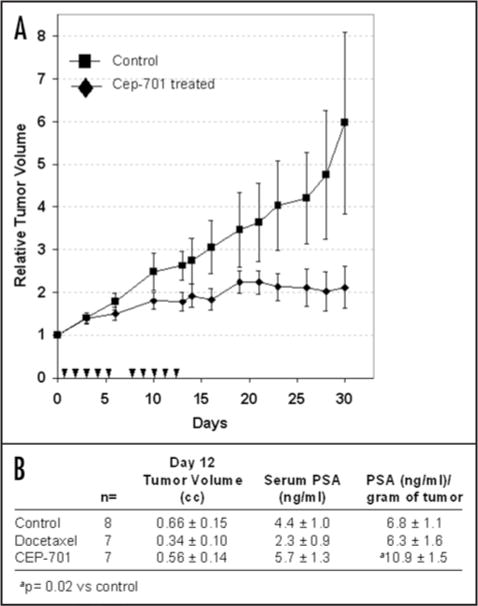
(A) Growth of LNCaP xenografts treated subcutaneously with either vehicle control (n = 8) or CEP-701 (n = 7) at a dose of 20 mg/kg per day. Treatment days indicated by triangles. Relative change in tumor volume determined by ratio of tumor measurement of given day/tumor measurement at day 0 for each animal. Data presented as average relative volume ± standard error. (B) Tumor volume, serum PSA and PSA/gram of tumor on Day 12 following initiation of therapy with either vehicle control × 10 doses, docetaxel 12.5 mg/kg every 3 days × 3 doses or CEP-701 20 mg/kg × 10 doses.
DISCUSSION
On the basis of CEP-701’s effects against human prostate cancer xenografts this agent entered clinical development as therapy for prostate cancer. Initial Phase I studies documented that patients could tolerate an oral dose of CEP-701 of 40 mg twice daily. On the basis of observed significant plasma protein binding, we conducted the preprostatectomy study reported and documented prostate tissue levels of CEP-701 of ~220 nM. These levels were well above the ~5 nM IC50 for inhibition of purified trks (i.e., A–C) but only ~2-fold higher than a dose that was consistently effective against prostate cancer cell lines in vitro.
On the basis of the preprostatectomy data and data from the AML trials with CEP-701, we initiated the phase II study in men with hormone refractory prostate cancer with rising PSA levels to determine effects of treatment with higher doses of CEP-701. Like the larger earlier Phase II study, PSA response was the sole therapeutic endpoint in this study. As described, none of the nine men treated in this study met response criteria of >50 % PSA decline. In contrast, the majority of men exhibited an increase in PSA slope while on study. Such an increase in PSA slope was also observed in ~75% of the 19 evaluable patients treated with 40 mg bid in the large phase II study. Based on these clinical observations, we returned to the laboratory and demonstrated that the profile of PSA production by PSA-producing cells in vitro and in vivo following exposure and then withdrawal of CEP-701 mimicked some of the features of PSA levels observed clinically in CEP-701 treated patients.
This increase in PSA levels following treatment with signal transduction inhibitors has been noted previously. For example, Rao et al. performed a phase II study with imatinib,20 which is used as a treatment for chronic myelogenous leukemia due to its ability to inhibit the fusion kinase bcr-abl.21 Based on its ability to also inhibit the PDGF receptor is being tested in patients with prostate cancer. This study was terminated early when five of 21 patients exhibited an inordinately fast rise in PSA.20 In a similar study, Mathew et al. evaluated the combination of imatinib and docetaxel in a phase II study.22 This study incorporated a lead in period in which patients were treated with imatinib alone for 30 days to assess response. While this study did not report changes in PSA slopes, the investigators noted that 67% of the patients had a greater than 50% increase in PSA over the 30 day lead-in period with PSA increases of 1.03 to 7.8 fold from baseline noted.22 Finally, in a recent reported study, Bajaj et al. evaluated efficacy of imatinib in a Phase II study in 27 patients with biochemical relapse after prostatectomy or radiotherapy. In this study, patients with PSA progression during the study (20/27) experienced, according to the authors, “a dramatic rise in the global PSA slope during the study.”23
Increased PSA levels following treatment with other signal transduction modifiers has also been observed in other preclinical models. For example, while HER2/neu has been demonstrated to induce AR transactivation, stabilize AR protein levels and optimize binding of AR to promoter/enhancer regions,24,25 the HER2/neu inhibitor Herceptin, was shown to markedly increase PSA levels/gram of tumor in LNCaP and CWR22R prostate cancer xenografts.26 In addition, Korkmaz et al. demonstrated that the histone deacetylase inhibitors trichostatin A, depsipeptide and sodium butyrate can enhance AR transcriptional activity in LNCaP cells leading to increased production of PSA.27 In our study the mechanism whereby CEP-701 may enhance PSA production in the androgen sensitive LNCaP cell line and in these castrated patients with presumed androgen independent prostate cancer is unknown. The preclinical and clinical results run contrary to preclinical data demonstrating that agents that inhibit signal transduction proteins may have a negative effect on normal AR function.28
While PSA response remains an important endpoint for evaluation of many new therapies for prostate cancer, the above noted studies along with our data from preclinical and the phase II studies with CEP-701 suggest that the use of PSA response as the sole endpoint in evaluating signal transduction inhibitors in prostate cancer may not be appropriate. This is of particular importance given the increasing number of signal transduction inhibitors, including multi-kinase inhibitors such as sunitinib and sorafenib, entering clinical evaluation as treatment for prostate cancer.29 The preclinical data with PSA-producing cell lines and the phase II data support the conclusion that the evaluation of PSA response alone is an inadequate indicator of clinical response in CEP-701 treated prostate cancer patients. Since the only two studies testing CEP-701 as treatment for prostate cancer have relied solely on PSA response, we conclude that the effectiveness of CEP-701 as treatment for prostate cancer has not been adequately tested to date. Based on the strong preclinical rationale presented here and in earlier studies, further clinical studies are indicated using alternative endpoints or patient populations to assess the efficacy of CEP-701 as treatment for men with prostate cancer. While no single validated biomarker has emerged as an alternative to PSA for following disease response, there is data to suggest that changes in markers of bone resorption [carboxyterminal telopeptide of type-I collagen (N-telopeptide)] or bone formation (e.g., bone alkaline phosphatase) may have some utility in assessing disease response in patients with known bone metastases. In addition, endpoints such as objective response in patients with nonbony metastases or subjective response secondary to pain relief might also be considered in the design of future trials.
Acknowledgments
Prostate Cancer Foundation and the Howard Hopkins Initiative (S.R. Denmeade) NIH Grants 1R01 DK52645 (J.T. Isaacs).
References
- 1.Huggins C, Stephens RC, Hodges CV. Studies on prostatic cancer: 2. The effects of castration on advanced carcinoma of the prostate gland. Arch Surg. 1941;43:209. [Google Scholar]
- 2.Huggins CB, Hodges CV. Studies on prostate cancer: 1. The effects of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. Cancer Res. 1941;1:203. [Google Scholar]
- 3.Matuo Y, Nishi N, Matsui S-I, Sandberg AA, Isaacs JT, Wada F. Heparin binding affinity of rat prostatic growth factor in normal and cancerous prostates: partial purification and characterization of rat prostatic growth factor in the Dunning tumor. Cancer Res. 1987;47:188–92. [PubMed] [Google Scholar]
- 4.Matuo Y, Nishi N, Tanaka H, Sasaki I, Isaacs JT, Wada F. Production of IGF-II-related peptide by an anaplastic cell line (AT-3) established from the Dunning prostatic carcinoma of rats. In Vitro Cell Dev Biol. 1988;24:1053–56. doi: 10.1007/BF02620881. [DOI] [PubMed] [Google Scholar]
- 5.Jarrard DF, Bussemakers MJG, Bova GS, Isaacs WB. Regional loss of imprinting of the insulin-like growth factor II gene occurs in human prostate tissues. Clin Cancer Res. 1995;1:1471–78. [PubMed] [Google Scholar]
- 6.Weeraratna AT, Arnold JT, George DJ, De Marzo A, Isaacs JT. Rational basis for trk inhibition therapy for prostate cancer. Prostate. 2000;45:145–53. doi: 10.1002/1097-0045(20001001)45:2<140::aid-pros8>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 7.Eisenberger MA, Blumenstein BA, Crawford ED, Miller G, McLeod DG, Loehrer PJ, Wilding G, Sears K, Culkin DJ, Thompson IM, Bueschen AJ, Lowe BA. Bilateral orchiectomy with or without flutamide for metastatic prostate cancer. N Engl J Med. 1998;339:1036–42. doi: 10.1056/NEJM199810083391504. [DOI] [PubMed] [Google Scholar]
- 8.Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, Oudard S, Théodore C, James ND, Turesson I, Rosenthal MA, Eisenberger MA. TAX 327 Investigators. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. N Engl J Med. 2004;351:1502–12. doi: 10.1056/NEJMoa040720. [DOI] [PubMed] [Google Scholar]
- 9.Petrylak DP, Tangen CM, Hussain MH, Lara PN, Jones JA, Taplin ME, Burch PA, Berry D, Moinpour C, Kohli M, Benson MC, Small EJ, Raghavan D, Crawford ED. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N Engl J Med. 2004;351:1513–20. doi: 10.1056/NEJMoa041318. [DOI] [PubMed] [Google Scholar]
- 10.Berges RR, Vukanovic J, Epstein JI, CarMichel M, Cisek L, Johnson DE, Veltri RW, Walsh PC, Isaacs JT. Implication of the cell kinetic changes during the progression of human prostatic cancer. Clin Cancer Res. 1995;1:473–80. [PMC free article] [PubMed] [Google Scholar]
- 11.Uzgare AR, Isaacs JT. Enhanced redundancy in Akt and mitogen-activated protein kinase-induced survival of malignant versus normal prostate epithelial cells. Cancer Res. 2004;64:6190–9. doi: 10.1158/0008-5472.CAN-04-0968. [DOI] [PubMed] [Google Scholar]
- 12.Dionne CA, Camoratto AM, Jani JP, Emerson E, Neff N, Vaught JL, Murakata C, Djakiew D, Lamb J, Bova S, George D, Isaacs JT. Cell cycle-independent death of prostate adenocarcinoma is induced by the trk tyrosine kinase inhibitor CEP-751 (KT6587) Clin Cancer Res. 1998;4:1887–98. [PubMed] [Google Scholar]
- 13.George DJ, Dionne CA, Jani J, Angeles T, Murakata C, Lamb J, Isaacs JT. Sustained in vivo regression of Dunning H rat prostate cancers treated with combinations of androgen ablation and trk tyrosine kinase inhibitors, CEP-751 (KT-6587) or CEP-701 (KT-5555) Cancer Res. 1999;59:2395–401. [PubMed] [Google Scholar]
- 14.Segal RA, Greenburg ME. Intracellular signaling pathways activated by neurotrophic factors. Ann Rev Neurosci. 1996;19:463–89. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 15.Simon R. Optimal two-stage designs for phase II clinical trials. Control Clin Trials. 1989;10:1–10. doi: 10.1016/0197-2456(89)90015-9. [DOI] [PubMed] [Google Scholar]
- 16.Sausville EA, Lush RD, Headlee D, Smith AC, Figg WD, Arbuck SG, Senderowicz AM, Fuse E, Tanii H, Kuwabara T, Kobayashi S. Clinical pharmacology of UCN-01: initial observations and comparison to preclinical models. Cancer Chemother Pharmacol. 1998;42:S54–9. doi: 10.1007/s002800051080. [DOI] [PubMed] [Google Scholar]
- 17.Marshall JL, Kindler H, Deeken J, Bhargava P, Vogelzang NJ, Rizvi N, Luhtala T, Boylan S, Dordal M, Robertson P, Hawkins MJ, Ratain MJ. Phase I trial of orally administered CEP-701, a novel neurotrophin receptor-linked tyrosine kinase inhibitor. Invest New Drugs. 2005;23:31–7. doi: 10.1023/B:DRUG.0000047103.64335.b0. [DOI] [PubMed] [Google Scholar]
- 18.Levis M, Allebach J, Tse KF, Zheng R, Baldwin BR, Smith BD, Jones-Bolin S, Ruggeri B, Dionne C, Small D. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood. 2002;99:3885–91. doi: 10.1182/blood.v99.11.3885. [DOI] [PubMed] [Google Scholar]
- 19.Smith BD, Levis M, Beran M, Giles F, Kantarjian H, Berg K, Murphy KM, Dauses T, Allebach J, Small D. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004;103:3669–76. doi: 10.1182/blood-2003-11-3775. [DOI] [PubMed] [Google Scholar]
- 20.Rao K, Goodin S, Levitt MJ, Dave N, Shih WJ, Lin Y, Capanna T, Doyle-Lindrud S, Juvidian P, DiPaola RS. A phase II trial of imatinib mesylate in patients with prostate specific antigen progression after local therapy for prostate cancer. Prostate. 2005;62:115–22. doi: 10.1002/pros.20130. [DOI] [PubMed] [Google Scholar]
- 21.Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–7. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 22.Mathew P, Thall PF, Jones D, Perez C, Bucana C, Troncoso P, Kim SJ, Fidler IJ, Logothetis C. Platelet-derived growth factor receptor inhibitor imatinib mesylate and docetaxel: a modular phase I trial in androgen-independent prostate cancer. J Clin Oncol. 2004;22:3323–9. doi: 10.1200/JCO.2004.10.116. [DOI] [PubMed] [Google Scholar]
- 23.Bajaj GK, Zhang Z, Garrett-Mayer E, Drew R, Sinibaldi V, Pili R, Denmeade SR, Carducci MA, Eisenberger MA, DeWeese TL. Phase II study of imatinib mesylate in patients with prostate cancer with evidence of biochemical relapse after definitive radical retropubic pros-tatectomy or radiotherapy. Urology. 2007;69:526–31. doi: 10.1016/j.urology.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 24.Mellinghoff IK, Vivanco I, Kwon A, Tran C, Wongvipat J, Sawyers CL. HER2/neu kinase-dependent modulation of androgen receptor function through effects on DNA binding and stability. Cancer Cell. 2004;6:517–27. doi: 10.1016/j.ccr.2004.09.031. [DOI] [PubMed] [Google Scholar]
- 25.Yeh S, Lin HK, Kang HY, Thin TH, Lin MF, Chang C. From HER2/Neu signal cascade to androgen receptor and its coactivators: a novel pathway by induction of androgen target genes through MAP kinase in prostate cancer cells. Proc Natl Acad Sci USA. 1999;96:5458–63. doi: 10.1073/pnas.96.10.5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agus DB, Scher HI, Higgins B, Fox WD, Heller G, Fazzari M, Cordon-Cardo C, Golde DW. Response of prostate cancer to anti-Her-2/neu antibody in androgen-dependent and -independent human xenograft models. Cancer Res. 1999;59:4761–4. [PubMed] [Google Scholar]
- 27.Korkmaz CG, Fronsdal K, Zhang Y, Lorenzo PI, Saatcioglu F. Potentiation of androgen receptor transcriptional activity by inhibition of histone deacetylation–rescue of transcriptionally compromised mutants. J Endocrinol. 2004;182:377–89. doi: 10.1677/joe.0.1820377. [DOI] [PubMed] [Google Scholar]
- 28.Singh P, Uzgare A, Litvinov I, Denmeade SR, Isaacs JT. Combinatorial androgen receptor targeted therapy for prostate cancer. Endocr Relat Cancer. 2006;13:653–66. doi: 10.1677/erc.1.00797. [DOI] [PubMed] [Google Scholar]
- 29.Pantuck AJ, Zomorodian N, Belldegrun AS. Phase I, open-label, single-center, multiple-dose, dose-escalation clinical study of SUO11248 (sunitinib) in subjects with high-risk prostate cancer who have elected to undergo radical prostatectomy. Curr Urol Rep. 2007;8:3–4. doi: 10.1007/s11934-007-0014-8. [DOI] [PubMed] [Google Scholar]
- 30.Nelson JB, Nabulsi AA, Vogelzang NJ, Breul J, Zonnenberg BA, Daliani DD, Schulman CC, Carducci MA. Suppression of prostate cancer induced bone remodeling by the endothelin receptor A antagonist atrasentan. J Urol. 2003;169:1143–9. doi: 10.1097/01.ju.0000042162.08938.27. [DOI] [PubMed] [Google Scholar]
- 31.Lein M, Wirth M, Miller K, Eickenberg HU, Weißbach L, Schmidt K, Haus U, Stephan C, Meissner S, Loening SA, Jung K. Serial Markers of Bone Turnover in Men with Metastatic Prostate Cancer Treated with Zoledronic Acid for Detection of Bone Metastases Progression. Eur Urol. 2007 doi: 10.1016/j.eururo.2007.02.033. Feb; in press. [DOI] [PubMed] [Google Scholar]