

Abstract
Hypertension is a complex condition and is the most common cardiovascular risk factor, contributing to widespread morbidity and mortality. Approximately 90% of hypertension cases are classified as essential hypertension, where the precise cause is unknown. Hypertension is associated with inflammation; however, whether inflammation is a cause or effect of hypertension is not well understood. The purpose of this review is to describe evidence from human and animal studies that inflammation leads to the development of hypertension, as well as the evidence for involvement of oxidative stress and endothelial dysfunction—both thought to be key steps in the development of hypertension. Other potential proinflammatory conditions that contribute to hypertension—such as activation of the sympathetic nervous system, aging, and elevated aldosterone—are also discussed. Finally, we consider the potential benefit of anti-inflammatory drugs and statins for antihypertensive therapy. The evidence reviewed suggests that inflammation can lead to the development of hypertension and that oxidative stress and endothelial dysfunction are involved in the inflammatory cascade. Aging and aldosterone may also both be involved in inflammation and hypertension. Hence, in the absence of serious side effects, anti-inflammatory drugs could potentially be used to treat hypertension in the future.
1. Introduction
Hypertension is the most common cardiovascular risk factor [1] and contributes to widespread morbidity and mortality worldwide [2]. Hypertension is a complex condition, and about 90% of cases are classified as essential hypertension, where the precise cause is unknown [3]. A small minority of hypertensive patients have secondary hypertension, in which a known factor is specifically responsible for raising blood pressure. Many secondary causes of hypertension include primary aldosteronism, obstructive sleep apnea, and renovascular disease [4]. An association between hypertension and inflammation has now been clearly demonstrated; however, it is presently unclear whether inflammation is predominately a cause or effect of hypertension. This brief review will describe evidence that inflammation might lead to the development of hypertension (Figure 1).
Figure 1.
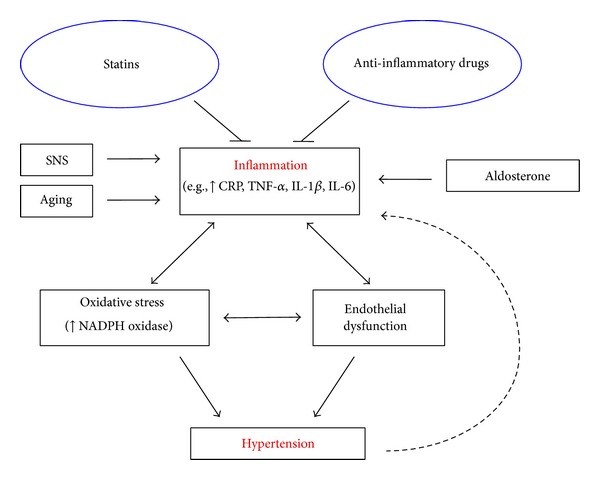
Schematic diagram illustrating the relationship between inflammation and hypertension and the contributing factors involved. Anti-inflammatory drugs and statins may be effective antihypertensive due to their anti-inflammatory properties.
2. Inflammation in Human Hypertension
Current therapies for human hypertension include angiotensin II (Ang II) type 1 receptor (AT1R) inhibitors, angiotensin converting enzyme (ACE) inhibitors, diuretics, calcium channel antagonists, and β-blockers. Treatment with commonly used antihypertensives reduces the risk of total major cardiovascular events, and importantly, it appears that the larger the reduction in blood pressure, the larger the reduction in cardiovascular risk [5]. Insulin resistance contributes causally toward the pathogenesis of hypertension [6, 7]. Indeed, hypertension has been found to be associated with hyperinsulinemia and insulin resistance in humans [8]. Yet, while the above-mentioned therapies successfully lower blood pressure in most individuals, there are a group of patients who are resistant to such treatments. Furthermore, even when blood pressure targets are achieved, many hypertensive patients remain at risk for a cardiovascular event, which may be due to underlying inflammation.
Inflammation is a protective response to injury or infection. It is a complex process that involves inflammatory cells first identifying the affected tissue, leukocyte recruitment into tissue, elimination of the offending agent, and repair of the site of injury. Inflammation requires interactions between cell surfaces, extracellular matrix, and proinflammatory mediators [9]. Excessive inflammation can have detrimental effects and contribute to the progression of chronic and/or prolonged diseases such as atherosclerosis [10], rheumatoid arthritis [11], and systemic lupus erythematosus [12].
The acute phase protein, C-reactive protein (CRP), is involved in innate immune responses and has roles that include activating the complement system and enhancing phagocytosis [13]. CRP can stimulate monocytes to release proinflammatory cytokines such as interleukin-6 (IL-6), interleukin-1 beta (IL-1β), and tumour necrosis factor alpha (TNF-α) [14] and also endothelial cells to express intracellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 [15], effects which will further promote inflammation.
CRP is considered the inflammatory marker with the strongest association with hypertension. It has been demonstrated in numerous clinical trials that hypertensive patients commonly have increased plasma CRP levels [16–21]. Both males and females participated in these studies and the ages of these patients varied from young [16] to middle aged [17]. Prehypertensive patients generally have higher plasma CRP levels than normotensive patients [22], and higher baseline CRP levels are reportedly associated with a higher risk of developing overt hypertension [23–25], consistent with the concept that systemic low-grade inflammation may precede hypertension. Systemic low-grade inflammation can be defined as a 2- to 3-fold increase in plasma levels of cytokines and acute phase proteins [26]. As discussed below, hypertensive patients have been reported to have higher plasma concentrations of proinflammatory cytokines. Nonhypertensive offspring of hypertensive parents tend to have higher serum CRP levels than offspring of nonhypertensive parents [27]. Studies have also demonstrated higher plasma IL-6 [28–30], IL-1β [31, 32], and TNF-α [28, 33, 34] levels in hypertensive patients compared to normotensive patients.
There is also evidence for involvement of immune cells in human hypertension. Patients with hypertensive nephrosclerosis have higher renal infiltration of CD4+ and CD8+ T cells than normotensive control patients [35]. Furthermore, C-X-C chemokine receptor type 3 (CXCR3) chemokines are well-known tissue-homing chemokines for T cells, and circulating levels of CXCR3 chemokines have been reported to be elevated in hypertensive patients [35]. Acquired immunodeficiency syndrome (AIDS) patients have reduced CD4+ T cells and the incidence of hypertension has been reported to be lower in AIDS patients than in HIV-negative participants. The highly active antiretroviral therapy (HAART) which can raise T cell levels increases the incidence of hypertension in AIDS patients similar to HIV-negative participants after treatment for less than 2 years, and the incidence of hypertension in AIDS patients is higher than in HIV-negative participants after treatment for 2 to 5 years [36]. In addition, it is becoming increasingly recognized that both neonatal and childhood health and disease are linked to the prenatal environment. Indeed, infants born following intrauterine inflammation are at increased risk of perinatal morbidity and mortality than infants born to healthy mothers [37–39].
3. Inflammation in Experimental Hypertension
Findings from animal studies have also suggested a role for inflammation in the pathophysiology of hypertension. Spontaneously hypertensive rats (SHR) are a genetic model of essential hypertension that develop hypertension as they age. SHR at 3 weeks of age are not yet hypertensive, yet their kidneys have higher levels of infiltrating lymphocytes and macrophages and activation of nuclear factor-kappaB (NF-κB) than in Wistar Kyoto (WKY) normotensive control rats. These inflammatory changes in the kidneys continue to increase, together with blood pressure, with age in the SHR [40]. Junctional adhesion molecule (JAM)-1 is involved in leukocyte binding to the endothelium and has been found to be upregulated in the brainstem of SHR compared to WKY rats. Overexpression of JAM-1 in WKY rats resulted in elevated systolic blood pressure [41].
Macrophage colony-stimulating factor (m-CSF) acts as a chemotactic factor for monocytes and regulates both effector functions of mature monocytes and macrophages, and production of cytokines. Mice deficient in m-CSF [42], IL-6 [43], TNF-α [44], or interleukin-17 (IL-17) [45] develop a lower blood pressure in response to a hypertensive dose of Ang II compared with control mice. RNA interference knockdown of IL-6 in rats has also been shown to inhibit hypertension [46]. Suppression of NF-κB reportedly inhibits the increase in blood pressure that normally occurs in SHR [47]. Lipopolysaccharide (LPS)- endotoxin from gram-negative bacteria elicits a strong immune response and intraperitoneal injection of LPS is a well-characterised model of systemic inflammation in rodents. In rats, LPS-induced increases in plasma levels of CRP, TNF-α, and IL-1β are associated with an increase in blood pressure [48]. Inhibition of Cox-2 in LPS-treated rats inhibited the increase in blood pressure, suggesting that inflammation in response to LPS treatment contributed to the hypertensive effect [48]. Offspring with prenatal exposure to LPS [49, 50] or IL-6 [51] have higher blood pressure than control offspring.
Immune cells have been implicated to play a role in the development of hypertension. RAG-1 deficient mice, which lack T and B cells, do not develop hypertension in response to Ang II [52], DOCA-salt [52], or chronic stress [53]. Adoptive transfer of T cells but not B cells restored the hypertensive effect in RAG-deficient mice [52]. An analogous effect has been reported to occur in macrophage-depleted mice in response to Ang II infusion [54].
T cells have been demonstrated to express AT1R (the main target receptor of Ang II), ACE, angiotensinogen, renin, and the renin receptor, all important components of the renin angiotensin aldosterone system (RAAS) [55]. The RAAS is a key regulator of blood pressure, and excessive stimulation of this system can cause hypertension. While T cells are capable of producing Ang II [56], which can cause vasoconstriction, stimulate inflammation, and aldosterone production [57], the role of T cell-derived Ang II in such effects is uncertain. There is some evidence that inflammation can overstimulate the RAAS. Inflammatory markers such as CRP [58], TNF-α, and IL-1β [59] have been found to upregulate AT1R. Furthermore, central administration of LPS to rats increases AT1R mRNA expression in the hypothalamus [60].
Given the importance of this recent concept that immune cells are involved in hypertension, we will briefly discuss a hypothesis presented in the literature by Harrison's group regarding how immune cells are activated in hypertension. Hypertensive stimuli, including salt, overactivity of the RAAS, oxidative stress, and inflammation lead to an initial elevation in blood pressure (mainly because of central actions but also due to endogenous hormones such as Ang II and aldosterone), which results in protein modifications. These altered proteins are no longer recognized as self (i.e., they serve as neoantigens), and T cells are activated. T cell-derived signals promote entry of macrophages (and other inflammatory cells) into the vasculature and kidney which results in cytokine release. In the vasculature, activated T cells promote vasoconstriction and remodeling. Together with the promotion of sodium and water retention in the kidney, more severe hypertension can result [61, 62].
4. Inflammation and Endothelial Dysfunction in Hypertension
One potential mechanism by which inflammation may promote hypertension is by causing endothelial dysfunction. The endothelium is a single cell layer that lines the luminal surface of blood vessels and is involved in regulation of vascular tone and structure. Nitric oxide (NO) derived from endothelial nitric oxide synthase (eNOS) is a signalling molecule important in regulating vascular tone. When NO is released from endothelial cells it causes smooth muscle relaxation and subsequent vasodilation (Figure 2) [63]. Endothelial dysfunction may contribute to increased systemic vascular resistance and thus lead to the development of hypertension and is commonly manifested as impaired endothelium-dependent vasodilation due to an imbalance between vasoconstrictors and vasodilators [64]. Inflammation can alter the rates of synthesis and degradation of vasoconstrictors and vasodilators including NO, and impaired NO bioactivity is associated with hypertension. Rats chronically treated with the NO synthase inhibitor, N-nitro-L-arginine methyl ester, have higher blood pressure than controls [65]. Inflammation has been shown to downregulate NO synthase activity. For example, CRP [66] and TNF [67] have both been demonstrated to attenuate NO production by destabilising eNOS mRNA, which reduces NOS protein expression, and inhibition of TNF restores endothelial-dependent vasodilation in humans [68] (Figure 2). IL-17 has been reported to cause endothelial dysfunction by activating Rho-kinase, which leads to phosphorylation of the inhibitory eNOS residue, threonine 495 [69]. Inhibition of eNOS increases vascular tone [70] and consistent with this, Rho-kinase has been shown to contribute to cerebral vascular tone in vivo and this is enhanced during chronic hypertension. In contrast, protein kinase C was not found to contribute to cerebral vascular tone in either normotensive or hypertensive animals [71]. Increased serum CRP levels also correlate with reduced NO bioavailability in coronary artery disease patients [72]. Mice deficient in m-CSF were found to develop less endothelial dysfunction and vascular oxidative stress in response to Ang II compared to wild type mice [42].
Figure 2.
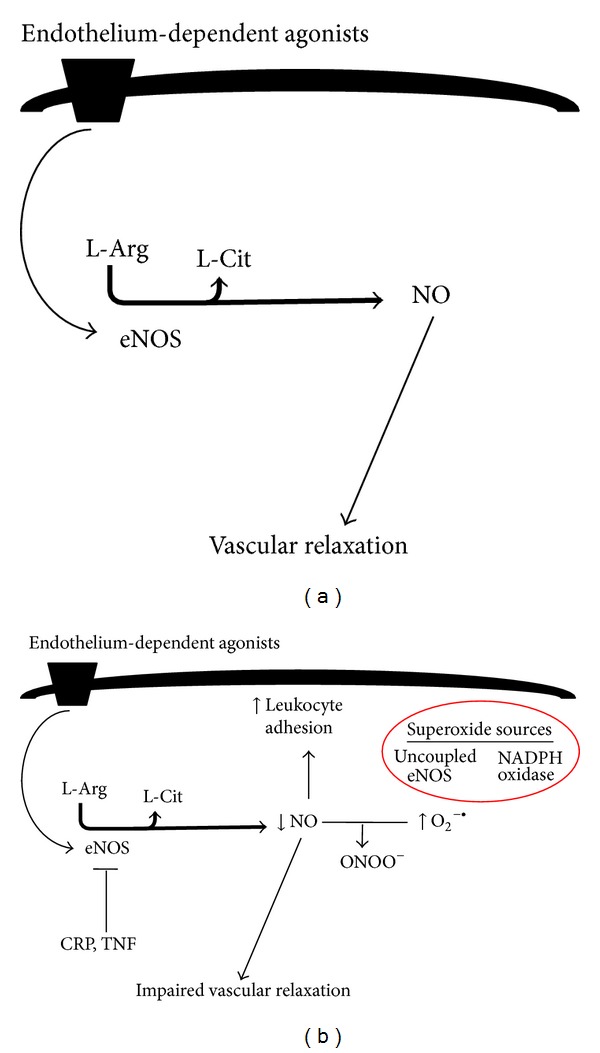
(a) Diagram illustrating endothelium-dependent relaxation during health. (b) Diagram illustrating mechanism by which inflammation and oxidative stress cause endothelial dysfunction. Inflammatory mediators such as CRP and TNF destabilise eNOS mRNA (thus inhibiting eNOS). NO protects endothelium by inhibiting leukocyte adhesion; thus, impaired NO function results in increased leukocyte adhesion. Increased superoxide (derived from NADPH oxidase or uncoupled eNOS) impairs NO bioavailability and leads to impaired vascular relaxation. The figure is based on text.
Importantly, normal healthy endothelium exerts anti-inflammatory effects such as NO-dependent inhibition of leukocyte adhesion [73]. Inhibition of eNOS activity augments expression of leukocyte adhesion molecules and chemokines such as monocyte chemotactic protein 1 (MCP-1) [74, 75]. Furthermore, gene therapy to augment vascular NO synthase activity attenuates hypercholesterolemia-induced leukocyte adhesion molecule expression and monocyte infiltration [76]. Therefore, endothelial dysfunction can further exacerbate vascular inflammation, which may in turn contribute to the development of hypertension.
5. Inflammation and Oxidative Stress in Hypertension
Chronic inflammation can also trigger oxidative stress, which has been associated with hypertension [77]. As mentioned, inflammation is the primary immune response to eliminate pathogens or to repair tissue damage. Innate immune cells, such as neutrophils and macrophages, produce reactive oxygen species (ROS) such as superoxide and hydrogen peroxide in order to kill pathogens [77]. Nicotinamide adenine dinucleotide phosphate-oxidase (NADPH) oxidase is a major source of ROS in immune cells and also in the vasculature [78]. Inflammatory processes continue until the pathogens are destroyed or the tissue repair process has been completed. However, sustained inflammation can lead to an overproduction of ROS. Oxidative stress (defined as an imbalance between the production and breakdown of ROS) is a major cause of endothelial dysfunction, primarily through reducing NO bioavailability via the direct chemical reaction of superoxide with NO, resulting in the formation of peroxynitrite [79]. The reaction between superoxide and NO is faster [80] than the breakdown of superoxide via superoxide dismutase [81]. Furthermore, peroxynitrite formation may result in further impairment of NO levels and enhanced oxidative stress by inhibiting eNOS activity through oxidation of 4-tetrahydrobiopterin (BH4), a cofactor of eNOS. This leads to eNOS uncoupling, where eNOS produces superoxide instead of NO [82]. Excessive ROS levels can also induce cellular damage by interacting with DNA, lipids, and proteins [83], which may further impair vascular structure and function. Immune cells such as T cells, macrophages, and neutrophils express NADPH oxidase subunits and produce ROS. In the setting of Ang II-induced hypertension, T cells express higher levels of p47phox, p22phox, and NOX2, components of NOX2 oxidase. Furthermore, adoptive transfer of T cells deficient in NADPH oxidase results in lower superoxide production and hypertension in response to Ang II [52]. Oxidative stress can promote inflammatory processes by activating transcription factors such as NF-κB [84]. CRP levels have been shown to correlate with the level of oxidative stress in inflammatory cells from hypertensive patients [85].
The kidney is an important organ involved in regulating blood pressure, and chronic kidney disease is one of the most common causes of secondary hypertension [86]. Elevated renal oxidative stress can be seen in the early stages of chronic kidney disease [87], and inflammation [88] and oxidative stress [87] increase as renal dysfunction progresses. Prehypertensive SHR from 2-3 weeks of age have elevated renal inflammation and oxidative stress compared to age-matched WKY rats [89]. Renal artery stenosis results in reduced renal perfusion and pressure at the afferent arteriole, thus stimulating the release of renin and, hence, activation of the RAAS [90]. Renal artery stenosis can cause renovascular hypertension, a secondary form of hypertension due to kidney dysfunction. Indeed, reduced blood flow to the kidney decreases renal function and can lead to chronic kidney disease [91]. Renal artery stenosis is the most common primary disease of renal arteries, and up to 90% of cases are caused by atherosclerosis [92], a chronic inflammatory disease that predominately occurs in large arteries, and oxidative stress plays a major role in its development [93].
Overall, with oxidative stress being able to act as a key trigger of both inflammation and hypertension, it remains unclear whether inflammation is predominately a cause or effect of hypertension, with evidence to support either scenario in a likely vicious cycle.
6. The Sympathetic Nervous System and Inflammation in Hypertension
Sympathetic nervous system (SNS) activation is a common feature of hypertension and can contribute to the development of hypertension [94]. Essential hypertension patients are reported to have increased renal sympathetic outflow [95]. Autonomic dysfunction is characterised by increased sympathetic and decreased parasympathetic activity, and the SHR has been shown to be a good rodent model of human autonomic dysfunction [96]. The SNS innervates primary and secondary lymphoid organs and most immune cells express receptors for catecholamines such as noradrenaline [97]. The SNS can enhance inflammatory responses. For example, deletion of extracellular superoxide dismutase in the circumventricular organs of mice increased sympathetic outflow, modestly elevated blood pressure and increased T cell activation [98]. Renal sympathetic nerves have been suggested to play a role in kidney inflammation. Rats that have undergone renal sympathetic denervation have reduced renal macrophage levels and cortical TNF expression [99]. Neuroinflammation has been associated with increased sympathetic drive during cardiovascular disease. Inflammation may also promote SNS activation. Central administration of LPS to rats is reported to increase renal sympathetic drive and blood pressure [60]. Blood-borne pro-inflammatory cytokines may also act on receptors in the microvasculature of the brain to induce Cox-2 activity and the production of prostaglandins which penetrate the blood brain barrier to activate the SNS [100]. Overall, these studies suggest a potentially important link between the SNS and inflammation in the development of hypertension.
Increased sympathetic drive to the kidneys causes the release of renin and subsequently raises blood pressure [101]. Catheter-based renal denervation is a promising therapeutic approach to treat resistant hypertension [102]. Catheter-based renal denervation involves the application of radiofrequency energy in short bursts along the main renal arteries to disrupt the renal nerves. However, the recent SYMPLICITY HTN-3 clinical trial has reported that renal denervation does not result in a significant reduction in systolic blood pressure in resistant hypertensive patients with systolic blood pressure readings above 160 mmHg when compared to sham control [103]. The previous SYMPLICITY HTN clinical trials [102, 104] had reported that renal denervation can cause large reductions in blood pressure; however, these clinical trials were unblinded, had small sample sizes, and lacked sham controls as opposed to SYMPLICITY HTN-3. This does not necessarily mean that renal denervation is ineffective in lowering blood pressure as SYMPLICITY HTN-3 used the Medtronic catheter and there are other catheters available; hence, future clinical trials could study the use of other catheters to lower blood pressure. Currently there is a clinical trial examining Covidien's OneShot device [105].
7. Aging and Chronic Inflammation
More than half of the elderly (above 65 years of age) have hypertension [106], and the prevalence of hypertension increases with age [107]. Secondary causes of hypertension such as obstructive sleep apnea, chronic kidney disease, and renal artery stenosis, which are all associated with inflammation, are highly prevalent in the elderly [108]. Chronic low-grade inflammation commonly occurs with aging and this has been termed “inflammaging” [109]. Inflammaging is characterised by an imbalance of proinflammatory markers and anti-inflammatory markers. Levels of proinflammatory markers such as IL-6, TNF-α, and CRP are elevated, while anti-inflammatory cytokines such as interleukin-10 are reduced [110]. Inflammaging is believed to be caused by continuous lifelong exposure to antigens, due either to infection and/or nonpathogenic antigens.
8. Aldosterone and Inflammation
Patients with primary aldosteronism have elevated aldosterone levels, and as mentioned earlier, primary aldosteronism is a common secondary cause of hypertension. More than 10% of hypertensive patients have raised aldosterone levels [111], and drugs that block the mineralocorticoid receptor (MR), the main target receptor of aldosterone, are used to treat hypertension that is resistant to ACE inhibition and AT1R antagonism [112]. Aldosterone is involved in the RAAS whereby a fall in blood pressure under physiological conditions leads to Ang II generation which, through its action on the AT1R in the adrenal zona glomerulosa, stimulates the release of the mineralocorticoid, aldosterone. Aldosterone activates the MR in the distal renal tubule of the kidney to increase sodium and water retention, and potassium excretion, leading to an increase in blood volume and thus blood pressure [113]. Actions of aldosterone were, until recently, believed to be restricted to the kidney, but it is now understood that aldosterone can target other tissues relevant to blood pressure control, including the brain [114], vasculature [115], and heart [116].
Aldosterone has been reported to exert proinflammatory effects that appear to be MR-mediated. Administration of exogenous aldosterone to experimental animals results in elevated levels of ICAM-1, MCP-1, and TNF-α in coronary arteries [117], and increased vascular infiltration of macrophages and lymphocytes [118]. In the heart, aldosterone can increase vascular expression of ICAM-1, MCP-1, osteopontin, and COX-2, which can be blocked by the MR antagonist, eplerenone [116], indicating involvement of the MR. Proinflammatory effects of aldosterone have also been reported to occur in the kidney, where aldosterone and salt treatment caused MR-dependent leukocyte infiltration and elevation of osteopontin, IL-6, IL-1β, and MCP-1 levels [119]. Association between aldosterone and inflammation has been reported in essential hypertensive patients, where high plasma aldosterone levels were correlated with high levels of circulating CRP and leukocytes [120].
9. Anti-Inflammatory Drugs and Hypertension
Currently, anti-inflammatory drugs are not used to treat hypertension. Of the different classes of anti-inflammatory agents, immunosuppressant drugs could potentially be used to treat hypertension. Mycophenolate mofetil, which blocks T cell and B cell proliferation by inhibiting inosine monophosphate dehydrogenase, has been demonstrated to reduce hypertension in SHR [121], in Dahl salt-sensitive rats (another rodent model of hypertension) [122, 123], and in psoriasis and rheumatoid arthritis patients [124]. Another immunosuppressant, tacrolimus, a calcineurin inhibitor which blocks T cell activation, is reported to reduce hypertension in Dahl salt-sensitive rats [125]. Furthermore, chronic kidney disease patients with hypertension and who are on immunosuppressant drugs were found to require less antihypertensive medication than patients who were not taking immunosuppressant drugs [126]. Based on these studies, T cells may be a potential target in treating hypertension. T cells are an important component of the immune system and are involved in various aspects such as regulating immune responses by secreting anti- and proinflammatory cytokines. Ang II and aldosterone have proinflammatory effects; hence, targeting the RAAS could also target inflammation in hypertension. Ang II has been demonstrated to stimulate T cell proliferation and T cells deficient in the AT1R proliferate much less than T cells from wild type mice [127]. Vaccines that target the RAAS are currently in development to treat hypertension. The CYT006-AngGb vaccine which targets Ang II has been reported to reduce blood pressure in patients with mild to moderate hypertension with no serious safety issues in a phase IIa clinical trial [128]. Recently, the ATRQβ-001 vaccine which targets the AT1R has been found to be successful in lowering blood pressure in Ang II-induced hypertensive mice and SHR [129]. However, further studies are required to demonstrate the long-term safety of vaccines which target the RAAS, as one of the earlier vaccines developed which targeted renin was found to cause fatal autoimmunity [130].
Immunosuppressants can have serious side effects [131]; hence, their potential use to treat hypertension should be carefully considered and is not currently justified. Moreover, it is noteworthy that nonsteroidal anti-inflammatory drugs can increase blood pressure by causing sodium retention [132]. The use of immunosuppressants to treat treatment-resistant hypertension could possibly be justified. Treatment-resistant hypertension is defined as blood pressure that is above the patient's goal, despite treatment with at least three different classes of antihypertensive drugs, including a diuretic, at optimal doses. Patients with controlled blood pressure, and taking at least four antihypertensive medications, are also considered to be treatment-resistant [133]. It is difficult to determine the exact prevalence of treatment-resistant hypertension, but clinical trials have suggested that this may include up to 30% of hypertensive patients [134]. Currently, there are few therapeutic options available to treat resistant hypertension.
Statins were developed to inhibit cholesterol synthesis by blocking HMG-CoA reductase, but these drugs may also have anti-inflammatory effects and can cause a small reduction in systolic blood pressure in hypercholesterolemic patients, an effect that is greater in patients with a higher baseline blood pressure [135]. Statins can reduce levels of proinflammatory cytokines such as IL-1, IL-6, TNF- α, ICAM-1, and CRP [136]. Even in participants without hyperlipidemia, rosuvastatin has been shown to reduce CRP levels in association with a lower incidence of cardiovascular events [137]. The anti-inflammatory effects of statins may occur through the mevalonate pathway (i.e., HMG-CoA reductase pathway), which is responsible for cholesterol synthesis [138]. Pitavastatin has been shown to reduce IL-6 and IL-8 secretions and mRNA expression of IL-6, IL-8, and granulocyte macrophage colony-stimulating factor (GM-CSF) in LPS-stimulated human bronchial epithelium cells. These effects were inhibited by treatment with mevalonate, the initial product of HMG-CoA reductase [139]. The anti-inflammatory effects of statins may also occur through NO. Statins have been shown to enhance mRNA and protein expression of eNOS in human endothelial cells by inhibiting Rho-kinase geranylgeranyl-phosphorylation [140], and as discussed earlier NO has protective anti-inflammatory effects. Statins may also improve eNOS coupling by reducing plasma asymmetrical dimethylarginine (ADMA) [141], as ADMA has been associated with eNOS uncoupling [142].
10. Conclusion
It is unclear whether inflammation is a cause or effect of hypertension, but as discussed in this review there is evidence from human and animal studies suggesting that inflammation can lead to the development of hypertension. Oxidative stress and endothelial dysfunction are known to be associated with inflammation and can contribute to hypertension, at least in part, by exacerbating the inflammatory response. Other factors that contribute to hypertension such as SNS activation, aging, or aldosterone are also associated with inflammation (summarized in Figure 1). Future studies should focus on whether anti-inflammatory drugs are beneficial in reversing hypertension, oxidative stress, and endothelial dysfunction in experimental models of hypertension. Hence, in the absence of serious side effects, anti-inflammatory drugs could potentially be used not only to treat hypertension in the future but also to treat other cardiovascular diseases by minimizing the impact of oxidative stress and endothelial dysfunction.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Ventura HO, Taler SJ, Strobeck JE. Hypertension as a hemodynamic disease: the role of impedance cardiography in diagnostic, prognostic, and therapeutic decision making. The American Journal of Hypertension. 2005;18(2):26S–43S. doi: 10.1016/j.amjhyper.2004.11.002. [DOI] [PubMed] [Google Scholar]
- 2.Kearney PM, Whelton M, Reynolds K, Muntner P, Whelton PK, He J. Global burden of hypertension: Analysis of worldwide data. The Lancet. 2005;365(9455):217–223. doi: 10.1016/S0140-6736(05)17741-1. [DOI] [PubMed] [Google Scholar]
- 3.Messerli FH, Williams B, Ritz E. Essential hypertension. The Lancet. 2007;370(9587):591–603. doi: 10.1016/S0140-6736(07)61299-9. [DOI] [PubMed] [Google Scholar]
- 4.Onusko E. Diagnosing secondary hypertension. The American Family Physician. 2003;67(1):67–74. [PubMed] [Google Scholar]
- 5.Turnbull F. Effects of different blood-pressure-lowering regimens on major cardiovascular events: results of prospectively-designed overviews of randomised trials. The Lancet. 2003;362(9395):1527–1535. doi: 10.1016/s0140-6736(03)14739-3. [DOI] [PubMed] [Google Scholar]
- 6.Bhanot S, McNeill JH. Insulin and hypertension: a causal relationship? Cardiovascular Research. 1996;31(2):212–221. [PubMed] [Google Scholar]
- 7.Tran LT, Yuen VG, McNeill JH. The fructose-fed rat: a review on the mechanisms of fructose-induced insulin resistance and hypertension. Molecular and Cellular Biochemistry. 2009;332(1-2):145–159. doi: 10.1007/s11010-009-0184-4. [DOI] [PubMed] [Google Scholar]
- 8.DeFronzo RA, Ferrannini E. Insulin resistance: a multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care. 1991;14(3):173–194. doi: 10.2337/diacare.14.3.173. [DOI] [PubMed] [Google Scholar]
- 9.Keane MP, Strieter RM. Chemokine signaling in inflammation. Critical Care Medicine. 2000;28(4):N13–N26. doi: 10.1097/00003246-200004001-00003. [DOI] [PubMed] [Google Scholar]
- 10.Lusis AJ. Atherosclerosis. Nature. 2000;407(6801):233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sweeney SE, Firestein GS. Rheumatoid arthritis: regulation of synovial inflammation. International Journal of Biochemistry and Cell Biology. 2004;36(3):372–378. doi: 10.1016/s1357-2725(03)00259-0. [DOI] [PubMed] [Google Scholar]
- 12.Asanuma YF. Accelerated atherosclerosis and inflammation in systemic lupus erythematosus. Japanese Journal of Clinical Immunology. 2012;35(6):470–480. doi: 10.2177/jsci.35.470. [DOI] [PubMed] [Google Scholar]
- 13.Mortensen RF, Zhong W. Regulation of phagocytic leukocyte activities by C-reactive protein. Journal of Leukocyte Biology. 2000;67(4):495–500. doi: 10.1002/jlb.67.4.495. [DOI] [PubMed] [Google Scholar]
- 14.Ballou SP, Lozanski G. Induction of inflammatory cytokine release from cultured human monocytes by C-reactive protein. Cytokine. 1992;4(5):361–368. doi: 10.1016/1043-4666(92)90079-7. [DOI] [PubMed] [Google Scholar]
- 15.Pasceri V, Willerson JT, Yeh ETH. Direct proinflammatory effect of C-reactive protein on human endothelial cells. Circulation. 2000;102(18):2165–2168. doi: 10.1161/01.cir.102.18.2165. [DOI] [PubMed] [Google Scholar]
- 16.Stumpf C, Jukic J, Yilmaz A, et al. Elevated VEGF-plasma levels in young patients with mild essential hypertension. European Journal of Clinical Investigation. 2009;39(1):31–36. doi: 10.1111/j.1365-2362.2008.02056.x. [DOI] [PubMed] [Google Scholar]
- 17.Bautista LE, Atwood JE, O'Malley PG, Taylor AJ. Association between C-reactive protein and hypertension in healthy middle-aged men and women. Coronary Artery Disease. 2004;15(6):331–336. doi: 10.1097/00019501-200409000-00006. [DOI] [PubMed] [Google Scholar]
- 18.Blake GJ, Rifai N, Buring JE, Ridker PM. Blood pressure, C-reactive protein, and risk of future cardiovascular events. Circulation. 2003;108(24):2993–2999. doi: 10.1161/01.CIR.0000104566.10178.AF. [DOI] [PubMed] [Google Scholar]
- 19.Stuveling EM, Bakker SJL, Hillege HL, et al. C-reactive protein modifies the relationship between blood pressure and microalbuminuria. Hypertension. 2004;43(4):791–796. doi: 10.1161/01.HYP.0000120125.08867.42. [DOI] [PubMed] [Google Scholar]
- 20.Sung KC, Suh JY, Kim BS, et al. High sensitivity C-reactive protein as an independent risk factor for essential hypertension. American Journal of Hypertension. 2003;16(6):429–433. doi: 10.1016/s0895-7061(03)00566-1. [DOI] [PubMed] [Google Scholar]
- 21.Xu T, Ju Z, Tong W, et al. Relationship of C-reactive protein with hypertension and interactions between increased C-reactive protein and other risk factors on hypertension in Mongolian people, China. Circulation Journal. 2008;72(8):1324–1328. doi: 10.1253/circj.72.1324. [DOI] [PubMed] [Google Scholar]
- 22.Chrysohoou C, Pitsavos C, Panagiotakos DB, Skoumas J, Stefanadis C. Association between prehypertension status and inflammatory markers related to atherosclerotic disease: the ATTICA Study. The American Journal of Hypertension. 2004;17(7):568–573. doi: 10.1016/j.amjhyper.2004.03.675. [DOI] [PubMed] [Google Scholar]
- 23.Sesso HD, Buring JE, Rifai N, Blake GJ, Gaziano JM, Ridker PM. C-reactive protein and the risk of developing hypertension. Journal of the American Medical Association. 2003;290(22):2945–2951. doi: 10.1001/jama.290.22.2945. [DOI] [PubMed] [Google Scholar]
- 24.Dauphinot V, Roche F, Kossovsky MP, et al. C-reactive protein implications in new-onset hypertension in a healthy population initially aged 65 years: the Proof study. Journal of Hypertension. 2009;27(4):736–743. doi: 10.1097/HJH.0b013e328326f801. [DOI] [PubMed] [Google Scholar]
- 25.Niskanen L, Laaksonen DE, Nyyssönen K, et al. Inflammation, abdominal obesity, and smoking as predictors of hypertension. Hypertension. 2004;44(6):859–865. doi: 10.1161/01.HYP.0000146691.51307.84. [DOI] [PubMed] [Google Scholar]
- 26.Tkacova R. Systemic inflammation in chronic obstructive pulmonary disease: may adipose tissue play a role? Review of the literature and future perspectives. Mediators of Inflammation. 2010;2010 doi: 10.1155/2010/585989.585989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lieb W, Pencina MJ, Wang TJ, et al. Association of parental hypertension with concentrations of select biomarkers in nonhypertensive offspring. Hypertension. 2008;52(2):381–386. doi: 10.1161/HYPERTENSIONAHA.108.113589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension. Journal of Human Hypertension. 2005;19(2):149–154. doi: 10.1038/sj.jhh.1001785. [DOI] [PubMed] [Google Scholar]
- 29.Chae CU, Lee RT, Rifai N, Ridker PM. Blood pressure and inflammation in apparently healthy men. Hypertension. 2001;38(3):399–403. doi: 10.1161/01.hyp.38.3.399. [DOI] [PubMed] [Google Scholar]
- 30.Fernandez-Real J-M, Vayreda M, Richart C, et al. Circulating interleukin 6 levels, blood pressure, and insulin sensitivity in apparently healthy men and women. The Journal of Clinical Endocrinology & Metabolism. 2001;86(3):1154–1159. doi: 10.1210/jcem.86.3.7305. [DOI] [PubMed] [Google Scholar]
- 31.Zhao S, Li Q, Liu L, Xu Z, Xiao J. Simvastatin reduces interleukin-1beta secretion by peripheral blood mononuclear cells in patients with essential hypertension. Clinica Chimica Acta. 2004;344(1-2):195–200. doi: 10.1016/j.cccn.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 32.Dalekos GN, Elisaf M, Bairaktari E, Tsolas O, Siamopoulos KC. Increased serum levels of interleukin-1β in the systemic circulation of patients with essential hypertension: additional risk factor for atherogenesis in hypertensive patients? Journal of Laboratory and Clinical Medicine. 1997;129(3):300–308. doi: 10.1016/s0022-2143(97)90178-5. [DOI] [PubMed] [Google Scholar]
- 33.Yu X, Yang Z, Yu M. Correlation of tumor necrosis factor alpha and interleukin 6 with hypertensive renal damage. Renal Failure. 2010;32(4):475–479. doi: 10.3109/08860221003664280. [DOI] [PubMed] [Google Scholar]
- 34.Furumoto T, Saito N, Dong J, Mikami T, Fujii S, Kitabatake A. Association of cardiovascular risk factors and endothelial dysfunction in Japanese hypertensive patients: implications for early atherosclerosis. Hypertension Research. 2002;25(3):475–480. doi: 10.1291/hypres.25.475. [DOI] [PubMed] [Google Scholar]
- 35.Youn J, Yu HT, Lim BJ, et al. Immunosenescent CD8+ T clls and C-X-C cemokine receptor type 3 chemokines are increased in human hypertension. Hypertension. 2013;62(1):126–133. doi: 10.1161/HYPERTENSIONAHA.113.00689. [DOI] [PubMed] [Google Scholar]
- 36.Seaberg EC, Muñoz A, Lu M, et al. Association between highly active antiretroviral therapy and hypertension in a large cohort of men followed from 1984 to 2003. AIDS. 2005;19(9):953–960. doi: 10.1097/01.aids.0000171410.76607.f8. [DOI] [PubMed] [Google Scholar]
- 37.Hodyl NA, Stark MJ, Osei-Kumah A, Clifton VL. Prenatal programming of the innate immune response following in utero exposure to inflammation: a sexually dimorphic process? Expert Review of Clinical Immunology. 2011;7(5):579–592. doi: 10.1586/eci.11.51. [DOI] [PubMed] [Google Scholar]
- 38.Msall ME, Buck GM, Rogers BT, et al. Multivariate risks among extremely premature infants. Journal of Perinatology. 1994;14(1):41–47. [PubMed] [Google Scholar]
- 39.Mwanyumba F, Inion I, Gaillard P, Mandaliya K, Praet M, Temmerman M. Placental inflammation and perinatal outcome. European Journal of Obstetrics Gynecology and Reproductive Biology. 2003;108(2):164–170. doi: 10.1016/s0301-2115(02)00438-4. [DOI] [PubMed] [Google Scholar]
- 40.Rodríguez-Iturbe B, Quiroz Y, Ferrebuz A, Parra G, Vaziri ND. Evolution of renal interstitial inflammation and NF-κB activation in spontaneously hypertensive rats. American Journal of Nephrology. 2004;24(6):587–594. doi: 10.1159/000082313. [DOI] [PubMed] [Google Scholar]
- 41.Waki H, Liu B, Miyake M, et al. Junctional adhesion molecule-1 is upregulated in spontaneously hypertensive rats: evidence for a prohypertensive role within the brain stem. Hypertension. 2007;49(6):1321–1327. doi: 10.1161/HYPERTENSIONAHA.106.085589. [DOI] [PubMed] [Google Scholar]
- 42.De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL. Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II-infused macrophage colony-stimulating factor-deficient mice: evidence for a role in inflammation in angiotensin-induced vascular injury. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25(10):2106–2113. doi: 10.1161/01.ATV.0000181743.28028.57. [DOI] [PubMed] [Google Scholar]
- 43.Zhang W, Wang W, Yu H, et al. Interleukin 6 underlies angiotensin II-induced hypertension and chronic renal damage. Hypertension. 2012;59(1):136–144. doi: 10.1161/HYPERTENSIONAHA.111.173328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sriramula S, Haque M, Majid DSA, Francis J. Involvement of tumor necrosis factor-α in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension. 2008;51(5):1345–1351. doi: 10.1161/HYPERTENSIONAHA.107.102152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Madhur MS, Lob HE, McCann LA, et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010;55(2):500–507. doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crosswhite P, Sun Z. Ribonucleic acid interference knockdown of interleukin 6 attenuates cold-induced hypertension. Hypertension. 2010;55(6):1484–1491. doi: 10.1161/HYPERTENSIONAHA.109.146902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rodríguez-Iturbe B, Ferrebuz A, Vanegas V, Quiroz Y, Mezzano S, Vaziri ND. Early and sustained inhibition of nuclear factor-κB prevents hypertension in spontaneously hypertensive rats. Journal of Pharmacology and Experimental Therapeutics. 2005;315(1):51–57. doi: 10.1124/jpet.105.088062. [DOI] [PubMed] [Google Scholar]
- 48.Wu KLH, Chan SHH, Chan JYH. Neuroinflammation and oxidative stress in rostral ventrolateral medulla contribute to neurogenic hypertension induced by systemic inflammation. Journal of Neuroinflammation. 2012;9(1, article 212) doi: 10.1186/1742-2094-9-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wei YL, Li XH, Zhou JZ. Prenatal exposure to lipopolysaccharide results in increases in blood pressure and body weight in rats. Acta Pharmacologica Sinica. 2007;28(5):651–656. doi: 10.1111/j.1745-7254.2007.00593.x. [DOI] [PubMed] [Google Scholar]
- 50.Hao X-Q, Zhang H-G, Yuan Z-B, Yang D, Hao L, Li X. Prenatal exposure to lipopolysaccharide alters the intrarenal renin-angiotensin system and renal damage in offspring rats. Hypertension Research. 2010;33(1):76–82. doi: 10.1038/hr.2009.185. [DOI] [PubMed] [Google Scholar]
- 51.Samuelsson A, Öhrn I, Dahlgren J, et al. Prenatal exposure to interleukin-6 results in hypertension and increased hypothalamic-pituitary-adrenal axis activity in adult rats. Endocrinology. 2004;145(11):4897–4911. doi: 10.1210/en.2004-0742. [DOI] [PubMed] [Google Scholar]
- 52.Guzik TJ, Hoch NE, Brown KA, et al. Role of the T cell in the genesis of angiotensin II-induced hypertension and vascular dysfunction. Journal of Experimental Medicine. 2007;204(10):2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marvar PJ, Vinh A, Thabet S, et al. T lymphocytes and vascular inflammation contribute to stress-dependent hypertension. Biological Psychiatry. 2012;71(9):774–782. doi: 10.1016/j.biopsych.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wenzel P, Knorr M, Kossmann S, et al. Lysozyme M-positive monocytes mediate angiotensin ii-induced arterial hypertension and vascular dysfunction. Circulation. 2011;124(12):1370–1381. doi: 10.1161/CIRCULATIONAHA.111.034470. [DOI] [PubMed] [Google Scholar]
- 55.Jurewicz M, McDermott DH, Sechler JM, et al. Human T and natural killer cells possess a functional renin-angiotensin system: further mechanisms of angiotensin II-induced inflammation. Journal of the American Society of Nephrology. 2007;18(4):1093–1102. doi: 10.1681/ASN.2006070707. [DOI] [PubMed] [Google Scholar]
- 56.Hoch NE, Guzik TJ, Chen W, et al. Regulation of T-cell function by endogenously produced angiotensin II. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology. 2009;296(2):R208–R216. doi: 10.1152/ajpregu.90521.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim S, Iwao H. Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacological Reviews. 2000;52(1):11–34. [PubMed] [Google Scholar]
- 58.Wang C, Li S, Weisel RD, et al. C-reactive protein upregulates angiotensin type 1 receptors in vascular smooth muscle. Circulation. 2003;107(13):1783–1790. doi: 10.1161/01.CIR.0000061916.95736.E5. [DOI] [PubMed] [Google Scholar]
- 59.Gurantz D, Cowling RT, Varki N, Frikovsky E, Moore CD, Greenberg BH. IL-1β and TNF-α upregulate angiotensin II type 1 (AT 1) receptors on cardiac fibroblasts and are associated with increased AT1 density in the post-MI heart. Journal of Molecular and Cellular Cardiology. 2005;38(3):505–515. doi: 10.1016/j.yjmcc.2004.12.015. [DOI] [PubMed] [Google Scholar]
- 60.Zhang ZH, Yu Y, Wei S, Felder RB. Centrally administered lipopolysaccharide elicits sympathetic excitation via NAD(P)H oxidase-dependent mitogen-activated protein kinase signaling. Journal of Hypertension. 2010;28(4):806–816. doi: 10.1097/HJH.0b013e3283358b6e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trott DW, Harrison DG. The immune system in hypertension. Advances in Physiology Education. 2014;38(1):20–24. doi: 10.1152/advan.00063.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harrison DG, Guzik TJ, Lob HE, et al. Inflammation, immunity, and hypertension. Hypertension. 2011;57(2):132–140. doi: 10.1161/HYPERTENSIONAHA.110.163576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chrissobolis S, Miller AA, Drummond GR, Kemp-Harper BK, Sobey CG. Oxidative stress and endothelial dysfunction in cerebrovascular disease. Frontiers in Bioscience. 2011;16(5):1733–1745. doi: 10.2741/3816. [DOI] [PubMed] [Google Scholar]
- 64.Chrissobolis S, Faraci FM. The role of oxidative stress and NADPH oxidase in cerebrovascular disease. Trends in Molecular Medicine. 2008;14(11):495–502. doi: 10.1016/j.molmed.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Baylis C, Mitruka B, Deng A. Chronic blockade of nitric oxide synthesis in the rat produces systemic hypertension and glomerular damage. Journal of Clinical Investigation. 1992;90(1):278–281. doi: 10.1172/JCI115849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Verma S, Wang C, Li S, et al. A self-fulfilling prophecy: C-reactive protein attenuates nitric oxide production and inhibits angiogenesis. Circulation. 2002;106(8):913–919. doi: 10.1161/01.cir.0000029802.88087.5e. [DOI] [PubMed] [Google Scholar]
- 67.Yan G, You B, Chen S, Liao JK, Sun J. Tumor necrosis factor-α downregulates endothelial nitric oxide synthase mRNA stability via translation elongation factor 1-α 1. Circulation Research. 2008;103(6):591–597. doi: 10.1161/CIRCRESAHA.108.173963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mäki-Petäjä KM, Hall FC, Booth AD, et al. Rheumatoid arthritis is associated with increased aortic pulse-wave velocity, which is reduced by anti-tumor necrosis factor-α therapy. Circulation. 2006;114(11):1185–1192. doi: 10.1161/CIRCULATIONAHA.105.601641. [DOI] [PubMed] [Google Scholar]
- 69.Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovascular Research. 2013;97(4):696–704. doi: 10.1093/cvr/cvs422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fleming I, Bauersachs J, Schafer A, Scholz D, Aldershvile J, Busse R. Isometric contraction induces the Ca2+-independent activation of the endothelial nitric oxidesynthase. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(3):1123–1128. doi: 10.1073/pnas.96.3.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chrissobolis S, Sobey CG. Evidence that Rho-kinase activity contributes to cerebral vascular tone in vivo and is enhanced during chronic hypertension: comparison with protein kinase C. Circulation Research. 2001;88(8):774–779. doi: 10.1161/hh0801.090441. [DOI] [PubMed] [Google Scholar]
- 72.Fichtlscherer S, Breuer S, Schächinger V, Dimmeler S, Zeiher AM. C-reactive protein levels determine systemic nitric oxide bioavailability in patients with coronary artery disease. European Heart Journal. 2004;25(16):1412–1418. doi: 10.1016/j.ehj.2004.06.026. [DOI] [PubMed] [Google Scholar]
- 73.Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(11):4651–4655. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zeiher AM, Fisslthaler B, Schray-Utz B, Busse R. Nitric oxide modulates the expression of monocyte chemoattractant protein 1 in cultured human endothelial cells. Circulation Research. 1995;76(6):980–986. doi: 10.1161/01.res.76.6.980. [DOI] [PubMed] [Google Scholar]
- 75.Tomita H, Egashira K, Kubo-Inoue M, et al. Inhibition of NO synthesis induces inflammatory changes and monocyte chemoattractant protein-1 expression in rat hearts and vessels. Arteriosclerosis, Thrombosis, and Vascular Biology. 1998;18(9):1456–1464. doi: 10.1161/01.atv.18.9.1456. [DOI] [PubMed] [Google Scholar]
- 76.Qian H, Neplioueva V, Shetty GA, Channon KM, George SE. Nitric oxide synthase gene therapy rapidly reduces adhesion molecule expression and inflammatory cell infiltration in carotid arteries of cholesterol-fed rabbits. Circulation. 1999;99(23):2979–2982. doi: 10.1161/01.cir.99.23.2979. [DOI] [PubMed] [Google Scholar]
- 77.Crowley SD. The cooperative roles of inflammation and oxidative stress in the pathogenesis of hypertension. Antioxidants & Redox Signaling. 2014;20(1):102–120. doi: 10.1089/ars.2013.5258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nature Reviews Drug Discovery. 2011;10(6):453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Touyz RM. Reactive oxygen species and angiotensin II signaling in vascular cells—implications in cardiovascular disease. Brazilian Journal of Medical and Biological Research. 2004;37(8):1263–1273. doi: 10.1590/s0100-879x2004000800018. [DOI] [PubMed] [Google Scholar]
- 80.Huie RE, Padmaja S. The reaction of no with superoxide. Free Radical Research Communications. 1993;18(4):195–199. doi: 10.3109/10715769309145868. [DOI] [PubMed] [Google Scholar]
- 81.Cudd A, Fridovich I. Electrostatic interactions in the reaction mechanism of bovine erythrocyte superoxide dismutase. Journal of Biological Chemistry. 1982;257(19):11443–11447. [PubMed] [Google Scholar]
- 82.Vásquez-Vivar J, Kalyanaraman B, Martásek P, et al. Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(16):9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hassoun EA, Li F, Abushaban A, Stohs SJ. Production of superoxide anion, lipid peroxidation and DNA damage in the hepatic and brain tissues of rats after subchronic exposure to mixtures of TCDD and its congeners. Journal of Applied Toxicology. 2001;21(3):211–219. doi: 10.1002/jat.744. [DOI] [PubMed] [Google Scholar]
- 84.Canty TG, Jr., Boyle EM, Jr., Farr A, Morgan EN, Verrier ED, Pohlman TH. Oxidative stress induces NF-κB nuclear translocation without degradation of IκBα. Circulation. Circulation. 1999;100(19):II-361–II-364. doi: 10.1161/01.cir.100.suppl_2.ii-361. [DOI] [PubMed] [Google Scholar]
- 85.Yasunari K, Maeda K, Nakamura M, Yoshikawa J. Oxidative stress in leukocytes is a possible link between blood pressure, blood glucose, and C-reacting protein. Hypertension. 2002;39(3):777–780. doi: 10.1161/hy0302.104670. [DOI] [PubMed] [Google Scholar]
- 86.Foley RN, Collins AJ. End-stage renal disease in the United States: an update from the United States Renal Data System. Journal of the American Society of Nephrology. 2007;18(10):2644–2648. doi: 10.1681/ASN.2007020220. [DOI] [PubMed] [Google Scholar]
- 87.Dounousi E, Papavasiliou E, Makedou A, et al. Oxidative stress is progressively enhanced with advancing stages of CKD. American Journal of Kidney Diseases. 2006;48(5):752–760. doi: 10.1053/j.ajkd.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 88.Stuveling EM, Hillege HL, Bakker SJL, Gans ROB, De Jong PE, De Zeeuw D. C-reactive protein is associated with renal function abnormalities in a non-diabetic population. Kidney International. 2003;63(2):654–661. doi: 10.1046/j.1523-1755.2003.00762.x. [DOI] [PubMed] [Google Scholar]
- 89.Biswas SK, de Faria JBL. Which comes first: renal inflammation or oxidative stress in spontaneously hypertensive rats? Free Radical Research. 2007;41(2):216–224. doi: 10.1080/10715760601059672. [DOI] [PubMed] [Google Scholar]
- 90.Lao D, Parasher PS, Cho KC, Yeghiazarians Y. Atherosclerotic renal artery stenosis—diagnosis and treatment. Mayo Clinic Proceedings. 2011;86(7):649–657. doi: 10.4065/mcp.2011.0181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Khatami MR. Ischemic nephropathy: more than a simple renal artery narrowing. Iranian Journal of Kidney Diseases. 2013;7(2):82–100. [PubMed] [Google Scholar]
- 92.Hansen KJ, Edwards MS, Craven TE, et al. Prevalence of renovascular disease in the elderly: a population-based study. Journal of Vascular Surgery. 2002;36(3):443–451. doi: 10.1067/mva.2002.127351. [DOI] [PubMed] [Google Scholar]
- 93.Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nature Medicine. 2011;17(11):1410–1422. doi: 10.1038/nm.2538. [DOI] [PubMed] [Google Scholar]
- 94.Guyenet PG. The sympathetic control of blood pressure. Nature Reviews Neuroscience. 2006;7(5):335–346. doi: 10.1038/nrn1902. [DOI] [PubMed] [Google Scholar]
- 95.Schlaich MP, Lambert E, Kaye DM, et al. Sympathetic augmentation in hypertension: role of nerve firing, norepinephrine reuptake, and angiotensin neuromodulation. Hypertension. 2004;43(2 I):169–175. doi: 10.1161/01.HYP.0000103160.35395.9E. [DOI] [PubMed] [Google Scholar]
- 96.Tan Z-Y, Lu Y, Whiteis CA, et al. Chemoreceptor hypersensitivity, sympathetic excitation, and overexpression of ASIC and TASK channels before the onset of hypertension in SHR. Circulation Research. 2010;106(3):536–545. doi: 10.1161/CIRCRESAHA.109.206946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nance DM, Sanders VM. Autonomic innervation and regulation of the immune system (1987–2007) Brain, Behavior, and Immunity. 2007;21(6):736–745. doi: 10.1016/j.bbi.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lob HE, Marvar PJ, Guzik TJ, et al. Induction of hypertension and peripheral inflammation by reduction of extracellular superoxide dismutase in the central nervous system. Hypertension. 2010;55(2):277–283. doi: 10.1161/HYPERTENSIONAHA.109.142646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Veelken R, Vogel E, Hilgers K, et al. Autonomic renal denervation ameliorates experimental glomerulonephritis. Journal of the American Society of Nephrology. 2008;19(7):1371–1378. doi: 10.1681/ASN.2007050552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yu Y, Zhang Z, Wei S, Serrats J, Weiss RM, Felder RB. Brain perivascular macrophages and the sympathetic response to inflammation in rats after myocardial infarction. Hypertension. 2010;55(3):652–659. doi: 10.1161/HYPERTENSIONAHA.109.142836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bunag RD, Page IH, McCubbin JW. Neural stimulation of release of renin. Circulation Research. 1966;19(4):851–858. doi: 10.1161/01.res.19.4.851. [DOI] [PubMed] [Google Scholar]
- 102.Esler MD, Krum H, Schlaich M, Schmieder RE, Böhm M, Sobotka PA. Renal sympathetic denervation for treatment of drug-resistant hypertension: one-year results from the symplicity htn-2 randomized, controlled trial. Circulation. 2012;126(25):2976–2982. doi: 10.1161/CIRCULATIONAHA.112.130880. [DOI] [PubMed] [Google Scholar]
- 103.Bhatt DL, Kandzari DE, O'Neill WW, et al. A controlled trial of renal denervation for resistant hypertension. The New England Journal of Medicine. 2014;370(15):1393–1401. doi: 10.1056/NEJMoa1402670. [DOI] [PubMed] [Google Scholar]
- 104.Krum H. Catheter-based renal sympathetic denervation for resistant hypertension: durability of blood pressure reduction out to 24 months. Hypertension. 2011;57(5):911–917. doi: 10.1161/HYPERTENSIONAHA.110.163014. [DOI] [PubMed] [Google Scholar]
- 105.Verheye S. TCT-62 preliminary result of the rapid renal sympathetic denervation for resistant hypertension using the maya medical OneShot(tm) Ablation System (RAPID) study. Journal of the American College of Cardiology. 2013;62(18, supplement 1):p. B20. [Google Scholar]
- 106.Lloyd-Sherlock P, Beard J, Minicuci N, Ebrahim S, Chatterji S. Hypertension among older adults in low- and middle-income countries: prevalence, awareness and control. International Journal of Epidemiology. 2014;43(1):116–128. doi: 10.1093/ije/dyt215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.van Rossum CTM, van de Mheen H, Witteman JCM, Hofman A, Mackenbach JP, Grobbee DE. Prevalence, treatment, and control of hypertension by sociodemographic factors among the Dutch elderly. Hypertension. 2000;35(3):814–821. doi: 10.1161/01.hyp.35.3.814. [DOI] [PubMed] [Google Scholar]
- 108.Rafey MA. Resistant hypertension in the elderly. Clinics in Geriatric Medicine. 2009;25(2):289–301. doi: 10.1016/j.cger.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 109.Franceschi C, Capri M, Monti D, et al. Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans. Mechanisms of Ageing and Development. 2007;128(1):92–105. doi: 10.1016/j.mad.2006.11.016. [DOI] [PubMed] [Google Scholar]
- 110.Bartlett DB, Firth CM, Phillips AC, et al. The age-related increase in low-grade systemic inflammation (Inflammaging) is not driven by cytomegalovirus infection. Aging Cell. 2012;11(5):912–915. doi: 10.1111/j.1474-9726.2012.00849.x. [DOI] [PubMed] [Google Scholar]
- 111.Funder JW, Carey RM, Fardella C, et al. Case detection, diagnosis, and treatment of patients with primary aldosteronism: an endocrine society clinical practice guideline. Journal of Clinical Endocrinology and Metabolism. 2008;93(9):3266–3281. doi: 10.1210/jc.2008-0104. [DOI] [PubMed] [Google Scholar]
- 112.Struthers A, Krum H, Williams GH. A comparison of the aldosterone-blocking agents eplerenone and spironolactone. Clinical Cardiology. 2008;31(4):153–158. doi: 10.1002/clc.20324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Luft FC. Mendelian forms of human hypertension and mechanisms of disease. Clinical Medicine & Research. 2003;1(4):291–300. doi: 10.3121/cmr.1.4.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zhang Z, Yu Y, Kang Y, Wei S, Felder RB. Aldosterone acts centrally to increase brain renin-angiotensin system activity and oxidative stress in normal rats. The American Journal of Physiology: Heart and Circulatory Physiology. 2008;294(2):H1067–H1074. doi: 10.1152/ajpheart.01131.2007. [DOI] [PubMed] [Google Scholar]
- 115.Leibovitz E, Ebrahimian T, Paradis P, Schiffrin EL. Aldosterone induces arterial stiffness in absence of oxidative stress and endothelial dysfunction. Journal of Hypertension. 2009;27(11):2192–2200. doi: 10.1097/HJH.0b013e328330a963. [DOI] [PubMed] [Google Scholar]
- 116.Rocha R, Rudolph AE, Frierdich GE, et al. Aldosterone induces a vascular inflammatory phenotype in the rat heart. American Journal of Physiology: Heart and Circulatory Physiology. 2002;283(5):H1802–H1810. doi: 10.1152/ajpheart.01096.2001. [DOI] [PubMed] [Google Scholar]
- 117.Sun Y, Zhang J, Lu L, Chen SS, Quinn MT, Weber KT. Aldosterone-induced inflammation in the rat heart: role of oxidative stress. The American Journal of Pathology. 2002;161(5):1773–1781. doi: 10.1016/S0002-9440(10)64454-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kasal DA, Barhoumi T, Li MW, et al. T regulatory lymphocytes prevent aldosterone-induced vascular injury. Hypertension. 2012;59(2):324–330. doi: 10.1161/HYPERTENSIONAHA.111.181123. [DOI] [PubMed] [Google Scholar]
- 119.Blasi ER, Rocha R, Rudolph AE, Blomme EAG, Polly ML, McMahon EG. Aldosterone/salt induces renal inflammation and fibrosis in hypertensive rats. Kidney International. 2003;63(5):1791–1800. doi: 10.1046/j.1523-1755.2003.00929.x. [DOI] [PubMed] [Google Scholar]
- 120.Tzamou V, Vyssoulis G, Karpanou E, Kyvelou S-, Gialernios T, Stefanadis C. Aldosterone levels and inflammatory stimulation in essential hypertensive patients. Journal of Human Hypertension. 2013;27(9):535–538. doi: 10.1038/jhh.2013.13. [DOI] [PubMed] [Google Scholar]
- 121.Rodríguez-Iturbe B, Quiroz Y, Nava M, et al. Reduction of renal immune cell infiltration results in blood pressure control in genetically hypertensive rats. The American Journal of Physiology: Renal Physiology. 2002;282(2):F191–F201. doi: 10.1152/ajprenal.0197.2001. [DOI] [PubMed] [Google Scholar]
- 122.Tian N, Gu J-W, Jordan S, Rose RA, Hughson MD, Manning RD., Jr. Immune suppression prevents renal damage and dysfunction and reduces arterial pressure in salt-sensitive hypertension. The American Journal of Physiology—Heart and Circulatory Physiology. 2007;292(2):H1018–H1025. doi: 10.1152/ajpheart.00487.2006. [DOI] [PubMed] [Google Scholar]
- 123.Mattson DL, James L, Berdan EA, Meister CJ. Immune suppression attenuates hypertension and renal disease in the Dahl salt-sensitive rat. Hypertension. 2006;48(1):149–156. doi: 10.1161/01.HYP.0000228320.23697.29. [DOI] [PubMed] [Google Scholar]
- 124.Herrera J, Ferrebuz A, MacGregor EG, Rodriguez-Iturbe B. Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. Journal of the American Society of Nephrology. 2006;17(12) supplement 3:S218–S225. doi: 10.1681/ASN.2006080918. [DOI] [PubMed] [Google Scholar]
- 125.de Miguel C, Guo C, Lund H, Feng D, Mattson DL. Infiltrating T lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease. The American Journal of Physiology—Renal Physiology. 2011;300(3):F734–F742. doi: 10.1152/ajprenal.00454.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ferro CJ, Edwards NC, Hutchison C, et al. Does immunosuppressant medication lower blood pressure and arterial stiffness in patients with chronic kidney disease? An observational study. Hypertension Research. 2011;34(1):113–119. doi: 10.1038/hr.2010.193. [DOI] [PubMed] [Google Scholar]
- 127.Nataraj C, Oliverio MI, Mannon RB, et al. Angiotensin II regulates cellular immune responses through a calcineurin-dependent pathway. Journal of Clinical Investigation. 1999;104(12):1693–1701. doi: 10.1172/JCI7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Tissot AC, Maurer P, Nussberger J, et al. Effect of immunisation against angiotensin II with CYT006-AngQb on ambulatory blood pressure: a double-blind, randomised, placebo-controlled phase IIa study. The Lancet. 2008;371(9615):821–827. doi: 10.1016/S0140-6736(08)60381-5. [DOI] [PubMed] [Google Scholar]
- 129.Chen X, Qiu Z, Yang S, et al. Effectiveness and safety of a therapeutic vaccine against angiotensin II receptor type 1 in hypertensive animals. Hypertension. 2013;61(2):408–416. doi: 10.1161/HYPERTENSIONAHA.112.201020. [DOI] [PubMed] [Google Scholar]
- 130.Michel J-B, Guettier C, Philippe M, Galen FX, Corvol P, Ménard J. Active immunization against renin in normotensive marmoset. Proceedings of the National Academy of Sciences of the United States of America. 1987;84(12):4346–4350. doi: 10.1073/pnas.84.12.4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Tepperman E, Ramzy D, Prodaer J, et al. Surgical biology forthe clinician: vascular effects of immunosuppression. Canadian Journal of Surgery. 2010;53(1):57–63. [PMC free article] [PubMed] [Google Scholar]
- 132.Johnson AG, Nguyen TV, Day RO. Do nonsteroidal anti-inflammatory drugs affect blood pressure? A meta-analysis. Annals of Internal Medicine. 1994;121(4):289–300. doi: 10.7326/0003-4819-121-4-199408150-00011. [DOI] [PubMed] [Google Scholar]
- 133.Myat A, Redwood SR, Qureshi AC, Spertus Director JA, Williams B. Resistant hypertension. BMJ. 2012;345(7884) doi: 10.1136/bmj.e7473.e7473 [DOI] [PubMed] [Google Scholar]
- 134.Pimenta E, Calhoun DA. Resistant hypertension: incidence, prevalence, and prognosis. Circulation. 2012;125(13):1594–1596. doi: 10.1161/CIRCULATIONAHA.112.097345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Strazzullo P, Kerry SM, Barbato A, Versiero M, D'Elia L, Cappuccio FP. Do statins reduce blood pressure? A meta-analysis of randomized, controlled trials. Hypertension. 2007;49(4):792–798. doi: 10.1161/01.HYP.0000259737.43916.42. [DOI] [PubMed] [Google Scholar]
- 136.Kinlay S, Schwartz GG, Olsson AG, et al. High-dose atorvastatin enhances the decline in inflammatory markers in patients with acute coronary syndromes in the MIRACL study. Circulation. 2003;108(13):1560–1566. doi: 10.1161/01.CIR.0000091404.09558.AF. [DOI] [PubMed] [Google Scholar]
- 137.Ridker PM, Danielson E, Fonseca FAH, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. The New England Journal of Medicine. 2008;359(21):2195–2207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 138.Tobert JA. Lovastatin and beyond: the history of the HMG-CoA reductase inhibitors. Nature Reviews Drug Discovery. 2003;2(7):517–526. doi: 10.1038/nrd1112. [DOI] [PubMed] [Google Scholar]
- 139.Iwata A, Shirai R, Ishii H, et al. Inhibitory effect of statins on inflammatory cytokine production from human bronchial epithelial cells. Clinical and Experimental Immunology. 2012;168(2):234–240. doi: 10.1111/j.1365-2249.2012.04564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. Journal of Biological Chemistry. 1998;273(37):24266–24271. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- 141.Lu T-M, Ding Y-A, Leu H-B, Yin W-H, Sheu WH-H, Chu K-M. Effect of rosuvastatin on plasma levels of asymmetric dimethylarginine in patients with hypercholesterolemia. The American Journal of Cardiology. 2004;94(2):157–161. doi: 10.1016/j.amjcard.2004.03.052. [DOI] [PubMed] [Google Scholar]
- 142.Antoniades C, Shirodaria C, Leeson P, et al. Association of plasma asymmetrical dimethylarginine (ADMA) with elevated vascular superoxide production and endothelial nitric oxide synthase uncoupling: implications for endothelial function in human atherosclerosis. European Heart Journal. 2009;30(9):1142–1150. doi: 10.1093/eurheartj/ehp061. [DOI] [PubMed] [Google Scholar]