

Abstract
Neuroinflammation mediated by microglial cells in the brain has been commonly associated with neurodegenerative diseases. Whether this microglia-mediated neuroinflammation is cause or consequence of neurodegeneration is still a matter of controversy. However, it is unequivocal that chronic neuroinflammation plays a role in disease progression and halting that process represents a potential therapeutic strategy. The neuromodulator adenosine emerges as a promising targeting candidate based on its ability to regulate microglial proliferation, chemotaxis, and reactivity through the activation of its G protein coupled A2A receptor (A2AR). This is in striking agreement with the ability of A2AR blockade to control several brain diseases. Retinal degenerative diseases have been also associated with microglia-mediated neuroinflammation, but the role of A2AR has been scarcely explored. This review aims to compare inflammatory features of Parkinson's and Alzheimer's diseases with glaucoma and diabetic retinopathy, discussing the therapeutic potential of A2AR in these degenerative conditions.
1. Introduction
1.1. Role of Microglia in Brain Physiology
In the central nervous system (CNS), microglial cells participate in innate immunity; microglia can respond to different types of signals, namely the presence of pathogens (extrinsic signals) or to intrinsic signals, namely diffusible mediators released by stressed neurons, astrocytes or microglia (reviewed in [1]). Although the present review mainly focuses on the contribution of microglia to the pathophysiology of neurodegeneration in the brain and the retina, any attempt to interfere with microglia in pathological conditions also needs to take into account the role of microglia in physiological conditions.
In the healthy brain, the majority of microglial cells exhibit a ramified phenotype, compatible with a surveillance function of the surrounding environment. This crucial sensor ability is supported by the constant extension and retraction of cellular processes [2, 3]. This dynamics is not random but instead instructed by increased neuronal activity, that activates pannexin-1 hemichannels, triggering the diffusion of signals, namely, ATP, that drive process motility towards that specific neuron [4]. The interconversion between the so-called “surveying” phenotype (considered more adequate, as compared to the old terminology “resting” phenotype) and the “alerted” phenotype can be driven either by external stimuli (e.g., pathogens) or by neural signals. The latter is achieved by direct neuron-microglia contact or by diffusible mediators (reviewed, e.g., in [1]). This activation of microglia drives some immediate responses that mainly consist in (1) production/release of rectifier mediators and (2) phagocytosis of neurons or subcellular components (mainly dendritic spines and synapses). Microglial phagocytosis of neurons or neuronal structures has been mostly studied in pathological conditions (e.g., [5–8]), but it also takes place in nonpathological conditions. In fact, it is a process of particular importance during neurodevelopment, as shown by Tremblay and coworkers [9] in the visual system: light deprivation and the subsequent decrease in the workload of neuronal circuits involved in visual processing lead to the engulfment of synaptic elements by microglia. This physiological process, termed synaptic pruning, is regulated by the immune system; synapses and axons to be phagocytosed are labeled by the complement components C1q and C3, which prompt their selective recognition by microglial cells [10–12]. Synaptic pruning is crucial to normal brain wiring and function and any impairment of this process may impact on neurodevelopment. For instance, this was recently associated with deficits in synaptic transmission, which are paralleled by behavioral abnormalities characteristic of disorders of the autism spectrum and other neuropsychiatric conditions [13]. This process also occurs during adulthood, particularly in neurogenic niches of the brain, such as the hippocampus, where microglia phagocytose apoptotic newborn neurons [14].
Intriguingly, as part of their physiological role, microglia also actively shape their neuronal environment thanks to their ability to trigger neuronal death [15–17]. Again, such a role has a particular relevance during brain development, namely, during the first postnatal week, as heralded by the observation that microglia accumulate in regions of developmental cell death in the embryonic cerebral cortex [18]; furthermore, in the spinal cord, the cell death of motor neurons correlates temporally with the arrival of microglia [19].
In addition to their role in synaptic pruning, microglia also regulate synapse formation [20–22]. This function has been shown to be dependent on the production and release of mediators, such as brain-derived neurotrophic factor [20] or interleukin- (IL-) 10 [22], although other diffusible mediators are likely to be involved. This critical function of microglia must be strictly preserved in order to prevent neurodevelopmental deficits, as suggested by a recent in vitro study showing that activation of microglia by an inflammatory stimulus may impact on the presynaptic differentiation of immature neurons [23].
Microglial support to synapse formation/elimination is tightly associated with the newly recognized role of microglia as active partners in the transmission of information within synapses [24]. Thus, recent studies show that microglia also monitor the functional state of synapses and respond to changes in synaptic activity [25, 26]. Accordingly, the highly motile processes of microglia contact with synapses and regulate synaptic transmission in nonpathological conditions [9, 10, 27–30].
1.2. Role of Microglia in Retinal Physiology
In the adult retina, the presence of microglia has been described in several mammals species, including rabbits [31–33], mice [34], rats [31, 35, 36], monkeys [37, 38], and humans [39–41]. Microglial cells in the adult normal retina are mainly located in the inner vascularized regions, that is, the nerve fiber and ganglion cell layers and in plexiform layers, whereas they are scarce in the inner nuclear layer and absent in the outer nuclear layer (Figure 1).
Figure 1.
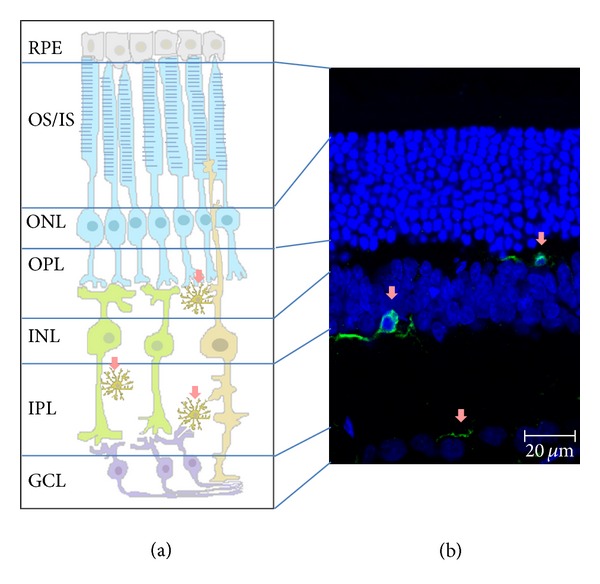
Microglial localization in the retina. Microglial cells in a “surveying” state (pink arrows) in nonpathological conditions are mainly located in the plexiform layers. Retinal layers: OS/IS, outer and inner segments of rods and cones; ONL, outer nuclear layer; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL, ganglion cell layer. Schematic draw of the retinal layers (a) and confocal image from a retinal section where the different layers are depicted (b): nuclear layers (in blue) and microglia cells (in green).
In the healthy retina, microglial cells represent a self-renewing population of innate immune cells, which constantly survey their microenvironment, as occurs in the brain. Retinal microglia can also phagocytose pyknotic cells generated upon neural remodeling of the retina [42]. A more recent study performed in zebrafish showed that microglial cells not only have a “cleaning” role in the developing retina, but also are required for normal retinal growth and neurogenesis [43]. Microglia may also play a role in the formation of blood vessels in the developing retina, since microglia depletion during retinal development reduces vascularization, an effect restored by intravitreal injection of microglia [44]. This is in agreement with the origin of retinal microglial cells that originate from cells of mesodermal lineage [45] and populate the retina before vascularization and along with the onset of vasculogenesis [46].
1.3. A2AR Regulation of Microglia Physiology
Adenosine is a neuromodulator, which also exerts important functions in the immune-inflammatory system [47]. Microglial cells express all subtypes of adenosine receptors, A1, A2A, A2B, and A3 receptors [48]. Although a large body of evidence highlights the ability of A1 and A3 receptors to regulate microglia responses, such as proliferation, morphological phenotype, and release of mediators [49–52], particular attention has been paid to A2AR, considered to have a central role in the pathophysiology of degeneration [53–55].
It is claimed that A2AR modulation (both activation and blockade) interferes with microglia-mediated inflammation in degenerative conditions (see below). Of note, in physiological conditions, important functions operated by microglia, namely, the release of mediators, such as trophic factors [56] or nitric oxide (NO) [57], as well as the extension and retraction of processes that govern the surveying activity of microglia [58], are apparently out of A2AR control, until a pathologic insult triggers a gain-of-function of A2AR [56, 57, 59, 60]. However, the milestone study by Davalos et al. [2] shows that the baseline motility of microglial processes in the healthy brain is governed by ATP (and prevented by ATP degradation), as occurs in pathological-like conditions. This observation raises the unanswered question whether the activation of A2AR by ATP-derived adenosine regulates the dynamics of microglial processes in physiological conditions.
1.4. Role of Microglia in Degenerative Conditions of the Brain
The main physiologic roles operated by microglia (release of mediators that control synaptic transmission, synapse formation, and phagocytosis of cells or cellular elements) are strictly dependent upon their sensor ability. Any interference at this functional level may create conditions favoring the development of degenerative processes, which are bolstered by abnormal synaptic transmission, aberrant synapse formation and/or elimination, and abnormal phagocytosis (Figure 2). Therefore, the identification of molecular systems able to modulate microglial functions may help defining new pharmacological targets to interfere with the progression of neurodegenerative diseases. Indeed, microglia-driven neuroinflammation is associated with a broad spectrum of neurodegenerative diseases and has been more detailed in Alzheimer's disease (AD) and Parkinson's disease (PD).
Figure 2.
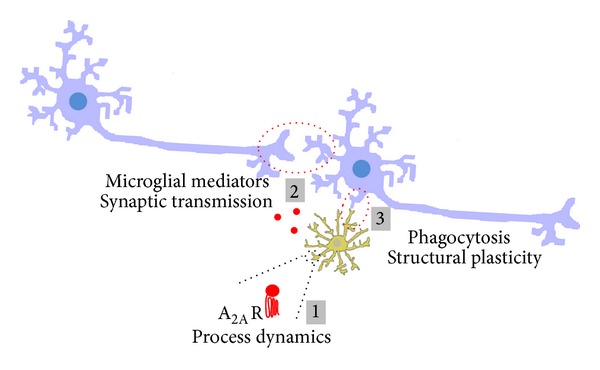
Microglia in the healthy brain/retina. Schematic representation of the main functions exerted by microglia (in yellow) under physiological conditions: surveying the environment by constant extension and retraction of processes (it remains to clarify if A2AR regulate this process, as occurs in pathology) (1); regulation of basal synaptic transmission and plasticity through the release of mediators (red circles), some of them being also important mediators of inflammation (2); regulation of spine/synapse structural plasticity, mainly by phagocytosis, a process regulated by inflammatory mediators, according to the neuronal workload (3).
The accumulation of misfolded β-amyloid-containing proteins (Abeta) and alpha-synuclein are histopathological hallmarks of established AD and PD, respectively [61–67]. Protein aggregates can directly exert neurotoxicity [68–70] and can trigger parallel maladaptive changes of glial cells; in fact, animal models of AD and PD and postmortem examination of the brain of AD or PD patients frequently reveal increased numbers of activated microglia in degenerated brain regions [71–76]. Moreover, in vivo studies using PET with a radiotracer for activated microglia in AD and PD patients have provided evidence for increased levels of activated microglia in brain regions that are affected by the disease [75–79]. Importantly, protein aggregates may be sufficient causative factors for microglial activation and release of inflammatory mediators [80], which, in turn, amplify neuroinflammation and further exacerbate neurodegeneration [73]. Such a scenario prompts the idea that microglia-induced neuroinflammation may play a critical role in the progression of neurodegenerative conditions [65–67, 81, 82].
Indeed, several microglia-derived inflammatory mediators have been shown to be involved in neuronal damage in neurodegenerative diseases. Thus, one possible causative factor for neuronal death in AD is Aβ-induced NO production by microglia [83]. Furthermore, Aβ and interferon-gamma (IFN-γ) can activate microglia to produce reactive nitrogen intermediates and tumor necrosis factor (TNF), contributing to neuronal degeneration observed in AD [84]. Additional proof-of-concept for the role of microglia in the progression of neuronal damage in AD was derived from the observation that drugs preventing microglial activation indeed delay the emergence of an AD-like phenotype in animal models [85]. Similarly, increased expression of inflammatory mediators is also found in PD animal models [51, 80, 86] and in postmortem PD brains [87, 88], including proinflammatory cytokines, such as IFN-γ, IL-1β, TNF, IL-2, and IL-6, released by microglia [89–91]. The microglial overactivation and the release of proinflammatory cytokines and reactive oxygen species (ROS) are associated with neuronal loss in PD [72, 73]; further evidence for the key role of these microglia-derived mediators in the evolution of neuronal damage in PD was obtained by showing that the inactivation of microglia-derived mediators counteracts neurodegeneration in the MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) animal model of PD [92–95].
In addition to the direct neurotoxic impact of these microglia-derived inflammatory mediators, the deregulation of the phagocytic activity of microglia also contributes to the progression of neuronal damage. This is heralded by the observations of an increased number of phagocytic microglia close to damaged neurons in PD [96, 97]; furthermore, blocking microglial activation attenuates neurodegeneration, further supporting the role of microglia in the evolution of the pathological process [98]. Increased phagocytosis of neuronal elements seems to be a selective process since in vitro studies have suggested that microglia may paradoxically reduce its ability to degrade Aβ-containing aggregates, and their intracellular accumulation leads to dysfunctional/dystrophic microglia [99–101]. In animal models of AD it has been shown in late stages of cerebral amyloidosis that the phagocytic capacity of microglia is impaired [102], and this impairment was described to accelerate pathology progression [103].
In summary, microglial functions, from the release of inflammatory mediators to the ability to phagocytose, are deregulated in neurodegenerative diseases. This implies that the identification of regulatory systems able to rebalance microglial function may be of therapeutic interest to manage the progression of neurodegenerative diseases.
1.5. Control of Microglia-Driven Neuroinflammation by A2AR in Brain Diseases
The ability of adenosine and A2AR activation to control the activation of different inflammatory cell types has been consistently documented by different groups [47]. Likewise, several in vitro and in vivo studies clearly demonstrate that A2AR controls several facets of microglia dynamics [56–58, 104, 105], such as (1) the proliferation, (2) the levels of inflammatory enzymes such as cyclooxygenase-2, and (3) the synthesis and release of inflammatory mediators. Furthermore, studies carried out in several models of brain disorders have found that pharmacological blockade or genetic inactivation of A2AR affords a robust neuroprotection [53, 54], and increasing evidence suggests this neuroprotection involves the control of microglia-mediated neuroinflammation [54, 106, 107]. Furthermore, different brain insults triggering neuroinflammation also cause an upregulation of A2AR [56, 60], namely, in microglial cells [56, 57, 59, 108], which is in line with the described ability of cytokines to upregulate A2AR (reviewed by [53]). Finally, A2AR seem to have an additional ability to protect neurons from proinflammatory priming neurodegeneration [109, 110]. This has bolstered the interest to exploit A2AR as a promising pharmacological target to control the neuroinflammatory component of neurodegenerative diseases, allowing the slowdown of their evolution [47, 56, 106, 107].
The clinical interest of the adenosine modulation system in the control of memory dysfunction in AD first arose from epidemiological studies showing an inverse correlation between the consumption of moderate doses of caffeine (a nonselective adenosine receptor antagonist) and the deterioration of memory performance upon aging and AD [111]. This was in notable agreement with animal studies showing that the chronic consumption of caffeine reduces cognitive impairment and decreases Aβ levels in the brain of transgenic mouse models of AD [112–114], as well as in mice exposed to Aβ [104, 115], a purported causative factor of AD [64]. Animal studies were paramount to identify A2AR as the likely targets of caffeine [116], since the pharmacological or genetic blockade of A2AR mimics the neuroprotective effects of caffeine [104, 117]. In accordance with the involvement of neuroinflammatory features in AD, the exposure of rodents to lipopolysaccharide (LPS), which is present in the cell wall of gram-negative bacteria and used as a prototypical activator of microglia, triggers the activation of microglia, a proinflammatory status in the brain parenchyma, and deterioration of synaptic plasticity and memory performance [105]. Notably, this LPS-induced neuroinflammation can be prevented both by the caffeine [118] and by the selective blockade of A2AR [60], which abrogates the LPS-induced dampening of hippocampal synaptic plasticity, the purported neurophysiological basis of learning and memory [119]. Further supporting this role of microglial A2AR in AD, the analysis of postmortem human cortex from AD patients revealed an increased density of A2AR [60] that is more prominent in microglia [120].
As in AD, there is also solid evidence for a role of A2AR in the control of PD, as testified by the recent introduction of A2AR antagonists as coadjuvants in the management of PD [121]. Thus, A2AR antagonists improve PD symptoms in different rodent and primate models of the disease and also in PD patients enrolled in clinical trials (for a review see [122]). Besides the control of motor function, A2AR blockade also dampens microglial activation in the striatum [108] and substantia nigra [123] in animal models of PD. Furthermore, caffeine downregulates microglia-driven neuroinflammatory responses and decreases NO production in animal models of PD [124]. Although caffeine acts on both A1R and A2AR, the neuroprotective properties of caffeine in PD are mediated through A2AR blockade [125, 126]. In fact, caffeine consumption has been associated with lower risk of PD in several case-control and cohort studies [127–132]. Interestingly, the association between coffee consumption and PD is strongest among subjects that slowly metabolize caffeine and are homozygous carriers of the CYP1A2 polymorphisms, the gene encoding for cytochrome P450 1A2 [133] which is the main enzyme involved in the metabolism of caffeine.
A recent ex vivo study (brain slices from MPTP-treated mice modeling PD) showed that a selective A2AR antagonist restores the ability of microglia to respond to tissue damage [134]. This A2AR-mediated control of neuroinflammation is argued to be critical for the neuroprotection afforded by A2AR blockade in PD since the inhibition of microglial function has been shown to be sufficient to decrease the dopaminergic neurodegeneration characteristic of PD.
These two examples of neurodegenerative diseases support the working hypothesis that the beneficial effects resulting from A2AR blockade may involve their ability to attenuate microglial activation and associated chronic neuroinflammatory status, which would interrupt the vicious cross amplifying cycle of degeneration and inflammation leading to a slower development of neurodegenerative disorders (Figure 3).
Figure 3.
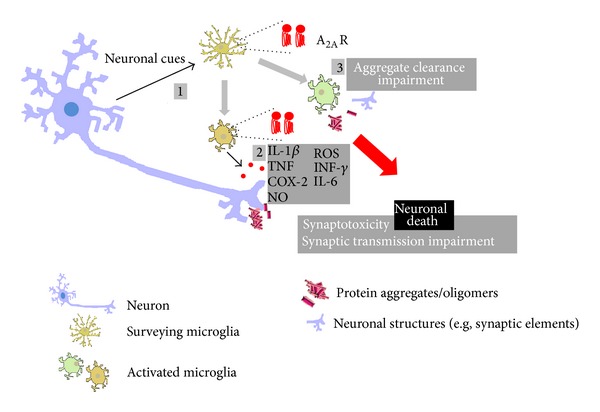
Microglia and neuroinflammation in the brain/retina. Schematic representation of the main inflammatory responses mediated by microglial cells (in yellow) in neurodegenerative conditions. Environment surveillance allows the detection of “pathological” events affecting neurons (in blue-purple); note that appropriate detection of danger signals may also be compromised under these conditions; one of the microglial changes consists in the upregulation of the expression/density of A2AR, as described in several degenerative disorders (1), usually paralleled by morphologic changes and by the release of inflammatory mediators (red circles), both anti- and proinflammatory molecules, that may impact on synaptic transmission, ultimately leading to synaptotoxicity (2); the ability of microglia to phagocytose subcellular components of damaged neurons or protein aggregates, typically present in some degenerative diseases, may also be impaired, further amplifying the cascade of events that lead to cell death/degeneration (3).
1.6. Neuroinflammation Is a Common Feature between Retinal and Brain Degenerative Diseases
The combined effect of an ageing population and increasing life expectancy will increase the prevalence of chronic diseases [135], which encompass not only neurodegenerative brain diseases, but also retinal degenerative conditions amongst others. Indeed, the demographic evolution, with an increasing elderly population in western countries, exponentially augments the number of people at risk of age-related visual impairment caused by age-related retinal degenerative diseases [136]. Glaucoma and diabetic retinopathy are leading causes of blindness worldwide. Glaucoma is the second cause of irreversible blindness [137], affecting 70 million people worldwide and approximately 2% of the population over the age of 40 [138]. Diabetic retinopathy is a frequent complication of diabetes and may lead to blindness, making it one of the most feared complications of diabetes. Indeed, diabetic retinopathy is the leading cause of vision loss in working age adults [139]. Since the number of people affected by diabetes is expected to increase significantly in the next 25 years, from the actual 382 million to beyond 592 million [139], the number of people affected by diabetic retinopathy is expected to greatly expand.
The similarities between AD pathology and retinal degenerative diseases have been described elsewhere [140, 141], and neuroinflammation is a common feature between brain and retinal degenerative diseases. It is, thus, plausible to speculate that therapeutic agents and strategies used for brain neurodegeneration could also be considered for retinal diseases with an underlying chronic inflammation process. Retinal microglia cells express A2AR [142], opening the possibility that the control of microglia-mediated neuroinflammation through A2AR modulation might also be an attractive approach to manage retinal diseases.
1.7. Glaucoma Has a Neuroinflammatory Component
Glaucoma is defined as a group of ocular disorders of multifactorial etiology characterized by progressive optic neuropathy [143] and gradual loss of retinal ganglion cells and optic nerve (retinal ganglion cell axons) damage. Elevated intraocular pressure (IOP) is one of the major risk factors for developing glaucoma or glaucomatous neuropathy [144]. The current therapeutic approach in glaucoma is focused on lowering IOP by pharmacological means, surgically, or with laser treatment. However, patients continue to lose vision despite successful IOP control, and it is becoming clear that the exclusive management of IOP is not sufficient, and neuroprotection of retinal ganglion cells has been proposed as a potential alternative therapy [145].
Several studies have reported that the progressive degeneration of optic nerve axons and retinal ganglion cells in glaucoma is accompanied by chronic alterations in structural and functional characteristics of glial cells in the optic nerve head and retina [146, 147], where an abnormal microglial reactivity and redistribution take place [148]. TNF, IL-6, and IL-18 levels are increased in the retina and optic nerve head in both glaucomatous patients and animal models of glaucoma [149–151] and recent studies demonstrate that microglial activation is an early event in experimental models of glaucoma, which coincides with the onset of RGC death, potentially contributing to disease onset and/or progression [152–154]. Also, the treatment with minocycline, a tetracycline derivative known to reduce microglial activation [155], was able to improve retinal ganglion cell axonal transport and integrity in a mouse model of glaucoma [156].
1.8. Diabetic Retinopathy: A Low-Grade Inflammatory Disease
Diabetic retinopathy is one of the most common complications of diabetes and the most frequent cause of new cases of blindness among adults aged 20–74 years. After 20 years of diabetes, nearly all patients with type 1 and more than 60% of patients with type 2 diabetes have some degree of retinopathy [157]. Diabetic retinopathy has been considered a microvascular disease, but growing evidence demonstrates that retinal neurodegeneration also occurs [158–160], and diabetic retinopathy is now more accurately defined as a neurovascular disease.
Diabetic retinopathy exhibits characteristics of a chronic inflammatory process: increased levels of cytokines, such as IL-1β, IL-6, and TNF, have been found in the vitreous fluid of diabetic patients [161–163]; retinal TNF levels are also increased in diabetic patients, particularly in those with proliferative diabetic retinopathy [164–166]. The inflammatory profile of diabetic retinopathy has been confirmed in animal models of diabetes, where an increase was found in the levels of IL-1β [167–170] and TNF [170–172] in the retina. Therefore, the role of inflammation is unequivocal in diabetic retinopathy, from the leukocyte adhesion [173, 174] to the increase in inflammatory mediators, such as TNF, which exerts a crucial role in blood retinal barrier breakdown [175], as well as the death of retinal neurons [176]. As occurs in neurodegenerative brain diseases, microglial activation in the retina is also present in different stages of human diabetic retinopathy [177] and further reported in animals models of type 1 [170, 178–180] and type 2 [181] diabetes.
1.9. Is There a Role for A2AR in Retinal Degenerative Diseases?
Retinal ischemia is a common cause of visual impairment and blindness (reviewed in [182]). Retinal degeneration after ischemia-reperfusion injury by transient elevation of IOP in rats exhibits an extensive damage at the level of the retinal ganglion cell layer [183], similarly to that reported in human glaucoma [184]. Therefore, IOP-induced retinal ischemia has been extensively used as an animal model of acute glaucoma [185], in which activation of microglia has also been observed [36]. The role of A2AR in retinal ischemia-reperfusion injury is still controversial. On one hand, the treatment with a selective A2AR antagonist protects retinal function and structure in a model of retinal ischemia [186, 187]. On the other hand, it was reported that administration of an A2AR agonist prevents retinal thinning induced by ischemia-reperfusion damage [188].
Traumatic optic neuropathy is an important cause of severe vision loss in 0.5 to 5% of patients with closed head trauma [189]. Trauma is known to cause immediate mechanical damage to the axons of retinal ganglion cells, leading to degeneration. The death of retinal ganglion cells after optic nerve damage seems to be related to the local production of ROS and inflammatory mediators from activated microglial cells [190]. Increased phagocytic and proliferative microglia have been reported after optic nerve injury [191–193]. In the optic nerve crush injury mouse model, an important experimental disease model for traumatic optic neuropathy, a selective A2AR agonist decreased microglial activation, retinal cell death, and release of ROS and proinflammatory cytokines [190]. Moreover, levels of TNF and Iba-1 (a marker of cells from the myeloid lineage, including microglia) are increased in A2AR-knockout mice with optic nerve crush. In a different model of retinal degeneration, diabetic retinopathy, it was recently shown that A2AR mRNA transcripts and protein levels increase in the retina of type 1 diabetes models and also in retinal cell cultures exposed to elevated glucose concentration, used to mimic hyperglycemic conditions [194, 195]. A2AR-knockout diabetic mice exhibit increased cell death and TNF levels as compared with diabetic wild-type mice [179]. Accordingly, the administration of a selective A2AR agonist resulted in opposite effects upon cell death and TNF levels [179].
Experiments performed in vitro emphasize the controversial role played by A2AR in the control of retinal neuroinflammation. While some authors reported that the activation of A2AR attenuates LPS-induced release of TNF in retinal microglia [190], others found that A2AR blockade prevents LPS-induced increase in NO [196]. Moreover, A2AR blockade inhibits the LPS-induced increase in TNF expression and phagocytosis. In a more complex system, the retinal organotypic culture, A2AR blockade inhibits the expression of inducible NO synthase [196].
In summary, it remains to be clarified whether A2AR activation or blockade is the best approach to pharmacologically control neuroinflammation in the retina. This dual neuroprotective ability of A2AR modulation seems to be related with the specific inflammatory profile of different pathologies or pathologic conditions, as well as with the temporal window of neuroinflammation where the exposure to A2AR agonists or antagonists occurs. Although the controversy exists, most studies in brain pathology point towards a neuroprotective effect of A2AR blockade, in line with the ability of selective and nonselective A2AR antagonists to decrease most microglial functions.
2. Concluding Remarks
Brain degenerative diseases, such as AD and PD, are associated with microglial activation and chronic neuroinflammation. In both pathologies, the blockade of A2AR emerges as a candidate mechanism of neuroprotection, through the control of microglial reactivity. Glaucoma and diabetic retinopathy are retinal degenerative diseases, in which neuroinflammation also plays a crucial role. In the retina, microglial cells are also equipped with A2AR. Therefore, it is plausible to assume that A2AR modulation may also have a potential protective effect upon inflammation underlying degenerative processes of the retina (Figure 4). It remains to be clarified whether A2AR modulation has a net positive effect in the control of clinical features and progression of retinal degenerative diseases.
Figure 4.
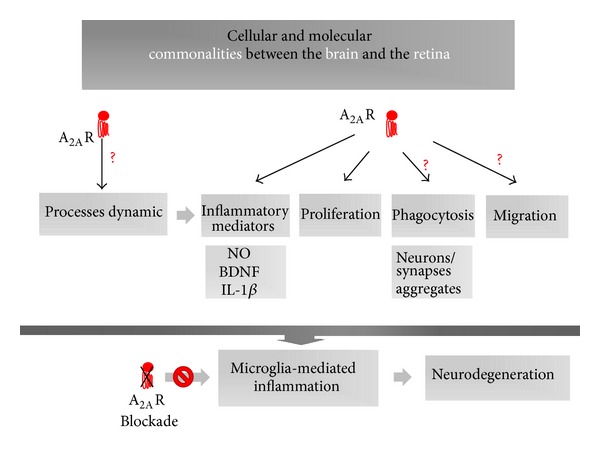
Cellular and molecular commonalities between the brain and the retina. Scheme identifying main microglial functions under the control of A2AR: release of inflammatory mediators and cellular proliferation. It remains to clarify if process extension/retraction (which supports the homeostatic surveying role of microglia), phagocytosis, and cellular migration are directly regulated by A2AR modulation (question marks). A2AR modulation is proposed as a promising pharmacological tool in the control of the chronic inflammatory process underlying degenerative conditions of the retina, based on similarities with microglia-mediated inflammation in brain disorders.
Acknowledgments
This work was supported by the Foundation for Science and Technology and COMPETE-FEDER (SFRH/BPD/86830/2012, SFRH/BPD/63013/2009, PTDC/BIM-MEC/0913/2012, PEst-C/SAU/LA0001/2013-2014, and PEst-C/SAU/UI3282/2011–2013) and AIBILI, Portugal.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nature Reviews Immunology. 2011;11(11):775–787. doi: 10.1038/nri3086. [DOI] [PubMed] [Google Scholar]
- 2.Davalos D, Grutzendler J, Yang G, et al. ATP mediates rapid microglial response to local brain injury in vivo . Nature Neuroscience. 2005;8(6):752–758. doi: 10.1038/nn1472. [DOI] [PubMed] [Google Scholar]
- 3.Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308(5726):1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- 4.Li Y, Du X, Liu C, Wen Z, Du J. Reciprocal regulation between resting microglial dynamics and neuronal activity in vivo. Developmental Cell. 2012;23(6):1189–1202. doi: 10.1016/j.devcel.2012.10.027. [DOI] [PubMed] [Google Scholar]
- 5.Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. The Journal of Neuroscience. 2009;29(13):3974–3980. doi: 10.1523/JNEUROSCI.4363-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu SM, Tremblay ME, King IL, et al. HIV-1 Tat-induced microgliosis and synaptic damage via interactions between peripheral and central myeloid cells. PLoS ONE. 2011;6(9) doi: 10.1371/journal.pone.0023915.e23915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kraft AD, Kaltenbach LS, Lo DC, Harry GJ. Activated microglia proliferate at neurites of mutant huntingtin-expressing neurons. Neurobiology of Aging. 2012;33(3):621.e17–621.e33. doi: 10.1016/j.neurobiolaging.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chugh D, Nilsson P, Afjei SA, Bakochi A, Ekdahl CT. Brain inflammation induces post-synaptic changes during early synapse formation in adult-born hippocampal neurons. Experimental Neurology. 2013;250:176–188. doi: 10.1016/j.expneurol.2013.09.005. [DOI] [PubMed] [Google Scholar]
- 9.Tremblay M, Lowery RL, Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLoS Biology. 2010;8(11) doi: 10.1371/journal.pbio.1000527.e1000527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paolicelli RC, Bolasco G, Pagani F, et al. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333(6048):1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- 11.Linnartz B, Kopatz J, Tenner AJ, Neumann H. Sialic acid on the neuronal glycocalyx prevents complement c1 binding and complement receptor-3-mediated removal by microglia. The Journal of Neuroscience. 2012;32(3):946–952. doi: 10.1523/JNEUROSCI.3830-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schafer DP, Lehrman EK, Kautzman AG, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhan Y, Paolicelli RC, Sforazzini F, et al. Deficient neuron-microglia signaling results in impaired functional brain connectivity and social behavior. Nature Neuroscience. 2014;17(3):400–406. doi: 10.1038/nn.3641. [DOI] [PubMed] [Google Scholar]
- 14.Sierra A, Encinas JM, Deudero JJP, et al. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7(4):483–495. doi: 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marín-Teva JL, Dusart I, Colin C, Gervais A, Van Rooijen N, Mallat M. Microglia promote the death of developing Purkinje cell. Neuron. 2004;41(4):535–547. doi: 10.1016/s0896-6273(04)00069-8. [DOI] [PubMed] [Google Scholar]
- 16.Wakselman S, Béchade C, Roumier A, Bernard D, Triller A, Bessis A. Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. The Journal of Neuroscience. 2008;28(32):8138–8143. doi: 10.1523/JNEUROSCI.1006-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tyler CM, Boulanger LM. Complement-mediated microglial clearance of developing retinal ganglion cell axons. Neuron. 2012;74(4):597–599. doi: 10.1016/j.neuron.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swinnen N, Smolders S, Avila A, et al. Complex invasion pattern of the cerebral cortex bymicroglial cells during development of the mouse embryo. Glia. 2013;61(2):150–163. doi: 10.1002/glia.22421. [DOI] [PubMed] [Google Scholar]
- 19.Rigato C, Buckinx R, Le-Corronc H, Rigo JM, Legendre P. Pattern of invasion of the embryonic mouse spinal cord by microglial cells at the time of the onset of functional neuronal networks. GLIA. 2011;59(4):675–695. doi: 10.1002/glia.21140. [DOI] [PubMed] [Google Scholar]
- 20.Parkhurst CN, Yang G, Ninan I. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 2013;155(7):1596–1609. doi: 10.1016/j.cell.2013.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ueno M, Fujita Y, Tanaka T, et al. Layer v cortical neurons require microglial support for survival during postnatal development. Nature Neuroscience. 2013;16(5):543–551. doi: 10.1038/nn.3358. [DOI] [PubMed] [Google Scholar]
- 22.Lim SH, Park E, You B, et al. Neuronal synapse formation induced by microglia and interleukin 10. PLoS One. 2013;8(11) doi: 10.1371/journal.pone.0081218.e81218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cristovao G, Pinto MJ, Cunha RA, Almeida RD, Gomes CA. Activation of microglia bolsters synapse formation. Frontiers in Cellular Neuroscience. 2014;8(article 153) doi: 10.3389/fncel.2014.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schafer DP, Lehrman EK, Stevens B. The “quad-partite” synapse: microglia-synapse interactions in the developing and mature CNS. Glia. 2013;61(1):24–36. doi: 10.1002/glia.22389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kettenmann H, Kirchhoff F, Verkhratsky A. Microglia: new roles for the synaptic stripper. Neuron. 2013;77(1):10–18. doi: 10.1016/j.neuron.2012.12.023. [DOI] [PubMed] [Google Scholar]
- 26.Tremblay ME, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A. The role of microglia in the healthy brain. Journal of Neuroscience. 2011;31(45):16064–16069. doi: 10.1523/JNEUROSCI.4158-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moriguchi S, Mizoguchi Y, Tomimatsu Y, et al. Potentiation of NMDA receptor-mediated synaptic responses by microglia. Molecular Brain Research. 2003;119(2):160–169. doi: 10.1016/j.molbrainres.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 28.Hayashi Y, Ishibashi H, Hashimoto K, Nakanishi H. Potentiation of the NMDA receptor-mediated responses through the activation of the glycine site by microglia secreting soluble factors. GLIA. 2006;53(6):660–668. doi: 10.1002/glia.20322. [DOI] [PubMed] [Google Scholar]
- 29.Rogers JT, Morganti JM, Bachstetter AD, et al. CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. The Journal of Neuroscience. 2011;31(45):16241–16250. doi: 10.1523/JNEUROSCI.3667-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ji K, Miyauchi J, Tsirka SE. Microglia: an active player in the regulation of synaptic activity. Neural Plasticity. 2013;2013:9 pages. doi: 10.1155/2013/627325.627325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ashwell KW, Holländer H, Streit W, Stone J. The appearance and distribution of microglia in the developing retina of the rat. Visual Neuroscience. 1989;2(5):437–448. doi: 10.1017/s0952523800012335. [DOI] [PubMed] [Google Scholar]
- 32.Schnitzer J. Enzyme-histochemical demonstration of microglial cells in the adult and postnatal rabbit retina. Journal of Comparative Neurology. 1989;282(2):249–263. doi: 10.1002/cne.902820207. [DOI] [PubMed] [Google Scholar]
- 33.Humphrey MF, Moore SR. Microglial responses to focal lesions of the rabbit retina: correlation with neural and macroglial reactions. Glia. 1996;16(4):325–341. doi: 10.1002/(SICI)1098-1136(199604)16:4<325::AID-GLIA5>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 34.Zhang C, Shen JK, Lam TT, et al. Activation of microglia and chemokines in light-induced retinal degeneration. Molecular Vision. 2005;11:887–895. [PubMed] [Google Scholar]
- 35.Harada T, Harada C, Kohsaka S, et al. Microglia-Müller glia cell interactions control neurotrophic factor production during light-induced retinal degeneration. Journal of Neuroscience. 2002;22(21):9228–9236. doi: 10.1523/JNEUROSCI.22-21-09228.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang C, Lam TT, Tso MO. Heterogeneous populations of microglia/macrophages in the retina and their activation after retinal ischemia and reperfusion injury. Experimental Eye Research. 2005;81(6):700–709. doi: 10.1016/j.exer.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 37.Vrabec F. Microglia in the monkey and rabbit retina. Journal of Neuropathology and Experimental Neurology. 1970;29(2):217–224. [PubMed] [Google Scholar]
- 38.Boycott BB, Hopkins JM. Microglia in the retina of monkey and other mammals: its distinction from other types of glia and horizontal cells. Neuroscience. 1981;6(4):679–688. doi: 10.1016/0306-4522(81)90151-2. [DOI] [PubMed] [Google Scholar]
- 39.Penfold PL, Madigan MC, Provis JM. Antibodies to human leucocyte antigens indicate subpopulations of microglia in human retina. Visual neuroscience. 1991;7(4):383–388. doi: 10.1017/s0952523800004879. [DOI] [PubMed] [Google Scholar]
- 40.Yang P, Das PK, Kijistra A. Localization and characterization of immunocompetent cells in the human retina. Ocular Immunology and Inflammation. 2000;8(3):149–157. [PubMed] [Google Scholar]
- 41.Provis JM, Diaz CM, Penfold PL. Microglia in human retina: a heterogeneous population with distinct ontogenies. Perspectives on Developmental Neurobiology. 1996;3(3):213–222. [PubMed] [Google Scholar]
- 42.Hume DA, Perry VH, Gordon S. Immunohistochemical localization of a macrophage-specific antigen in developing mouse retina: phagocytosis of dying neurons and differentiation in microglial cells to form a regular array in the plexiform layers. Journal of Cell Biology. 1983;97(1):253–257. doi: 10.1083/jcb.97.1.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang T, Cui J, Li L, Hitchcock PF, Li Y. The role of microglia in the neurogenesis of zebrafish retina. Biochemical and Biophysical Research Communications. 2012;421(2):214–220. doi: 10.1016/j.bbrc.2012.03.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Checchin D, Sennlaub F, Levavasseur E, Leduc M, Chemtob S. Potential role of microglia in retinal blood vessel formation. Investigative Ophthalmology and Visual Science. 2006;47(8):3595–3602. doi: 10.1167/iovs.05-1522. [DOI] [PubMed] [Google Scholar]
- 45.Santos AM, Calvente R, Tassi M, et al. Embryonic and postnatal development of microglial cells in the mouse retina. Journal of Comparative Neurology. 2008;506(2):224–239. doi: 10.1002/cne.21538. [DOI] [PubMed] [Google Scholar]
- 46.Diaz-Araya CM, Provis JM, Penfold PL, Billson FA. Development of microglial topography in human retina. Journal of Comparative Neurology. 1995;363(1):53–68. doi: 10.1002/cne.903630106. [DOI] [PubMed] [Google Scholar]
- 47.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414(6866):916–920. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 48.Fredholm BB, Ijzerman AP, Jacobson KA, Klotz K-, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacological Reviews. 2001;53(4):527–552. [PMC free article] [PubMed] [Google Scholar]
- 49.Luongo L, Guida F, Imperatore R, et al. The A1 adenosine receptor as a new player in microglia physiology. Glia. 2014;62(1):122–132. doi: 10.1002/glia.22592. [DOI] [PubMed] [Google Scholar]
- 50.Haselkorn ML, Shellington DK, Jackson EK, et al. Adenosine A1 receptor activation as a brake on the microglial response after experimental traumatic brain injury in mice. Journal of Neurotrauma. 2010;27(5):901–910. doi: 10.1089/neu.2009.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Choi D, Pennathur S, Perier C, et al. Ablation of the inflammatory enzyme myeloperoxidase mitigates features of Parkinson’s disease in mice. Journal of Neuroscience. 2005;25(28):6594–6600. doi: 10.1523/JNEUROSCI.0970-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee JY, Jhun BS, Oh YT, et al. Activation of adenosine A3 receptor suppresses lipopolysaccharide-induced TNF-α production through inhibition of PI 3-kinase/Akt and NF-κB activation in murine BV2 microglial cells. Neuroscience Letters. 2006;396(1):1–6. doi: 10.1016/j.neulet.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 53.Cunha RA. Neuroprotection by adenosine in the brain: from A1 receptor activation to A2A receptor blockade. Purinergic Signalling. 2005;1(2):111–134. doi: 10.1007/s11302-005-0649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gomes CV, Kaster MP, Tomé AR, Agostinho PM, Cunha RA. Adenosine receptors and brain diseases: neuroprotection and neurodegeneration. Biochimica et Biophysica Acta. 2011;1808(5):1380–1399. doi: 10.1016/j.bbamem.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 55.Chen J, Sonsalla PK, Pedata F, et al. Adenosine A2A receptors and brain injury: broad spectrum of neuroprotection, multifaceted actions and “fine tuning” modulation. Progress in Neurobiology. 2007;83(5):310–331. doi: 10.1016/j.pneurobio.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 56.Gomes C, Ferreira R, George J, et al. Activation of microglial cells triggers a release of brain-derived neurotrophic factor (BDNF) inducing their proliferation in an adenosine A2A receptor-dependent manner: A2A receptor blockade prevents BDNF release and proliferation of microglia. Journal of Neuroinflammation. 2013;10, article 16 doi: 10.1186/1742-2094-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Saura J, Angulo E, Ejarque A, et al. Adenosine A2A receptor stimulation potentiates nitric oxide release by activated microglia. Journal of Neurochemistry. 2005;95(4):919–929. doi: 10.1111/j.1471-4159.2005.03395.x. [DOI] [PubMed] [Google Scholar]
- 58.Orr AG, Orr AL, Li X, Gross RE, Traynelis SF. Adenosine A2A receptor mediates microglial process retraction. Nature Neuroscience. 2009;12(7):872–878. doi: 10.1038/nn.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dai SS, Zhou YG, Li W, et al. Local glutamate level dictates adenosine A2A receptor regulation of neuroinflammation and traumatic brain injury. The Journal of Neuroscience. 2010;30(16):5802–5810. doi: 10.1523/JNEUROSCI.0268-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Albasanz JL, Perez S, Barrachina M, Ferrer I, Martín M. Up-regulation of adenosine receptors in the frontal cortex in Alzheimer's disease. Brain Pathology. 2008;18(2):211–219. doi: 10.1111/j.1750-3639.2007.00112.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holtzman DM, Morris JC, Goate AM. Alzheimer’s disease: the challenge of the second century. Science Translational Medicine. 2011;3(77) doi: 10.1126/scitranslmed.3002369.77sr1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Research Reviews. 2000;33(1):95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- 63.Gendron TF, Petrucelli L. The role of tau in neurodegeneration. Molecular Neurodegeneration. 2009;4(1, article 13) doi: 10.1186/1750-1326-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Selkoe DJ, Schenk D. Alzheimer's disease: molecular understanding predicts amyloid-based therapeutics. Annual Review of Pharmacology and Toxicology. 2003;43:545–584. doi: 10.1146/annurev.pharmtox.43.100901.140248. [DOI] [PubMed] [Google Scholar]
- 65.McGeer PL, McGeer EG. Glial reactions in Parkinson’s disease. Movement Disorders. 2008;23(4):474–483. doi: 10.1002/mds.21751. [DOI] [PubMed] [Google Scholar]
- 66.Hirsch EC, Hunot S. Neuroinflammation in Parkinson's disease: a target for neuroprotection? The Lancet Neurology. 2009;8(4):382–397. doi: 10.1016/S1474-4422(09)70062-6. [DOI] [PubMed] [Google Scholar]
- 67.Nagatsu T, Sawada M. Inflammatory process in Parkinson’s disease: role for cytokines. Current Pharmaceutical Design. 2005;11(8):999–1016. doi: 10.2174/1381612053381620. [DOI] [PubMed] [Google Scholar]
- 68.Gómez-Tortosa E, Newell K, Irizarry MC, Albert M, Growdon JH, Hyman BT. Clinical and quantitative pathologic correlates of dementia with Lewy bodies. Neurology. 1999;53(6):1284–1291. doi: 10.1212/wnl.53.6.1284. [DOI] [PubMed] [Google Scholar]
- 69.Masliah E, Rockenstein E, Veinbergs I, et al. Dopaminergic loss and inclusion body formation in α-synuclein mice: implications for neurodegenerative disorders. Science. 2000;287(5456):1265–1269. doi: 10.1126/science.287.5456.1265. [DOI] [PubMed] [Google Scholar]
- 70.Yankner BA, Lu T. Amyloid β-protein toxicity and the pathogenesis of Alzheimer disease. The Journal of Biological Chemistry. 2009;284(8):4755–4759. doi: 10.1074/jbc.R800018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McGeer PL, Itagaki S, Tago H, McGeer EG. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neuroscience Letters. 1987;79(1-2):195–200. doi: 10.1016/0304-3940(87)90696-3. [DOI] [PubMed] [Google Scholar]
- 72.McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology. 1988;38(8):1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- 73.More SV, Kumar H, Kim IS, Song SY, Choi DK. Cellular and molecular mediators of neuroinflammation in the pathogenesis of Parkinson's disease. Mediators of Inflammation. 2013;2013:12 pages. doi: 10.1155/2013/952375.952375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Su X, Federoff HJ, Maguire-Zeiss KA. Mutant α-synuclein overexpression mediates early proinflammatory activity. Neurotoxicity Research. 2009;16(3):238–254. doi: 10.1007/s12640-009-9053-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ouchi Y, Yoshikawa E, Sekine Y, et al. Microglial activation and dopamine terminal loss in early Parkinson's disease. Annals of Neurology. 2005;57(2):168–175. doi: 10.1002/ana.20338. [DOI] [PubMed] [Google Scholar]
- 76.Gerhard A, Pavese N, Hotton G, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson's disease. Neurobiology of Disease. 2006;21(2):404–412. doi: 10.1016/j.nbd.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 77.Schuitemaker A, Kropholler MA, Boellaard R, et al. Microglial activation in Alzheimer's disease: an (R)-[11C]PK11195 positron emission tomography study. Neurobiology of Aging. 2013;34(1):128–136. doi: 10.1016/j.neurobiolaging.2012.04.021. [DOI] [PubMed] [Google Scholar]
- 78.Okello A, Edison P, Archer HA, et al. Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology. 2009;72(1):56–62. doi: 10.1212/01.wnl.0000338622.27876.0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Edison P, Archer HA, Gerhard A, et al. Microglia, amyloid, and cognition in Alzheimer’s disease: an [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiology of Disease. 2008;32(3):412–419. doi: 10.1016/j.nbd.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 80.Zhang W, Wang T, Pei Z, et al. Aggregated α-synuclein activates microglia: a process leading to disease progression in Parkinson's disease. The FASEB Journal. 2005;19(6):533–542. doi: 10.1096/fj.04-2751com. [DOI] [PubMed] [Google Scholar]
- 81.Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE. Inflammatory mechanisms in Alzheimer's disease: inhibition of β-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARγ agonists. Journal of Neuroscience. 2000;20(2):558–567. doi: 10.1523/JNEUROSCI.20-02-00558.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Qin L, Liu Y, Cooper C, Liu B, Wilson B, Hong J. Microglia enhance β-amyloid peptide-induced toxicity in cortical and mesencephalic neurons by producing reactive oxygen species. Journal of Neurochemistry. 2002;83(4):973–983. doi: 10.1046/j.1471-4159.2002.01210.x. [DOI] [PubMed] [Google Scholar]
- 83.Ii M, Sunamoto M, Ohnishi K, Ichimori Y. β-Amyloid protein-dependent nitric oxide production from microglial cells and neurotoxicity. Brain Research. 1996;720(1-2):93–100. doi: 10.1016/0006-8993(96)00156-4. [DOI] [PubMed] [Google Scholar]
- 84.Meda L, Cassatella MA, Szendrei GI, et al. Activation of microglial cells by β-amyloid protein and interferon-γ . Nature. 1995;374(6523):647–650. doi: 10.1038/374647a0. [DOI] [PubMed] [Google Scholar]
- 85.McCarty MF. Down-regulation of microglial activation may represent a practical strategy for combating neurodegenerative disorders. Medical Hypotheses. 2006;67(2):251–269. doi: 10.1016/j.mehy.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 86.Wu D, Teismann P, Tieu K, et al. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(10):6145–6150. doi: 10.1073/pnas.0937239100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hunot S, Boissière F, Faucheux B, et al. Nitric oxide synthase and neuronal vulnerability in Parkinson's disease. Neuroscience. 1996;72(2):355–363. doi: 10.1016/0306-4522(95)00578-1. [DOI] [PubMed] [Google Scholar]
- 88.Knott C, Stern G, Wilkin GP. Inflammatory regulators in Parkinson’s disease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2. Molecular and Cellular Neuroscience. 2000;16(6):724–739. doi: 10.1006/mcne.2000.0914. [DOI] [PubMed] [Google Scholar]
- 89.Lee Mosley R, Benner EJ, Kadiu I, et al. Neuroinflammation, oxidative stress, and the pathogenesis of Parkinson's disease. Clinical Neuroscience Research. 2006;6(5):261–281. doi: 10.1016/j.cnr.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.de Lella Ezcurra AL, Chertoff M, Ferrari C, Graciarena M, Pitossi F. Chronic expression of low levels of tumor necrosis factor-α in the substantia nigra elicits progressive neurodegeneration, delayed motor symptoms and microglia/macrophage activation. Neurobiology of Disease. 2010;37(3):630–640. doi: 10.1016/j.nbd.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 91.Nagatsu T, Mogi M, Ichinose H, Togari A. Changes in cytokines and neurotrophins in Parkinson's disease. Journal of Neural Transmission, Supplement. 2000;(60):277–290. doi: 10.1007/978-3-7091-6301-6_19. [DOI] [PubMed] [Google Scholar]
- 92.Teismann P, Tieu K, Choi DK, et al. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(9):5473–5478. doi: 10.1073/pnas.0837397100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hunot S, Vila M, Teismann P, et al. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson's disease. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(2):665–670. doi: 10.1073/pnas.0307453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sánchez-Pernaute R, Ferree A, Cooper O, Yu M, Brownell A, Isacson O. Selective COX-2 inhibition prevents progressive dopamine neuron degeneration in a rat model of Parkinson's disease. Journal of Neuroinflammation. 2004;1, article 6 doi: 10.1186/1742-2094-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liberatore GT, Jackson-Lewis V, Vukosavic S, et al. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nature Medicine. 1999;5(12):1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- 96.Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson's disease brains. Acta Neuropathologica. 2003;106(6):518–526. doi: 10.1007/s00401-003-0766-2. [DOI] [PubMed] [Google Scholar]
- 97.Croisier E, Moran LB, Dexter DT, Pearce RKB, Graeber MB. Microglial inflammation in the parkinsonian substantia nigra: relationship to α-synuclein deposition. Journal of Neuroinflammation. 2005;2, article 14 doi: 10.1186/1742-2094-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wu DC, Jackson-Lewis V, Vila M, et al. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. Journal of Neuroscience. 2002;22(5):1763–1771. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Paresce DM, Chung H, Maxfield FR. Slow degradation of aggregates of the Alzheimer's disease amyloid β- protein by microglial cells. The Journal of Biological Chemistry. 1997;272(46):29390–29397. doi: 10.1074/jbc.272.46.29390. [DOI] [PubMed] [Google Scholar]
- 100.Walker DG, Lue L-F. Investigations with cultured human microglia on pathogenic mechanisms of Alzheimer's disease and other neurodegenerative diseases. Journal of Neuroscience Research. 2005;81(3):412–425. doi: 10.1002/jnr.20484. [DOI] [PubMed] [Google Scholar]
- 101.Majumdar A, Cruz D, Asamoah N, et al. Activation of microglia acidifies lysosomes and leads to degradation of Alzheimer amyloid fibrils. Molecular Biology of the Cell. 2007;18(4):1490–1496. doi: 10.1091/mbc.E06-10-0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Krabbe G, Halle A, Matyash V, et al. Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS ONE. 2013;8(4) doi: 10.1371/journal.pone.0060921.e60921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.El Khoury J, Toft M, Hickman SE, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nature Medicine. 2007;13(4):432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- 104.Canas PM, Porciúncula LO, Cunha GMA, et al. Adenosine A2A receptor blockade prevents synaptotoxicity and memory dysfunction caused by β-amyloid peptides via p38 mitogen-activated protein kinase pathway. Journal of Neuroscience. 2009;29(47):14741–14751. doi: 10.1523/JNEUROSCI.3728-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rebola N, Simões AP, Canas PM, et al. Adenosine A2A receptors control neuroinflammation and consequent hippocampal neuronal dysfunction. Journal of Neurochemistry. 2011;117(1):100–111. doi: 10.1111/j.1471-4159.2011.07178.x. [DOI] [PubMed] [Google Scholar]
- 106.Minghetti L, Greco A, Potenza RL, et al. Effects of the adenosine A2A receptor antagonist SCH 58621 on cyclooxygenase-2 expression, glial activation, and brain-derived neurotrophic factor availability in a rat model of striatal neurodegeneration. Journal of Neuropathology and Experimental Neurology. 2007;66(5):363–371. doi: 10.1097/nen.0b013e3180517477. [DOI] [PubMed] [Google Scholar]
- 107.Di Virgilio F, Ceruti S, Bramanti P, Abbracchio MP. Purinergic signalling in inflammation of the central nervous system. Trends in Neurosciences. 2009;32(2):79–87. doi: 10.1016/j.tins.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 108.Yu L, Shen H, Coelho JE, et al. Adenosine A2A receptor antagonists exert motor and neuroprotective effects by distinct cellular mechanisms. Annals of Neurology. 2008;63(3):338–346. doi: 10.1002/ana.21313. [DOI] [PubMed] [Google Scholar]
- 109.Simões AP, Duarte JA, Agasse F, et al. Blockade of adenosine A2A receptors prevents interleukin-1β-induced exacerbation of neuronal toxicity through a p38 mitogen-activated protein kinase pathway. Journal of Neuroinflammation. 2012;9(article 204) doi: 10.1186/1742-2094-9-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Stone TW, Behan WMH. Interleukin-1β but not tumor necrosis factor-α potentiates neuronal damage by quinolinic acid: protection by an adenosine A2A receptor antagonist. Journal of Neuroscience Research. 2007;85(5):1077–1085. doi: 10.1002/jnr.21212. [DOI] [PubMed] [Google Scholar]
- 111.Fredholm BB, Chen JF, Cunha RA, Svenningsson P, Vaugeois JM. Adenosine and brain function. International Review of Neurobiology. 2005;63:191–270. doi: 10.1016/S0074-7742(05)63007-3. [DOI] [PubMed] [Google Scholar]
- 112.Arendash GW, Mori T, Cao C, et al. Caffeine reverses cognitive impairment and decreases brain amyloid-β levels in aged Alzheimer's disease mice. Journal of Alzheimer's Disease. 2009;17(3):661–680. doi: 10.3233/JAD-2009-1087. [DOI] [PubMed] [Google Scholar]
- 113.Cao C, Cirrito JR, Lin X, et al. Caffeine suppresses amyloid-β levels in plasma and brain of Alzheimerls disease transgenic mice. Journal of Alzheimer's Disease. 2009;17(3):681–697. doi: 10.3233/JAD-2009-1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chu YF, Chang WH, Black RM, et al. Crude caffeine reduces memory impairment and amyloid β(1–42) levels in an Alzheimer's mouse model. Food Chemistry. 2012;135(3):2095–2102. doi: 10.1016/j.foodchem.2012.04.148. [DOI] [PubMed] [Google Scholar]
- 115.Dall’Igna OP, Fett P, Gomes MW, Souza DO, Cunha RA, Lara DR. Caffeine and adenosine A2a receptor antagonists prevent β-amyloid (25–35)-induced cognitive deficits in mice. Experimental Neurology. 2007;203(1):241–245. doi: 10.1016/j.expneurol.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 116.Cunha RA, Agostinho PM. Chronic caffeine consumption prevents memory disturbance in different animal models of memory decline. Journal of Alzheimer's Disease. 2010;20(supplement 1):S95–S116. doi: 10.3233/JAD-2010-1408. [DOI] [PubMed] [Google Scholar]
- 117.Dall'Igna OP, Porciuncula LO, Souza DO, Cunha RA, Lara DR. Neuroprotection by caffeine and adenosine A2A receptor blockade of beta-amyloid neurotoxicity. British Journal of Pharmacology. 2003;138(7):1207–1209. doi: 10.1038/sj.bjp.0705185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Brothers HM, Marchalant Y, Wenk GL. Caffeine attenuates lipopolysaccharide-induced neuroinflammation. Neuroscience Letters. 2010;480(2):97–100. doi: 10.1016/j.neulet.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lynch MA. Long-term potentiation and memory. Physiological Reviews. 2004;84(1):87–136. doi: 10.1152/physrev.00014.2003. [DOI] [PubMed] [Google Scholar]
- 120.Angulo E, Casadó V, Mallol J, et al. A1 adenosine receptors accumulate in neurodegenerative structures in Alzheimer disease and mediate both amyloid precursor protein processing and tau phosphorylation and translocation. Brain Pathology. 2003;13(4):440–451. doi: 10.1111/j.1750-3639.2003.tb00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chen W, Wang H, Wei H, Gu S. Istradefylline, an adenosine A2A receptor antagonist, for patients with Parkinson's disease: a meta-analysis. Journal of the Neurological Sciences. 2013;324(1):21–28. doi: 10.1016/j.jns.2012.08.030. [DOI] [PubMed] [Google Scholar]
- 122.Morelli M, Carta AR, Jenner P. Adenosine A2A receptors and Parkinson's disease. Handbook of Experimental Pharmacology. 2009;193:589–615. doi: 10.1007/978-3-540-89615-9_18. [DOI] [PubMed] [Google Scholar]
- 123.Pierri M, Vaudano E, Sager T, Englund U. KW-6002 protects from MPTP induced dopaminergic toxicity in the mouse. Neuropharmacology. 2005;48(4):517–524. doi: 10.1016/j.neuropharm.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 124.Yadav S, Gupta SP, Srivastava G, Srivastava PK, Singh MP. Role of secondary mediators in caffeine-mediated neuroprotection in maneb- and paraquat-induced Parkinson's disease phenotype in the mouse. Neurochemical Research. 2012;37(4):875–884. doi: 10.1007/s11064-011-0682-0. [DOI] [PubMed] [Google Scholar]
- 125.Stayte S, Vissel B. Advances in non-dopaminergic treatments for Parkinson's disease. Frontiers in Neuroscience. 2014;8:p. 113. doi: 10.3389/fnins.2014.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chen JF, Xu K, Petzer JP, et al. Neuroprotection by caffeine and A2A adenosine receptor inactivation in a model of Parkinson’s disease. Journal of Neuroscience. 2001;21(10):p. RC143. doi: 10.1523/JNEUROSCI.21-10-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hernán MA, Takkouche B, Caamaño-Isorna F, Gestal-Otero JJ. A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson's disease. Annals of Neurology. 2002;52(3):276–284. doi: 10.1002/ana.10277. [DOI] [PubMed] [Google Scholar]
- 128.Costa J, Lunet N, Santos C, Santos J, Vaz-Carneiro A. Caffeine exposure and the risk of Parkinson's disease: a systematic review and meta-analysis of observational studiess. Journal of Alzheimer's Disease. 2010;20(supplement 1):S221–S238. doi: 10.3233/JAD-2010-091525. [DOI] [PubMed] [Google Scholar]
- 129.Liu R, Guo X, Park Y, et al. Caffeine intake, smoking, and risk of parkinson disease in men and women. American Journal of Epidemiology. 2012;175(11):1200–1207. doi: 10.1093/aje/kwr451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.van der Mark M, Nijssen PC, Vlaanderen J, et al. A case-control study of the protective effect of alcohol, coffee, and cigarette consumption on Parkinson disease risk: time-since-cessation modifies the effect of tobacco smoking. PLoS ONE. 2014;9(4) doi: 10.1371/journal.pone.0095297.e95297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sääksjärvi K, Knekt P, Rissanen H, Laaksonen MA, Reunanen A, Männistö S. Prospective study of coffee consumption and risk of Parkinson's disease. European Journal of Clinical Nutrition. 2008;62(7):908–915. doi: 10.1038/sj.ejcn.1602788. [DOI] [PubMed] [Google Scholar]
- 132.Ross GW, Abbott RD, Petrovitch H, et al. Association of coffee and caffeine intake with the risk of Parkinson disease. Journal of the American Medical Association. 2000;283(20):2674–2679. doi: 10.1001/jama.283.20.2674. [DOI] [PubMed] [Google Scholar]
- 133.Popat RA, Van Den Eeden SK, Tanner CM, et al. Coffee, ADORA2A, and CYP1A2: the caffeine connection in Parkinson's disease. European Journal of Neurology. 2011;18(5):756–765. doi: 10.1111/j.1468-1331.2011.03353.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gyoneva S, Shapiro L, Lazo C, et al. Adenosine A2A receptor antagonism reverses inflammation-induced impairment of microglial process extension in a model of Parkinson's disease. Neurobiology of Disease. 2014;67:191–202. doi: 10.1016/j.nbd.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.World Population Ageing 2013. New York, NY, USA: Department of Economic and Social Affairs, Population Division; 2013. [Google Scholar]
- 136.Dagnelie G. Age-related psychophysical changes and low vision. Investigative Ophthalmology & Visual Science. 2013;54(14):ORSF88–ORSF93. doi: 10.1167/iovs.13-12934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Resnikoff S, Pascolini D, Etya'ale D, et al. Global data on visual impairment in the year 2002. Bulletin of the World Health Organization. 2004;82(11):844–851. [PMC free article] [PubMed] [Google Scholar]
- 138.Cheung W, Guo L, Cordeiro MF. Neuroprotection in glaucoma: drug-based approaches. Optometry and Vision Science. 2008;85(6):E406–E416. doi: 10.1097/OPX.0b013e31817841e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.International Diabetes Federation. IDF Diabetes Atlas. 6th edition. Brussels, Belgium: International Diabetes Federation; 2013. [Google Scholar]
- 140.McKinnon SJ. The cell and molecular biology of glaucoma: common neurodegenerative pathways and relevance to glaucoma. Investigative Ophthalmology and Visual Science. 2012;53(5):2485–2487. doi: 10.1167/iovs.12-9483j. [DOI] [PubMed] [Google Scholar]
- 141.Sivak JM. The aging eye: common degenerative mechanisms between the Alzheimer's brain and retinal disease. Investigative Ophthalmology and Visual Science. 2013;54(1):871–880. doi: 10.1167/iovs.12-10827. [DOI] [PubMed] [Google Scholar]
- 142.Liou GI, Auchampach JA, Hillard CJ, et al. Mediation of cannabidiol anti-inflammation in the retina by equilibrative nucleoside transporter and A2A adenosine receptor. Investigative Ophthalmology and Visual Science. 2008;49(12):5526–5531. doi: 10.1167/iovs.08-2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Casson RJ, Chidlow G, Wood JPM, Crowston JG, Goldberg I. Definition of glaucoma: clinical and experimental concepts. Clinical and Experimental Ophthalmology. 2012;40(4):341–349. doi: 10.1111/j.1442-9071.2012.02773.x. [DOI] [PubMed] [Google Scholar]
- 144.Kersey T, Clement CI, Bloom P, Cordeiro MF. New trends in glaucoma risk, diagnosis & management. Indian Journal of Medical Research. 2013;137(4):659–668. [PMC free article] [PubMed] [Google Scholar]
- 145.Cordeiro MF, Levin LA. Clinical evidence for neuroprotection in glaucoma. American Journal of Ophthalmology. 2011;152(5):715–716. doi: 10.1016/j.ajo.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tezel G. The role of glia, mitochondria, and the immune system in glaucoma. Investigative Ophthalmology and Visual Science. 2009;50(3):1001–1012. doi: 10.1167/iovs.08-2717. [DOI] [PubMed] [Google Scholar]
- 147.Baltmr A, Duggan J, Nizari S, Salt TE, Cordeiro MF. Neuroprotection in glaucoma: is there a future role? Experimental Eye Research. 2010;91(5):554–566. doi: 10.1016/j.exer.2010.08.009. [DOI] [PubMed] [Google Scholar]
- 148.Naskar R, Wissing M, Thanos S. Detection of early neuron degeneration and accompanying microglial responses in the retina of a rat model of glaucoma. Investigative Ophthalmology and Visual Science. 2002;43(9):2962–2968. [PubMed] [Google Scholar]
- 149.Sappington RM, Chan M, Calkins DJ. Interleukin-6 protects retinal ganglion cells from pressure-induced death. Investigative Ophthalmology and Visual Science. 2006;47(7):2932–2942. doi: 10.1167/iovs.05-1407. [DOI] [PubMed] [Google Scholar]
- 150.Tezel G. TNF-α signaling in glaucomatous neurodegeneration. Progress in Brain Research. 2008;173:409–421. doi: 10.1016/S0079-6123(08)01128-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zhou X, Li F, Kong L, Tomita H, Li C, Cao W. Involvement of inflammation, degradation, and apoptosis in a mouse model of glaucoma. The Journal of Biological Chemistry. 2005;280(35):31240–31248. doi: 10.1074/jbc.M502641200. [DOI] [PubMed] [Google Scholar]
- 152.Bosco A, Steele MR, Vetter ML. Early microglia activation in a mouse model of chronic glaucoma. Journal of Comparative Neurology. 2011;519(4):599–620. doi: 10.1002/cne.22516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Taylor S, Calder CJ, Albon J, Erichsen JT, Boulton ME, Morgan JE. Involvement of the CD200 receptor complex in microglia activation in experimental glaucoma. Experimental Eye Research. 2011;92(5):338–343. doi: 10.1016/j.exer.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.de Hoz R, Gallego BI, Ramirez AI, et al. Rod-like microglia are restricted to eyes with laser-induced ocular hypertension but absent from the microglial changes in the contralateral untreated eye. PLoS ONE. 2013;8(12) doi: 10.1371/journal.pone.0083733.e83733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. Journal of Immunology. 2001;166(12):7527–7533. doi: 10.4049/jimmunol.166.12.7527. [DOI] [PubMed] [Google Scholar]
- 156.Bosco A, Inman DM, Steele MR, et al. Reduced retina microglial activation and improved optic nerve integrity with minocycline treatment in the DBA/2J mouse model of glaucoma. Investigative Ophthalmology and Visual Science. 2008;49(4):1437–1446. doi: 10.1167/iovs.07-1337. [DOI] [PubMed] [Google Scholar]
- 157.Fong DS, Aiello LP, Ferris FL, III, Klein R. Diabetic retinopathy. Diabetes Care. 2004;27(10):2540–2553. doi: 10.2337/diacare.27.10.2540. [DOI] [PubMed] [Google Scholar]
- 158.Santiago AR, Cristóvão AJ, Santos PF, Carvalho CM, Ambrósio AF. High glucose induces caspase-independent cell death in retinal neural cells. Neurobiology of Disease. 2007;25(3):464–472. doi: 10.1016/j.nbd.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 159.Barber AJ. A new view of diabetic retinopathy: a neurodegenerative disease of the eye. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2003;27(2):283–290. doi: 10.1016/S0278-5846(03)00023-X. [DOI] [PubMed] [Google Scholar]
- 160.Barber AJ, Lieth E, Khin SA, Antonetti DA, Buchanan AG, Gardner TW. Neural apoptosis in the retina during experimental and human diabetes: early onset and effect of insulin. Journal of Clinical Investigation. 1998;102(4):783–791. doi: 10.1172/JCI2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.El Asrar AMA, Maimone D, Morse PH, Gregory S, Reder AT. Cytokines in the vitreous of patients with proliferative diabetic retinopathy. American Journal of Ophthalmology. 1992;114(6):731–736. doi: 10.1016/s0002-9394(14)74052-8. [DOI] [PubMed] [Google Scholar]
- 162.Yuuki T, Kanda T, Kimura Y, et al. Inflammatory cytokines in vitreous fluid and serum of patients with diabetic vitreoretinopathy. Journal of Diabetes and Its Complications. 2001;15(5):257–259. doi: 10.1016/s1056-8727(01)00155-6. [DOI] [PubMed] [Google Scholar]
- 163.Patel JI, Saleh GM, Hykin PG, Gregor ZJ, Cree IA. Concentration of haemodynamic and inflammatory related cytokines in diabetic retinopathy. Eye. 2008;22(2):223–228. doi: 10.1038/sj.eye.6702584. [DOI] [PubMed] [Google Scholar]
- 164.Schram MT, Chaturvedi N, Schalkwijk CG, Fuller JH, Stehouwer CDA. Markers of inflammation are cross-sectionally associated with microvascular complications and cardiovascular disease in type 1 diabetes—the EURODIAB Prospective Complications Study. Diabetologia. 2005;48(2):370–378. doi: 10.1007/s00125-004-1628-8. [DOI] [PubMed] [Google Scholar]
- 165.Demircan N, Safran BG, Soylu M, Ozcan AA, Sizmaz S. Determination of vitreous interleukin-1 (IL-1) and tumour necrosis factor (TNF) levels in proliferative diabetic retinopathy. Eye. 2006;20(12):1366–1369. doi: 10.1038/sj.eye.6702138. [DOI] [PubMed] [Google Scholar]
- 166.Gustavsson C, Agardh E, Bengtsson B, Agardh CD. TNF-α is an independent serum marker for proliferative retinopathy in type 1 diabetic patients. Journal of Diabetes and Its Complications. 2008;22(5):309–316. doi: 10.1016/j.jdiacomp.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 167.Carmo A, Cunha-Vaz JG, Carvalho AP, Lopes MC. L-arginine transport in retinas from streptozotocin diabetic rats: Correlation with the level of IL-1β and NO synthase activity. Vision Research. 1999;39(23):3817–3823. doi: 10.1016/s0042-6989(99)00117-0. [DOI] [PubMed] [Google Scholar]
- 168.Kowluru RA, Odenbach S. Role of interleukin-1β in the pathogenesis of diabetic retinopathy. British Journal of Ophthalmology. 2004;88(10):1343–1347. doi: 10.1136/bjo.2003.038133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Gerhardinger C, Costa MB, Coulombe MC, Toth I, Hoehn T, Grosu P. Expression of acute-phase response proteins in retinal Müller cells in diabetes. Investigative Ophthalmology and Visual Science. 2005;46(1):349–357. doi: 10.1167/iovs.04-0860. [DOI] [PubMed] [Google Scholar]
- 170.Krady JK, Basu A, Allen CM, et al. Minocycline reduces proinflammatory cytokine expression, microglial activation, and caspase-3 activation in a rodent model of diabetic retinopathy. Diabetes. 2005;54(5):1559–1565. doi: 10.2337/diabetes.54.5.1559. [DOI] [PubMed] [Google Scholar]
- 171.Joussen AM, Poulaki V, Mitsiades N, et al. Nonsteroidal anti-inflammatory drugs prevent early diabetic retinopathy via TNF-alpha suppression. The FASEB Journal. 2002;16(3):438–440. doi: 10.1096/fj.01-0707fje. [DOI] [PubMed] [Google Scholar]
- 172.Behl Y, Krothapalli P, Desta T, DiPiazza A, Roy S, Graves DT. Diabetes-enhanced tumor necrosis factor-α production promotes apoptosis and the loss of retinal microvascular cells in type 1 and type 2 models of diabetic retinopathy. The American Journal of Pathology. 2008;172(5):1411–1418. doi: 10.2353/ajpath.2008.071070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Gustavsson C, Agardh E, Bengtsson B, Agardh CD. TNF-α is an independent serum marker for proliferative retinopathy in type 1 diabetic patients. Journal of Diabetes and Its Complications. 2003;44(5):2184–2191. doi: 10.1016/j.jdiacomp.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 174.Leal EC, Manivannan A, Hosoya K, et al. Inducible nitric oxide synthase isoform is a key mediator of leukostasis and blood-retinal barrier breakdown in diabetic retinopathy. Investigative Ophthalmology and Visual Science. 2007;48(11):5257–5265. doi: 10.1167/iovs.07-0112. [DOI] [PubMed] [Google Scholar]
- 175.Aveleira CA, Lin CM, Abcouwer SF, Ambrósio AF, Antonetti DA. TNF-α signals through PKCζ/NF-κB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes. 2010;59(11):2872–2882. doi: 10.2337/db09-1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Costa GN, Vindeirinho J, Cavadas C, Ambrósio AF, Santos PF. Contribution of TNF receptor 1 to retinal neural cell death induced by elevated glucose. Molecular and Cellular Neuroscience. 2012;50(1):113–123. doi: 10.1016/j.mcn.2012.04.003. [DOI] [PubMed] [Google Scholar]
- 177.Zeng H, Green WR, Tso MOM. Microglial activation in human diabetic retinopathy. Archives of Ophthalmology. 2008;126(2):227–232. doi: 10.1001/archophthalmol.2007.65. [DOI] [PubMed] [Google Scholar]
- 178.Barber AJ, Antonetti DA, Kern TS, et al. The Ins2Akita mouse as a model of early retinal complications in diabetes. Investigative Ophthalmology and Visual Science. 2005;46(6):2210–2218. doi: 10.1167/iovs.04-1340. [DOI] [PubMed] [Google Scholar]
- 179.Ibrahim AS, El-Remessy AB, Matragoon S, et al. Retinal microglial activation and inflammation induced by amadori-glycated albumin in a rat model of diabetes. Diabetes. 2011;60(4):1122–1133. doi: 10.2337/db10-1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Zeng XX, Ng YK, Ling EA. Neuronal and microglial response in the retina of streptozotocin-induced diabetic rats. Visual Neuroscience. 2000;17(3):463–471. doi: 10.1017/s0952523800173122. [DOI] [PubMed] [Google Scholar]
- 181.Omri S, Behar-Cohen F, de Kozak Y, et al. Microglia/macrophages migrate through retinal epithelium barrier by a transcellular route in diabetic retinopathy: role of PKCzeta in the Goto Kakizaki rat model. The American Journal of Pathology. 2011;179(2):942–953. doi: 10.1016/j.ajpath.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Osborne NN, Casson RJ, Wood JPM, Chidlow G, Graham M, Melena J. Retinal ischemia: mechanisms of damage and potential therapeutic strategies. Progress in Retinal and Eye Research. 2004;23(1):91–147. doi: 10.1016/j.preteyeres.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 183.Lam TT, Abler AS, Tso MOM. Apoptosis and caspases after ischemia-reperfusion injury in rat retina. Investigative Ophthalmology and Visual Science. 1999;40(5):967–975. [PubMed] [Google Scholar]
- 184.Quigley HA. Neuronal death in glaucoma. Progress in Retinal and Eye Research. 1999;18(1):39–57. doi: 10.1016/s1350-9462(98)00014-7. [DOI] [PubMed] [Google Scholar]
- 185.Osborne NN, Melena J, Chidlow G, Wood JPM. A hypothesis to explain ganglion cell death caused by vascular insults at the optic nerve head: possible implication for the treatment of glaucoma. The British Journal of Ophthalmology. 2001;85(10):1252–1259. doi: 10.1136/bjo.85.10.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ghiardi GJ, Gidday JM, Roth S. The purine nucleoside adenosine in retinal ischemia-reperfusion injury. Vision Research. 1999;39(15):2519–2535. doi: 10.1016/s0042-6989(99)00038-3. [DOI] [PubMed] [Google Scholar]
- 187.Li B, Rosenbaum PS, Jennings NM, Maxwell KM, Roth S. Differing roles of adenosine receptor subtypes in retinal ischemia- reperfusion injury in the rat. Experimental Eye Research. 1999;68(1):9–17. doi: 10.1006/exer.1998.0573. [DOI] [PubMed] [Google Scholar]
- 188.Konno T, Sato A, Uchibori T, Nagai A, Kogi K, Nakahata N. Adenosine A2A receptor mediated protective effect of 2-(6-cyano-1-hexyn-1-yl)adenosine on retinal ischaemia/reperfusion damage in rats. British Journal of Ophthalmology. 2006;90(7):900–905. doi: 10.1136/bjo.2006.091496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Steinsapir KD, Goldberg RA. Traumatic optic neuropathy. Survey of Ophthalmology. 1994;38(6):487–518. doi: 10.1016/0039-6257(94)90145-7. [DOI] [PubMed] [Google Scholar]
- 190.Ahmad S, Fatteh N, El-Sherbiny NM, et al. Potential role of A2A adenosine receptor in traumatic optic neuropathy. Journal of Neuroimmunology. 2013;264(1-2):54–64. doi: 10.1016/j.jneuroim.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 191.Garcia-Valenzuela E, Sharma SC. Laminar restriction of retinal macrophagic response to optic nerve axotomy in the rat. Journal of Neurobiology. 1999;40(1):55–66. [PubMed] [Google Scholar]
- 192.Zhang C, Tso MOM. Characterization of activated retinal microglia following optic axotomy. Journal of Neuroscience Research. 2003;73(6):840–845. doi: 10.1002/jnr.10713. [DOI] [PubMed] [Google Scholar]
- 193.Wohl SG, Schmeer CW, Witte OW, Isenmann S. Proliferative response of microglia and macrophages in the adult mouse eye after optic nerve lesion. Investigative Ophthalmology and Visual Science. 2010;51(5):2686–2696. doi: 10.1167/iovs.09-4537. [DOI] [PubMed] [Google Scholar]
- 194.Vindeirinho J, Costa GN, Correia MB, Cavadas C, Santos PF. Effect of diabetes/hyperglycemia on the rat retinal adenosinergic system. PLoS ONE. 2013;8(6) doi: 10.1371/journal.pone.0067499.e67499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Elsherbiny NM, Ahmad S, Naime M, et al. ABT-702, an adenosine kinase inhibitor, attenuates inflammation in diabetic retinopathy. Life Sciences. 2013;93(2-3):78–88. doi: 10.1016/j.lfs.2013.05.024. [DOI] [PubMed] [Google Scholar]
- 196.Madeira M, Elvas F, Martins J, Ambrósio AF, Santiago AR. denosine A2AR blockade prevents retinal microglial activation. Proceedings of the 22nd IUBMB 37th FEBS Congress; 2012; Seville, Spain. Blackwell; [Google Scholar]