

Abstract
Objective
We evaluated the association of single nucleotide polymorphisms (SNPs) in TLRs with infant HIV-1 acquisition and viral control.
Design
Infant HIV-1 outcomes were assessed in a Kenyan perinatal HIV-1 cohort.
Methods
Infants were genotyped for six candidate and 118 haplotype-tagging polymorphisms in TLRs 2, 3, 4, 7, 8, and 9, MYD88 and TIRAP. Cox proportional hazards and linear regression were performed to assess associations with time to HIV-1 acquisition, time to infant mortality, and peak viral load (VL).
Results
Among 368 infants, 56 (15%) acquired HIV-1 by month 1 and 17 (4.6%) between 1 and 12 months. Infants with the TLR9 1635A (rs352140) variant were more likely to acquire HIV-1 by 1 month (HR=1.81, 95% confidence interval [CI] =1.05-3.14, p=0.033) and by 12 months (HR=1.62, CI=1.01-2.60, p=0.044) in dominant models adjusted for maternal plasma HIV-1 RNA level and genetic ancestry. Among 56 infants infected at ≤1 month of age, ≥1 copy of the TLR9 1635A allele was associated with a 0.58 log10 c/ml lower peak VL (p=0.002). Female infants with ≥1 copy of the TLR8 1G (rs3764880) variant had a 0.78 log10 c/ml higher peak VL (p=0.0009) and having ≥1 copy of the C allele for a haplotype tagging TLR7 variant (rs1634319) was associated with a 0.80 log10 c/ml higher peak VL in female infants (p=0.0003).
Conclusions
In this African perinatal cohort, we found several TLR polymorphisms associated with HIV-1 acquisition and progression. Defining mechanisms for these TLR associations may inform HIV-1 prevention strategies that leverage innate responses.
Keywords: pediatric HIV, mother-to-child transmission, genetic epidemiology, HIV genetics, innate immunity, single nucleotide polymorphisms, toll-like receptors, TLRs
Background
Recent studies demonstrate that innate immune responses play a critical role in HIV-1 acquisition and control [1-3]. This may be especially true in infants whose adaptive responses are still under development. Pattern Recognition Receptors (PRRs) are key effectors of the innate response that also bridge to adaptive immune response pathways. PRRs recognize evolutionarily conserved pathogen-associated molecular patterns (PAMPs) and PAMP recognition triggers activation of signal transduction pathways and downstream effector responses [4].
Toll-like receptors (TLRs) were the first mammalian PRRs discovered and 10 TLR genes have been identified in humans [5, 6]. Each TLR recognizes specific PAMPs characteristic of fungi, bacteria, viruses and/or parasites; TLRs 1, 2, and 4-6 preferentially recognize bacterial and fungal PAMPs while TLRs 3 and 7-9 preferentially recognize viral nucleic acids [2, 7]. Although TLR2 and TLR4 predominantly recognize bacterial motifs, they may also recognize viral components [5, 7]. TLR3 recognizes double-stranded RNA; TLRs 7 and 8 bind single-stranded RNA, and TLR9 recognizes unmethylated cytidine-phosphate-guanine (CpG) DNA motifs of bacteria and viruses [6].
All TLRs contain an extracellular leucine-rich repeat domain and an intracellular Toll/IL-1 Receptor homology (TIR) domain that binds to adaptor molecules involved in TLR-associated signaling cascades [4, 6, 8]. Biologic responses to TLR PAMP recognition are dependent on which TIR domain containing adaptor molecules are recruited and which signaling pathways are initiated. The TIR domain containing adapter-inducing interferon-β (TRIF)-dependent pathway induces the production of type I interferons and results in antiviral and immunoregulatory responses [4, 9] while the myeloid differentiation factor 88 (MyD88)-dependent pathway induces the production of proinflammatory cytokines and chemokines and induction of genes involved in antiviral response [8]. TIR domain containing adaptor protein (TIRAP), another TLR-adaptor molecule, functions mainly in TLR4 and TLR2 signaling either independently or in combination with MyD88 in the MyD88-dependent signaling pathway to upregulate NF-κB and MAPKs [4, 8].
Because of their sentinel role in pathogen recognition and initiation of antiviral response, genetic variation in TLR and TLR-associated genes may influence HIV-1 acquisition and progression. Previous studies evaluating polymorphisms in TLR2-TLR4 and TLR7-TLR9 have shown that single nucleotide polymorphisms (SNPs) in TLR genes may contribute to differences in HIV-1 disease progression and acquisition [10-17]. Most notably, the TLR9 1635A/G variant has been associated with HIV-1 progression [12-14, 16]. However, the direction and strength of associations of this variant with HIV-1 progression differ between studies. The only study to date to evaluate associations between variants in TLR9 and HIV-1 acquisition found a higher risk of HIV-1 acquisition in European children carrying a haplotype that included TLR9 1635A/G [17]. Studies in adults have also reported differences in HIV-1 disease progression or HIV-1 virus levels associated with SNPs in TLR2 (597T/C), TLR4 (896A/G and 1196C/T), TLR7 (32A/T) and TLR8 (1A/G) [10, 11, 13, 14]. Studies evaluating TLR variant associations with the presence of HIV-1 infection found that the TLR7 32A/T variant was detected more frequently in HIV-1 infected women compared to uninfected women [10] and the TLR3 1234C/T variant was significantly overrepresented in HIV-1-exposed seronegative (HESN) individuals when compared to healthy controls [15]. Other studies have shown correlations between levels of TLR mRNA, TLR protein expression, and TLR protein function with HIV-1 disease progression and acquisition in adult cohorts, further supporting the potential importance of TLR genetic variations for HIV-1 outcomes [11, 15, 18, 19].
The role of the innate immune system generally, and PRRs specifically, in pediatric HIV-1 infection remain largely unstudied. Furthermore, few HIV genetic studies have been conducted in African populations. We tested whether polymorphisms in TLR and TLR-associated genes are associated with altered risk of infant HIV-1 acquisition or disease progression in a perinatal African cohort.
Methods
Study Population and Sample
Our study used biological samples and phenotypic data collected from a cohort of mother-infant pairs recruited and followed between 1999 and 2005. As previously described, this cohort included 510 HIV-1-infected pregnant women who were enrolled at ~32 weeks gestation and mother-infant pairs were followed up to 2 years postpartum [20-23]. Briefly, HIV-1 seropositive pregnant women received standard antenatal care and short course zidovudine (ZDV) from 34-36 weeks gestation through delivery for the prevention of mother-to-child transmission (MTCT) [24]. Women were counseled on safe infant-feeding practices and elected to either breastfeed or formula feed their infants. Infants were evaluated monthly during the first year of life. Infant HIV-1 status was determined by DNA PCR assays at 48 hours of birth and at 2 weeks and 1, 3, 6, 9, and 12 months of age. We limited the analysis to DNA samples linked to infants of known sex and for whom time to HIV-1 acquisition could be determined. All participants provided written informed consent for the primary research study and for use of samples and data in future research. The Kenyatta National Hospital Ethical Review Committee (ERC) and the University of Washington Institutional Review Board (IRB) specifically approved use of these biological samples and phenotypic data for this study.
Data Collection
Infant CD4 and CD8 percentages and lymphocyte counts were determined using a FACScan flow cytometer (Becton Dickinson, Franklin Lakes, NJ) at the University of Nairobi, Kenya. Viral load was measured at the Fred Hutchinson Cancer Research Center in Seattle Washington, USA. Specifically, plasma HIV-1 RNA levels were quantified using the Gen-Probe transcription-mediated amplification assay. Previous studies have demonstrated that this assay is appropriate for quantifying the HIV-1 A, C, and D subtypes that are prevalent in Kenya [25]. Infant peripheral blood mononuclear cells (PBMCs) were isolated from EDTA anti-coagulated blood using a Ficoll gradient (Lymphocyte Separation Medium; Organon, Teknika), washed in RPMI 1640 medium (Sigma-Aldrich), and counted using trypan blue staining under a hemocytometer. Cells were cryopreserved at the University of Nairobi and sent to the United States for DNA extraction.
Cryopreserved cells were thawed and washed in R-10 media (RPMI with 10% FCS) and 1% PBS (Sigma-Aldrich). DNA was extracted using the Gentra Puregene Blood Kit (Qiagen, Valencia, CA) and eluted in 20, 25, and 50μl of hydration buffer depending on estimated cell pellet size. DNA was quantified using a Spectra Max Gemina (Molecular Devices, Sunnyvale, CA) fluorimeter and the Quant-iT PicoGreen dsDNA Assay (Invitrogen, New York). Samples lacking 1μg of DNA (N=45) were whole genome amplified (WGA) using the REPLI-g Whole Genome Amplification Kit (Qiagen, Valencia, CA). Samples were genotyped using an Illumina Custom Oligo Pooled Assay (OPA) microarray platform (Illumina, Inc., San Diego, CA) designed for this study.
SNP Selection
We genotyped 124 SNPs in six TLR genes (TLRs 2, 3, 4, 7, 8 and 9) and two TLR-associated genes (MYD88 and TIRAP). SNPs were selected using haplotype tagging and candidate SNP approaches. Haplotype tagging SNPs (TagSNPs) were selected using the LDSelect algorithm available through the University of Washington’s genome variation server (http://www.gvs.gs.washington.edu/GVS/) using the r2 threshold value of 0.8 and a minor allele frequency (MAF) cutoff value of 5% in the Yoruba (YRI) HapMap population [26, 27]. TagSNPs were augmented with six candidate SNPs, selected based on previously identified associations between variants in TLRs and HIV-1 outcomes [10-14] (Supplemental Table 1). To assess population stratification, we genotyped 144 ancestry informative markers (AIMS) distinguishing Asian, European and African (West and East) ancestry.
Quality Control
We excluded samples in which there was a discrepancy between the reported and experimentally determined sex (n=5) or which had >10% missingness (n=24). SNPs were excluded from the analysis if they were monomorphic (n=2), had >10% missingness (n=8), or violated Hardy-Weinberg equilibrium (p<0.001) (n=1). TagSNPs with a MAF <5% in our sample population were also excluded (n=18). Overall, 29 of 397 infants and 29 of 118 SNPs did not meet quality control criteria and were excluded from this analysis (Supplemental Figure 1). In addition, five samples were genotyped in duplicate to estimate concordance rates in our genotyping platform. The genotype concordance rate was 100% for all successfully genotyped markers. Post-genotyping quality of WGA samples was evaluated and genotype frequencies for WGA and non-WGA samples were similar.
Data Analyses
Cox proportional hazards regression was performed to assess TLR polymorphism associations with time to HIV-1 acquisition by month 1 and month 12. Because viral load, CD4 count and ZDV use are collinear measures of maternal HIV-1 disease progression, genetic acquisition analyses were only adjusted for maternal viral load at 32 weeks gestation. Adjustment for population stratification was performed using the first principle component defined by EIGENSTRAT [28] analysis of genotyped AIMS.
Cox proportional hazards regression was performed to assess TLR polymorphism associations with time to infant mortality by 24 months in infants infected by month 1. Linear regression was performed to assess peak HIV-1 RNA levels in infected infants for infants infected by month 1 and month 12. All progression analyses were adjusted for genetic ancestry using the same method as the acquisition analysis.
All regression analyses were performed using Intercooled STATA 11.0 (College Station, USA). Sex-stratified analyses of the X chromosome genes, TLR7 and TLR8, were conducted. For analysis with candidate TLR SNPs previously shown to be associated with HIV-1-specific outcomes, we did not adjust for multiple comparisons. All TagSNPs were adjusted for multiple comparisons using a Bonferroni correction to account for the 89 quality-controlled SNPs. All analyses were first performed assuming an additive model of inheritance. All candidate SNP analyses and TagSNPs showing uncorrected significant (p<.05) associations in additive models were rerun using dominant and recessive models of inheritance and the model best representing the association was selected.
Results
Cohort characteristics
Among 510 mother-infant pairs initially enrolled in a perinatal HIV-1 transmission cohort, 368 had samples available, passed quality control procedures, and were included in this analysis. Characteristics of these mother-infant pairs are provided in Table 1. Among the infants, 56 (15%) acquired HIV-1 by 1 month of age and 17 (4.6%) acquired HIV-1 between 1 and 12 months of age. Maternal CD4 counts at 32 weeks gestation were significantly lower in those who transmitted HIV-1 compared to those who did not (406 vs. 483 cells/μl, p =0.011). Plasma HIV-1 RNA levels were significantly higher among women who transmitted HIV-1 compared to those who did not, both at 32 weeks gestation (5.11 vs. 4.56 log10 copies/ml, p <0.001) and at delivery (4.66 vs. 3.93 log10 copies/ml, p <0.001). Most women (89%) reported use of zidovudine for the prevention of mother-to-child transmission. Women who transmitted HIV-1 before one month were significantly less likely to report zidovudine use than those who did not transmit (OR=0.338, 95% confidence interval [CI]: 0.159, 0.717; p-value=0.005). A similar association was noted for 12-month transmission (OR=0.43, 95% CI: 0.2, 0.89; p=0.023).
Table 1.
Cohort Characteristics
| Characteristic | Total Cohort | Positive by M1 | Positive by M12 | |||
|---|---|---|---|---|---|---|
| N | Median (IQR) or Number (%) | N | Median (IQR) or Number (%) | N | Median (IQR) or Number (%) | |
| Maternal (Prenatal) | ||||||
| Maternal CD4+ T Cell Count (32 weeks gestation)* | 364 | 433 (306-619) | 56 | 420 (237-597) | 72 | 371 (237-558) |
| Maternal HIV-1 Plasma RNA Level (32 weeks gestation)** | 355 | 4.72 (4.19-5.24) | 54 | 5.20 (4.72-5.44) | 71 | 5.19 (4.72-5.54) |
| Maternal (Delivery and Postpartum) | ||||||
| Maternal CD4+ T Cell Count (1 month postpartum)* | 323 | 540 (370-731) | 49 | 469 (319-629) | 62 | 466 (311-604) |
| Maternal HIV-1 Plasma RNA Level (time of delivery)** | 293 | 4.12 (3.51-4.69) | 45 | 4.52 (4.02-5.09) | 61 | 4.65 (4.06-5.26) |
| ZDV During Pregnancy | 361 | 322 (89) | 54 | 42 (78) | 70 | 57 (81) |
| Infant | ||||||
| HIV Positive | 368 | 73 (20)* | 56 | 56 (100) | 73 | 73 (100) |
| Female | 368 | 178 (48) | 56 | 29 (52) | 73 | 33 (45) |
| Completed 12 Months of Follow-up | 368 | 296 (80) | 56 | 28 (50) | 73 | 42 (58) |
| Completed 24 Months of Follow-up | 368 | 41 (11) | 56 | 20 (35) | 73 | 28 (38) |
| Infant deaths (12 months) | 368 | 48 (13) | 56 | 25 (45) | 73 | 28 (38) |
| Infant deaths (24 months) | 368 | 62 (17) | 56 | 33 (59) | 73 | 39 (53) |
| Whole Genome Amplification | 368 | 45 (12) | 56 | 9 (16) | 73 | 11 (15) |
| Peak HIV-1 plasma RNA level** | - | - | 56 | 6.77 (6.43-7.41) | 73 | 6.71 (6.41-7.40) |
CD4+ T cell counts measured in cells/μl
HIV-1 plasma RNA levels measured in log10 copies/ml
Infants positive by 12 months of age
HIV-1 infected children experienced high rates of mortality with 45% of infants infected by 1 month of age dying before 12 months and 18% of infants infected between months 1 and 12 dying before reaching 12 months. Among the 56 infants HIV-1 infected at ≤1 month, the mean peak plasma HIV-1 RNA level was 6.8 log10 copies/ml (log10 c/mL) (Table 1).
HIV-1 Acquisition
The primary analysis endpoint was HIV-1 acquisition in infants. Infants with one or more copies of the candidate variant TLR9 1635A (rs352140) were more likely to acquire HIV-1 by 1 month (HR=1.81, 95% CI: 1.05, 3.14; p=0.033) and 12 months (HR=1.62, 95% CI: 1.01, 2.60; p=0.044) in dominant models adjusted for maternal plasma HIV-1 RNA levels and genetic ancestry (Figure 1). The TLR9 1635A allele association with time to HIV-1 acquisition remained significant at both time-points when evaluated using an additive model of inheritance and showed a trend for association at both acquisition time-points when assessed using a recessive model of inheritance. No other candidate or TLR, TIRAP or MyD88 haplotype tagging SNPs were significantly associated with HIV-1 acquisition in analyses adjusted for multiple comparisons (Supplemental Table 2).
Figure 1.
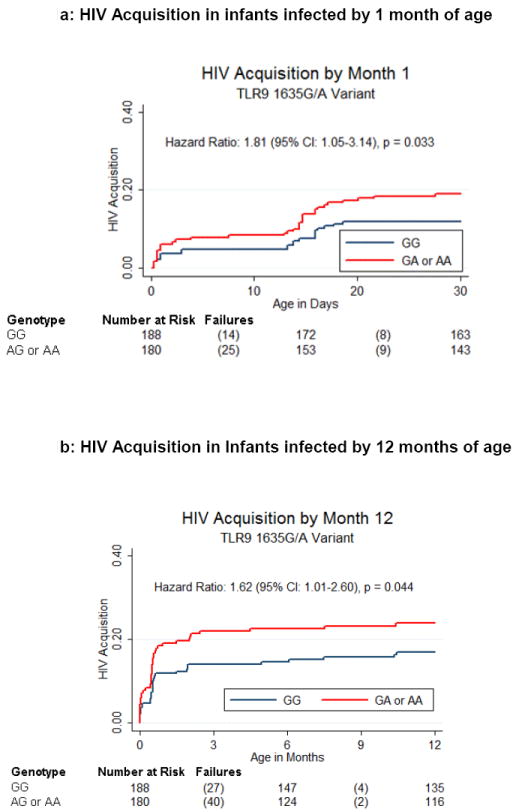
Time to HIV-1 Acquisition by TLR9 Genotype
Peak Viral Load
We found associations between TLR9 1635G/A (rs352140), TLR8 1A/G (rs3764880), and TLR7 rs1634319 variants and peak plasma HIV-1 RNA levels in HIV-1 infected infants (Figure 2 and Table 2). The presence of one or more copies of the TLR9 1635A allele was associated with 0.58 log10 c/ml lower peak plasma HIV-1 RNA levels (95% CI: -0.95, -0.22; p=0.002) in infants infected by month 1 and with 0.49 log10 c/ml lower peak plasma HIV-1 RNA levels (95% CI: -0.85, -0.12; p=0.009) in infants infected by month 12 using dominant models of inheritance. The association remained significant for both time points in additive models of inheritance and showed a trend for association with a recessive model of inheritance in infants infected by 1 month of age.
Figure 2.

SNP Variations Associated with Peak Plasma HIV-1 RNA Levels (log10 copies/ml) in Infants Infected with HIV-1 by 1 and 12 Months of Age*
NOTE: Log10 peak plasma HIV-1 RNA levels are displayed separately for infants who acquired HIV-1 by 1 month of age and all infants who acquired HIV-1 by 12 months of age. Each graph presents the mean and standard deviation of peak plasma HIV-1 RNA level stratified by genotype and the p-value corresponding to the mean difference. The number of individuals in each genotypic category is specified on the x-axis next to the genotype.
* Uncorrected p-values are displayed for the TagSNP TLR7 rs1634319
Table 2.
SNP Variations Associated with Peak Plasma HIV-1 RNA Levels in HIV-1 Infected Infants
| SNP | Model | Genotype | N (%) | Median Peak Viral Load (IQR) | Mean (SD) | Mean Difference (95% CI) | p-value |
|---|---|---|---|---|---|---|---|
| Transmission by Month 1 | |||||||
| rs352140 | Dominant | GG | 22 (39) | 7.12 (6.71-7.60) | 7.18 (0.58) | -0.581 (-0.946, -0.217) | 0.0023 |
| GA of AA | 34 (61) | 6.54 (6.28-7.00) | 6.60 (0.68) | ||||
| rs3764880 | Dominant -Female Only | AA | 13 (45) | 6.52 (6.28-6.60) | 6.38 (0.47) | 0.783 (0.351, 1.214) | 0.0009 |
| AG or GG | 16 (55) | 7.27 (6.65-7.57) | 7.16 (0.61) | ||||
| rs1634319 | Dominant -Female Only | TT | 14 (48) | 6.50 (6.30-6.75) | 6.43 (0.47) | 0.746 (0.302, 1.189) | 0.0019* |
| TC or CC | 15 (52) | 7.40 (6.56-7.58) | 7.17 (0.64) | ||||
| Transmission by Month 12 | |||||||
| rs352140 | Dominant | GG | 31 | 7.09 (6.57 -7.60) | 7.04 (0.72) | -0.485 (-0.847, -0.123) | 0.0094 |
| GA or AA | 42 | 6.54 (6.28-7.00) | 6.56 (0.78) | ||||
| rs3764880 | Dominant -Female Only | AA | 14 | 6.49 (6.22-6.60) | 6.36 (0.46) | 0.652 (0.208, 1.095) | 0.0054 |
| AG or GG | 19 | 7.02 (6.46-7.56) | 7.01 (0.69) | ||||
| rs1634319 | Dominant -Female Only | TT | 17 | 6.46 (6.06-6.71) | 6.34 (0.47) | 0.7999 (0.400, 1.200) | 0.0003** |
| TC or CC | 16 | 7.27 (6.58-7.57) | 7.14 (0.63) | ||||
Bonferroni corrected p-value = 0.17
Bonferroni corrected p-value = 0.027
Female infants infected by 1 month of age and having one or more copies of the TLR8 1G allele (rs3764880) had a 0.78 log10 c/ml higher peak viral load (95% CI: 0.35, 1.21; p<0.001); a similar association was seen in female infants infected by 12 months (0.65 log10 c/ml higher peak viral load; 95% CI: 0.21, 1.10; p=0.005). The association remained significant at both infection time-points in additive models but not recessive models of inheritance. There was no significant association between peak plasma HIV-1 RNA levels and the TLR8 1A/G variant in male infants (Supplemental Table 3).
We also found an association between a haplotype tagging intronic variant in TLR7 (rs1634319) and peak plasma HIV-1 RNA levels. Female infants infected by 1 year of age with ≥1 copy of the TLR7 rs1634319 C allele had a 0.80 log10 c/ml higher peak viral load (95% CI: 0.40, 1.20; corrected p=0.027 assuming a dominant model of inheritance and controlling for genetic ancestry. A similar association was noted among female infants infected by ≤1 month (mean difference=0.75; 95% CI: 0.30, 1.19; corrected p=0.17), but this association was not significant after controlling for multiple comparisons. This variant showed no significant associations with peak plasma HIV-1 RNA levels in male infants. No other candidate or TLR, TIRAP or MyD88 haplotype tagging SNPs were significantly associated with peak plasma HIV-1 RNA levels in analyses adjusted for multiple comparisons (Supplemental Table 3).
Mortality
Because of potential survival bias, we evaluated time to death only in infants infected by 1 month of age. We found no significant associations with time to death by any TLR, TIRAP or MyD88 haplotype tagging SNP included in this analysis after adjusting for multiple comparisons and found no significant associations with candidate SNPs; however we had limited statistical power to evaluate associations with mortality. We noted a trend for association with mortality for two candidate SNPs (TLR2 1350T/C and TLR7 32A/T) (Supplemental Table 4). In infants infected by 1 month of age, the TLR2 1350CC (rs3804100) genotype had a trend for association with increased risk of mortality (HR=6.36; 95% CI: 0.97, 51.24; p=0.082) using a recessive model of inheritance and controlling for genetic ancestry. However the minor allele frequency for this polymorphism was low (4.92%) and only 1 infected infant had this genotype. We also saw a trend for association between each additional copy of the TLR7 32T (rs179008) allele and time to mortality in female infants infected by month 1 (HR=2.84, 95% CI: 0.87, 9.23; p=0.082).
Discussion
Few studies have evaluated the role of TLR genetic variation in infant HIV-1 acquisition and progression [16, 17] and our study is the first to evaluate the influence of TLR polymorphisms in a perinatal African cohort. We found that TLR9 1635G/A was associated with both HIV-1 acquisition and peak HIV-1 viral load, and TLR8 1A/G and the haplotype tagging TLR7 variant (rs1634319) were associated with peak plasma HIV-1 RNA level.
We found that TLR9 1635G/A was associated with a higher risk of HIV-1 acquisition early (<1 month of age), and by the end of the first year of life. This supports a trend observed in a previous study evaluating TLR9 genetic variations in HIV-1 MTCT [17]. However, in contrast, this variant was also reported to be associated with protection from HIV-1 acquisition in an African heterosexual serodiscordant couples cohort suggesting that this variant may exert different effects in heterosexual and vertical transmission [29]. While this seeming discrepancy in findings could reflect population differences in LD, it is notable that the two cohorts that had the greatest likelihood for differences in population stratification with one African and the other European, shared a common biological mode of HIV-1 transmission and had similar TLR9 association results. Conversely, the African heterosexual couples and our vertical transmission cohorts share a common population background (Eastern African) but were distinct in the biological mode of HIV-1 transmission. These findings could suggest that the source of the discrepancy is in the functional role that TLR9 plays in heterosexual HIV-1 acquisition compared to mother-to-child HIV-1 acquisition.
We also found that the TLR9 1635A allele was associated with a decrease in peak VL. The protective effect we found associated with the 1635A allele is consistent with one previous study in adults of European ancestry [13] but contradicts the findings of two other HIV-1 progression studies in adults of European ancestry [12, 14] and one study in HIV-1 infected children of European ancestry [16]. Differences between these cohorts, including HAART use, population ancestry, and progression measures observed could have led to differences in TLR associations. Specifically, the timing during HIV disease course when these variants are being evaluated may have an impact in the observed phenotype for specific TLR variants. A differential effect of TLR variants on HIV-1 set point was noted in acute verses chronically infected African adults [29], and in a functional evaluation of TLR signaling in acute versus chronic infection [30]. In early HIV-1 infected infants, there is delayed containment or non-containment of HIV-1, making it difficult to evaluate set-point comparably to adults [31] and may contribute to the different effect we observed of TLR9 1635A on HIV-1 progression.
Our observation that TLR9 1635G/A was associated both with higher HIV-1 acquisition risk and lower peak viral load in HIV-1 infected infants may be explained by potentially distinct effects of TLR9 on HIV-1 acquisition versus progression. In the context of HIV-1 uninfected infants, this response could increase recruitment of activated CD4+ T cells that serve as targets for HIV-1 replication. We could speculate that the TLR9 variant was associated with recruitment of activated immune cells to infant mucosal environments where they serve as HIV-1 susceptible targets. However, TLR9 is primarily expressed on plasmacytoid dendritic cells and mediates interferon-α release and antigen presentation thereby linking the innate and adaptive immune responses [12]. Thus, TLR9-mediated pro-inflammatory cytokine release could also initiate anti- HIV mechanisms that limit viral replication in already infected infants and decrease peak HIV-1 RNA levels.
The specific function of the TLR9 1635G/A variant in HIV-1 disease remains unknown. The G to A variation is a synonomous change in exon 2, making it difficult to infer any specific functional outcome. However, an intronic TLR9 1174A/G variant associated with decreased TLR9 transcriptional activity [32] is found in high linkage disequilibrium (LD) (r2=0.98) with 1635G/A in European but not African cohorts. This could explain the observed differences in association of the TLR9 1635G/A variant and HIV-1 disease progression in these distinct ancestral populations. Thus, it is possible that, in our sampled African cohort, the TLR9 1635G/A variant is in high LD with a different currently unidentified causal SNP. Alternatively, TLR9 1625G/A could alter mRNA splicing or stability, or protein expression or function, which has been documented for other synonymous changes [33].
We found that TLR8 1A/G was associated with peak HIV-1 RNA level. We found that one or more copies of the TLR8 1G allele was associated with a 0.78 log10 c/ml higher peak HIV-1 RNA level in female but not male infants. In contrast, Oh et al. found that the TLR8 1G variant was protective against rapid CD4+ T-cell depletion in males of European ancestry [11]. This difference in associations could be due to differences between adult and pediatric HIV-1 progression, gender differences, or differences in outcome measure assessed [34, 35]. Infants have much more rapid HIV-1 disease progression than adults which may be due to the relatively weaker adaptive immune response in infants [35]. We also identified a novel TLR7 variant (rs1634319) associated with higher peak plasma HIV-1 RNA levels in female infected infants. TLR7 viral RNA recognition can lead either towards immune activation and viral replication or an effective antiviral response [8]. The rs1634319 variant we identified is intronic but could influence TLR7 expression or be in linkage disequilibrium with another causal variant.
While previous studies evaluating genetic variations in TLR2 and TLR4 have found positive associations with HIV-1 progression outcomes, our study did not confirm these associations or identify new associations with TLR2 or TLR4, nor did our study confirm a previously observed association between a TLR7 variant (32A/T) and HIV-1 progression in adult males [10]. Our study found no associations between genetic variations in TIRAP or MYD88 and HIV-1 acquisition or progression. We may not have detected specific associations because of relatively limited statistical power, unique properties of vertical HIV-1 transmission that differ from sexual HIV-1 transmission, or differences in allele frequencies in our population compared to other previously published cohorts.
In summary, this study is the first to evaluate the association between TLR polymorphisms and HIV-related outcomes in a perinatal African cohort and confirms that variations in TLRs may contribute to pediatric HIV-1 outcomes. Further studies are needed to define mechanisms underlying these associations and the specific functions of TLRs during the course of HIV-1 disease.
Supplementary Material
Acknowledgments
We would like to thank the women and children who participated in the study, the University of Washington Northwest Genomics Center, the Center for Clinical Genomics at the University of Washington and K. Buckingham for technical assistance.
Sponsorship: This research was supported by National Institutes of Health (NIH) research grant R21 AI073115 and R01 HD023412. KBS was supported by the University of Washington Institute of Translational Health Sciences Multidisciplinary Clinical Research Training Program (TL1 TR 000422) from the National Center for Advancing Translational Sciences, National Institutes of Health and the Fogarty International Clinical Research Scholars Program (Grant Number 5 R24 TW007988) from National Institutes of Health, Fogarty International Center through Vanderbilt University. AWB was supported by a training fellowship from the NIH/National Human Genome Research Institute (T32 HG00035).
Footnotes
Contributions: K.M.B, A.W.B, M.J.B, J.R.L and G.C.J conceived of the project. K.M.B. conducted the analysis with help from A.W.B, R.D.M and B.A.R. K.M.B. and A.W.B prepared samples. K.M.B. wrote the manuscript and all authors contributed to the final manuscript. D.W., E.O. and G.C.J participated in the larger MTCT cohort study. G.C.J. and J.R.L co-supervised the project and provided financial support for the study.
References
- 1.Ploquin MJ, Jacquelin B, Jochems SP, Barre-Sinoussi F, Muller-Trutwin MC. Innate immunity in the control of HIV/AIDS: recent advances and open questions. AIDS. 2012;26(10):1269–79. doi: 10.1097/QAD.0b013e328353e46b. [DOI] [PubMed] [Google Scholar]
- 2.Mogensen TH, Melchjorsen J, Larsen CS, Paludan SR. Innate immune recognition and activation during HIV infection. Retrovirology. 2010;7:54. doi: 10.1186/1742-4690-7-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang JJ, Altfeld M. Innate immune activation in primary HIV-1 infection. J Infect Dis. 2010;202(Suppl 2):S297–301. doi: 10.1086/655657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124(4):783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 5.Kawai T, Akira S. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int Immunol. 2009;21(4):317–37. doi: 10.1093/intimm/dxp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–76. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 7.Murphy K, Travers P, Walport M. Immuno Biology. 7. New York, New York: Garland Science; 2008. [Google Scholar]
- 8.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11(5):373–84. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 9.Bowie AG, Haga IR. The role of Toll-like receptors in the host response to viruses. Mol Immunol. 2005;42(8):859–67. doi: 10.1016/j.molimm.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 10.Oh DY, Baumann K, Hamouda O, Eckert JK, Neumann K, Kucherer C, et al. A frequent functional toll-like receptor 7 polymorphism is associated with accelerated HIV-1 disease progression. AIDS. 2009;23(3):297–307. doi: 10.1097/QAD.0b013e32831fb540. [DOI] [PubMed] [Google Scholar]
- 11.Oh DY, Taube S, Hamouda O, Kucherer C, Poggensee G, Jessen H, et al. A functional toll-like receptor 8 variant is associated with HIV disease restriction. J Infect Dis. 2008;198(5):701–9. doi: 10.1086/590431. [DOI] [PubMed] [Google Scholar]
- 12.Soriano-Sarabia N, Vallejo A, Ramirez-Lorca R, Rodriguez Mdel M, Salinas A, Pulido I, et al. Influence of the Toll-like receptor 9 1635A/G polymorphism on the CD4 count, HIV viral load, and clinical progression. J Acquir Immune Defic Syndr. 2008;49(2):128–35. doi: 10.1097/QAI.0b013e318184fb41. [DOI] [PubMed] [Google Scholar]
- 13.Bochud PY, Hersberger M, Taffe P, Bochud M, Stein CM, Rodrigues SD, et al. Polymorphisms in Toll-like receptor 9 influence the clinical course of HIV-1 infection. AIDS. 2007;21(4):441–6. doi: 10.1097/QAD.0b013e328012b8ac. [DOI] [PubMed] [Google Scholar]
- 14.Pine SO, McElrath MJ, Bochud PY. Polymorphisms in toll-like receptor 4 and toll-like receptor 9 influence viral load in a seroincident cohort of HIV-1-infected individuals. AIDS. 2009;23(18):2387–95. doi: 10.1097/QAD.0b013e328330b489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sironi M, Biasin M, Cagliani R, Forni D, De Luca M, Saulle I, et al. A common polymorphism in TLR3 confers natural resistance to HIV-1 infection. J Immunol. 2012;188(2):818–23. doi: 10.4049/jimmunol.1102179. [DOI] [PubMed] [Google Scholar]
- 16.Freguja R, Gianesin K, Del Bianco P, Malacrida S, Rampon O, Zanchetta M, et al. Polymorphisms of innate immunity genes influence disease progression in HIV-1-infected children. AIDS. 2012;26(6):765–8. doi: 10.1097/QAD.0b013e3283514350. [DOI] [PubMed] [Google Scholar]
- 17.Ricci E, Malacrida S, Zanchetta M, Mosconi I, Montagna M, Giaquinto C, et al. Toll-like receptor 9 polymorphisms influence mother-to-child transmission of human immunodeficiency virus type 1. J Transl Med. 2010;8:49. doi: 10.1186/1479-5876-8-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sanders CM, Cruse JM, Lewis RE. Toll-like receptors, cytokines and HIV-1. Exp Mol Pathol. 2008;84(1):31–6. doi: 10.1016/j.yexmp.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 19.Lester RT, Yao XD, Ball TB, McKinnon LR, Kaul R, Wachihi C, et al. Toll-like receptor expression and responsiveness are increased in viraemic HIV-1 infection. AIDS. 2008;22(6):685–94. doi: 10.1097/QAD.0b013e3282f4de35. [DOI] [PubMed] [Google Scholar]
- 20.Farquhar C, Rowland-Jones S, Mbori-Ngacha D, Redman M, Lohman B, Slyker J, et al. Human leukocyte antigen (HLA) B*18 and protection against mother-to-child HIV type 1 transmission. AIDS Res Hum Retroviruses. 2004;20(7):692–7. doi: 10.1089/0889222041524616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Farquhar C, VanCott TC, Mbori-Ngacha DA, Horani L, Bosire RK, Kreiss JK, et al. Salivary secretory leukocyte protease inhibitor is associated with reduced transmission of human immunodeficiency virus type 1 through breast milk. J Infect Dis. 2002;186(8):1173–6. doi: 10.1086/343805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Otieno PA, Brown ER, Mbori-Ngacha DA, Nduati RW, Farquhar C, Obimbo EM, et al. HIV-1 disease progression in breast-feeding and formula-feeding mothers: a prospective 2-year comparison of T cell subsets, HIV-1 RNA levels, and mortality. J Infect Dis. 2007;195(2):220–9. doi: 10.1086/510245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lohman BL, Slyker J, Mbori-Ngacha D, Bosire R, Farquhar C, Obimbo E, et al. Prevalence and magnitude of human immunodeficiency virus (HIV) type 1-specific lymphocyte responses in breast milk from HIV-1-seropositive women. J Infect Dis. 2003;188(11):1666–74. doi: 10.1086/379374. [DOI] [PubMed] [Google Scholar]
- 24.Shaffer N, Chuachoowong R, Mock PA, Bhadrakom C, Siriwasin W, Young NL, et al. Short-course zidovudine for perinatal HIV-1 transmission in Bangkok, Thailand: a randomised controlled trial. Bangkok Collaborative Perinatal HIV Transmission Study Group. Lancet. 1999;353(9155):773–80. doi: 10.1016/s0140-6736(98)10411-7. [DOI] [PubMed] [Google Scholar]
- 25.Emery S, Bodrug S, Richardson BA, Giachetti C, Bott MA, Panteleeff D, et al. Evaluation of performance of the Gen-Probe human immunodeficiency virus type 1 viral load assay using primary subtype A, C, and D isolates from Kenya. J Clin Microbiol. 2000;38(7):2688–95. doi: 10.1128/jcm.38.7.2688-2695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437(7063):1299–320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.International HapMap Consortium. The International HapMap Project. Nature. 2003;426(6968):789–96. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 28.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 29.Mackelprang RD, Bigham AW, Celum C, Bruyn Gd, Beima-Sofie K, John-Stewart G, et al. Polymorphisms in TLR2 and TLR7 are associated with plasma HIV-1 RNA set point in an African heterosexual cohort. 19th Internation AIDS Conference; Washington, DC. 2012. [Google Scholar]
- 30.Chang JJ, Lacas A, Lindsay RJ, Doyle EH, Axten KL, Pereyra F, et al. Differential regulation of toll-like receptor pathways in acute and chronic HIV-1 infection. AIDS. 2012;26(5):533–41. doi: 10.1097/QAD.0b013e32834f3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Obimbo EM, Wamalwa D, Richardson B, Mbori-Ngacha D, Overbaugh J, Emery S, et al. Pediatric HIV-1 in Kenya: pattern and correlates of viral load and association with mortality. J Acquir Immune Defic Syndr. 2009;51(2):209–15. doi: 10.1097/qai.0b013e31819c16d8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tao K, Fujii M, Tsukumo S, Maekawa Y, Kishihara K, Kimoto Y, et al. Genetic variations of Toll-like receptor 9 predispose to systemic lupus erythematosus in Japanese population. Ann Rheum Dis. 2007;66(7):905–9. doi: 10.1136/ard.2006.065961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sauna ZE, Kimchi-Sarfaty C. Understanding the contribution of synonymous mutations to human disease. Nat Rev Genet. 2011;12(10):683–91. doi: 10.1038/nrg3051. [DOI] [PubMed] [Google Scholar]
- 34.Matt C, Roger M. Genetic determinants of pediatric HIV-1 infection: vertical transmission and disease progression among children. Mol Med. 2001;7(9):583–9. [PMC free article] [PubMed] [Google Scholar]
- 35.Tiemessen CT, Kuhn L. Immune pathogenesis of pediatric HIV-1 infection. Curr HIV/AIDS Rep. 2006;3(1):13–9. doi: 10.1007/s11904-006-0003-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.